

Abstract
Rapid evolutionary radiations are among the most challenging phylogenetic problems, wherein different types of data (e.g., morphology and molecular) or genetic markers (e.g., nuclear and organelle) often yield inconsistent results. The tribe Arundinarieae, that is, the temperate bamboos, is a clade of tetraploid originated 22 Ma and subsequently radiated in East Asia. Previous studies of Arundinarieae have found conflicting relationships and/or low support. Here, we obtain nuclear markers from ddRAD data for 213 Arundinarieae taxa and parallel sampling of chloroplast genomes from genome skimming for 147 taxa. We first assess the feasibility of using ddRAD-seq data for phylogenetic estimates of paleopolyploid and rapidly radiated lineages, optimize clustering thresholds, and analysis workflow for orthology identification. Reference-based ddRAD data assembly approaches perform well and yield strongly supported relationships that are generally concordant with morphology-based taxonomy. We recover five major lineages, two of which are notable (the pachymorph and leptomorph lineages), in that they correspond with distinct rhizome morphologies. By contrast, the phylogeny from chloroplast genomes differed significantly. Based on multiple lines of evidence, the ddRAD tree is favored as the best species tree estimation for temperate bamboos. Using a time-calibrated ddRAD tree, we find that Arundinarieae diversified rapidly around the mid-Miocene corresponding with intensification of the East Asian monsoon and the evolution of key innovations including the leptomorph rhizomes. Our results provide a highly resolved phylogeny of Arundinarieae, shed new light on the radiation and reticulate evolutionary history of this tribe, and provide an empirical example for the study of recalcitrant plant radiations. [Arundinarieae; ddRAD; paleopolyploid; genome skimming; rapid diversification; incongruence.]
Many of the most diverse clades of organisms are characterized by rapid evolutionary radiations. Studies of such clades have helped elucidate the evolutionary mechanisms underlying species diversification; famous examples include Darwin’s finches (Grant and Grant 2002; Lamichhaney et al. 2015), African lake cichlids (Seehausen 2006; Wagner et al. 2012) and representatives of the Andean flora (Pennington et al. 2010; Hughes and Atchison 2015). Robust phylogenetic estimates contribute to our understanding of evolutionary radiations, however, resolving relationships within rapidly diverged clades is challenging. Moreover, the conflicting genealogies reconstructed from different types of genetic data can complicate our understanding of evolutionary radiations (Morales-Briones et al. 2018; Platt II et al. 2018).
In recent years, phylogenomics has become increasingly affordable owing to rapid advances in next-generation sequencing (NGS) techniques. Methods for subsampling genomes, such as restriction-site-associated DNA sequencing (RAD-seq) methods are particularly promising to resolve the phylogenetic problems for shallow taxonomic scales. RAD-seq methods, which sequence regions adjacent to restriction enzyme cut sites, gives a good representation of the genome. This makes it a powerful method for phylogeographic and systematic studies (Eaton and Ree 2013; Cruaud et al. 2014; Massatti et al. 2016), even for species-rich radiations (Wagner et al. 2013; Vargas et al. 2017, Hipp et al. 2018). Newer variants of RAD-seq have made it more cost and time-effective while mitigating some of the issues around missing data (Baird et al. 2008; Peterson et al. 2012).
Polyploidization is a prevalent driving force during the evolution of flowering plants (One Thousand Plant Transcriptomes Initiative 2019). Phylogenetic studies on polyploid taxa, however, are challenging due to the complexity of their genome composition, which requires distinguishing orthologs from duplicated paralogs produced via whole-genome duplication (WGD) (Glover et al. 2016). Once duplicated, the duplicated loci were followed by cycles from polyploidization to diploidization, including gene loss, biased fractionation, genome downsizing, and chromosome rearrangement that happened over time (Wendel 2000, 2015). It is important to be aware of if any consideration should be taken for the polyploids in question, such as the degree of homozygosity of the polyploids (autopolyploids or allopolyploids), how recent the polyploid is (paleopolyploids, mesopolyploids, or neopolyploids), and the proportion of duplicate remnants within the genome. Variation in the rate of the diploidization process may be also of interest, as the perennial habit and long flowering cycles may delay diploidization in some lineages such as Arundinarieae (from a few up to 120 years; Janzen 1976) more than others. Although polyploidy can affect phylogenetic reconstruction, few bioinformatic methods have been developed to address this problem (Blischak et al. 2018). Current approaches are largely focused on genotyping crops with well-studied polyploid histories (Chen et al. 2013; Clevenger et al. 2018), including comparison of closely related diploid parents and their allopolyploid progeny (Brandrud et al. 2020). Most NGS approaches to dealing with paleoallopolyploids involve the use of diploid base callers, and excluding sites that do not appear biallelic and allow for differentiating subgenomes (Brandrud et al. 2017; Ye et al. 2019).
Awareness of the implications that the bioinformatic processing has on the outcome of the data analysis is now being put much emphasis on like in Shafer et al. (2017). There is, however, still much to be learned about the bioinformatic impact on the phylogenetic inference. For RAD-seq data, many studies have demonstrated the power of de novo assembled RAD-seq data to resolve intractable plant radiations, including Iberian Linaria (Fernández-Mazuecos et al. 2017), Petalidium (Tripp et al. 2017), and Diplostephium (Vargas et al. 2017). However, conclusions drawn in these studies may have limited validity when applying to polyploids, as the confounding effect of gene duplications on the de novo assembly may be prevalent because it relies only on sequence similarity to identify homology. This was demonstrated by Wang et al. (2017), where different values for some assembly parameters (e.g., the threshold of coverage depth required to create a stack, “ m”) considerably influence de novo locus discovery and phylogeny inference. Methods for reference-based assembly can greatly improve the ability to identify orthologs, filter paralogs, and remove reads mapping to multiple regions of the genome (Cornille et al. 2016), since its homology identification takes into account both sequence similarity and mapping position of reads with regard to the reference. Andrews et al. (2016) indicated that high coverage is required for confident de novo locus discovery, whereas reference-based assembly performs well even with low coverage data. Although reference-based methods are increasingly being used for the RAD-seq data assembly of diploids (Rubin et al. 2012; Hipp et al. 2014; McCluskey and Postlethwait 2015; Andrews et al. 2016), it can present trade-offs if it yields many fewer overall loci due to a reliance on a single reference genome (Stetter and Schmid 2017; Tripp et al. 2017). Another critical parameter affecting assembly results is the sequence similarity threshold used to identify homology (Harvey et al. 2016). The use of a threshold that is too low can incorrectly combine paralogs as a single locus (undersplitting), whereas overly stringent thresholds may divide some part of the true homologous alleles into separate alignments (oversplitting) (Harvey et al. 2015). Although previous studies have drawn some conclusions on the selection of assembly parameters to mitigate the effects of oversplitting and undersplitting based on simulated and empirical RAD-seq data (Ilut et al. 2014; Harvey et al. 2015; Paetzold et al. 2019), they have mainly focused on diploid species. Whether these conclusions are compatible with polyploids remains poorly known.
m”) considerably influence de novo locus discovery and phylogeny inference. Methods for reference-based assembly can greatly improve the ability to identify orthologs, filter paralogs, and remove reads mapping to multiple regions of the genome (Cornille et al. 2016), since its homology identification takes into account both sequence similarity and mapping position of reads with regard to the reference. Andrews et al. (2016) indicated that high coverage is required for confident de novo locus discovery, whereas reference-based assembly performs well even with low coverage data. Although reference-based methods are increasingly being used for the RAD-seq data assembly of diploids (Rubin et al. 2012; Hipp et al. 2014; McCluskey and Postlethwait 2015; Andrews et al. 2016), it can present trade-offs if it yields many fewer overall loci due to a reliance on a single reference genome (Stetter and Schmid 2017; Tripp et al. 2017). Another critical parameter affecting assembly results is the sequence similarity threshold used to identify homology (Harvey et al. 2016). The use of a threshold that is too low can incorrectly combine paralogs as a single locus (undersplitting), whereas overly stringent thresholds may divide some part of the true homologous alleles into separate alignments (oversplitting) (Harvey et al. 2015). Although previous studies have drawn some conclusions on the selection of assembly parameters to mitigate the effects of oversplitting and undersplitting based on simulated and empirical RAD-seq data (Ilut et al. 2014; Harvey et al. 2015; Paetzold et al. 2019), they have mainly focused on diploid species. Whether these conclusions are compatible with polyploids remains poorly known.
Arundinarieae (Poaceae: Bambusoideae), known as the temperate bamboos, is a model lineage for understanding complex plant radiations. The entire tribe originated from an ancient allopolyploid event around 22 Ma, and due to the extinction of diploid progenitors (CC and DD), all extant species are now allotetraploids with base chromosome number 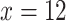 (CCDD, 2
(CCDD, 2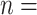 4X
4X 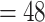 ) (Guo et al. 2019). The clade has experienced a rapid radiation (Hodkinson et al. 2010; Zhang et al. 2016) and now comprises approximately 550 species across 33 genera (Clark et al. 2015; Soreng et al. 2015; Zhang et al. 2018). This diversity is unevenly distributed globally, with over 95% of species endemic to East Asia, and the remaining 20 species distributed in Sri Lanka, southern India, Madagascar, and Africa, with only three species in eastern North America (BPG 2012). Many species are widely cultivated for both culm and edible shoot production (Ruiz-Pérez et al. 2001; Clark et al. 2015), including the economically important moso bamboo, Phyllostachys edulis (syn. P. heterocycla) (Peng et al. 2013). Certain species provide habitat and exclusive foods for wildlife, including giant pandas, and many species are dominant plants of alpine and subalpine forests in the mountains of Southwest China (Li et al. 2006; Ye et al. 2019).
) (Guo et al. 2019). The clade has experienced a rapid radiation (Hodkinson et al. 2010; Zhang et al. 2016) and now comprises approximately 550 species across 33 genera (Clark et al. 2015; Soreng et al. 2015; Zhang et al. 2018). This diversity is unevenly distributed globally, with over 95% of species endemic to East Asia, and the remaining 20 species distributed in Sri Lanka, southern India, Madagascar, and Africa, with only three species in eastern North America (BPG 2012). Many species are widely cultivated for both culm and edible shoot production (Ruiz-Pérez et al. 2001; Clark et al. 2015), including the economically important moso bamboo, Phyllostachys edulis (syn. P. heterocycla) (Peng et al. 2013). Certain species provide habitat and exclusive foods for wildlife, including giant pandas, and many species are dominant plants of alpine and subalpine forests in the mountains of Southwest China (Li et al. 2006; Ye et al. 2019).
As one of the major grass lineages that have diversified in forests, Arundinarieae has evolved unique and complex morphologies that have also made it a taxonomically difficult group. These traits include semelauctant and iterauctant inflorescences, pachymorph and leptomorph rhizomes, and growth habits from solitary to multiple branches (Supplementary Fig. S1 available on Dryad at https://doi.org/10.5061/dryad.rr4xgxd6m, Li et al. 2006). Bamboos have a compound inflorescence where the basic terminal unit of the inflorescence is a spikelet, as in other grasses (Kellogg 2015). Each spikelet is analogous to flower and is composed of one to several florets. According to the different manner of development (McClure 1966), two types of inflorescences are recognized in bamboos. Semelauctant inflorescences represent a simple form where the primary axis develops only once, and no meristem remains afterward in the form of lateral buds; while iterauctant inflorescences are more complex, containing a jointed axis and lateral buds at the base of spikelets which can further develop into spikelet-like structures (pseudospikelets) (Soderstrom and Ellis 1987). The bamboo rhizome is a horizontal underground culm system, which contains shortened internodes and nodes that bear roots and buds. Different rhizomes are thought to underlie adaptations to diverse habitats (Wen 1983). The pachymorph rhizomes are characterized by thick, highly compressed internodes, in which lateral buds of mother culms produce vertically curved rhizomes with culms eventually arising from the apex to above ground. The leptomorph rhizomes feature slender, longer internodes, in which lateral buds of mother culms only grow into horizontal rhizomes. The apical buds of the leptomorph rhizomes are persistent, and the axillary buds can be further developed into new rhizomes or new culms (McClure 1966; Stapleton 1998).
The problem of resolving relationships within Arundinarieae has recently been called “phylogenetically intractable” (Ma et al. 2014), due to the clades’ history of a polyploidization event followed by a rapid radiation. Previous molecular analyses based on chloroplast loci divided Arundinarieae into 12 well-supported clades (clades I–XII) (Triplett and Clark 2010; Zeng et al. 2010; Yang et al. 2013; Attigala et al. 2014). However, several relationships among and within these clades are poorly resolved even when using whole chloroplast genome data (Ma et al. 2014; Attigala et al. 2016). Compared with the chloroplast, nuclear sequences can be more effective in resolving phylogeny (Wickett et al. 2014; Zeng et al. 2014); however, large nuclear data sets have been limited in Arundinarieae largely due to the difficulties of identifying sufficient unambiguous single or low copy nuclear loci and their orthologs in tetraploid plants. Most previous phylogenetic studies have used either a few nuclear markers with broad sampling (Guo and Li 2004; Zhang et al. 2012; Yang et al. 2013) or large NGS data sets sampled across only a subset of taxa (Wang et al. 2017; Ye et al. 2019; Zhang et al. 2019). Large genome-scale data have yet to be applied across a comprehensive sampling of Arundinarieae.
Previous phylogenomic studies in Arundinarieae conflict in many major ways but agree in several interesting respects as well. For example, the newly described genus Hsuehochloa (Zhang et al. 2018) was consistently found as sister to the rest of Arundinarieae (Yang et al. 2013; Ma et al. 2014). Examples of conflict include nuclear support for the establishment of an early-diverging lineage of Ampelocalamus 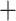 Drepanostachyum
Drepanostachyum 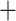 Himalayacalamus (ADH) (Yang et al. 2013), which also shares morphological and biogeographic affinity (Li et al. 1996), but was not supported in chloroplast phylogenies (Ma et al. 2014). Similarly, the monotypic genus Gaoligongshania, which is uniquely distinguished by its epiphytic habit (Li et al. 1995), falls in very different parts of the nuclear and chloroplast phylogenies (Ma et al. 2014; Wang et al. 2017). Recent nuclear and chloroplast topologies both deviate substantially from the current taxonomy based on diagnostic morphological characters (i.e., rhizome types, inflorescence and spikelet structures, and branching patterns).
Himalayacalamus (ADH) (Yang et al. 2013), which also shares morphological and biogeographic affinity (Li et al. 1996), but was not supported in chloroplast phylogenies (Ma et al. 2014). Similarly, the monotypic genus Gaoligongshania, which is uniquely distinguished by its epiphytic habit (Li et al. 1995), falls in very different parts of the nuclear and chloroplast phylogenies (Ma et al. 2014; Wang et al. 2017). Recent nuclear and chloroplast topologies both deviate substantially from the current taxonomy based on diagnostic morphological characters (i.e., rhizome types, inflorescence and spikelet structures, and branching patterns).
In this study, we present Arundinarieae as a model empirical system, by providing extensive sampling of taxa representing all recognized clades and genera, to explore an effective approach for resolving phylogenetic relationships in a rapid radiation of polyploids. Using a combination of large data sets including more than 200 taxa from ddRAD-seq, 147 chloroplast genomes, and 92 ribosomal DNAs assembled from genome skimming, our primary objective is to disentangle phylogenetic relationships within Arundinarieae. Considering its radiation and allopolyploid history, we assess whether RAD-seq data are suitable for phylogenetic study of such lineages, and investigate to find a proper and precise workflow to identify orthologs by adopting the de novo and reference-based assemblies and comparing their impact on phylogeny inference. Given our best resolution of the phylogeny, we also examine patterns of morphological character evolution and explore temporal diversification patterns with respect to palaeoenvironmental changes using a time-calibrated phylogeny. Comparisons among nuclear ddRAD, nuclear ribosomal DNA, and chloroplast genomic data make it possible to track the reticulate evolution in this rapidly radiated bamboo tribe. It is our hope that the comprehensive approach and multifaceted verification of methodologies employed in our analysis of the Arundinarieae will provide general insights into the studies of other rapidly radiated lineages with polyploid origin.
Materials and Methods
Taxon Sampling
For ddRAD, we sampled a total of 213 taxa in 200 species from 32 genera representing 36% of species and 97% of genera (Soreng et al. 2015; Zhang et al. 2018; Zhang et al. 2020) and all 12 clades in Arundinarieae. Two paleotropical woody bamboos and three neotropical woody bamboos were selected as outgroups. Samples of Arundinarieae and neotropical woody bamboos are all allotetraploids, and two paleotropical woody bamboos are allohexaploids. We newly sequenced 129 Arundinarieae plastomes, which we combined with 18 published plastomes of Arundinarieae (Burke et al. 2012; Ma et al. 2014), for a total of 147 taxa in 142 species from 31 genera sampled, once again including five tropical woody bamboos as outgroups. Voucher information for samples included in this study is available in Supplementary Table S1 available on Dryad. The voucher specimens were deposited in the herbarium of Kunming Institute of Botany, Chinese Academy of Sciences (KUN).
DNA Extraction, Library Preparation, and Data Generation
Total genomic DNAs were extracted from fresh or silica-dried leaf tissues using a modified CTAB method (Doyle 1987). The ddRAD sequencing library was prepared following the modified ddRAD protocol known as MiddRAD (Yang et al. 2016) with AvaII 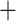 MspI enzyme pair. Following the library protocol, 40 or 80 samples were pooled into a library which was then sequenced on one or two lanes to achieve similar coverage. A total of 15 sequencing lanes were prepared, including 342 bamboo samples from different projects. Paired-end reads (150 bp) were generated on an Illumina HiSeq X Ten System (San Diego, CA, USA). Total genomic DNAs were also sent to BGI (Shenzhen, China) for library (500 bp) preparation for genome skimming sequencing. Paired-end (150 bp) sequencing was conducted on an Illumina HiSeq 2000, generating
MspI enzyme pair. Following the library protocol, 40 or 80 samples were pooled into a library which was then sequenced on one or two lanes to achieve similar coverage. A total of 15 sequencing lanes were prepared, including 342 bamboo samples from different projects. Paired-end reads (150 bp) were generated on an Illumina HiSeq X Ten System (San Diego, CA, USA). Total genomic DNAs were also sent to BGI (Shenzhen, China) for library (500 bp) preparation for genome skimming sequencing. Paired-end (150 bp) sequencing was conducted on an Illumina HiSeq 2000, generating  2 Gb data per sample.
2 Gb data per sample.
Workflow for ddRAD-seq Analysis of Polyploid Lineages
In silico digestion and clustering threshold optimization.—We performed a series of analyses on empirical and in silico digested genome data to investigate the feasibility of RAD-seq data for the phylogenetic study of polyploid lineages. We first selected four published genome sequences in Poaceae, to investigate the proportion of ddRAD-seq loci that are likely to represent duplicated gene regions. This included the diploid rice genome, a temperate bamboo (Phyllostachys edulis) allotetraploid genome, a tropical woody bamboo (Guadua angustifolia) allotetraploid genome, and a diploid herbaceous bamboo (Olyra latifolia) genome. Comparison of markers sampled from within the P. edulis, G. angustifolia, and O. latifolia genomes allowed us to see how many duplicated loci are found in the allotetraploid bamboo species compared to diploid ones. We generated ddRAD loci by implementing an in silico double enzyme digestion with AvaII and MspI enzymes for each reference genome corresponding to our MiddRAD protocol. Paired reads were generated from the digested fragments and clustered under different sequence similarity thresholds to identify the proportion of single-copy loci within each genome. Detailed descriptions of this workflow are available in the Supplementary Methods available on Dryad.
An optimal clustering threshold was identified from the in silico digested experiments that offered a balance between undersplitting and oversplitting loci. This optimal clustering threshold was used in our empirical data assembly. To assess the extent of paralogy in the final assembled data, trimmed ddRAD data from nine individuals were randomly selected and applied in the following protocol. First, we clustered exactly matching reads into nonredundant stacks by allowing zero mismatches and gaps using the ustacks program in Stacks v1.41 (Catchen et al. 2013). Then, we extracted consensus sequences from each stack, performed all-by-all blast searches, and filtered the top six hits which met the following condition as candidate orthologs: e-value higher than 1e-5, alignment length longer than 130 bp, identity higher than 85%, and only matched with another consensus sequence once. For each of nine experimental samples, we then tested the impact of the sequence similarity threshold parameter on orthology detection by repeating all-by-all blast clustering across a series of values (88–98%) and examining the size of resulting clusters.
ddRAD data assembly.— Paired-end raw reads were demultiplexed, trimmed, and filtered using the process_radtags program of Stacks v1.41 to 140 bp long. The sequence quality was assessed using FASTQC v.0.11.5 (Andrews 2016) which revealed low quality over the restriction overhang regions and a portion of the R2 reads. Therefore, we excluded R2 reads from subsequent analyses. To compare the effect of de novo versus reference-based assembly methods on the determination of orthologs, three assemblies were performed in Stacks. Each locus was allowed to have only two alleles, and thus our final filtered data sets appear diploid-like. De novo assembly was performed in Stacks v1.41. Clean reads were passed through the ustacks, cstacks, and sstacks programs following the standard workflow of de novo assembly for diploids. In the de novo pipeline (Supplementary Fig. S2 available on Dryad), reads were clustered into stacks using a minimum depth of coverage (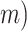 ranging from 5 to 18 by considering varied sequencing depth for different samples, and loci were then filtered only by the clustering similarity thresholds, which were set up to 96% similarity (
ranging from 5 to 18 by considering varied sequencing depth for different samples, and loci were then filtered only by the clustering similarity thresholds, which were set up to 96% similarity (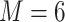 ) for intraspecies clustering and 90% (
) for intraspecies clustering and 90% (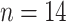 ) for intertaxa clustering based on our evaluation, respectively (see Results and discussion for more details).
) for intertaxa clustering based on our evaluation, respectively (see Results and discussion for more details).
For the reference-based assembly method, sequenced reads were required not only to map with a minimum of 96% similarity but also must map uniquely to the reference genome of P. edulis (Peng et al. 2013). Bowtie2-2.2.7 (Langmead and Salzberg 2012) was used to align clean reads to the reference genome. Two different strategies were employed to filter the mapped reads before subsequent assembly. The first strategy, which we refer to as bowtie2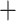 mapq
mapq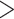 10, filtered and obtained mapped reads with mapping quality scores higher than 10 using SAMtools v1.3 (Li et al. 2008). The second strategy, called bowtie2
10, filtered and obtained mapped reads with mapping quality scores higher than 10 using SAMtools v1.3 (Li et al. 2008). The second strategy, called bowtie2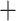 stacks m
stacks m 3, discarded those mapped reads with depth lower than 3 (
3, discarded those mapped reads with depth lower than 3 (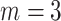 ). After that, filtered reads which aligned to the same position of the reference genome were extracted and clustered into loci within each sample using pstacks, and the resulting loci from different taxa were further clustered under 90% similarity threshold (
). After that, filtered reads which aligned to the same position of the reference genome were extracted and clustered into loci within each sample using pstacks, and the resulting loci from different taxa were further clustered under 90% similarity threshold (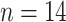 ) using cstacks. For both de novo and reference-based approaches, clustered loci were then filtered and single nucleotide polymorphism (SNPs) were identified using the sstacks and populations programs. Finally, different data sets were created by requiring filtered loci to be shared by at least 75–140 taxa (
) using cstacks. For both de novo and reference-based approaches, clustered loci were then filtered and single nucleotide polymorphism (SNPs) were identified using the sstacks and populations programs. Finally, different data sets were created by requiring filtered loci to be shared by at least 75–140 taxa (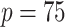 –140 in populations) among the whole 218 samples for the three assembly strategies. These data sets had different proportions of missing data, the higher minimum samples threshold retained fewer loci but contained less missing data. The detailed process and parameters of our assembly workflow are shown in Supplementary Figure S2 available on Dryad.
–140 in populations) among the whole 218 samples for the three assembly strategies. These data sets had different proportions of missing data, the higher minimum samples threshold retained fewer loci but contained less missing data. The detailed process and parameters of our assembly workflow are shown in Supplementary Figure S2 available on Dryad.
Genome Skimming Data Assembly
Genome skimming data were assembled using GetOrganelle v1.9.77 (https://github.com/Kinggerm/GetOrganelle, Jin et al. 2020) with a range of k-mers of 65, 75, 85, 95, and 105 for chloroplasts and 75, 85, 95, 105, and 115 for ribosomal data, respectively. Four chloroplast genomes, Chimonocalamus longiusculus (NC_024714.1), Fargesia nitida (NC_024715.1), Ferrocalamus rimosivaginus (NC_015831.1), and Phyllostachys edulis (NC_015817.1), and ribosomal DNA sequence of Oryza sativa cultivar TN1 (KM036285.1) were used as references for assembly of all other accessions, respectively. Contigs were visualized and extracted using Bandage v0.8.0 (Wick et al. 2015). Gaps produced by nonoverlapping contigs were filled using GapCloser from SOAPdenovo2 (https://sourceforge.net/projects/soapdenovo2/files/GapCloser). Chloroplasts and ribosomes were checked by aligning the contigs to the reference with the pairwise align option and scanned by eye to confirm appropriate mapping using Geneious v.9.1.4 (Kearse et al. 2012). Finally, whole plastome sequences from all 152 species and ribosomal sequences of 92 species were aligned with MAFFT v.7.222 (Katoh and Standley 2013) using the default settings. A few poorly aligned regions were manually adjusted in Geneious.
Phylogenetic Analyses
Concatenated SNP matrices from three assemblies of the ddRAD data and the chloroplast and ribosomal data sets were used to infer maximum likelihood phylogenies in RAxML-HPC2 (v8.2.10) under the GTRGAMMA model as recommended by jModelTest v2.1.6 (Darriba et al. 2012). Maximum likelihood (ML) trees were inferred with the combined rapid bootstrap (100 replicates) and search for the best-scoring ML tree. Since strong contentions have arisen at the relationships among three early-diverged lineages in different ddRAD data sets (see Results), we further employed the Quartet Sampling (QS) method to dissect this phylogenetic discordance. Unlike other commonly used branch support metric such as bootstrap (BS) and Bayesian posterior probability (PP), the QS method assesses the confidence, consistency, and informativeness for each branch in a given topology by calculating Quartet Concordance (QC), Quartet Differential (QD), and Quartet Informativeness (QI) scores (Pease et al. 2018). It adopts a quartet-based approach and is a viable method to distinguish strongly conflicting signals from low information in large-sparse and genome-wide data sets. The total of 18 concatenated alignments from three ddRAD assembly approaches and the resulting ML tree topologies (in Fig. 1) were used as input for the QS analysis. The number of replicate quartet searches per branch was set to 500 and the log-likelihood cutoff was set to 2.
Figure 1.

A summary of alternative topologies in results from three different assembly approaches for ddRAD data. There are three lineages with conflicting results in our analyses, illustrated in (a). Topologies and bootstrap support values from ML analysis and Quartet Sampling (QS) analysis of two nodes (s1, s2) are summarized in (b). The number of each topology in (a) corresponds to those in (b). Interpretations of QS scores are: strong support refers to QC 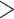 0.2, QD
0.2, QD 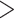 0.3, and QI
0.3, and QI 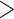 0.3; moderate support refers to 0.2
0.3; moderate support refers to 0.2 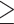 QC
QC 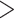 0, QD
0, QD 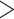 0.3, and QI
0.3, and QI  0.3; variable support (0
0.3; variable support (0 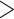 QC
QC 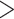
 0.05, QD
0.05, QD  0.3, and QI
0.3, and QI 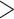 0.3); counter support (QC
0.3); counter support (QC 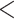 -0.05, QD
-0.05, QD 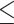 0.3, and QI
0.3, and QI 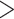 0.3); and poorly informed (QC
0.3); and poorly informed (QC 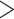 0.2, QD
0.2, QD 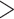 0.3, and QI
0.3, and QI 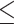 0.3). Different
0.3). Different 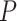 values of matrices indicate that they were created by requiring loci to be shared by at least 75–140 taxa among the total 218 samples. For the p100 column, each method is represented using the matrices created by those loci shared by at least 100 taxa, except for the bowtie2
values of matrices indicate that they were created by requiring loci to be shared by at least 75–140 taxa among the total 218 samples. For the p100 column, each method is represented using the matrices created by those loci shared by at least 100 taxa, except for the bowtie2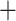 stacks m
stacks m
 approach, which was represented using the matrix p101.
approach, which was represented using the matrix p101.
Congruence Assessment among ddRAD and Chloroplast Phylogenies
We investigated conflicting relationships between the ddRAD and chloroplast phylogenies using the Procrustean Approach to Cophylogeny (PACo) tool (Balbuena et al. 2013) executed as in Pérez-Escobar et al. (2016). This approach assumes that the chloroplast phylogeny is dependent on the nuclear phylogeny and transforms the topologies into matrices of patristic distances, which are then transformed into principal coordinates (PCo) matrices. From these data, a global goodness-of-fit statistic of congruence between the two trees is computed, and taxa with significantly incongruent phylogenetic positions, called outlier associations, can then be identified. For this approach, we used ML trees inferred from the alignment of loci to be shared with more than 101 taxa (p101) in the reference-based bowtie2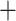 stacks m
stacks m 3 assembly, which was selected by multiple lines of evidence (see Results for details), and chloroplast data.
3 assembly, which was selected by multiple lines of evidence (see Results for details), and chloroplast data.
Ancestral Character Reconstruction
Morphological characters were obtained from the Flora of China (http://www.iplant.cn/foc/), Stapleton (2013), Attigala et al. (2014), Zhang et al. (2017), and Zhang et al. (2020). Three rhizome types were scored: 0, leptomorph; 1, long-necked pachymorph; 2, short-necked pachymorph. Two inflorescence types were scored: 0, semelauctant inflorescence; 1, iterauctant inflorescence. Four branch types were scored: 0, solitary branch; 1, two branches; 2, three branches; 3, multiple branches. All the morphologies analyzed here were presented in Supplementary Figure S1 available on Dryad. The ancestral characters were analyzed using Maximum Parsimony method in Mesquite v3.2 (Maddison 2017) based on the topology of the same RAD matrix for congruence assessment.
Divergence Time Estimation
Divergence times were estimated with a subset of SNPs (139,468 SNPs) randomly selected from the p101 data set assembled from the bowtie2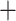 stacks m
stacks m 3 approach using BEAST v.1.8.4 (Drummond and Rambaut 2007). Due to a lack of credible fossils for Arundinarieae, we used three secondary calibration points to perform our dating analysis, as defined by Zhang et al. (2016), a large phylogenetic study of Arundinarieae and relatives with three fossil calibration points: 1) Arundinarieae (6.88–20.96 Ma, median age
3 approach using BEAST v.1.8.4 (Drummond and Rambaut 2007). Due to a lack of credible fossils for Arundinarieae, we used three secondary calibration points to perform our dating analysis, as defined by Zhang et al. (2016), a large phylogenetic study of Arundinarieae and relatives with three fossil calibration points: 1) Arundinarieae (6.88–20.96 Ma, median age  12.72 Ma); 2) paleotropical woody bamboo clade (15.02–31.02 Ma, median age
12.72 Ma); 2) paleotropical woody bamboo clade (15.02–31.02 Ma, median age  22.40 Ma); and 3) neotropical woody bamboo clade (13.88–34.04 Ma, median age
22.40 Ma); and 3) neotropical woody bamboo clade (13.88–34.04 Ma, median age  23.96 Ma). Each calibration point was implemented as a uniform distribution between the minimal and maximal age of the constraint. We used a Yule process tree prior, the uncorrelated lognormal relaxed clock model and GTR
23.96 Ma). Each calibration point was implemented as a uniform distribution between the minimal and maximal age of the constraint. We used a Yule process tree prior, the uncorrelated lognormal relaxed clock model and GTR 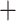 G substitution model. In addition, we fixed the tree topology based on the ML topology 5 of the p101 data set. Ten independent runs of 200,000,000 generations were carried out, with sampling every 1000 generations, and effective sample sizes (ESSs) of all parameters were checked by Tracer v1.6. Independent tree files were combined using the LogCombiner package after removing 20% as burn-in to construct the maximum clade credibility (MCC) chronogram.
G substitution model. In addition, we fixed the tree topology based on the ML topology 5 of the p101 data set. Ten independent runs of 200,000,000 generations were carried out, with sampling every 1000 generations, and effective sample sizes (ESSs) of all parameters were checked by Tracer v1.6. Independent tree files were combined using the LogCombiner package after removing 20% as burn-in to construct the maximum clade credibility (MCC) chronogram.
Diversification Analyses
We examined diversification rates using the time-calibrated trees based on topology 5 of the reference-based p101 data set (see Results) with outgroups pruned. We generated a lineage through time (LTT) plots using APE v.3.5 (Paradis et al. 2004). Analyses were run using the maximum clade credibility chronogram together with 1000 random BEAST trees.
Shifts in diversification were examined using Bayesian analysis of macroevolutionary mixtures (BAMM v.2.5) (Rabosky 2014). We set the prior for the “ExpectedNumberofShifts” to 1, 2, 3, 4, 5, 6, 8, and 10 to determine whether our results were affected by different settings of the parameter. We performed BAMM with 10,000,000 Markov chain Monte Carlo (MCMC) generations and sampled every 1000 generations using the MCC tree of BEAST. We used the R package BAMMtools to analyze the output data. The mean phylorate plot was generated to show the mean of the marginal posterior density of speciation rates along each branch of the phylogeny. We examined the 95% credible set of distinct shift configurations using the “credibleShiftSet” function and show the number and location of the rate shifts as well as their posterior probability. The “plotRateThroughTime” function was used to plot speciation, extinction, and net diversification rates through time for all of Arundinarieae.
Results
Genome Characteristics of Bamboo Polyploids and Clustering Threshold Optimization
We simulated and clustered RAD reads from four reference genomes and further calculated the proportion of single haplotype clusters (singletons) for simulated RAD reads of each reference genome across a range of clustering similarity threshold series (Supplementary Fig. S3 available on Dryad). The proportions of singletons of the two allotetraploid bamboo genomes (G. angustifolia and P. edulis) were similar across all the similarity thresholds we tested, which reveal a similar extent of divergence among their simulated RAD reads. In general, the degree of divergence of simulated RAD reads within diploid rice and diploid bamboo (O. latifolia) genomes was higher than that of tetraploid bamboos when using clustering thresholds between 0.9 and 0.95, likely due to the clustering of duplicated loci in tetraploid bamboos. At thresholds above 0.96, this divergence of the two tetraploid bamboo genomes exceeds that of diploid rice and becomes similar to diploid bamboo. These results suggest that the number of paralogs can be effectively minimized by increasing the similarity threshold when performing clustering. For our data assembly, when the similarity threshold was set to 96% (which allow less than six mismatches) or higher, the paralogous sequences in allopolyploid bamboos reached a similar level with diploids and could be controlled within an acceptable fraction (less than 15%).
For nine empirical samples, the average proportions of five types of clusters are shown in Supplementary Fig. S4 available on Dryad, with results summarized in the Supplementary Text available on Dryad. These data indicate that as clustering similarity thresholds decrease from 98% to 88%, the proportions of single haplotype clusters (putative homozygous loci) and two-haplotype clusters (putative heterozygous loci) decreases, while three- and four-haplotype clusters (putative paralogous loci) increases. The number of single-allele clusters and two-allele clusters steeply increases when the threshold is set to 96% or higher, indicating oversplitting, which is concordant with the threshold from the simulated in silico experiment. As similarity thresholds decrease the proportion of oversplit clusters also decreases until approximately 90%, at which point the collapsing of paralogous alleles together becomes more common. Thus, considering the different genetic distances between alleles within and across species, we selected to perform our empirical analyses at 96% and 90% similarity thresholds (allowing no more than 6 or 14 mismatches) for intraspecies and interspecies clustering separately.
ddRAD Provides a Well-resolved Arundinarieae Phylogeny
Of the 18 ddRAD data sets that we assembled under different combinations of programs and locus filtering parameters, maximum likelihood (ML) phylogenetic results consistently recovered the same five major lineages within Arundinarieae (Supplementary Fig. S5 available on Dryad). Among these phylogenies, most nodes have full statistical support (100% bootstrap support), showing that ddRAD provides significant information at both deep and shallow divergences, as is expected for large phylogenetic data sets (Eaton et al. 2017). The order of relationships among three early-diverged lineages (Hsuehochloa, the Ampelocalamus–Drepanostachyum–Himalayacalamus [ADH] lineage, and Gaoligongshania) was variable. Conflicts among data sets with regard to these relationships are summarized into five alternative topologies (Fig. 1). Regardless of those conflicts, two newly recognized lineages (including 175 sampled taxa) were consistently recovered matching to taxa with pachymorph versus leptomorph rhizome morphologies.
To choose the best tree hypothesis among our five proposed trees (Fig. 1), we investigated further the effects of assembly methods on our results, focusing on total information content, the proportion of missing data, as well as consistency and statistical support (see Supplementary Text available on Dryad for more details). The supermatrix alignments produced by different assemblies varied in their proportions of missing data and numbers of recovered loci. When using stringent filtering options that require loci to be shared across 100 taxa or more, the bowtie2 stacks m
stacks m 3 approach constructed the most complete supermatrix. Of the 18 data sets summarized in Figure 1, topology 5 inferred from the reference-based method was supported by the most data sets (10 out of 18) and received the highest bootstrap support at the two equivocal backbone nodes among the five topologies (average BS
3 approach constructed the most complete supermatrix. Of the 18 data sets summarized in Figure 1, topology 5 inferred from the reference-based method was supported by the most data sets (10 out of 18) and received the highest bootstrap support at the two equivocal backbone nodes among the five topologies (average BS  100%, 81%).
100%, 81%).
Results from quartet-based analyses (QS) highlight the instability of nodes that contribute most to low support values in ML analyses (Fig. 1, Supplementary Table S2 available on Dryad). For the three de novo assembled matrices that support topology 1, the Quartet Concordance (QC) values of branch s1 are all negative ( 0.21 to
0.21 to  0.29), indicating a substantial number of sampled quartets support discordant relationships different from the input tree, and thus not supporting the earliest-diverging relationship of the ADH lineage. The low or negative QC scores (
0.29), indicating a substantial number of sampled quartets support discordant relationships different from the input tree, and thus not supporting the earliest-diverging relationship of the ADH lineage. The low or negative QC scores ( 0.065 to 0.19) of branch s2 indicate moderate or conflicting support for the sister relationship of Hsuehochloa and Gaoligongshania. A similar pattern was found in topology 2 and topology 3 derived from three de novo alignments, with negative QC values (
0.065 to 0.19) of branch s2 indicate moderate or conflicting support for the sister relationship of Hsuehochloa and Gaoligongshania. A similar pattern was found in topology 2 and topology 3 derived from three de novo alignments, with negative QC values ( 0.3 to
0.3 to  0.35) for branch s1, and low Quartet Informativeness (QI) (0.035–0.21) for branch s2. These results led us to a consensus QS interpretation for the conflicting support for the position of Hsuehochloa and low phylogenetic information of Gaoligongshania in these matrices. For topology 4 from a reference-based assembly, strong QC values (0.82–0.85) for branch s1 support Hsuehochloa as the earliest-diverging lineage of Arundinarieae, and low QI values (0.056–0.085) for branch s2 indicate low information for the sister relationship of Gaoligongshania and the ADH lineage. For topology 5 derived from 10 matrices of reference-based assemblies, high QC values (0.45–0.88) consistently support s1, which supports the divergence of the ADH lineage after Hsuehochloa. Low QI values (0.056–0.15) were observed in 7 out of 10 data sets for branch s2, and the placement of Gaoligongshania was moderately supported with low positive QC scores (0.12–0.168) from the remaining three matrices. In summary, most matrices from de novo assemblies showed not just low information content, but rather conflicting support for the uncertain relationships of three lineages, while in all 12 data sets yielded from reference-based methods, the placement of Hsuehochloa was strongly maintained, with only moderate support or low information contributing to uncertainty for the position of Gaoligongshania. Taking into consideration the factors mentioned above, we decided to focus our remaining analyses on topology 5 (Fig. 2), which was inferred from the alignment of 323,802 SNPs from loci shared by more than 101 taxa in the bowtie2
0.35) for branch s1, and low Quartet Informativeness (QI) (0.035–0.21) for branch s2. These results led us to a consensus QS interpretation for the conflicting support for the position of Hsuehochloa and low phylogenetic information of Gaoligongshania in these matrices. For topology 4 from a reference-based assembly, strong QC values (0.82–0.85) for branch s1 support Hsuehochloa as the earliest-diverging lineage of Arundinarieae, and low QI values (0.056–0.085) for branch s2 indicate low information for the sister relationship of Gaoligongshania and the ADH lineage. For topology 5 derived from 10 matrices of reference-based assemblies, high QC values (0.45–0.88) consistently support s1, which supports the divergence of the ADH lineage after Hsuehochloa. Low QI values (0.056–0.15) were observed in 7 out of 10 data sets for branch s2, and the placement of Gaoligongshania was moderately supported with low positive QC scores (0.12–0.168) from the remaining three matrices. In summary, most matrices from de novo assemblies showed not just low information content, but rather conflicting support for the uncertain relationships of three lineages, while in all 12 data sets yielded from reference-based methods, the placement of Hsuehochloa was strongly maintained, with only moderate support or low information contributing to uncertainty for the position of Gaoligongshania. Taking into consideration the factors mentioned above, we decided to focus our remaining analyses on topology 5 (Fig. 2), which was inferred from the alignment of 323,802 SNPs from loci shared by more than 101 taxa in the bowtie2 stacks m
stacks m 3 assembly.
3 assembly.
Figure 2.

Phylogeny of Arundinarieae inferred from maximum likelihood (ML) analysis based on the ddRAD p101 data set yielded from the bowtie2 stacks m
stacks m
 assembly method. Full phylogeny with heat map coloration of branches by Quartet Concordance (QC) scores of Quartet Sampling analyses for internal branches was presented as: dark blue (QC
assembly method. Full phylogeny with heat map coloration of branches by Quartet Concordance (QC) scores of Quartet Sampling analyses for internal branches was presented as: dark blue (QC 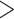 0.2), light blue (0.2
0.2), light blue (0.2 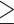 QC
QC 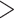 0), light orange (0
0), light orange (0 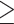 QC
QC 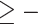 0.05), or dark orange (QC
0.05), or dark orange (QC 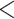
 0.05). Numbers associated with nodes indicate QC/Quartet Differential/Quartet Informativeness scores. Inferences of character state change for rhizome type of Arundinarieae were indicated by different colors of clades: black represents short-necked pachymorph rhizomes, green represents long-necked pachymorph rhizomes, and pink represents leptomorph rhizomes. Images of three rhizome types were demonstrated on the left, from top to bottom are short-necked pachymorph rhizome of Fargesia Franchet: this type of rhizome is short, thick, with axillary buds that develop directly into new shoots and then grow into new culms; long-necked pachymorph rhizome of Yushania P. C. Keng: this type of rhizome bears long necks with no root or buds on each node; leptomorph rhizome of Acidosasa C. D. Chu and C. S. Chao ex P. C. Keng: this type of rhizome is long, slender with root and buds on each node, and buds grow into new shoots or new rhizomes which extend horizontally underground for long distance.
0.05). Numbers associated with nodes indicate QC/Quartet Differential/Quartet Informativeness scores. Inferences of character state change for rhizome type of Arundinarieae were indicated by different colors of clades: black represents short-necked pachymorph rhizomes, green represents long-necked pachymorph rhizomes, and pink represents leptomorph rhizomes. Images of three rhizome types were demonstrated on the left, from top to bottom are short-necked pachymorph rhizome of Fargesia Franchet: this type of rhizome is short, thick, with axillary buds that develop directly into new shoots and then grow into new culms; long-necked pachymorph rhizome of Yushania P. C. Keng: this type of rhizome bears long necks with no root or buds on each node; leptomorph rhizome of Acidosasa C. D. Chu and C. S. Chao ex P. C. Keng: this type of rhizome is long, slender with root and buds on each node, and buds grow into new shoots or new rhizomes which extend horizontally underground for long distance.
Chloroplast Phylogenomic Analyses
Phylogenetic analyses of chloroplast genomes recovered 12 well-supported clades within Arundinarieae (Supplementary Fig. S6 available on Dryad) with relationships among them generally strongly supported. These results are congruent with previous studies except for clades I and X, which were poorly supported previously (Ma et al. 2017) but received moderate BS support (97%, 84%) in this study. This improvement may reflect our increased sampling. Several deep nodes are weakly supported with short internodes, which suggests a lack of phylogenetic information in chloroplast genomes for resolving some parts of the tree in this radiation.
Incongruence among Nuclear and Chloroplast Genomic Data Sets
As noticed in previous studies, the cpDNA topologies conflict significantly with the nuclear ddRAD results (Fig. 3). Some of these major conflicts are driven by four lineages (I, VIII, IX, and X) that are each represented by only one extant species. Besides those lineages, only three clades (II, VII, and XII) are recovered consistently across both data sets, and their positions relative to other clades are still incongruent. Three large chloroplast clades, IV (Shibataea), V (Phyllostachys), and VI (Arundinaria), were separated into several different clades in the ddRAD phylogenies (see Supplementary Text available on Dryad for more details). From genome skimming, we further examined nuclear ribosomal data for 92 species, as a separate nuclear data set from the ddRAD data. The ribosomal data set recovered several monophyletic clades consistent with the ddRAD phylogenies (Supplementary Fig. S7 available on Dryad); however, the relationships for deep nodes were poorly resolved due to insufficient informative variation.
Figure 3.

A simplified version of Figure 2 (left) and Supplementary Figure S6 available on Dryad (right), which denotes the major incongruence of topologies between the nuclear ddRAD and chloroplast trees. The clade names correspond to those in Figure 2 and Supplementary Figure S6 available on Dryad. Asterisks (*) indicate 100% support values.
Topological conflicts among our selected ddRAD data sets and the chloroplast data were visualized in the results of PACo analyses (Supplementary Fig. S8 available on Dryad). The cophylogenetic analysis in PACo accepts the hypothesis that at least some parts of the clades in the chloroplast tree are dependent on clades in the ddRAD tree (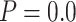 ). However, the normalized squared residuals obtained by PACo identified 65 of the 147 tips (44%) as conflicting taxa (Supplementary Fig. S9 available on Dryad).
). However, the normalized squared residuals obtained by PACo identified 65 of the 147 tips (44%) as conflicting taxa (Supplementary Fig. S9 available on Dryad).
Ancestral Characters based on the ddRAD Tree as the Most Likely True Topology
We reconstructed ancestral states of three major morphological characters in the taxonomy of Arundinarieae (Stapleton 1997) based on the topology 5 reconstructed from the reference-based p101 matrix. The pachymorph rhizome with a short neck is inferred to be the ancestral type present in many species as a symplesiomorphy (Fig. 2). The more derived types evolved during the diversification of the tribe. Pachymorph rhizomes with an elongated neck originated at least twice in the pachymorph lineage, in subclades II and V-Yushania, respectively. Leptomorph rhizomes also evolved independently twice within this tribe, first in the crown of the leptomorph lineage and second in the V-Alpine Bashania subclade in the pachymorph lineage.
The other two characters examined show less phylogenetic signal. Semelauctant inflorescences and a multiple branching pattern were inferred to be ancestral states. Pseudospikelets with iterauctant inflorescence evolved at least three times in the leptomorph lineage (Supplementary Fig. S10 available on Dryad). The pattern of evolution in branching patterns is more complex with multiple origins and reversions of each state, except for two-branching, which appears to be a synapomorphy of Phyllostachys (Supplementary Fig. S11 available on Dryad).
Age Estimation and Diversification Analysis
The stem age of Arundinarieae was estimated to the early Oligocene (30.05 Ma with 95% highest posterior density (HPD): 20.22–51.55) with a crown age estimate of 18.73 Ma (95% HPD: 11.52–20.96). Divergence of the four major lineages was estimated to have occurred in a short geological period (13.65–11.05 Ma) in the Miocene (Supplementary Table S3, Fig. S12 available on Dryad).
Lineage through time plots of extant species detected an increased accumulation of lineages during the mid-Miocene (13–9 Ma) by showing a steeper slope of curves (Fig. 4a). BAMM was used to reconstruct and visualize the dynamics of species diversification on a time-scaled phylogenetic tree. The accuracy of this method has been the subject of debate (Moore et al. 2016; Rabosky et al. 2017), thus we ran several separate analyses, following an approach that was used in Theaceae (Yu et al. 2017). Differences among results under different priors in BAMM analyses were quite small (Supplementary Figs. S13–S15 and Supplementary Table S4 available on Dryad). They are consistent with two core shifts occurring at the crown of the (leptomorph 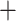 pachymorph
pachymorph  Gaoligongshania
Gaoligongshania 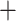 ADH) lineage, and the crown of (Arundinaria
ADH) lineage, and the crown of (Arundinaria 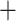 Pleioblastus
Pleioblastus 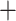 Pseudosasa)
Pseudosasa) 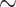 (Phyllostachys
(Phyllostachys 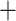 Shibataea) subclade. The phylorate plot indicated diversification heterogeneity within Arundinarieae, with a range from 0.18 to 0.96 (sp/Myr) (Fig. 4d). The rate-through-time plots show that global speciation and net diversification rates of Arundinarieae were significantly accelerated and reached a peak near the mid-Miocene, followed by slight fluctuation and a soft uptick, then a steady decrease from the late Miocene towards the present (Fig. 4b).
Shibataea) subclade. The phylorate plot indicated diversification heterogeneity within Arundinarieae, with a range from 0.18 to 0.96 (sp/Myr) (Fig. 4d). The rate-through-time plots show that global speciation and net diversification rates of Arundinarieae were significantly accelerated and reached a peak near the mid-Miocene, followed by slight fluctuation and a soft uptick, then a steady decrease from the late Miocene towards the present (Fig. 4b).
Figure 4.

Diversification analyses in Arundinarieae based on the maximum clade credibility (MCC) tree of BEAST analyses. a) Lineage-through-time plot, the red plot represents the maximum clade credibility tree, and the grey plots denote 1000 trees. b) Rate-through-time plots for speciation (red), extinction (black) and net diversification (blue) with 95% confidence interval indicated by shaded areas, obtained from the BAMM analysis conducted under the “ExpectedNumberofShifts” as 2. c) Past fluctuations of East Asian monsoons from the mid-Miocene, plotted from data of Clift et al. (2014). The dots are modified and redrawn from Fig. 5B in Clift et al. (2014), which represent the hematite/goethite proxy calculated from color spectral data from the Ocean Drilling Program (ODP) Site 1148 in the South China Sea. And the dynamic curve represents CRAT proxy from ODP Site 1148 (Clift et al. 2008) which shown in Fig. 5A of Clift et al. (2014). d) Mean phylorate plot of Arundinarieae, clade colors indicate the mean evolutionary rate across all shift configurations sampled during simulation of the posterior (cool colors  slow, warm
slow, warm  fast). The locations of two distinct shift configurations with the highest posterior probability rate shifts are shown as circles with numbers.
fast). The locations of two distinct shift configurations with the highest posterior probability rate shifts are shown as circles with numbers.
Discussion
Clustering Threshold Optimization and De novo Versus Reference-based Assemblies for RAD-seq Data in Radiated Polyploids
Polyploid lineages present unique challenges for phylogenetic inference that require closer examination than for diploids. To address this, we used in silico digestion of published genomes from two paleotetraploid bamboo species, a diploid bamboo species and a diploid grass species to simulate RAD loci and assess the proportion of duplicated loci within each genome, with emphasis on comparing the difference between paleotetraploids and diploids. Our results revealed that, for species with highly divergent subgenome composition, and generally few transposable elements, like for instance the temperate bamboos, the number and impact of paralogs can be effectively minimized by increasing the similarity threshold during assembly. Based on the time elapsed since the WGD event, and the level of diploidization in Arundinarieae (Mandakova and Lysak 2018; Guo et al. 2019; Huang et al. 2020), species are treated as paleopolyploids, exhibiting diploid-like characteristics (Peng et al. 2013), which can make orthology assessment relatively easier to handle.
Previous studies suggested that liberal clustering thresholds should be preferred to reduce the effect of oversplitting since undersplitting is a minor issue in RAD-seq data for many diploid species (Ilut et al. 2014; Harvey et al. 2015), but these studies did not consider differences in genetic divergence between subgenomes within and between species. Thus, we further explore a procedure to estimate proper clustering thresholds for loci assembly of temperate bamboos. Based on the results of our in silico simulation analyses, we agree with Ilut et al. (2014) that oversplitting is only a problem at very stringent similarity thresholds (higher than 96%; Supplementary Fig. S4 available on Dryad). The average proportions of putative homozygous and heterozygous loci reach an asymptote at approximately 90% similarity, suggesting this threshold is an optimum value to prevent oversplitting (Ilut et al. 2014). The proportions of putative paralogs in temperate bamboos decrease with the increasing similarity threshold and are slightly higher than in diploids (Ilut et al. 2014), but typically smaller when compared with the true orthologs, and can be eliminated by ploidy-based filtering. Based on our simulations, the similarity values adopted for intraspecies and interspecies clustering on a given data set can be properly handled through prior investigations like the present analyses.
In the present study, three Stacks-based assembly strategies were explored to demonstrate the negative impact of de novo assembly on phylogenetic inference in polyploids, and further to compare the validity of de novo versus reference-based assemblies in the determination of orthologs for polyploid plants. Our results revealed that, although the size of data sets assembled under different methods appeared quite sufficient, they resulted in different tree estimates (Fig. 1). Three alternative topologies (topology 1–3) were obtained from the de novo assembly, in which low bootstrap support values and negative/low QC scores were commonly observed for nodes of the three early-diverging lineages. In contrast, only two topologies were recovered from reference-based assemblies, and most nodes on these trees were strongly supported by ML analyses and QS analyses. These results highlight that, despite our efforts to optimize assembly parameters for polyploids, and the fact that interspecies divergences are greater than within-species allelic divergence in Arundinarieae, de novo assembly may still introduce a substantial number of paralogs. Their impact on the backbone relationships of the five major lineages acts to confound these splits that are otherwise well resolved. Given relatively ancient divergences among deep nodes in Arundinarieae, and their polyploid history (paleopolyploidization), it reveals that reference-based assembly approaches perform better at the tribe level.
Here, we compared the de novo and reference-based assembly approaches to analyze genomic data for Arundinarieae, a complex allotetraploid plant lineage that underwent WGD and subsequent radiation. Our workflow (Supplementary Fig. S2 available on Dryad) to identify orthologs in RAD-seq data in allotetraploids included simulating RAD-seq data from related diploid and allotetraploid genomes to estimate the divergence of allopolyploids, optimizing clustering thresholds, and assembling orthologous loci and filtering paralogs by using a reference genome. Based on our results, we recommend estimating the proportion of duplicated loci in the genome before performing phylogenetic studies of polyploid species using RAD-seq or other NGS data. Simulations based on these data can be used to optimize clustering or mapping thresholds to reduce the negative effect of oversplitting and undersplitting on locus assembly. Finally, anchoring RAD loci on a reference genome is essential to minimize the conflation of orthology/paralogy.
New Insights into Phylogeny of Arundinarieae using ddRAD Data
Our phylogenetic analyses provide strong support for most nodes in Arundinarieae across multiple strategies of data assembly (Supplementary Fig. S5 available on Dryad). Trees inferred from 18 assemblies of the data recovered five proposals of the backbone topology that all support a division of Arundinarieae into five major lineages, as summarized in Figure 1. Based on multiple lines of evidence we select topology 5, obtained from the reference-based p101 data set, as the best-resolved topology (Fig. 2). The dramatic differences between our ddRAD and plastome results clearly illustrate the promise of ddRAD data for the resolution of plant radiations.
We propose that incongruence in the relationships of three early-diverging lineages among trees estimated from different assemblies is likely due to several analytic and biological factors. First, the assembly pipelines employ different clustering and alignment strategies (Wang et al. 2017), which may affect the recovery of accurately assembled loci and downstream tree inferences (see our detailed discussion above). Second, within the same assembly method, such as bowtie2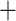 mapq
mapq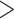 10, the interactions between sequence divergence and the loci filtering parameters (more conserved loci along with increasing
10, the interactions between sequence divergence and the loci filtering parameters (more conserved loci along with increasing 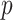 value in population program) showed large effects on the position of Gaoligongshania. Gaoligongshania possesses more than 95% missing data (SNPs) and received low QI scores from QS analyses among most of the data sets generated across different assembly approaches, which may be related to its genome size (1C
value in population program) showed large effects on the position of Gaoligongshania. Gaoligongshania possesses more than 95% missing data (SNPs) and received low QI scores from QS analyses among most of the data sets generated across different assembly approaches, which may be related to its genome size (1C  6.23 pg, which is about three times of moso bamboo) (Zhang 2013), probably the largest in Arundinarieae, and its effect on fragments generated during library preparations. Third, long-terminal branches in the phylogeny have a higher susceptibility to the problems of substitution saturation and long-branch attraction in the case of rapid divergences (Whitfield and Lockhart 2007). Therefore, the inconsistency of positions of the three long-branched lineages may be partly caused by long-branch attraction, as the case for the ADH lineage and Gaoligongshania in topology 4. Moreover, we suspect introgression and gene duplications have contributed to a reticulate evolutionary history in Arundinarieae as conflicting signals were observed in network analyses among three early-diverged lineages (Supplementary Method, Text, and Fig. S16 available on Dryad).
6.23 pg, which is about three times of moso bamboo) (Zhang 2013), probably the largest in Arundinarieae, and its effect on fragments generated during library preparations. Third, long-terminal branches in the phylogeny have a higher susceptibility to the problems of substitution saturation and long-branch attraction in the case of rapid divergences (Whitfield and Lockhart 2007). Therefore, the inconsistency of positions of the three long-branched lineages may be partly caused by long-branch attraction, as the case for the ADH lineage and Gaoligongshania in topology 4. Moreover, we suspect introgression and gene duplications have contributed to a reticulate evolutionary history in Arundinarieae as conflicting signals were observed in network analyses among three early-diverged lineages (Supplementary Method, Text, and Fig. S16 available on Dryad).
Resolving phylogenetic relationships of Arundinarieae has evaded previous attempts despite substantial effort (Zeng et al. 2010; Ma et al. 2014; Zeng et al. 2014; Ma et al. 2017). Our ddRAD phylogeny, and its comparison with alternative data sets, represents the most comprehensive and robustly resolved phylogenetic species tree in Arundinarieae to date. Relationships among the major lineages here roughly agree with previous studies, but with great improvements. Hsuehochloa is placed as the earliest-diverging lineage of Arundinarieae, consistent with previous analysis based on nuclear LEAFY and GBSSI genes (Yang et al. 2013), followed by the ADH lineage. Our analysis identified two new lineages, that is, the pachymorph lineage and the leptomorph lineage, underpinning the evolutionary significance of the well-characterized rhizome types. Compared with the inferred phylogeny from chloroplast genomes, of which many relationships remain unresolved and identification of clades largely deviated from morphological taxonomy (Supplementary Fig. S6 available on Dryad, Attigala et al. 2016), the ddRAD tree has strong statistical support at both deep and shallow nodes and reveals that rhizome character states show clear phylogenetic signal (Fig. 2).
Conflicting Signals Imply Incomplete Lineage Sorting (ILS) and Hybridization Events
Empirical studies have demonstrated conflicting phylogenetic signals among markers with different patterns of inheritance (nuclear, mitochondrial, ribosomal, and chloroplast) both at the level of deep-scale phylogenetic relationships (e.g., the COM clade within Rosidae; Sun et al. 2015; Zhao et al. 2016) as well as among shallower-scale clades like the three tribes of the bamboo subfamily (Wysocki et al. 2015; Guo et al. 2019). As demonstrated here, substantial conflict between nuclear and chloroplast data in the temperate bamboos is conspicuous even among angiosperm lineages, and especially for analysis at the tribe level. Deep and strong incongruence among the topologies obtained from nuclear and chloroplast genomic data sets is also found at both the generic and species levels (Fig. 3; Supplementary Fig. S8 available on Dryad). Despite our efforts to identify credible nuclear and chloroplast trees, we were unable to recover overlapping topologies between marker types, suggesting that nuclear and the chloroplast genomes of Arundinarieae have distinct evolutionary histories. Several factors can drive such conflict, including phylogenetic uncertainty and/or biological factors such as ILS and hybridization (Maddison 1997), which are hypothesized to play an important role in adaptive radiations (Seehausen 2004). To rule out phylogenetic uncertainty, we performed a PACo analysis to identify conflicting taxa while incorporating phylogenetic uncertainty. This showed that 44% of samples are outliers (Supplementary Fig. S9 available on Dryad) indicating that the strong conflict in our analysis is caused by biological factors—ILS and/or hybridization—and not simply a lack of resolution.
Previous studies have proposed that hybridization has played a significant and recurrent role in bamboo evolution (Triplett and Clark 2010; Yang et al. 2013; Triplett et al. 2014). In North American Arundinaria, Triplett et al. (2010) have identified hybrids among A. gigantea, A. tecta, and A. appalachiana in regions of geographic range overlap. In East Asia, Pseudosasa japonica, Hibanobambusa tranquillans, and Chimonobambusa sichuanensis have shown historical introgression during their divergence (Zhang et al. 2012; Triplett et al. 2014). In our study, Semiarundinaria has close relationships with Phyllostachys in the ddRAD tree and forms a well-supported clade with four species of Pleioblastus in the chloroplast tree. This observation provides additional evidence to the hypothesis that Semiarundinaria possesses both Phyllostachys-like and Pleioblastus-like alleles (Triplett et al. 2014); and that Pleioblastus may be the maternal parent of hybridization with Phyllostachys. The low QC scores ( 0.055, 0.38) and zero QD scores for the branches related to Shibataea kumasasa, S. chiangshanensis, and S. strigosa supported the hypothesis of our parallel study that introgression may have occurred among these species (Guo et al. 2019).
0.055, 0.38) and zero QD scores for the branches related to Shibataea kumasasa, S. chiangshanensis, and S. strigosa supported the hypothesis of our parallel study that introgression may have occurred among these species (Guo et al. 2019).
Incongruence caused by incomplete lineage sorting (ILS) may be common in Arundinarieae, as the time between speciation events is often short, and species tend to have high morphological diversity. In our study, Fargesia yunnanensis and Yushania complanata are identified as two outlier associations by the PACo analysis; they are sister species in the chloroplast tree but somewhat distantly related in the ddRAD tree. They have no geographic overlap, and so hybridization between the two species seems unlikely. Moreover, low QC scores (0.12–0.168) and high QD (0.77–0.9) scores for branch related Gaoligongshania from three reference-based alignments (Supplementary Table S2 available on Dryad) suggests ILS alone may be the cause of incongruence among this species.
Distinguishing between ILS and hybridization can be challenging (Buckley et al. 2006; Joly et al. 2009; Meyer et al. 2016), and further studies will be needed. Other possibilities should be concerned for the conflict, such as the emergence of recombination between the pairing homologs which may lead to the incongruent nuclear phylogenetic relationships for related species when using those recombined regions as markers. However, it is beyond our ability to address this issue with the current data set, so we may not excessively discuss it here.
Radiation of Arundinarieae with Leptomorph Rhizomes as a Key Innovative Character
The rapid radiation of Arundinarieae is supported by compelling evidence in our study. The four major lineages (the ADH, Gaoligongshania, leptomorph, and pachymorph lineages) are estimated to have diverged within a short interval of 2.6 myr. We detect a sharp increase in the diversification rate of Arundinarieae during the mid-Miocene (Fig. 4b,d), which is similarly reflected in a rapid accumulation of lineages during this period in LTT plots (Fig. 4a).
Given the uncertainty in node age estimates in Arundinarieae, the driving factor for this radiation has not yet been determined. Hodkinson et al. (2010) hypothesized the radiation coincided with the continental collision of the Indo-Australian and Eurasian Plates, while Zhang et al. (2016) suggested the radiation was triggered by the second intensification of the East Asian monsoons in the late Miocene. The timing of diversification in our results suggests the rapid diversification of Arundinarieae may be related to the changes in the East Asian summer monsoons (EASM) (Fig. 4b,c). The first increased rate shift within Arundinarieae is concordant with the proposed first strengthening of the EASM (15.5–13 Ma), when abundant rainfall was brought to East Asia (Clift et al. 2002; Sun and Wang 2005; Wan et al. 2007). Climate change is thought to have played a significant role in the diversification of many plant lineages (Erwin 2009; Hoffmann and Sgrò 2011). Temperate bamboos are common in the understory and often form the dominant elements in wet montane vegetation (Clark et al. 2015). Therefore, we extrapolate that the moist climate brought by the intensification of EASM during the mid-Miocene may be a pivotal factor in facilitating their expansion in Mainland East and Southeast Asia. Similar patterns have also been observed in other vascular plants in East Asia, such as ferns (Wang et al. 2012), orchids (Xiang et al. 2016), and Primulina (Gesneriaceae) of the karst area (Kong et al. 2017).
The differentiation of the pachymorph and leptomorph rhizomes happened shortly after the mid-Miocene, circa 11.05 Ma. The consequences of this morphological divergence in the ancestors of the pachymorph and leptomorph lineages likely facilitated their expansion into different habitats (Arber 1934; Wen 1983) and contributed to the rapid adaptive diversification of bamboos. Rhizomes are particularly important in bamboo taxonomy and development (Keng 1982; Arber 1934) because of their long dormancy between flowering times; rhizomes control when the culms develop, how they spread, and dictate vegetative propagation (McClure 1966). The ancestral rhizome type is the pachymorph rhizome, which is associated with a sessile living habit that limits the spread of these species. Because they are fixed in one place for a long time, pachymorph rhizomes may also limit adaptation to local habitats (Wen 1983). In the pachymorph lineage, some subclades evolved an elongated neck (in Yushania and Oldeania) in the middle or late Miocene. These bamboos can extend up to 1 m underground and spread more easily to new niches. This approach of spreading is relatively inefficient though, as there is no root and bud on the elongated neck; such bamboos are mainly limited to the Hengduan Mountain area (Yushania), with a few in Africa and Madagascar (Oldeania) (Li et al. 2006; Stapleton 2013; Zhang et al. 2017). Leptomorph rhizomes may represent a key innovation, as they allow for indeterminate extension and an interconnected underground nutrition network. Leptomorph rhizome species spread broadly and colonize different niches (Wen 1983; Clark et al. 2015). Under the combined effect of the strengthening of EASM and the divergence of varied rhizome types, temperate bamboos spread among various habitats which likely facilitated their rapid diversification (Stapleton 1998).
Many diverse lineages of angiosperms have experienced rapid radiations (Li et al. 2019), and understanding the dynamics of these diversifications can help us to better understand the origins of biological diversity (Gavrilets and Losos 2009). Many well-explored model groups from island and lake ecosystems (Seehausen 2004; Lamichhaney et al. 2015) have offered uniquely powerful insights into how and why species diverge. However, in most radiations, species-level phylogenies remain poorly resolved, especially in species-rich continental lineages (Losos 2010; Hughes and Atchison 2015). Using temperate bamboos as a model group, we used a comprehensive sampling of ddRAD and genome skimming data sets in parallel to resolve phylogenetic relationships of this challenging lineage. We demonstrated a practical procedure for optimizing RAD-seq assembly for polyploid species. Our exhaustive data sets provide a reference phylogenetic framework and a new diversification timescale for Arundinarieae. Together with insights into the evolution of key adaptive innovations like the leptomorph rhizomes, we explore mechanisms of radiative and reticulate evolution of this important bamboo tribe. The findings generated here will not only be important for further studies on the biogeography and taxonomic revision of this complex group but also serve as a case for research into other unresolved radiated polyploidy plant groups.
Acknowledgments
We would like to thank Nian-He Xia (South China Botanical Garden), Cheng Liu, Jia-Wei Sun, Ling Mao, Wen-Gen Zhang (Jiangxi Agricultural University) for assisting with sample collection. We thank Yu-Xiao Zhang, Cun-Xia Zeng, Jian-Jun Jin, Hong-Tao Li, and Yang Luo (all KIB unless specified), Pete M. Hollingsworth and Linda Neaves (Royal Botanic Garden Edinburgh), Alex D. Twyford (University of Edinburgh), Sandra L. Hoffberg (Columbia University), Patrick F. McKenzie (Columbia University) for assistance and advice on the data analyses. Laboratory work was facilitated at the Experimental Center of Molecular Biology of the Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences.
Supplementary Material
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.rr4xgxd6m.
Funding
This work was supported by the Strategic Priority Research Program of Chinese Academy of Sciences (CAS) (XDB31000000), the National Natural Science Foundation of China (31430011, 31470322, and 31670227), and the CAS Large-scale Scientific Facilities (2017-LSFGBOWS-02). The first author was supported by the University of Chinese Academy of Sciences Joint Ph.D. Training Program for her study at Columbia University.
References
- Andrews S. 2016. FastQC: a quality control tool for high throughput sequence data. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [Google Scholar]
- Andrews K.R., Good J.M., Miller M.R., Luikart G., Hohenlohe P.A.. 2016. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 17:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber A. 1934. Chapter IV. Bamboo: vegetative phase. In: Arber A., editor. The Gramineae: a study of cereal, bamboo and grass. Cambridge: Cambridge University Press. p. 58–80. [Google Scholar]
- Attigala L., Triplett J.K., Kathriarachchi H.S., Clark L.G.. 2014. A new genus and a major temperate bamboo lineage of the Arundinarieae (Poaceae: Bambusoideae) from Sri Lanka based on a multi-locus plastid phylogeny. Phytotaxa 174:187–205. [Google Scholar]
- Attigala L., Wysocki W.P., Duvall M.R., Clark L.G.. 2016. Phylogenetic estimation and morphological evolution of Arundinarieae (Bambusoideae: Poaceae) based on plastome phylogenomic analysis. Mol. Phylogenet. Evol. 101:111-121. [DOI] [PubMed] [Google Scholar]
- Baird N.A., Etter P.D., Atwood T.S., Currey M.C., Shiver A.L., Lewis Z.A., Selker E.U., Cresko W.A., Johnson E.A.. 2008. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:e3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbuena J.A., Míguez-Lozano R., Blasco-Costa I.. 2013. PACo: a novel procrustes application to cophylogenetic analysis. PLoS One 8:e61048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blischak P.D., Kubatko L.S., Wolfe A.D.. 2018. SNP genotyping and parameter estimation in polyploids using low-coverage sequencing data. Bioinformatics 34:407-415. [DOI] [PubMed] [Google Scholar]
- BPG. 2012. An updated tribal and subtribal classification of the bamboos (Poaceae: Bambusoideae). J. Am. Bamboo Soc. 24:1-10. [Google Scholar]
- Brandrud M.K., Baar J., Lorenzo M.T., Athanasiadis A., Bateman R.M., Chase M.W., Hedrén M., Paun O.. 2020. Phylogenomic relationships of diploids and the origins of allotetraploids in Dactylorhiza (Orchidaceae). Syst. Biol. 69:91-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandrud M.K., Paun O., Lorenzo M.T., Nordal I., Brysting A.K.. 2017. RADseq provides evidence for parallel ecotypic divergence in the autotetraploid Cochlearia officinalis in Northern Norway. Sci. Rep. 7:5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley T.R., Cordeiro M., Marshall D.C., Simon C.. 2006. Differentiating between hypotheses of lineage sorting and introgression in New Zealand alpine cicadas (Maoricicada Dugdale). Syst. Biol. 55:411-425. [DOI] [PubMed] [Google Scholar]
- Burke S.V., Grennan C.P., Duvall M.R.. 2012. Plastome sequences of two New World bamboos—Arundinaria gigantea and Cryptochloa strictiflora (Poaceae)—extend phylogenomic understanding of Bambusoideae. Am. J. Bot. 99:1951-1961. [DOI] [PubMed] [Google Scholar]
- Catchen J., Hohenlohe P.A., Bassham S., Amores A., Cresko W.A.. 2013. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22:3124-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Li X.M., Zhang B., Xu J.S., Wu Z.K., Wang B., Li H.T., Younas M., Huang L., Luo Y.F., Wu J.S., Hu S.N., Liu K.D.. 2013. Detection and genotyping of restriction fragment associated polymorphisms in polyploid crops with a pseudo-reference sequence: a case study in allotetraploid Brassica napus. BMC Genomics 14:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark L.G., Londoño X., Ruiz-Sanchez E.. 2015. Chapter 1, Bamboo taxonomy and habitat. In: Liese W., Koehl M., editors. Bamboo, the plant and its uses. Heidelberg: Tropical Forest Series, Springer Verlag. p. 1–30. [Google Scholar]
- Clevenger J.P., Korani W., Ozias-Akins P., Jackson S.. 2018. Haplotype-based genotyping in polyploids. Front. Plant. Sci. 9:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clift P.D., Lee J.I., Clark M.K., Blusztajn J.. 2002. Erosional response of South China to arc rifting and monsoonal strengthening; a record from the South China Sea. Mar. Geol. 184:207-226. [Google Scholar]
- Clift P.D., Wan S.M., Blusztajn J.. 2014. Reconstructing chemical weathering, physical erosion and monsoon intensity since 25Ma in the northern South China Sea: a review of competing proxies. Earth-Sci. Rev. 130:86-102. [Google Scholar]
- Cornille A., Salcedo A., Kryvokhyzha D., Glémin S., Holm K., Wright S.I., Lascoux M.. 2016. Genomic signature of successful colonization of Eurasia by the allopolyploid shepherd’s purse (Capsella bursa-pastoris). Mol. Ecol. 25:616-629. [DOI] [PubMed] [Google Scholar]
- Cruaud A., Gautier M., Galan M., Foucaud J., Sauné L., Genson G., Dubois E., Nidelet S., Deuve T., Rasplus J.Y.. 2014. Empirical assessment of RAD sequencing for interspecific phylogeny. Mol. Biol. Evol. 31:1272-1274. [DOI] [PubMed] [Google Scholar]
- Darriba D., Taboada G.L., Doallo R., Posada D.. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle J. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19:11-15. [Google Scholar]
- Drummond A.J., Rambaut A.. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton D.A., Ree R.H.. 2013. Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Syst. Biol. 62:689-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton D.A., Spriggs E.L., Park B., Donoghue M.J.. 2017. Misconceptions on missing data in RAD-seq phylogenetics with a deep-scale example from flowering plants. Syst. Biol. 66:399-412. [DOI] [PubMed] [Google Scholar]
- Erwin D.H. 2009. Climate as a driver of evolutionary change. Curr. Biol. 19:R575-R583. [DOI] [PubMed] [Google Scholar]
- Fernández-Mazuecos M., Mellers G., Vigalondo B., Llorenç S.Vargas P., Glover B.J.. 2017. Resolving recent plant radiations: power and robustness of genotyping-by-sequencing. Syst. Biol. 67:250–268. [DOI] [PubMed] [Google Scholar]
- Gavrilets S., Losos J.B.. 2009. Adaptive radiation: contrasting theory with data. Science 323:732-737. [DOI] [PubMed] [Google Scholar]
- Glover N.M., Redestig H., Dessimoz C.. 2016. Homoeologs: What are they and how do we infer them? Trends Plant Sci. 21:609-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant P.R., Grant B.R.. 2002. Unpredictable evolution in a 30-year study of Darwin’s finches. Science 296:707-711. [DOI] [PubMed] [Google Scholar]
- Guo C., Guo Z.H., Li D.Z.. 2019. Phylogenomic analyses reveal intractable evolutionary history of a temperate bamboo genus (Poaceae: Bambusoideae). Plant Divers. 41:213-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z.H., Li D.Z.. 2004. Phylogenetics of the Thamnocalamus group and its allies (Gramineae: Bambusoideae): inference from the sequences of GBSSI gene and ITS spacer. Mol. Phylogenet. Evol. 30:1-12. [DOI] [PubMed] [Google Scholar]
- Guo Z.H., Ma P.F., Yang G.Q., Hu J.Y., Liu Y.L., Xia E.H., Zhong M.C., Zhao L., Sun G.L., Xu Y.X., Zhao Y.J., Zhang Y.C., Zhang Y.X., Zhang X.M., Zhou M.Y., Guo Y., Guo C., Liu J.X., Ye X.Y., Chen Y.M., Yang Y., Han B., Lin C.S., Lu Y., Li D.Z.. 2019. Genome sequences provide insights into the reticulate origin and unique traits of woody bamboos. Mol. Plant 12:1353-1365. [DOI] [PubMed] [Google Scholar]
- Harvey M.G., Judy C.D., Seeholzer G.F., Maley J.M., Graves G.R., Brumfield R.T.. 2015. Similarity thresholds used in DNA sequence assembly from short reads can reduce the comparability of population histories across species. PeerJ 3:e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey M.G., Smith B.T., Glenn T.C., Faircloth B.C., Brumfield R.T.. 2016. Sequence capture versus restriction site associated DNA sequencing for shallow systematics. Syst. Biol. 65:910-924. [DOI] [PubMed] [Google Scholar]
- Hipp A.L., Eaton D.A., Cavender-Bares J., Fitzek E., Nipper R., Manos P.S.. 2014. A framework phylogeny of the American oak clade based on sequenced RAD data. PLoS One 9:e93975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp A.L., Manos P.S., González-Rodríguez A., Hahn M., Kaproth M., McVay J.D., Avalos S.V., Cavender-Bares J.. 2018. Sympatric parallel diversification of major oak clades in the Americas and the origins of Mexican species diversity. New Phytol. 217:439-452. [DOI] [PubMed] [Google Scholar]
- Hodkinson T.R., Ní Chonghaile G., Sungkaew S., Chase M.W., Salamin N., Stapleton C.M.. 2010. Phylogenetic analyses of plastid and nuclear DNA sequences indicate a rapid late Miocene radiation of the temperate bamboo tribe Arundinarieae (Poaceae, Bambusoideae). Plant Ecol. Divers. 3:109-120. [Google Scholar]
- Hoffmann A.A., Sgrò C.M.. 2011. Climate change and evolutionary adaptation. Nature 470:479-485. [DOI] [PubMed] [Google Scholar]
- Huang X.C., German D.A., Koch M.A.. 2020. Temporal patterns of diversification in Brassicaceae demonstrate decoupling of rate shifts and mesopolyploidization events. Ann. Bot. 125:29-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes C.E., Atchison G.W.. 2015. The ubiquity of alpine plant radiations: from the Andes to the Hengduan Mountains. New Phytol. 207:275-282. [DOI] [PubMed] [Google Scholar]
- Ilut D.C., Nydam M.L., Hare M.P.. 2014. Defining loci in restriction-based reduced representation genomic data from nonmodel species: sources of bias and diagnostics for optimal clustering. Biomed. Res. Int. 2014:675158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzen D. 1976. Why bamboos wait so long to flower. Annu. Rev. Ecol. Syst. 7:347-391. [Google Scholar]
- Jin J.J., Yu W.B., Yang J.B., Song Y., dePamphilis C.W., Yi T.S., Li D.Z.. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly S., McLenachan P.A., Lockhart P.J.. 2009. A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am. Nat. 174: E54-E70. [DOI] [PubMed] [Google Scholar]
- Katoh K., Standley D.M.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., Thierer T., Ashton B., Meintjes P., Drummond A.. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg E.A. 2015. Flowering plants. In: Kubitzki K., editor. Monocots: Poaceae, vol. XIII. Berlin: Springer. p. 25-38. [Google Scholar]
- Keng P.C. 1982. A revision of the genera of bamboos from the world (I). J. Bamboo Res. 1:1-19. [Google Scholar]
- Kong H., Condamine F.L., Harris A.J., Chen J., Pan B., Möller M., Hoang V.S., Kang M.. 2017. Both temperature fluctuations and East Asian monsoons have driven plant diversification in the karst ecosystems from southern China. Mol. Ecol. 26:6414-6429. [DOI] [PubMed] [Google Scholar]
- Lamichhaney S., Berglund J., Almén M.S., Maqbool K., Grabherr M., Martinez-Barrio A., Promerová M., Rubin C.J., Wang C., Zamani N., Grant B.R., Grant P.R., Webster M.T., Andersson L.. 2015. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518:371-375. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L.. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.Z., Hsueh C.J., Xia N.H.. 1995. Gaoligongshania, a new bamboo genus from Yunnan, China. Acta Phytotax. Sin. 33:597-601. [Google Scholar]
- Li D.Z., Stapleton C.M.A., Xue J.R.. 1996. A new combination in Ampelocalamus and notes on A. patellaris (Gramineae: Bambusoideae). Kew Bull. 51:809-813. [Google Scholar]
- Li D.Z., Wang Z.P., Zhu Z.D., Xia N.H., Jia L.Z., Guo Z.H., Yang G.Y., Stapleton C.M.A.. 2006. Bambuseae (Poaceae). In: Wu Z.Y., Raven P.H., Hong D.Y., editors. Flora of China, vol. 22. Beijing and St. Louis: Science Press and Missouri Botanical Garden Press. [Google Scholar]
- Li H., Ruan J., Durbin R.. 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18:1851-1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.T., Yi T.S., Gao L.M., Ma P.F., Zhang T., Yang J.B., Gitzendanner M.A., Fritsch P.W., Cai J., Luo Y., Wang H., Bank M., Zhang S.D., Wang Q.F., Wang J., Zhang Z.R., Fu C.N., Yang J., Hollingsworth P.M., Chase M.W., Soltis D.E., Soltis P.S., Li D.Z.. 2019. Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 5:461-470. [DOI] [PubMed] [Google Scholar]
- Losos Jonathan B. 2010. Adaptive radiation, ecological opportunity, and evolutionary determinism. Am. Nat. 175:623-639. [DOI] [PubMed] [Google Scholar]
- Ma P.F., Vorontsova M.S., Nanjarisoa O.P., Razanatsoa J., Guo Z.H., Haevermans T., Li D.Z.. 2017. Negative correlation between rates of molecular evolution and flowering cycles in temperate woody bamboos revealed by plastid phylogenomics. BMC Plant Biol. 17:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma P.F., Zhang Y.X., Zeng C.X., Guo Z.H., Li D.Z.. 2014. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 63:933-950. [DOI] [PubMed] [Google Scholar]
- Maddison W.P. 1997. Gene trees in species trees. Syst. Biol. 46:523-536. [Google Scholar]
- Maddison W.P. 2017. Mesquite: a modular system for evolutionary analysis. Version 3.2. Available from: http://mesquiteproject.org/. [Google Scholar]
- Mandáková T., Lysak M.A.. 2018. Post-polyploid diploidization and diversification through dysploid changes. Curr. Opin. Plant. Biol. 42:55-65. [DOI] [PubMed] [Google Scholar]
- Massatti R., Reznicek A.A., Knowles L.L.. 2016. Utilizing RADseq data for phylogenetic analysis of challenging taxonomic groups: a case study in Carex sect. Racemosae. Am. J. Bot. 103:337-347. [DOI] [PubMed] [Google Scholar]
- McClure F.A. 1966. The bamboos: a fresh perspective. Part I. The bamboo plant. Cambridge, MA: Harvard University Press, p.11-146. [Google Scholar]
- McCluskey B.M., Postlethwait J.H.. 2015. Phylogeny of zebrafish, a “model species,” within Danio, a “model genus”. Mol. Biol. Evol. 32:635-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B.S., Matschiner M., Salzburger W.. 2016. Disentangling incomplete lineage sorting and introgression to refine species-tree estimates for Lake Tanganyika cichlid fishes. Syst. Biol. 66:531-550. [DOI] [PubMed] [Google Scholar]
- Moore B.R., Höhna S., May M.R., Rannalaa B., Huelsenbeckb J.P.. 2016. Critically evaluating the theory and performance of Bayesian analysis of macroevolutionary mixtures. Proc. Natl. Acad. Sci. USA 113:9569-9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Briones D.F., Liston A., Tank D.C.. 2018. Phylogenomic analyses reveal a deep history of hybridization and polyploidy in the Neotropical genus Lachemilla (Rosaceae). New Phytol. 218:1668-1684. [DOI] [PubMed] [Google Scholar]
- One Thousand Plant Transcriptomes Initiative. 2019. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574:679-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paetzold C., Wood K.R., Eaton D.A.R., Wagner W.L., Appelhans M.S.. 2019. Phylogeny of Hawaiian Melicope (Rutaceae): RAD-seq resolves species relationships and reveals ancient introgression. Front. Plant Sci. 10:1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E., Claude J., Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289-290. [DOI] [PubMed] [Google Scholar]
- Pease J.B., Brown J.W., Walker J.F., Hinchliff C.E., Smith S.A.. 2018. Quartet sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am. J. Bot. 105:385-403. [DOI] [PubMed] [Google Scholar]
- Peng Z.H., Lu Y., Li L.B., Zhao Q., Feng Q., Gao Z.M., Lu H.Y., Hu T., Yao N., Liu K.Y., Li Y., Fan D.L., Guo Y.L., Li W.J., Lu Y.Q., Weng Q.J., Zhou C.C., Zhang L., Huang T., Zhao Y., Zhu C.R., Liu X.G., Yang X.W., Wang T., Miao K., Zhuang C.Y., Cao X.L., Tang W.L., Liu G.S., Liu Y.L., Chen J., Liu Z.J., Yuan L.C., Liu Z.H., Huang X.H., Lu T.T., Fei B.H., Ning Z.M., Han B., Jiang Z.H.. 2013. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 45:456-461. [DOI] [PubMed] [Google Scholar]
- Pennington R.T., Lavin M., Särkinen T., Lewis G.P., Klitgaard B.B., Hughes C.E.. 2010. Contrasting plant diversification histories within the Andean biodiversity hotspot. Proc. Natl. Acad. Sci. USA 107:13783-13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Escobar O.A., Balbuena J.A., Gottschling M.. 2016. Rumbling orchids: how to assess divergent evolution between chloroplast endosymbionts and the nuclear host. Syst. Biol. 65:51-65. [DOI] [PubMed] [Google Scholar]
- Peterson B.K., Weber J.N., Kay E.H., Fisher H.S., Hoekstra H.E.. 2012. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7: e37135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt II R.N., Faircloth B.C., Sullivan K.A.M., Kieran T.J., Glenn T.C., Vandewege M.W., Lee T.E. Jr., Baker R.J., Stevens R.D., Ray D.A.. 2018. Conflicting evolutionary histories of the mitochondrial and nuclear genomes in New World myotis bats. Syst. Biol. 67:236-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabosky D.L. 2014. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS One 9:e89543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabosky D.L., Mitchell J.S., Chang J.. 2017. Is BAMM flawed? Theoretical and practical concerns in the analysis of multi-rate diversification models. Syst. Biol. 66:477-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin B.E., Ree R.H., Moreau C.S.. 2012. Inferring phylogenies from RAD sequence data. PLoS One 7:e33394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Pérez M., Fu M.Y., Yang X.S., Belcher B.. 2001. Bamboo forestry in China: toward environmentally friendly expansion. J. For. 99:14-20. [Google Scholar]
- Seehausen O. 2004. Hybridization and adaptive radiation. Trends Ecol. Evol. 19:198-207. [DOI] [PubMed] [Google Scholar]
- Seehausen O. 2006. African cichlid fish: a model system in adaptive radiation research. Proc. Biol. Sci. 273:1987-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer A.B.A., Peart C.R., Tusso S., Maayan I., Brelsford A., Wheat C.W., Wolf J.B.W., Gilbert M.. 2017. Bioinformatic processing of RAD-seq data dramatically impacts downstream population genetic inference. Methods Ecol. Evol. 8:907-917. [Google Scholar]
- Soderstrom T.R., Ellis R.P.. 1987. The position of bamboo genera and allies in a system of grass classification. In: Soderstrom T.R., Hilu K.W., Campbell C.S., Barkworth M.E., editors. Grass systematics and evolution. Washington DC & London: Smithsonian Institution Press. [Google Scholar]
- Soreng R.J., Peterson P.M., Romaschenko K., Davidse G., Zuloaga F.O., Judziewicz E.J., Filgueiras T.S., Davis J.I., Morrone O.. 2015. A worldwide phylogenetic classification of the Poaceae (Gramineae). J. Syst. Evol. 53:117-137. [Google Scholar]
- Stapleton C.M. 2013. Bergbambos and Oldeania, new genera of African bamboos (Poaceae, Bambusoideae). PhytoKeys 87-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton C.M. 1998. Form and function in the bamboo rhizome. J. Am. Bamboo Soc. 12:21-29. [Google Scholar]
- Stapleton C.M. 1997. The morphology of woody bamboos. In: Chapman G.P., editor. The bamboos. Academic Press. p. 251-267. [Google Scholar]
- Stetter M.G., Schmid K.J.. 2017. Analysis of phylogenetic relationships and genome size evolution of the Amaranthus genus using GBS indicates the ancestors of an ancient crop. Mol. Phylogenet. Evol. 109:80-92. [DOI] [PubMed] [Google Scholar]
- Sun M., Soltis D.E., Soltis P.S., Zhu X., Burleigh J.G., Chen Z.. 2015. Deep phylogenetic incongruence in the angiosperm clade Rosidae. Mol. Phylogenet. Evol. 83:156-166. [DOI] [PubMed] [Google Scholar]
- Sun X.J., Wang P.X.. 2005. How old is the Asian monsoon system?—Palaeobotanical records from China. Palaeogeogr. Palaeoclimatol. Palaeoecol. 222:181-222. [Google Scholar]
- Triplett J.K., Clark L.G.. 2010. Phylogeny of the temperate bamboos (Poaceae: Bambusoideae: Bambuseae) with an emphasis on Arundinaria and allies. Syst. Bot. 35:102-120. [Google Scholar]
- Triplett J.K., Clark L.G., Fisher A.E., Wen J.. 2014. Independent allopolyploidization events preceded speciation in the temperate and tropical woody bamboos. New Phytol. 204:66-73. [DOI] [PubMed] [Google Scholar]
- Triplett J.K., Oltrogge K.A., Clark L.G.. 2010. Phylogenetic relationships and natural hybridization among the North American woody bamboos (Poaceae: Bambusoideae: Arundinaria). Am. J. Bot. 97:471-492. [DOI] [PubMed] [Google Scholar]
- Tripp E.A., Tsai Y.E., Zhuang Y., Dexter K.G.. 2017. RADseq dataset with 90% missing data fully resolves recent radiation of Petalidium (Acanthaceae) in the ultra-arid deserts of Namibia. Ecol. Evol. 7:7920-7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas O.M., Ortiz E.M., Simpson B.B.. 2017. Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high-Andean diversification (Asteraceae: Astereae: Diplostephium). New Phytol. 214:1736-1750. [DOI] [PubMed] [Google Scholar]
- Wagner C.E., Harmon L.J., Seehausen O.. 2012. Ecological opportunity and sexual selection together predict adaptive radiation. Nature 487:366-369. [DOI] [PubMed] [Google Scholar]
- Wagner C.E., Keller I., Wittwer S., Selz O.M., Mwaiko S., Greuter L., Sivasundar A., Seehausen O.. 2013. Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Mol. Ecol. 22:787-798. [DOI] [PubMed] [Google Scholar]
- Wan S.M., Li A.C., Clift P.D., Stuut J.B.W.. 2007. Development of the East Asian monsoon: mineralogical and sedimentologic records in the northern South China Sea since 20 Ma. Palaeogeogr. Palaeoclimatol. Palaeoecol. 254:561-582. [Google Scholar]
- Wang L., Schneider H., Zhang X.C., Xiang Q.P.. 2012. The rise of the Himalaya enforced the diversification of SE Asian ferns by altering the monsoon regimes. BMC Plant Biol. 12:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.Q., Ye X.Y., Zhao L., Li D.Z., Guo Z.H., Zhuang H.F.. 2017. Genome-wide RAD sequencing data provide unprecedented resolution of the phylogeny of temperate bamboos (Poaceae: Bambusoideae). Sci. Rep. 7:11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen T.H. 1983. Some ideas about the origin of bamboos. J. Bamboo Res. 2:1-10. [Google Scholar]
- Wendel J.F. 2000. Genome evolution in polyploids. Plant Mol. Biol. 42:225-249. [PubMed] [Google Scholar]
- Wendel J.F. 2015. The wondrous cycles of polyploidy in plants. Am. J. Bot. 102:1753-1756. [DOI] [PubMed] [Google Scholar]
- Whitfield J.B., Lockhart P.J.. 2007. Deciphering ancient rapid radiations. Trends Ecol. Evol. 22:258-265. [DOI] [PubMed] [Google Scholar]
- Wick R.R., Schultz M.B., Zobel J., Holt K.E.. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31:3350-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickett N.J., Mirarab S., Nguyen N., Warnow T., Carpenter E., Matasci N., Ruhfel B.R.. 2014. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 111:E4859-E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki W.P., Clark L.G., Attigala L., Ruiz-Sanchez E., Duvall M.R.. 2015. Evolution of the bamboos (Bambusoideae; Poaceae): a full plastome phylogenomic analysis. BMC Evol. Biol. 15:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang X.G., Mi X.C., Zhou H.L., Li J.W., Chung S.W., Li D.Z., Huang W.C., Jin W.T., Li Z.Y., Huang L.Q., Jin X.H.. 2016. Biogeographical diversification of mainland Asian Dendrobium (Orchidaceae) and its implications for the historical dynamics of evergreen broad-leaved forests. J. Biogeogr. 43:1310-1323. [Google Scholar]
- Yang G.Q., Chen Y.M., Wang J.P., Guo C., Zhao L., Wang X.Y., Guo Y., Li L., Li D.Z., Guo Z.H.. 2016. Development of a universal and simplified ddRAD library preparation approach for SNP discovery and genotyping in angiosperm plants. Plant Methods 12:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.M., Zhang Y.X., Yang J.B., Li D.Z.. 2013. The monophyly of Chimonocalamus and conflicting gene trees in Arundinarieae (Poaceae: Bambusoideae) inferred from four plastid and two nuclear markers. Mol. Phylogenet. Evol. 68:340-356. [DOI] [PubMed] [Google Scholar]
- Ye X.Y., Ma P.F., Yang G.Q., Guo C., Zhang Y.X., Chen Y.M., Guo Z.H., Li D.Z.. 2019. Rapid diversification of alpine bamboos associated with the uplift of the Hengduan Mountains. J. Biogeogr. 46:2678–2689. [Google Scholar]
- Yu X.Q., Gao L.M., Soltis D.E., Soltis P.S., Yang J.B., Fang L., Yang S.X., Li D.Z.. 2017. Insights into the historical assembly of East Asian subtropical evergreen broadleaved forests revealed by the temporal history of the tea family. New Phytol. 215:1235-1248. [DOI] [PubMed] [Google Scholar]
- Zeng C.X., Zhang Y.X., Triplett J.K., Yang J.B., Li D.Z.. 2010. Large multi-locus plastid phylogeny of the tribe Arundinarieae (Poaceae: Bambusoideae) reveals ten major lineages and low rate of molecular divergence. Mol. Phylogenet. Evol. 56:821-839. [DOI] [PubMed] [Google Scholar]
- Zeng L., Zhang Q., Sun R., Kong H., Zhang N., Ma H.. 2014. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat. Commun. 5:4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.N., Zhang X.Z., Zhang Y.X., Zeng C.X., Ma P.F., Zhao L., Guo Z.H., Li D.Z.. 2014. Identification of putative orthologous genes for the phylogenetic reconstruction of temperate woody bamboos (Poaceae: Bambusoideae). Mol. Ecol. Resour. 14:988-999. [DOI] [PubMed] [Google Scholar]
- Zhang L.N., Ma P.F., Zhang Y.X., Zeng C.X., Zhao L., Li D.Z.. 2019. Using nuclear loci and allelic variation to disentangle the phylogeny of Phyllostachys (Poaceae, Bambusoideae). Mol. Phylogenet. Evol. 137:222-235. [DOI] [PubMed] [Google Scholar]
- Zhang X.Z., Zeng C.X., Ma P.F., Haevermans T., Zhang Y.X., Zhang L.N., Guo Z.H., Li D.Z.. 2016. Multi-locus plastid phylogenetic biogeography supports the Asian hypothesis of the temperate woody bamboos (Poaceae: Bambusoideae). Mol. Phylogenet. Evol. 96:118-129. [DOI] [PubMed] [Google Scholar]
- Zhang Y.C. 2013. Estimation of nuclear DNA content of woody bamboos and related researches. The University of Chinese Academy of Sciences and Kunming Institute of Botany, Chinese Academy of Sciences, Kunming. Master’s Thesis. [Google Scholar]
- Zhang Y.X., Guo C., Li, D.Z.. 2020. A new subtribal classification of Arundinarieae (Poaceae, Bambusoideae) with the description of a new genus. Plant Divers. 42:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.X., Ma P.F., Havermans T., Vorontsova M.S., Zhang T., Nanjarisoa O.P., Li D.Z.. 2017. In search of the phylogenetic affinity of the temperate woody bamboos from Madagascar, with description of a new species (Bambusoideae, Poaceae). J. Syst. Evol. 55:453–465. [Google Scholar]
- Zhang Y.X., Ma P.F., Li D.Z.. 2018. A new genus of temperate woody bamboos (Poaceae, Bambusoideae, Arundinarieae) from a limestone montane area of China. PhytoKeys 109:67-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.X., Zeng C.X., Li D.Z.. 2012. Complex evolution in Arundinarieae (Poaceae: Bambusoideae): incongruence between plastid and nuclear GBSSI gene phylogenies. Mol. Phylogenet. Evol. 63:777-797. [DOI] [PubMed] [Google Scholar]
- Zhao L., Li X., Zhang N., Zhang S.D., Yi T.S., Ma H., Guo Z.H., Li D.Z.. 2016. Phylogenomic analyses of large-scale nuclear genes provide new insights into the evolutionary relationships within the rosids. Mol. Phylogenet. Evol. 105:166-176. [DOI] [PubMed] [Google Scholar]