

Abstract
Amongst the pleotropic roles of transforming growth factor β (TGFβ) signaling in cancer, its impact on genomic stability is least understood. Inhibition of TGFβ signaling increases use of alternative end-joining (alt-EJ), an error-prone DNA repair process that typically functions as a ‘backup’ pathway if double strand break repair by homologous recombination or non-homologous end-joining is compromised. However, the consequences of this functional relationship on therapeutic vulnerability in human cancer remain unknown. Here, we show that TGFβ broadly controls the DNA damage response and that it suppresses alt-EJ genes that are associated with genomic instability. Mechanistically-based TGFβ and alt-EJ gene expression signatures were anti-correlated in glioblastoma, squamous cell lung cancer, and serous ovarian cancer. Consistent with error-prone repair, more of the genome was altered in tumors classified as low TGFβ and high alt-EJ, and the corresponding patients had better outcomes. Pan-cancer analysis of solid neoplasms revealed that alt-EJ genes are coordinately expressed and anti-correlated with TGFβ competency in 16 of 17 cancer types tested. Moreover, regardless of cancer type, tumors classified as low TGFβ and high alt-EJ were characterized by an insertion-deletion mutation signature containing short microhomologies and were more sensitive to genotoxic therapy. Collectively, experimental studies revealed that loss or inhibition of TGFβ signaling compromises the DNA damage response, resulting in ineffective repair by alt-EJ. Translation of this mechanistic relationship into gene expression signatures identified a robust anti-correlation that predicts response to genotoxic therapies, thereby expanding the potential therapeutic scope of TGFβ biology.
One Sentence Summary:
The impact of TGFβ signaling on DNA repair competency is observed in pan-cancer analysis of survival after treatments that cause DNA damage.
Introduction
The cytokine transforming growth factor β (TGFβ) is considered a canonical tumor suppressor that exerts profound control upon epithelial proliferation. Although cancer must evade TGFβ growth regulation, complete loss of TGFβ signaling competency is not universal because autocrine TGFβ promotes malignant phenotypes, such as invasion, and paracrine TGFβ has pro-tumorigenic effects on the tumor microenvironment (reviewed in (1)). Some cancers, including colorectal cancer, pancreatic cancer, and head and neck squamous cell carcinoma (HNSC), exhibit genetic alterations of key pathway components, including somatic mutations of SMAD4 (mothers against decapentaplegic family member 4) and TGFBR2 (transforming growth factor beta receptor 2) (2). The conversion from tumor suppressor to tumor promoter is one of the paradoxes that have complicated the targeting of TGFβ in cancer therapy. A clearer understanding of its detrimental effects on cancer biology could provide an actionable rationale for TGFβ inhibition in cancer therapy.
One aspect of TGFβ biology that remains poorly understood is its role in genomic stability, which was initially recognized more than 25 years ago (3). Over the last decade it has been established that TGFβ regulates the expression or function of key DNA repair proteins, including ATM (ataxia telangiectasia mutated), BRCA1 (breast cancer 1 gene), and LIG4 (DNA ligase 4), which are necessary for maintenance of genomic integrity (reviewed in (4)). Faulty DNA repair is a hallmark of cancer, and specific repair defects can provide the basis for response to precise therapies (5). Moreover, key DNA repair effectors are attractive targets for drug development, which can be deployed in cancers with specific vulnerabilities, as evidenced by the success of poly(ADP-ribose) polymerase (PARP) inhibitors in BRCA1/2 mutant tumors (6).
Human papilloma virus (HPV) positive HNSC exhibits striking sensitivity to standard of care genotoxic therapy with cisplatin and radiotherapy (7). We demonstrated that loss of TGFβ competency in HPV-positive cancer in turn compromises the canonical DNA double strand break (DSB) repair pathways, homologous recombination repair (HR) and non-homologous end-joining (NHEJ) (8). Pharmaceutical TGFβ inhibition in HPV-negative cancer cells replicates the DNA repair defects exhibited by HPV-positive cancer cells and tumors. When classical DSB repair is defective, alternative end-joining (alt-EJ, also called microhomology-mediated end-joining) is thought to take over as a backup repair (9, 10). In support of this, we demonstrated that alt-EJ is increased in HPV-positive cells, and in HPV-negative cells in which TGFβ signaling is blocked (8). DSB repair by alt-EJ is highly error-prone because it generates frequent genomic deletions and insertions with microhomologies at processed ends (11, 12). Hence cells using alt-EJ are more sensitive to genotoxic chemotherapy or radiotherapy (8). Because radiosensitivity is increased when TGFβ signaling is inhibited (8, 13–15), defective TGFβ signaling may present a specific therapeutic opportunity.
The view that alt-EJ provides a survival mechanism in the face of classical DNA repair failure has spawned efforts to target its effector, polymerase θ (Pol θ, encoded by POLQ), an approach supported by the high POLQ expression in HR-deficient breast and ovarian tumors (16). More recently, experiments using alt-EJ and HR competition repair substrates demonstrated that alt-EJ can be used to repair 10–20% of DSB even in mammalian cells where both HR and NHEJ are available (17) and provide evidence that Pol θ deletion compromises cell survival even when canonical DSB repair pathways are intact (18). Thus alt-EJ function may be more complex than a simple back-up system (9), especially if alt-EJ is used as a fail-safe primarily by cancer cells, which would provide a considerable therapeutic opportunity if it could be identified prospectively.
Because TGFβ responsiveness is modulated by multiple genetic and epigenetic mechanisms and varies widely across human cancers, measuring TGFβ signaling status could be a strategy for selecting the most effective therapy for patients. Informed by this perspective, we identified TGFβ and alt-EJ gene signatures to further examine the consequences of this relationship using The Cancer Genome Atlas (TCGA) data. We determined that these signatures are anti-correlated in 16 of 17 solid malignancies tested, and that tumors exhibiting low TGFβ and high alt-EJ signatures have more mutations, a specific mutational signature, and better patient outcomes in response to genotoxic therapy. This research provides an avenue by which TGFβ impacts response to cancer therapy and a rationale for TGFβ inhibitors to sensitize previously unresponsive cancers to genotoxic therapies.
Results
Inhibition of TGFβ broadly compromises DNA damage response (DDR)
TGFβ affects the DDR by multiple mechanisms. Blocking TGFβ signaling decreases autophosphorylation of ATM protein kinase, which is a major mediator of the DDR (19). ATM autophosphorylation is a robust biomarker of activation of the ATM-centered signaling network that broadly initiates DDR by affecting critical effector signals (20). Inhibiting TGFβ decreases autophosphorylation ATM as well as phosphorylation of key effectors and increases radiosensitivity in breast, brain, lung, and squamous cell cancer cell lines (8, 13–15). In addition, TGFβ affects BRCA1 via suppression of miR-182, which directly degrades BRCA1 mRNA (21). MiR-182 also affects ATM kinase activity via FOXO3 (forkhead box O3) (8). Moreover, TGFβ facilitates NHEJ by increasing LIG4 expression (22).
To characterize TGFβ effects on ATM-dependent phospho-signaling in response to DNA damage, we irradiated TGFβ-competent HPV-negative HNSC cells (SAS cell line) in the presence of LY2157299 (galunisertib), a small molecule inhibitor of TGFβ type I receptor and quantified changes in the phosphoproteome using two well-established targeted mass spectrometry-based assays (23–26). The results show that radiation-induced ATM autophosphorylation at S2996 was blocked by TGFβ inhibition, as was phosphorylation of ATM targets BRCA1 S1524 and NBN S343 (Fig. 1A). Unsupervised clustering of the results of the multiple reaction monitoring (MRM) assays revealed that TGFβ inhibition compromised the radiation-induced phosphorylation of a block of proteins (Fig. 1B; table S1 in data file S1). Phosphorylation of three proteins, TP53 (tumor protein 53) at S315, NBN (nibrin) at S432, and UBE2T (ubiquitin-conjugating enzyme E2 T) at S184, was increased by TGFβ inhibition in the absence of radiation, and decreased TP53 phosphorylation was observed in irradiated cells (Fig. 1B). Where there was overlap of the two independent MRM-based assays, comparable results were observed (fig. S1A, table S2 in data file S1).
Fig. 1. Blockade of TGFβ signaling disrupts DDR and increases alt-EJ.

(A) SAS cells were treated with radiation (5 Gy, 1 hour), LY2157299 (2 μM, 24 hour), or combination, and cell lysates were prepared for proteomic analysis. Protein phosphorylation analyses was performed using two targeted, multiple reaction monitoring mass spectrometry (MRM-MS)-based assays (tables S1-S2). Representative phospho-analytes are plotted in the figure, including ATM phosphorylation at Ser2996, NBN phosphorylation at Ser343, and BRCA1 phosphorylation at Ser1524 quantified using the two independent assay panels that gave comparable results as shown in lower left and right panels. Precise quantification of the phospho-analytes relative to stable isotope labeled spiked-in standards are shown as peak area ratios. Fold changes of these proteins between irradiated cells and LY2157299 pretreated and irradiated cells are indicated. Data shown as means ± SEM of n = 3. Experiment was performed once. (B) Protein expression and phosphorylation in SAS cells treated with IR (5 Gy), LY2157299, or combination of both. Unsupervised clustering of Z-score data is shown as a heatmap. Representative proteins that are reciprocally regulated are indicated in red box and protein phosphorylations increased by LY2157299 are shown in blue box. (C) The frequency of HR measured by flow cytometry using reporter plasmid-transfected SAS cells that expressed miR-182, anti-miR-182, or scramble miRNA, and were treated with or without TGFβ receptor inhibitor LY2157299. (D) Alt-EJ repair frequency measured by flow cytometry of EJ2GFP reporter transfected SAS cells expressing anti-miR-182 or scrambled anti-miR and treated with or without LY2157299. (E) DNA repair efficiency measured by the PFGE assay after irradiation (IR, 20 Gy) of SAS cells pre-treated with DNA-dependent protein kinase inhibitor KU57788, TGFβ inhibitor LY364947, or both. Percentages of residual DNA damage at the indicated time points after IR are shown. Statistical significance is indicated according to Student’s t-test: *, P < 0.05; **, P < 0.01; ***, P < 0.005; n.s., P > 0.05.
HR and NHEJ are thought to be backed up by alt-EJ in that failure of either process can increase deployment of this alternative repair pathway (27). Thus, increased use of alt-EJ could be considered a consequence of defective HR. If so, restoration of BRCA1 should rescue HR and suppress alt-EJ. To test this, we antagonized miR-182 in TGFβ competent SAS cells, and measured the effect of TGFβ inhibition on HR and alt-EJ function by flow cytometry (fig. S1B) using pathway-specific reporters for HR (pDRGFP) and distal end-joining by either-NHEJ or alt-EJ (pimEJ5GFP and EJ2GFP, respectively) (28). Cells expressing the miR-182 antagomir were HR-competent when TGFβ was inhibited (Fig. 1C), consistent with the necessity of BRCA1 for HR competency; however, alt-EJ repair remained increased upon TGFβ inhibition (Fig. 1D). This observation demonstrates that HR deficiency is not required for alt-EJ to increase when TGFβ is inhibited.
We next sought to test whether TGFβ inhibition would increase alt-EJ in the context of NHEJ inhibition. Here we used pulsed-field gel electrophoresis to measure residual DSBs in SAS cells after irradiation with 20 Gy (fig. S1C). NHEJ was blocked by treating cells with the DNA-protein kinase inhibitor KU57788 for 1 hour before irradiation, and TGFβ signaling was blocked for 24 hours with LY364947, a small molecule inhibitor of TGFβ type I receptor kinase similar to LY2157299 (29). As expected, KU57788 inhibited repair of radiation-induced DSBs, which was partially rescued by pretreatment with LY364947 (Fig. 1E). This observation supports the idea that TGFβ inhibition promotes an alternative process of repair. Together, these data show that TGFβ signaling is essential for both the fundamental molecular mechanisms of DNA repair, as evidenced by ATM kinase activity, and the functional consequences such as DNA repair pathway choice and resolution of DSBs.
TGFβ regulates expression of DDR genes
To further investigate TGFβ impact on DDR, we evaluated the expression of DNA repair-associated genes using the NanoString DDR gene panel. Treatment of SAS cells for 24 hours with TGFβ plus or minus its inhibitor LY2157299 revealed striking reciprocal regulation of 180 DDR genes (Fig. 2A). According to KEGG pathway analyses, expression of genes implicated in HR and NHEJ was increased by TGFβ and reduced by its inhibition (table S3 in data file S1). Consistent with prior literature (30, 31), CDKN1A was strongly induced by TGFβ and blocked by LY2157299 (fig. S1D), even though SAS cells, like most cancer cells (32), are insensitive to TGFβ-mediated cell cycle control (8). BRCA1 expression was increased by TGFβ and suppressed by LY2157299, as were ABL1 (ABL proto-oncogene 1, non-receptor tyrosine kinase) and POLD4 (DNA polymerase delta 4, accessory subunit; table S4 in data file S1). In contrast, TGFβ decreased and LY2157299 increased expression of LIG1 (DNA ligase 1), PARP1, and POLQ (Fig. 2B), which are key genes involved in alt-EJ (33).
Fig. 2. TGFβ signaling regulates DDR gene expression.

(A) Gene expression measured in SAS cells that were treated with TGFβ, LY2157299, or combination of both for 24 hours using the NanoString panel. Unsupervised clustering of Z-score gene expression values is shown as a heatmap. Alt-EJ genes LIG1, PARP1, and POLQ are indicated. (B) Percentage of TGFβ-induced gene expression change versus LY2157299-induced gene expression, normalized to control, for SAS cells. Genes reciprocally regulated by TGFβ or LY2157299, including ABL1, CCND2, CDKN1A, LIG1, PARP1, POLD4, and POLQ, are indicated by red dots. (C) Percentage of TGFβ-induced gene expression in SAS cells overexpressing (OE) anti-miR-182 versus LY2157299-induced gene expression, normalized to control samples. Genes reciprocally regulated by TGFβ or LY2157299 independent of miR-182 are indicated by red dots. (D-E) qRT-PCR of POLQ, PARP1, and LIG1 in SAS cells treated with TGFβ (D) or LY2157299 (E) for 72 hours, normalized to untreated control. (F) Changes in gene expression in U251 GBM cells treated with 2 μM LY2157299 for 24 hours as measured using the NanoString panel. (G-H) Gene expression of PARP1, LIG1, and POLQ measured by qRT-PCR in U251 cells treated with TGFβ (G) or LY2157299 (H) for 72 hours. (I) Alt-EJ repair event frequency measured by EJ2GFP reporter in U251 cells in which TGFβ signaling was inhibited with either LY2157299 or LY364947. (J) Alt-EJ repair event frequency measured by EJ2GFP reporter in U251 cells transfected with anti-miR-182 or scramble anti-miR and treated with or without LY2157299, normalized to untreated control. Two-tailed Student’s t-test; *, P < 0.05; **, P < 0.01; ***, P < 0.005.
Given that miR-182 was essential for TGFβ-mediated control of HR and microRNAs can target hundreds of genes, we next determined whether miR-182 was involved in TGFβ-regulated DDR gene expression. SAS cells in which miR-182 was overexpressed or antagonized were treated as above and then analyzed using the NanoString panel (Fig. 2C). TGFβ-mediated changes in BRCA1 expression were miR-182-dependent, as previously reported (21), as were its effects on MRE11A (meiotic recombination 11 homolog A), MYD88 (myeloid differentiation primary response 88) and PARP3 expression (table S5 in data file S1). In contrast, changes in CCND2 (cyclin D2), CDKN1A (cyclin dependent kinase inhibitor 1A) and POLD4 expression were miR-182-independent, consistent with the presence of SMAD binding elements in these genes (34). Notably, expression of the alt-EJ genes, LIG1 (DNA Ligase 1), PARP1, and POLQ, was found to be miR-182-independent (Fig. 2B,C). Together these data confirm that TGFβ has a broad impact on DDR via expression and molecular regulation of many genes and via ATM kinase activity (8, 21). They also extend the range of TGFβ influence on expression of DDR-associated genes and show that this occurs through both miR-182-dependent and -independent mechanisms. Because neither alt-EJ execution (Fig. 1C, 1D) nor expression of critical genes in this process (Fig. 2B, 2C) are miR-182-dependent, these data mechanistically separate the effects of TGFβ on HR from those on alt-EJ.
LIG1and POLQ do not contain recognizable SMAD-binding elements (34), yet their expression was decreased upon exposure to TGFβ. To confirm this effect, we conducted quantitative gene expression measurements as a function of duration of TGFβ stimulation or small molecule receptor kinase inhibition in SAS cells. The expression of each of the three genes was reciprocally suppressed by TGFβ signaling (Fig. 2D) and increased by its inhibition (Fig. 2E). Although the early (6 hours) regulation of POLQ may be consistent with direct SMAD-mediated transcriptional regulation, the delayed impact on LIG1 and PARP1 suggests an indirect effect (35).
The above observations led us to hypothesize that TGFβ suppression of alt-EJ gene expression is a distinct mechanism that influences differential DNA repair pathway use. To test whether this biology is broadly observed beyond HNSC, we considered glioblastoma (GBM) because high TGFβ ligand and receptor expression correlate with poorer survival in this cancer type, and because TGFβ inhibition enhances radiosensitivity in GBM cell lines and primary tumor explants (14, 36). Consistent with this, GBM patients in TCGA with low TGFB1 (transforming growth factor beta1) expression had significantly better overall survival (OS; log-rank test, P = 0.045) and progression-free survival (PFS; log-rank test, P = 0.034) compared to those with high TGFB1 expression. We first evaluated the U251 human GBM cell line using the NanoString panel (table S6 in data file S1). As observed in HNSC SAS cells, TGFβ inhibition increased expression of the key alt-EJ genes, LIG1, PARP1, and POLQ, in GBM cells (Fig. 2F). These results were validated by quantitative expression assays in response to a time course of TGFβ treatment (Fig. 2G) and TGFβ signaling blockade with LY2157299 (Fig. 2H). As in HNSC cells, expression of LIG1, PARP1, and POLQ was decreased upon TGFβ treatment and increased when TGFβ signaling was blocked. We further established U251 reporter cells (EJ2GFP) to evaluate alt-EJ repair. Consistent with SAS data, LY2157299 and LY364947 markedly increased alt-EJ events (Fig. 2I), effects that were again independent of miR-182 status (Fig. 2J). Data file S2 contains primary data for experimental data in Figs. 1 and 2.
To interrogate more extensively the interplay between TGFβ and alt-EJ, we curated a 36-gene alt-EJ competency signature based on the literature and results of DDR-gene knockdown screen using the EJ2GFP reporter (11, 33). This signature was evaluated in concert with a previously described (8) 50-gene set that is induced by chronic TGFβ stimulation (Fig. 3A). There were no known targets of TGFβ in the alt-EJ signature gene list, nor vice versa. Unsupervised clustering of HNSC TCGA data using the chronic TGFβ signature had previously shown HPV-positive cancers to be TGFβ unresponsive (8). Here we found that they are also characterized by high alt-EJ gene expression (Fig. 3B). Given that HPV-positive cancers clustered with low expression of TGFβ target genes and high expression of alt-EJ genes, we conducted single specimen gene set enrichment analysis (ssGSEA) to determine the signature correspondence across TCGA HNSC tumors (Fig. 3C). Consistent with the biology described above, TGFβ and alt-EJ signatures were negatively correlated (Pearson’s correlation coefficient (PCC) = −0.4, P < 0.00001; fig. S2A). The negative correlation remained after removing HPV-positive cancers, indicating that the relationship is also present in HPV-negative cancers (fig. S2B). We next used ssGSEA scores of both signatures to examine unsupervised clustering of GBM TCGA microarray data. Patients with GBM clustered into two major groups that differed in their alt-EJ signature scores (Fig. 3D). This signature was negatively correlated with the TGFβ signature (PCC = −0.35, P < 0.00001). Consistent with the biology observed in cell lines, these analyses reveal a reciprocal relationship between TGFβ competency and alt-EJ gene expression in two human cancer types.
Fig. 3. TGFβ and alt-EJ gene expression signatures are anti-correlated in HNSC and GBM.
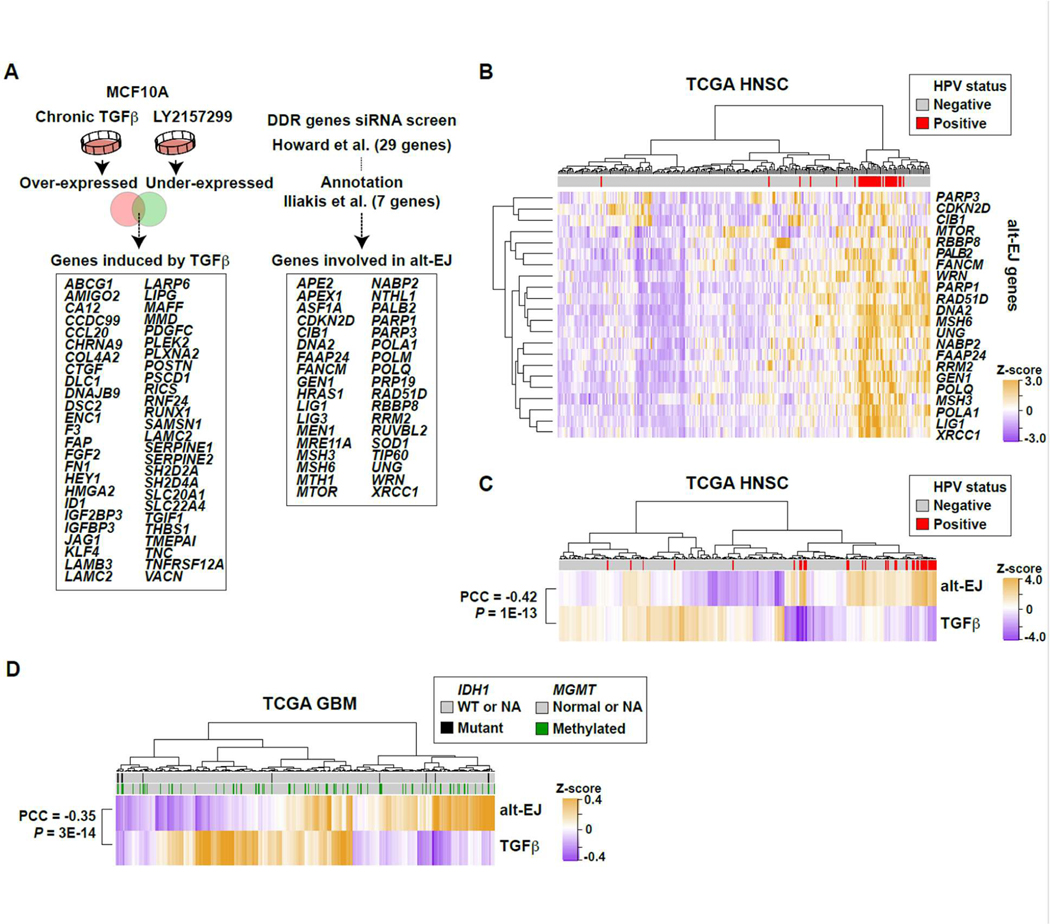
(A) Schematic illustration of compiled signatures for TGFβ-induced and alt-EJ-linked genes. The TGFβ signature was established from MCF10A cells that were treated with TGFβ or LY364947 for seven days. The 36 gene alt-EJ signature was curated from the literature (11, 33). (33)(B) Unsupervised clustering of TCGA HNSC primary tumors based on the expression profiles of all genes included in the alt-EJ signature. The dataset included 243 HPV-negative and 36 HPV-positive (red bars) cases. The HPV-positive ones were clustered by high expression of alt-EJ genes. (C) Unsupervised clustering of HNSC based on the ssGSEA scores of the alt-EJ or TGFβ signatures. HPV positivity indicated in red. The two signatures are significantly anti-correlated (PCC = −0.42, P < 0.00001). (D) Heatmap based on unsupervised clustering of ssGSEA scores for alt-EJ and TGFβ signatures in the TCGA GBM microarray dataset; IDH1 mutation (black) and MGMT methylation status (green) are indicated. The two signatures are significantly anti-correlated (PCC = −0.35, P < 0.00001).
Low TGFβ/high alt-EJ signature predicts better outcomes after genotoxic therapy
Compared to canonical NHEJ, alt-EJ repair is both more error-prone, resulting in more genome alterations, and less efficient, such that cells using this pathway have greater sensitivity to DSB-inducing agents (11). Consistent with this, Tgfb1-null murine cells are genomically unstable (3), as are human cells in which TGFβ signaling is inhibited (37). Loss of TGFβ signaling, whether through HPV infection, ligand neutralizing antibodies, or TGFβ receptor kinase inhibitors, increases sensitivity to DSB induced by ionizing radiation and platinum drugs (8, 13–15). Because genotoxic therapy is standard-of-care (SOC) for many cancers, we postulated that patients with tumors characterized by low TGFβ and high alt-EJ signatures would have more genome alterations and be more responsive to genotoxic therapy than those with high TGFβ and low alt-EJ signatures.
To classify patients according to their TGFβ and alt-EJ transcriptional profiles, we calculated a βAlt score as the difference between the alt-EJ and TGFβ normalized signature value in primary, untreated tumors, where a high βAlt score represents specimens in which expression of TGFβ target genes is low and expression of alt-EJ genes is high. We then tested the association between βAlt and the fraction of the tumor genome altered, defined as the percentage of altered copy number regions out of all measured regions, and between βAlt and OS and PFS or disease-free survival (DFS) for patients who were treated with genotoxic agents. We compared tumors in the upper and lower βAlt tertiles using TCGA pan-cancer clinical data resource (TCGA-CDR) (38).
To analyze the signatures in GBM, we first excluded tumors categorized as ‘neural’ because this subtype is now thought to represent samples contaminated by normal brain tissue (39), resulting in 442 cases. Again, TGFβ and alt-EJ signatures were significantly anti-correlated in GBM (PCC = −0.35, P < 0.00001; Fig. 4A). Although GBMs generally exhibit low somatic mutation burden (40), the fraction of genome altered was significantly associated with βAlt (Mann-Whitney test, P < 0.001; Fig. 4B). SOC treatment for newly diagnosed GBM consists of surgery, radiotherapy (RT) and chemotherapy (ChT) with temozolomide (41). To evaluate patient survival, datasets were curated to eliminate specimens from patients who were not treated with both RT and ChT, leaving a total of 274 cases. While OS (log-rank test, P = 0.096, Fig. 4C) was not different, PFS of patients with a high βAlt score was greater than those with a low score (log-rank test, P = 0.031; Fig. 4D). Hypermethylation of the MGMT promoter (40) and mutation of IDH1/2 (42) are known prognostic biomarkers in GBM, but few TCGA specimens had IDH1/2 mutations (n = 8). We conducted a multivariate Cox regression analysis that included MGMT status; the Cox regression coefficient (β) corresponding to the βAlt score was associated with OS (β = −0.36, P = 0.026) and PFS (β = −0.42, P= 0.006), indicating that TGFβ and alt-EJ signatures are independent of MGMT status. In a multivariate analysis that also included age, the βAlt score maintained significant association with PFS (β = −0.35, P = 0.027), although not with OS (β = −0.26, P = 0.12; table S7 in data file S1).
Fig. 4. TGFβ and alt-EJ signature status associated with differential clinical outcomes after genotoxic therapy.

(A) Negative correlation of TGFβ and alt-EJ scores of TCGA GBM cases excluding the neural samples (PCC = −0.35, P < 0.00001); orange dots indicate low βAlt score tertile and blue dots indicate high βAlt score tertile, here and in E and I. (B) Fraction of genomic alterations as a function of βAlt score tertiles (Mann Whitney test P < 0.0001). (C-D) Kaplan-Meier graphs corresponding to the (C) OS (P = 0.096) or (D) PFS (P = 0.031) for subpopulations of patients with GBM treated with chemoradiation classified by βAlt score tertiles as shown in panel A. (E) Negative correlation of TGFβ and alt-EJ scores of TCGA LUSC cases (PCC = −0.43, P < 0.00001). (F) Fraction of genomic alterations as a function of βAlt score tertiles (Mann Whitney test P < 0.0001). (G-H) Kaplan-Meier graphs corresponding to the (G) OS (P = 0.05) or (H) PFS (P = 0.02) for subpopulations of patients with LUSC treated with chemotherapy and/or radiotherapy classified by βalt score tertiles as shown in panel E. (I) OVCA tumors exhibit a negative correlation of the two signatures (PCC = −0.32, P < 0.00001). (J) Fraction of genomic alterations as a function of βAlt score tertiles for OVCA tumors (Mann Whitney test, P < 0.001). (K-L) Kaplan-Meier graphs corresponding to the (K) OS (P = 0.004) or (L) PFS (P = 0.0027) for TCGA patients with OVCA in subpopulations classified by βAlt score tertiles as shown in panel I.
Patients with lung squamous cell carcinoma (LUSC) are generally treated with surgery, RT, and/or ChT depending on tumor stage and lung function (43). TCGA specimens of LUSC (n = 502) also exhibited significant (PCC = −0.43, P < 0.00001; Fig. 4E) anti-correlation of TGFβ and alt-EJ signatures. The fraction of the genome altered was also significantly greater (Mann-Whitney test, P < 0.00001; Fig. 4F) in patients with high βAlt scores. To assess outcomes following ChT and/or RT, patients in whose treatment variables were null were excluded, as were stage I patients because they are usually treated with surgery alone. Based on SOC, the remaining patients (n = 231) were likely to have been treated with ChT, RT or both. The OS did not reach significance in patients with high βAlt scores (log-rank test P = 0.05; Fig. 4G), but PFS of these patients was significantly increased (log-rank test P = 0.025; Fig. 4H). In a multivariate Cox regression analysis including patient age and tumor stage, the βAlt score was significantly associated with OS (β = −0.62, P = 0.035) and PFS (β = −0.86, P = 0.010; table S7 in data file S1).
The anti-correlation of TGFβ and alt-EJ signatures in ovarian cancer (OVCA; n = 541) was also significant (PCC = −0.32, P < 0.00001; Fig. 4I) and the fraction of altered genome was greater in those with higher βAlt scores (Mann-Whitney test P < 0.00001, Fig. 4J). Patients with stage II-IV serous ovarian cancer in the TCGA data set were treated with surgical resection followed by systemic treatment with platinum and taxane genotoxic agents (44). Compared to those with low βAlt scores, both OS (log-rank test P = 0.004, Fig. 4K) and PFS (log-rank test P = 0.003; Fig. 4L) were significantly increased in patients with tumors characterized by high βAlt. As above, in a multivariate Cox regression analysis including patient age and tumor stage, the βAlt score maintained significant association with OS (β = −0.33, P = 0.035) and PFS (β = −0.43, P = 0.002; table S7 in data file S1). To further evaluate signature associations with therapeutic outcome, we analyzed an expression dataset from a randomized clinical trial that evaluated response to carboplatin in OVCA (CTCR-OV01, GSE15622; (45)). TGFβ and alt-EJ signatures were significantly anti-correlated (PCC= −0.83, P = 0.0001; fig. S3A) and in 15 cases treated with carboplatin, tumors that were sensitive to this drug had significantly higher alt-EJ signature scores (Mann-Whitney test, P < 0.01) and βAlt scores (P < 0.05) than those that were resistant (fig. S3B).
These patient data show that, despite different tissue origins and treatment regimens, a high βAlt score is consistently associated with better outcome for cancer patients treated with genotoxic agents. This provides evidence that the mechanisms by which TGFβ impacts alt-EJ repair have biological and clinical consequences, including associations with genomic alterations and response to cancer therapy.
TGFβ and alt-EJ genes are anti-correlated across solid cancer
The coordinated expression of alt-EJ genes in HNSC was unanticipated because these genes have not previously been identified as a network or pathway. To further evaluate this observation, we conducted consensus clustering of both gene sets across all solid cancers in TCGA (n = 10,848; Fig. 5A). As expected, subsets of TGFβ target genes clustered together, which is likely due to the pleiotropic actions of TGFβ in both cancer cells and the tumor microenvironment. A block containing 27 of the alt-EJ signature genes indicates that they are highly co-regulated.
Fig. 5. Pan-cancer analysis shows that TGFβ and alt-EJ gene expression are anti-correlated and associated with genomic alterations.

(A) Gene co-expression analyses for TGFβ and alt-EJ signature genes across solid tumors in TCGA database. Major clusters containing most of the alt-EJ and TGFβ signature genes are indicated: cluster A contains 27/32 (85%) of the alt-EJ signature genes, and cluster B contains 32/33 (97%) of the TGFβ signature genes. (B) Forest plot showing the PCC and 95% confidence interval (CI) in each cancer type (numbers of tumors included in each setting are indicated). A non-significant, negative PCC corresponds to pancreatic adenocarcinoma (PAAD, gray bar). (C) Forest plot showing the PCC and 95% CI for cell lines of each cancer type (numbers of each are indicated). Non-significant PCCs are indicated by gray bars. (D) Heatmap showing the PCC for each indel (ID) pattern versus the TGFβ and alt-EJ signatures. The signatures are reciprocally associated with ID6, ID10, and ID13.
Next, we asked whether specific cancers were driving this anti-correlation. Among the 17 cancer types analyzed, the TGFβ and alt-EJ signatures were anti-correlated in 16 (Fig. 5B). These data indicate that this relationship is broadly present in human solid cancers. Pancreatic adenocarcinoma (PAAD, n = 177) was the only cancer type in which the signatures were not significantly anti-correlated (PCC = −0.08).
TGFβ signaling in the tumor microenvironment affects diverse responses within and between tumor, immune, and stromal cells, any of which may contribute to the relationship between TGFβ and alt-EJ. To assess this, we used immune and stromal cell inference (46) to test the association of these factors with TGFβ/alt-EJ signatures across different cancer types. There were no specific associations of the signatures with inferred immune and stromal cell content (fig. S4), lending credence to the hypothesis that TGFβ suppression of an alt-EJ program is a cancer-cell autonomous feature.
To assess the cancer cell autonomy of this relationship further, we analyzed the TGFβ and alt-EJ signatures across multiple cancer cell lines (47). As observed in primary tumors, the overall negative correlation was strongly maintained (n = 966, PCC = −0.35, P < 0.00001) and negative correlations were observed in many cancer cell types (Fig. 5C), which included cell lines from GBM (n = 35, PCC = −0.43, P < 0.01), HNSC (n = 42, PCC = −0.57, P < 0.001), and LUSC (n = 15, PCC = −0.68, P < 0.001).
Pan-cancer TGFβ and alt-EJ signatures associate with specific microhomology indel mutation and survival after genotoxic therapy
Because the TGFβ and alt-EJ signatures frequently showed a negative correlation in solid cancers, we sought evidence of functional consequences. The alt-EJ process is inherently mutagenic because it uses sequence microhomology to facilitate DSB ligation (48, 49). Hence, we predicted that the TGFβ and alt-EJ signature relationship would be associated with the frequency of specific genomic alterations across cancers. To evaluate this, signature scores in tumors were assessed for their association with the somatic frequencies of small insertions and deletions, and with silent non-coding mutations of 11 malignancies cancers in which analysis of these mutations have been published (50). The alt-EJ signature was positively correlated with higher frequencies of these mutation types in several cancer settings; 26 positive correlations were identified with false discovery rate < 5%, while no negative correlations reached significance (fig. S5A). In contrast, the TGFβ signature was negatively correlated with these types of mutations in 15 instances, and only four positive correlations were detected. The alt-EJ signature was positively correlated with higher frequencies of these mutation types in most cancers (fig. S5B). The average distribution of the observed PCC for alt-EJ was significantly higher than 0 (t-test, P < 0.00001). In contrast, the TGFβ signature was negatively correlated with these types of mutations in several cancer settings (PCC average < 0, t-test, P = 0.009). Consistent with these results, the frequency of distinct types of somatic structural variants, including chromosomal translocations, was also positively correlated with the alt-EJ signature and negatively with the TGFβ signature (fig. S5C).
These results led us to consider a recent comprehensive analysis of mutational signatures of cancer genomes in which we focused on insertion-deletion (indel, ID) signatures (51). Indels designated ID6 and ID8 are characterized by >5-base pair deletions, but ID6 contains overlapping microhomology at deletion boundaries with a mode of 2-base pairs. This signature pattern is consistent with end-joining by either NHEJ or alt-EJ. Notably, a similar pattern can be experimentally induced in cells in a PolΘ-dependent manner (52). Therefore, we matched samples in the cancer genome study (51) to our TCGA analyses of TGFβ and alt-EJ expression signatures, and evaluated correlations for each gene set with ID pattern probabilities (table S8 in data file S1). The heatmap of PCCs shows that the gene signatures are differentially associated with ID patterns (Fig. 5D). ID6, which exhibited microhomology at deletion boundaries (51), was positively correlated with the alt-EJ signature and negatively correlated with the TGFβ signature. In contrast, ID10 and ID13, which are linked to a different DNA damage process (51), showed the opposite correlation with TGFβ and alt-EJ. The reciprocal correlation of alt-EJ and TGFβ with ID6 indicates that it is a genomic scar of alt-EJ, which further endorses their functional relationship.
Given the evidence that the biology represented in these signatures did not depend on cancer type, we conducted OS pan-cancer analysis for patients who were treated with RT (n = 1,737). The anti-correlation of signatures was comparable to the full set of specimens (Fig. 6A), which represented 17 malignancies (table S9 in data file S1). Patients with high βAlt scores fared significantly better (P < 0.0001, hazard ratio = 0.56, 95% CI 0.43–0.73) than those with low βAlt scores (Fig. 6B). Because chemotherapy is not reported in detail in TCGA, we then selected patients for whom SOC would include RT and/or genotoxic ChT, based on cancer type and stage (n = 3,577). The signature anti-correlation was comparable to the full set of specimens (Fig. 6C) and represented 17 malignancies (table S10 in data file S1). Patients with high βAlt scores again showed better survival (P < 0.0001; hazard ratio = 0.60, 95% CI 0.52–0.97; Fig. 6D). Although the βAlt score was significantly anti-correlated with age in all specimens (PCC = −0.040, P = 0.001; Fig. S6), a multivariate Cox regression analysis including age and tumor stage maintained the βAlt score association with OS in patients who had been treated with radiotherapy (β = −0.73, P = 0.003) as well as in patients who had been treated with radiotherapy and/or probably genotoxic chemotherapy (β = −1.11, P < 0.001; table S11 in data file S1). Thus, survival duration in response to genotoxic therapy associates with loss of TGFβ competency and implementation of alt-EJ.
Fig. 6. Pan-cancer βAlt signature status associates with clinical outcomes after genotoxic therapy.

(A) Negative correlation of TGFβ and alt-EJ scores of TCGA cases treated with RT (PCC = −0.234, P < 0.0001). Symbols indicate βAlt low (orange) and βAlt high (blue) tertiles here and in C. (B) Kaplan-Meier graphs corresponding to the OS subpopulations classified by βalt score tertiles as shown in panel A. The HR, 95% CI, cases (n) included in the analysis, and log-rank test P value are shown. (C) Negative correlation of TGFβ and alt-EJ scores of TCGA cases treated with RT and/or ChT (PCC = −0.159, P < 0.0001). (D) Kaplan-Meier graphs corresponding to the OS of subpopulations classified by βAlt score tertiles as shown in panel C. The HR, 95% CI, cases (n) included in the analysis, and log-rank test P value are shown.
Discussion
This study uncovered an unexpected reciprocal relationship between TGFβ signaling and alt-EJ-mediated DNA damage repair that has implications for understanding cancer therapeutic vulnerability. Proteomic, gene expression, and functional evidence from HNSC and GBM cancer cells demonstrates that TGFβ signaling has extensive influence on DNA damage responses. In contrast to the TGFβ-miR-182-BRCA1 axis that regulates HR, TGFβ inhibition increases the use of alt-EJ and expression of key components in this process in a miR-182-independent manner. DNA repair by alt-EJ is both inefficient and error-prone, which leads to more residual damage and cell death that we postulated would be reflected in human cancers by mutational burden and response to genotoxic therapy. To test this, alt-EJ and TGFβ competency signatures were analyzed in GBM, LUSC, and OVCA TCGA data, which revealed signature anti-correlation in each setting and, therefore, associations with both mutational burden and survival outcomes. Expanding on these cancer types, the signatures were frequently anticorrelated in solid cancers, reciprocally linked to a recently reported indel signature, ID6 (51), and associated with survival benefit from genotoxic therapies across all cancer patients studied.
Several insights are gained from this study. First, it substantiates that TGFβ pro-tumorigenic biology includes regulation of genomic instability, which was first reported in mice 25 years ago by Glick et al. (3) and extended to human cells in our initial research (37). Genomic instability due to defective DNA repair is a hallmark of cancers (53). Thus, both maintenance and loss of TGFβ signaling in cancer contribute to carcinogenesis, the former by promoting DDR to enable survival of malignant cells, and the latter by creating the genetic diversity that is a prerequisite for the evolution of cancer.
Second, although TGFβ inhibitors are currently being tested in clinical trials for cancer patients, their use in conjunction with DNA damaging agents has been limited. The key information predicting the utility of such combinations is the experiment of nature provided by HPV-positive HNSC. Analyses in this cancer type provide compelling evidence that loss of TGFβ signaling is not only pro-oncogenic, but also creates a vulnerability that can be exploited for clinical benefit. HPV-positive cancers are more sensitive to cytotoxic agents (54–56), exhibit decreased DNA repair capacity (57, 58), and confer a markedly better prognosis than those identified as HPV-negative (7). Although HPV degrades p53 and retinoblastoma protein, it also blocks TGFβ by multiple means (59). We demonstrated that loss of TGFβ signaling contributes to the striking sensitivity to cisplatin and radiotherapy of HPV-positive HNSC, and that TGFβ inhibition in HPV-negative cancer cells and tumors phenocopies HPV-associated sensitivity (8). We used small molecule inhibitors of the TGFβ type I receptor kinase, which specifically decreases phosphorylation of SMAD2, abrogating activation of the TGFβ canonical pathway. It may be that non-canonical signaling or crosstalk with other major signaling pathways, such as tyrosine kinase, G-protein-coupled, or cytokine receptors that are also mediators of gene regulation could contribute as well (60), which underscores the complex nature of TGFβ signaling. Nonetheless, our analysis of HPV-positive cancers in which TGFβ signaling is lost showed that they are characterized by high expression of an alt-EJ signature that we curated from the literature (11, 33), which indicates a functional network and its relationship with TGFβ.
Third, we identified a specific DNA repair deficit that provides a potential target for individualized cancer therapy (61). A prime example is the use of PARP inhibitors in HR-deficient cancers based on an initially unexpected synthetic lethality (6). Because alt-EJ critically depends on PARP function, TGFβ-unresponsive HPV-positive HNSC cell lines are more sensitive to the PARP inhibitor olaparib than TGFβ-responsive HPV-negative cancer cells; furthermore, TGFβ inhibition increased olaparib sensitivity in TGFβ-responsive cell lines (8). A case in point is GBM, in which there is an incontestable unmet need to identify and overcome mechanisms of resistance to radiation therapy and chemotherapy, which have failed to improve outcomes beyond 16 months median survival (62). Despite chemoradiation, 90% of tumors recur within the original tumor volume (63), an observation that has largely been attributed to efficient DNA damage repair, despite numerous trials of radiation dose escalation and alternative fractionation schedules (64, 65). TGFβ signaling is associated with poor prognosis (66) and the mRNAs of ligands, targets, and receptors are increased in recurrent tumors (36). Indeed, we noted that TCGA GBM patients with low TGFB1 expression had better OS and PFS compared to those with high TGFB1 expression. When adjusted for MGMT status, patients whose tumors were classified as TGFβ low and alt-EJ high had longer survival after radiotherapy and chemotherapy. Together with the PARP dependence of alt-EJ, our data suggest that βAlt scores might be useful to select GBM patients for optimal response to PARP inhibitors, which are currently being evaluated in combination with radiotherapy (67). Expanding the study to all cancers, our analyses found that patients classified as low TGFβ and high alt-EJ exhibit better response to SOC genotoxic therapy. The mechanism-based assays in vitro combined with extensive analysis of human cancer data provide a rationale for using this relationship between TGFβ and DDR to identify patients likely to benefit from specific therapies.
Fourth, we have added to understanding of the mechanisms by which TGFβ signaling affects DDR in cancer cells, through which loss or inhibition of this signaling compromises both HR and NHEJ. TGFβ inhibition impedes radiation-induced autophosphorylation of ATM (19), essential to both HR and NHEJ, as well as reducing expression of LIG4, a critical component of NHEJ (22). More recently, we have demonstrated that TGFβ suppresses miR-182, which in turn suppresses both BRCA1, a necessary component of HR, and FOXO3, which is required for ATM kinase activity (8). Blocking each of these steps via different strategies confirmed that TGFβ signaling was the critical regulator of DDR pathway choice. Indeed, TGFβ promotes resistance to chemotherapy (68) and genomic stability (37), whereas inhibition of TGFβ, either by neutralizing antibodies or small molecule inhibitors of its receptor kinase, increases clonogenic cell death and tumor control in response to ionizing radiation, cisplatin, and temozolomide in multiple preclinical models (8, 13–15, 69–71). Although the current studies used cell lines that are P53 mutant, this prior work showed that TGFβ inhibition radiosensitized T53 wildtype cells. The use of alt-EJ upon loss of TGFβ signaling creates a specific DDR deficit that is exploitable within the current cancer therapy repertoire, although further investigation may also provide additional targets through which to overcome resistance to standard of care genotoxic therapy.
Last, even though earlier studies implicated TGFβ in genomic instability and DNA damage repair fidelity (3, 37), the strong anti-correlation of TGFβ and alt-EJ signatures across multiple cancer types and cancer cell lines was unexpected. When classical DNA repair pathways are compromised, inefficient alternative repair processes are used, which are characterized by a distinct pattern of mutation (12). Alt-EJ acts on the same 5’ to 3’ resected DSBs as HR, but repairs using a synthesis-dependent mechanism that is directed by short tracts of flanking microhomology (18). Our finding of the positive correlation of alt-EJ signature with the ID6 mutation pattern is consistent with this knowledge. Most studies in cells report low frequencies of alt-EJ repair events under standard conditions, and an increase when HR or NHEJ are defective (28, 72). Such data are the basis for the prevailing model in which alt-EJ is a backup mechanism to repair DSB. Here, we were able to functionally dissociate the TGFβ-mediated HR deficit from alt-EJ use.
Conclusions from our studies are limited by several considerations. First, although the alt-EJ signature was correlated with specific indel mutations in tumors, a definitive test of this statistical correlation requires abrogating TGFβ signaling in cell lines with stable genomes for an extended period of culture and subsequent subcloning to determine the type of genomic alterations. In the absence of such studies, the implied relationship between specific mutation patterns and alt-EJ remain to be verified. Second, we restricted our analysis to solid carcinomas, without consideration of hematopoietic cancer or childhood cancers. The negative correlation of βAlt score with age in adult cancer suggests that the signature should be explored in childhood cancers, in which the most common cancers are leukemia and lymphoma. However, the impact of TGFβ in hematological cancers may differ from that of epithelia. Third, analysis of outcomes from TCGA is limited due to the retrospective collection of information and the minimal annotation of treatment, which led us to make assumptions based on current standard of care. We reported a reciprocal association of the βAlt score and outcome from one clinical trial dataset, but more extensive analysis of trial data with associated transcriptomic profiling is warranted. It may be possible to refine or extend the utility of our signatures upon publication of more gene expression data from clinical trials. Another possibility is retrospective analysis of βAlt association with outcomes by using a gene expression platform amenable to using RNA extracted from tissue blocks. Although our signatures were applicable for predicting patient responses to the combination of genotoxic chemotherapy and radiotherapy, they have not been extended to investigate predictive power for specific classes of chemotherapy or targeted therapy agents.
Although alt-EJ is not commonly defined or identified as a functional network, our data support the existence of such a network by reciprocal pattern of expression of TGFβ and alt-EJ genes in GBM, LUSC, and OVCA, and are strongly endorsed by consensus clustering of the genes across more than 10,000 cancer specimens represented in TCGA. Twenty-seven (75%) of the genes in the alt-EJ signature were identified in one consensus cluster, strongly suggestive of co-regulation. Correlation analysis of the signatures across TCGA demonstrated that a strong anti-correlation persists in the context of tumor heterogeneity in almost all (16/17) cancer types tested. Similarly, most cancer cell lines exhibit a similar pattern, consistent with a cancer cell-intrinsic program. Therefore, this feature of cancer reveals opportunities to improve the care of patients, from estimating prognosis to selection of therapeutic approaches that exploit this vulnerability.
Materials and Methods
Study Design
This study was designed to evaluate the effect of TGFβ on the DNA damage response to genotoxic therapies in cancer cell lines and primary patient tumors. Experiments were performed in biological replicates of three or more. Based on the initial results, gene signatures were used to evaluate the identified functional relationship in regard to cancer genomes and patient outcomes. Criteria for data exclusion are reported for each analysis. The researchers were not blinded.
Cell Culture
HNSC cell line SAS was obtained from RIKEN BRC (#RCB1974). Human GBM cell line U251 was a gift from Dr. Kevin Camphausen (National Cancer Institute). Both cell lines harbor homozygous TP53 mutations (SAS, CDS mutation: c.1006G>T; AA mutation: p.E336*; U251, CDS mutation: c.818G>A; AA mutation: p.R273H) according to COSMIC database (https://cancer.sanger.ac.uk/cell_lines). Cells were cultured as described in our previous publications (8, 14). Briefly, SAS and U251 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% bovine growth serum (HyClone), 100 IU/ml streptomycin-penicillin, and 1% GlutaMAX (all Gibco Thermo Fisher unless indicated otherwise). Cell lines were routinely tested and confirmed to be mycoplasma-free and authenticated according to the microsatellite markers in their genome (IDEXX). Cells in exponential growth were maintained in a humidified incubator at 37°C and 5% CO2 after resuscitation, and less than 10 passages were used for all experiments.
Treatments
The selective inhibitors for TGFβ receptor type I kinase, LY2157299and LY364947 (SelleckChem), were used at 2 μM. DNA-protein kinase inhibitor NU7441 (KU57788, 5 μM; SelleckChem) was added 1 hour before irradiation. Both small molecular inhibitors were dissolved and stocked in dimethyl sulfoxide (DMSO, Sigma-Aldrich) at more than 1000-fold concentrations than the final concentration on cells. Drugs were aliquoted and stored at −20°C for up to 6 months with protection from light. Recombinant human TGFβ (2 ng/ml as indicated; R&D Systems) was used to stimulate TGFβ signaling in cells. 220 kV X-ray radiation was generated by a small animal radiation research platform (Xstrahl Medical & Life Sciences).
qRT-PCR
Total RNA was prepared from the exponentially growing cells after treatments using the Qiazol reagent and miRNAeasy Mini Kit following the manufacturer’s manuals (Qiagen). First strand cDNA was synthesized on the Veriti Thermal Cycler (Applied Biosystems) from 1 μg of total RNA using the SuperScript IV First-Strand Synthesis System (Invitrogen). Quantitative PCR was performed on a QUANT STUDIO 5 system (Life Technologies) using SYBR Green Mix (Applied Biosystems). Primers validated by the vendor were used (Bio-Rad POLQ: qHsaCID0018136; PARP1: qHsaCED0045162; LIG1: qHsaCID0008449). GADPH primer sequences were forward: 5’-CAGCCTCCAGATCATCAGCA-3’ and reverse: 5’-TGTGGTCATGAGTCCTTCCA-3’). Comparative Ct method was used to calculate the relative expression of each gene to the expression of the housekeeping gene GADPH.
Gene expression analyses
Total RNA was extracted using the miRNAeasy Mini kit (Qiagen). Sample RNA (250 ng) was used for the DNA Damage Repair Gene Expression panel, consisting of 180 DDR genes and 12 housekeeping genes for normalization, according to manufacturer’s instructions (NanoString Technologies, Inc.). Hybridization efficiency and background signals were evaluated based on internal positive and negative control probes analyzed using nCounter Gene Expression. The raw counts, gene expression, and expression ratios between treated and untreated samples were analyzed by the nSolver software. Ratios from 2–3 biological repeats were used for further analysis and graphs.
DSB repair reporter assay
DSB repair reporters for HR (pDRGFP, 26475, Addgene) or Alt-EJ (EJ2GFP-puro, 44025; Addgene) were established in SAS and U251 cells as previously described, using plasmid constructs provided by Dr. J. Stark (City of Hope, CA) (28). Linearized plasmids were transfected by Lipofectamine 3000. Permanently transfected cells were then selected in 2 μg/ml puromycin-containing medium. Single cell clones were established by seeding at limiting dilution into 96-well plates. I-SceI expressing retrovirus was used to generate DSBs (73). Following the generation of DSB in the established reporter clones by expression of I-SceI endonuclease, the expression of the integrated GFP gene from the plasmid constructs pDRGFP (74) or EJ2GFP-puro (75) is indicative of DNA repair by HR or alt-EJ respectively. GFP-positive cells were quantified by flow cytometry.
Pulsed-field gel electrophoresis
SAS cells, treated and allowed to repair as indicated, were embedded in agarose blocks. Cells were lysed using a high temperature lysis protocol, where agarose blocks were placed in freshly prepared lysis buffer (10 mM Tris–HCl, pH 7.6, 50 mM NaCl, 100 mM EDTA, 2% N-lauryl sarcosyl, NLS, and 0.2 mg/ml protease) at 50°C for 18 h (76). Gels cast with 0.5% agarose (Bio-Rad) were used for electrophoresis in the presence of 0.5 μg/ml ethidium bromide. Gel images were obtained by “Typhoon” scan, and the fraction of DNA released from the well into the lane was quantified by Image Quant 5.2 (GE-Healthcare).
TCGA and Sanger database analysis
TCGA data were obtained from cBioPortal (http://www.cbioportal.org/public-portal/), from the Genomic Data commons (GDC) using the R package “TCGAbiolinks” from the University of California Santa Cruz Xena platform using the R package “UCSCXenaTools”, or from the corresponding TCGA publications. Somatic mutations at the individual level were obtained following approval by the dbGaP Data Access Committee (project #11689). The “fraction of genome altered” corresponded to the percentage of the genome that was detected affected by copy number gains or losses and was obtained from the Oncoprint tab of cBioPortal. The numbers of different types of genomic alterations in each primary tumor were taken from the corresponding TCGA publications (38, 50), and the ID mutational profiles from the PCAWG-ICGC results (51). Metastases and recurrent tumors were excluded from this study, making primary tumor samples the focus of the analyses. Cancer types are named using the corresponding TCGA study abbreviations (https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations). A chronic TGFβ signature was defined using data from non-malignant epithelial cell line, MCF10A, exposed to TGFβ or LY364947 for 7 days and analyzed using Affymetrix gene expression microarray (77). Genes involved in alt-EJ were identified from a functional study (72) and from a study of RNAi library screening against 238 DDR genes targeted by siRNA that decreased alt-EJ events with a cutoff of lower than 0.5 fold decrease compared to controls (28). The normalized gene signature scores were based on ssGSEA computed using the Gene Set Variation Analysis (GSVA) software package (78) and used for survival based on PCC analyses, or on the Kendall rank correlation coefficient when assessing somatic mutation profiles. Signature association with patient survival was computed in R software with survfit package using multivariate Cox regression analyses and log-rank tests comparing the survival curves of tertiles 1 versus 3 of patients. To calculate the hazard ratio between tertiles 1 and 3, univariate Cox regressions were performed. Multivariate Cox regressions between the βAlt score and OS/PFS were performed in TCGA-GBM including the MGMT status and in TCGA-HNSC including the HPV status. Unsupervised hierarchical clustering used ssGSEA scores of TGFβ and alt-EJ signatures (79). Euclidean distance was used as the similarity metric and the Ward.D2 method as the between-cluster distance metric. Hierarchical clustering based on Euclidean distance was used to generate heatmap plots. Consensus clustering analysis of TGFβ and alt-EJ signatures in 10,848 patients with solid cancers from TCGA Pan-Cancer dataset were computed with 1,000 resampling using ConsensusClusterPlus based on Spearman’s correlation (80). All data were downloaded from the TCGA PanCancer website (https://gdc.cancer.gov/about-data/publications/pancanatlas).
To classify patients according to their expression pattern of the TGFβ and alt-EJ gene signatures, a βAlt score was calculated for each sample in an open source package, genScore (https://github.com/pujana-lab/genScore). The βAlt score collapses in one dimension the position of TGFβ versus alt-EJ normalized signature ssGSEA scores as follows:
Multiplexed multiple reaction monitoring (MRM) mass spectrometry
Two well-established multiplexed targeted mass spectrometry-based assays were used for quantification of protein and protein phosphorylation in response to radiation (23–26). The immune-MRM assay targeted 97 peptides (46 modifications, 53 proteins), and the IMAC-MRM assay targeted 236 phosphorylated peptides (41 overlapping with the immuno-MRM panels) for 182 proteins. Briefly, after treatments cells were lysed by freshly-prepared ice-cold 6 M urea lysis buffer that contained 25 mM Tris (pH 8.0), 1 mM EDTA, 1 mM EGTA, 1% phosphatase inhibitor cocktail 2, 1% phosphatase inhibitor cocktail 3, and 1% protease inhibitor cocktail (all Sigma Aldrich unless otherwise noted). Cell lysates were sonicated for 2X 10 seconds using a Sonic Dismembrator Model 100 (Fisher Scientific). Lysates were transferred to microcentrifuge tubes, vortexed, and then cleared by collecting supernatants after centrifugation at 20,000 × g for 10 min at 4 °C. Supernatants were stored at −80 °C. Protein concentrations were measured in triplicate using Micro BCA Protein Assay Kit (Thermo # 23235). A negative control containing 100 μL of 1X PBS, 0.01% CHAPS, and 100 μl of lysis buffer was analyzed in parallel. Lysates were reduced, alkylated with iodoacetamide, and digested by the addition of Lys-C at a 1:50 enzyme:protein ratio (by mass). After 2 hours, a trypsin aliquot was added at a 1:50 enzyme:protein ratio and incubated overnight at 37°C with shaking. After 16 hours, the reaction was quenched with formic acid (final concentration 1% by volume). A mix of stable isotope-labeled peptide standards was added to the digest at 150 fmol/mg. Peptide immunoaffinity enrichment and MRM-MS were conducted as previously described (23, 81) using custom monoclonal antibodies coupled to Protein G agarose magnetic beads (GE Sepharose, #28–9513-79) in two panels enriched in a serial fashion. The elution plate was covered with sealing foil and stored at −80 °C until analysis with LC-MRM-MS. The eluted peptides were thawed and analyzed by an Eksigent Ultra nanoLC system with a nano autosampler and chipFLEX system (Eksigent Technologies) coupled to a 5500 QTRAP mass spectrometer (SCIEX). Peptides were loaded on a trap column (C18, 5 mm × 200 μm) at 5 μL/min for 3 minutes using mobile phase A (0.1% formic acid in water) and eluted at 300 nL/minute using a linear gradient of mobile phase B (90% acetonitrile and 0.1% formic acid in water) developed from 3–14% B in 1 minute, 14–34% B in 20 minutes, 34–90% B in 2 minutes on a 15 cm × 75 μm chip column (Reprosil AQ C18 particles, 3 μm). The mass spectrometer was operated in the positive ion MRM mode with optimized collision energy (CE) values. Scheduled MRM transitions used a retention time window of 100 seconds and a desired cycle time of 0.5 seconds, enabling enough points across a peak for quantitation. MRM data were analyzed by Skyline with manual review of peak integrations to confirm transitions from analyte peptides, and heavy stable isotope-labeled peptides had equivalent retention times and relative areas of transitions (82, 83). Transitions with detected interferences were not used in the data analysis. The data were shown as the peak area ratio (light:heavy).
Statistical analysis
All experiments were repeated more than three times unless otherwise noted. Results are presented as means ± SEM and considered significantly different at P < 0.05 based on two-tailed Student’s t-test or as otherwise indicated. * P< 0.05, **P< 0.01, *** P< 0.005. The data were analyzed by Prism 6 (GraphPad, Inc.)
Supplementary Material
Acknowledgements
The authors would like to thank Kevin Camphausen (National Cancer Institute) and Jeremy Stark (City of Hope) for reagents, Trevor Jones and William Chou at UCSF for scientific support and Jacob J. Kennedy and Lei Zhao at UW for data generation. The results presented here are partly based on data generated by the TCGA Research Network (https://www.cancer.gov/tcga), and we would like to express our gratitude to the TCGA consortia and their coordinators for the data provision and clinical information used in this study.
Funding
Work was supported by funding from the foundations DACMA, GINKGO, Sosciathlon, and “Viladecans Contra el Càncer”, grants from the Carlos III Institute of Health (ISCIII; Ministry of Science, Innovation and Universities; PI18/01029), Generalitat de Catalunya (SGR 2017-449), CERCA program, and the European Union (European Regional Development Fund (FEDER), “A way to make Europe”) to MAP, by National Institutes of Health (R01 CA239235) to MHBH, by grants from German Federal Ministry of Education and Research (02NUK043B-COLLAR and 02NUK037B-VERCHROMT) and the DFG GRK1739 to GI, and by NCI U01 CA 214114 and NCI R50CA211499 to AGP.
Footnotes
Competing Interests
MAP and AA are recipients of an unrestricted research grant from Roche Pharma for the development of the ProCURE ICO research program. MHBH receives research grants from Roche-Genentech, Varian, Inc. and Lilly, Inc. and consults for EMD-Serano, Varian, Inc., and Telos. MHBH, LQ and MAP have filed a provisional patent entitled Identification of DDR Defects for Cancer Treatment”. MHBH, QL and MAP are inventors on a provisional patent application UCSF088P submitted by the University of California, San Francisco that covers use of the gene signatures. AC receives research funding and has received honoraria from AstraZeneca. This funding did not support any of the work presented in this manuscript. All other authors declare that they have no competing interests.
Data and materials availability
All data associated with this study are available in the manuscript or the supplementary materials.
References and notes
- 1.Massague J, TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korkut A, Zaidi S, Kanchi RS, Rao S, Gough NR, Schultz A, Li X, Lorenzi PL, Berger AC, Robertson G, Kwong LN, Datto M, Roszik J, Ling S, Ravikumar V, Manyam G, Rao A, Shelley S, Liu Y, Ju Z, Hansel D, de Velasco G, Pennathur A, Andersen JB, O’Rourke CJ, Ohshiro K, Jogunoori W, Nguyen BN, Li S, Osmanbeyoglu HU, Ajani JA, Mani SA, Houseman A, Wiznerowicz M, Chen J, Gu S, Ma W, Zhang J, Tong P, Cherniack AD, Deng C, Resar L, Weinstein JN, Mishra L, Akbani R, A pan-cancer analysis reveals high-frequency genetic alterations in mediators of signaling by the TGF-beta superfamily. Cell Syst. 7, 422–437.e427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glick AB, Weinberg WC, Wu IH, Quan W, Yuspa SH, Transforming growth factor β1 suppresses genomic instability independent of a G1 arrest, p53, and Rb. Cancer Res. 56, 3645–3650 (1996). [PubMed] [Google Scholar]
- 4.Liu Q, Lopez K, Murnane J, Humphrey T, Barcellos-Hoff MH, Misrepair in context: TGFβ regulation of DNA repair. Front. Oncol. 9, 799 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ceccaldi R, Rondinelli B, D’Andrea AD, Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lord CJ, Ashworth A, PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, Westra WH, Chung CH, Jordan RC, Lu C, Kim H, Axelrod R, Silverman CC, Redmond KP, Gillison ML, Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 363, 24–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Q, Ma L, Jones T, Palomero L, Pujana MA, Martinez-Ruiz H, Ha PK, Murnane J, Cuartas I, Seoane J, Baumann M, Linge A, Barcellos-Hoff MH, Subjugation of TGFβ signaling by human papilloma virus in head and neck squamous cell carcinoma shifts DNA repair from homologous recombination to alternative end joining. Clin. Cancer Res. 24, 6001–6014 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Sfeir A, Symington LS, Microhomology-mediated end joining: A back-up survival mechanism or dedicated pathway? Trends Biochem. Sci. 40, 701–714 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wood RD, Doublie S, DNA polymerase theta (POLQ), double-strand break repair, and cancer. DNA Repair 44, 22–32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iliakis G, Murmann T, Soni A, Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 793, 166–175 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Sallmyr A, Tomkinson AE, Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 293, 10536–10546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bouquet SF, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, Babb JS, Lonning SM, DeWyngaert JK, Formenti S, Barcellos-Hoff MH, Transforming growth factor β1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin. Cancer Res. 17, 6754–6765 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM, Barcellos-Hoff MH, Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-β. Cancer Res. 72, 4119–4129 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du S, Bouquet F, Lo C-H, Pellicciotta I, Bolourchi S, Parry R, Barcellos-Hoff MH, Attenuation of the DNA damage response by TGFβ inhibitors enhances radiation sensitivity of NSCLC cells in vitro and in vivo. Int. J. Radiat. Oncol. Biol. Phys. 91, 91–99 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, Yusufzai T, D’Andrea AD, Homologous-recombination-deficient tumours are dependent on PolΘ-mediated repair. Nature 518, 258–262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, Wu X, Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 110, 7720–7725 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng W, Simpson DA, Carvajal-Garcia J, Price BA, Kumar RJ, Mose LE, Wood RD, Rashid N, Purvis JE, Parker JS, Ramsden DA, Gupta GP, Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 10, 4286–4286 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirshner J, Jobling MF, Pajares MJ, Ravani SA, Glick AB, Lavin MJ, Koslov S, Shiloh Y, Barcellos-Hoff MH, Inhibition of transforming growth factor-β1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res. 66, 10861–10869 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Shiloh Y, Ziv Y, The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210 (2013). [PubMed] [Google Scholar]
- 21.Martinez-Ruiz H, Illa-Bochaca I, Omene C, Hanniford D, Liu Q, Hernando E, Barcellos-Hoff MH, A TGFβ-miR-182-BRCA1 axis controls the mammary differentiation hierarchy. Sci. Signal. 9, ra118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim MR, Lee J, An YS, Jin YB, Park IC, Chung E, Shin I, Barcellos-Hoff MH, Yi JY, TGFβ1 protects cells from γ-IR by enhancing the activity of the NHEJ repair pathway. Mol. Cancer Res. 13, 319–329 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Whiteaker JR, Zhao L, Saul R, Kaczmarczyk JA, Schoenherr RM, Moore HD, Jones-Weinert C, Ivey RG, Lin C, Hiltke T, Reding KW, Whiteley G, Wang P, Paulovich AG, A multiplexed mass spectrometry-based assay for robust quantification of phosphosignaling in response to DNA Damage. Radiat. Res. 189, 505–518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whiteaker JR, Zhao L, Schoenherr RM, Kennedy JJ, Ivey RG, Paulovich AG, Peptide immunoaffinity enrichment with targeted mass spectrometry: Application to quantification of ATM kinase phospho-signaling. Methods Mol. Biol. 1599, 197–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones GN, Rooney C, Griffin N, Roudier M, Young LA, Garcia-Trinidad A, Hughes GD, Whiteaker JR, Wilson Z, Odedra R, Zhao L, Ivey RG, Howat WJ, Harrington EA, Barrett JC, Ramos-Montoya A, Lau A, Paulovich AG, Cadogan EB, Pierce AJ, pRAD50: A novel and clinically applicable pharmacodynamic biomarker of both ATM and ATR inhibition identified using mass spectrometry and immunohistochemistry. Br. J. Cancer 119, 1233–1243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kennedy JJ, Yan P, Zhao L, Ivey RG, Voytovich UJ, Moore HD, Lin C, Pogosova-Agadjanyan EL, Stirewalt DL, Reding KW, Whiteaker JR, Paulovich AG, Immobilized metal affinity chromatography coupled to multiple reaction monitoring enables reproducible quantification of phospho-signaling. Mol. Cell. Proteomics 15, 726–739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahrabi S, Sarkar S, Pfister SX, Pirovano G, Higgins GS, Porter AC, Humphrey TC, A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res. 44, 5743–5757 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennardo N, Cheng A, Huang N, Stark JM, Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, Cleverly AL, Desaiah D, Guba SC, Benhadji KA, Slapak CA, Lahn MM, Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 9, 4479–4499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulder KM, Morris SL, Activation of p21ras by transforming growth factor β in epithelial cells. J. Biol. Chem. 267, 5029–5031 (1992). [PubMed] [Google Scholar]
- 31.Li C, Suardet L, Little JB, Potential role of WAF/Cip1/p21 as a mediator of TGF-β cytoinhibitory effect. J. Biol. Chem. 270, 4971–4974 (1995). [DOI] [PubMed] [Google Scholar]
- 32.Fynan TM, Reiss M, Resistance to inhibition of cell growth by transforming growth factor-β and its role in oncogenesis. Crit. Rev. Onco. 4, 493–540 (1993). [PubMed] [Google Scholar]
- 33.Howard SM, Yanez DA, Stark JM, DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 11, e1004943 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma’ayan A, The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016:baw100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill CS, Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 8, a022079 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayin NS, Ma L, Thomas C, Baitalmal R, Sure A, Fansiwala K, Bustoros M, Golfinos JG, Pacione D, Snuderl M, Zagzag D, Barcellos-Hoff MH, Placantonakis D, Patient-specific screening using high-grade glioma explants to determine potential radiosensitization by a TGF-β small molecule inhibitor. Neoplasia 18, 795–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maxwell CA, Fleisch MC, Costes SV, Erickson AC, Boissiere A, Gupta R, Ravani SA, Parvin B, Barcellos-Hoff MH, Targeted and nontargeted effects of ionizing radiation that impact genomic instability. Cancer Res. 68, 8304–8311 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, Omberg L, Wolf DM, Shriver CD, Thorsson V, Cancer N Genome Atlas Research, H. Hu, An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416.e411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sidaway P, Glioblastoma subtypes revisited. Nat. Rev. Clin. Oncol. 14, 587–587 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Garg AD, Vandenberk L, Van Woensel M, Belmans J, Schaaf M, Boon L, De Vleeschouwer S, Agostinis P, Preclinical efficacy of immune-checkpoint monotherapy does not recapitulate corresponding biomarkers-based clinical predictions in glioblastoma. Oncoimmunology 6, e1295903-e1295903 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ajaz M, Jefferies S, Brazil L, Watts C, Chalmers A, Current and investigational drug strategies for glioblastoma. Clin. Oncol. 26, 419–430 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Weller M, Le Rhun E, Preusser M, Tonn J-C, Roth P, How we treat glioblastoma. ESMO Open 4, e000520-e000520 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ettinger DS, Wood DE, Aggarwal C, Aisner DL, Akerley W, Bauman JR, Bharat A, Bruno DS, Chang JY, Chirieac LR, D’Amico TA, Dilling TJ, Dobelbower M, Gettinger S, Govindan R, Gubens MA, Hennon M, Horn L, Lackner RP, Lanuti M, Leal TA, Lin J, Jr BWL, Martins RG, Otterson GA, Patel SP, Reckamp KL, Riely GJ, Schild SE, Shapiro TA, Stevenson J, Swanson SJ, Tauer KW, Yang SC, Gregory K, OCN M Hughes, NCCN guidelines insights: Non-small cell lung cancer, version 1.2020. 17, 1464 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Network CGAR, Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Earl H, Ahmed A, Vallier A, Hatcher H, Parkinson C, Iddawela M, Latimer J, Crawford R, Brenton J, Cambridge Translational Cancer Research Ovarian Study 01 (CTCR-OV01): Expression profiling of advanced epithelial ovarian cancer (EOC) to predict chemotherapy response. A randomised phase II trial design with prospective translational endpoints. J. Clin. Oncol. 24, 15018 (2006). [Google Scholar]
- 46.Jiménez-Sánchez A, Cast O, Miller ML, Comprehensive benchmarking and integration of tumour microenvironment cell estimation methods. Cancer Res. 79, 6238–6246 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H, Cokelaer T, Greninger P, van Dyk E, Chang H, de Silva H, Heyn H, Deng X, Egan RK, Liu Q, Mironenko T, Mitropoulos X, Richardson L, Wang J, Zhang T, Moran S, Sayols S, Soleimani M, Tamborero D, Lopez-Bigas N, Ross-Macdonald P, Esteller M, Gray NS, Haber DA, Stratton MR, Benes CH, Wessels LFA, Saez-Rodriguez J, McDermott U, Garnett MJ, A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chaudhuri RA, Nussenzweig A, The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bentley J, Diggle CP, Harnden P, Knowles MA, Kiltie AE, DNA double strand break repair in human bladder cancer is error prone and involves microhomology-associated end-joining. Nucleic Acids Res. 32, 5249–5259 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Yang L, Kucherlapati M, Chen F, Hadjipanayis A, Pantazi A, Bristow CA, Lee EA, Mahadeshwar HS, Tang J, Zhang J, Seth S, Lee S, Ren X, Song X, Sun H, Seidman J, Luquette LJ, Xi R, Chin L, Protopopov A, Li W, Park PJ, Kucherlapati R, Creighton CJ, A pan-cancer compendium of genes deregulated by somatic genomic rearrangement across more than 1,400 cases. Cell Rep. 24, 515–527 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, Islam SMA, Lopez-Bigas N, Klimczak LJ, McPherson JR, Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, Alexandrov LB, Bergstrom EN, Boot A, Boutros P, Chan K, Covington KR, Fujimoto A, Getz G, Gordenin DA, Haradhvala NJ, Huang MN, Islam SMA, Kazanov M, Kim J, Klimczak LJ, Lopez-Bigas N, Lawrence M, Martincorena I, McPherson JR, Morganella S, Mustonen V, Nakagawa H, Tian Ng AW, Polak P, Prokopec S, Roberts SA, Rozen SG, Sabarinathan R, Saini N, Shibata T, Shiraishi Y, Stratton MR, Teh BT, Vázquez-García I, Wheeler DA, Wu Y, Yousif F, Yu W, Getz G, Rozen SG, Stratton MR, Group PMSW, Consortium P, The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wyatt DW, Feng W, Conlin MP, Yousefzadeh MJ, Roberts SA, Mieczkowski P, Wood RD, Gupta GP, Ramsden DA, Essential roles for polymerase theta-mediated end joining in the repair of chromosome breaks. Mol. Cell 63, 662–673 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Negrini S, Gorgoulis VG, Halazonetis TD, Genomic instability — An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Ziemann F, Arenz A, Preising S, Wittekindt C, Klussmann JP, Engenhart-Cabillic R, Wittig A, Increased sensitivity of HPV-positive head and neck cancer cell lines to x-irradiation +/− Cisplatin due to decreased expression of E6 and E7 oncoproteins and enhanced apoptosis. Am. J. Cancer Res. 5, 1017–1031 (2015). [PMC free article] [PubMed] [Google Scholar]
- 55.Mirghani H, Amen F, Tao Y, Deutsch E, Levy A, Increased radiosensitivity of HPV-positive head and neck cancers: Molecular basis and therapeutic perspectives. Cancer Treat. Rev. 41, 844–852 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Kimple RJ, Smith MA, Blitzer GC, Torres AD, Martin JA, Yang RZ, Peet CR, Lorenz LD, Nickel KP, Klingelhutz AJ, Lambert PF, Harari PM, Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 73, 4791–4800 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weaver AN, Cooper TS, Rodriguez M, Trummell HQ, Bonner JA, Rosenthal EL, Yang ES, DNA double strand break repair defect and sensitivity to poly ADP-ribose polymerase (PARP) inhibition in human papillomavirus 16-positive head and neck squamous cell carcinoma. Oncotarget 6, 26995–27007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wallace NA, Khanal S, Robinson KL, Wendel SO, Messer JJ, Galloway DA, High-risk Alphapapillomavirus oncogenes impair the homologous recombination pathway. J. Virol. 91, pii: e01084–01017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nees M, Geoghegan JM, Munson P, Prabhu V, Liu Y, Androphy E, Woodworth CD, Human papillomavirus type 16 E6 and E7 proteins inhibit differentiation-dependent expression of transforming growth factor-β 2 in cervical keratinocytes. Cancer Res. 60, 4289–4298 (2000). [PubMed] [Google Scholar]
- 60.Moustakas A, Heldin CH, Non-smad TGF-β signals. J. Cell Sci. 118, 3573–3584 (2005). [DOI] [PubMed] [Google Scholar]
- 61.O’Connor MJ, Targeting the DNA damage response in cancer. Mol. Cell 60, 547–560 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, European Organisation for R, T. Treatment of Cancer Brain, G. Radiotherapy, G. National Cancer Institute of Canada Clinical Trials, Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Liang BC, Thornton AF Jr., Sandler HM, Greenberg HS, Malignant astrocytomas: Focal tumor recurrence after focal external beam radiation therapy. J. Neurosurg. 75, 559–563 (1991). [DOI] [PubMed] [Google Scholar]
- 64.Badiyan SN, Markovina S, Simpson JR, Robinson CG, DeWees T, Tran DD, Linette G, Jalalizadeh R, Dacey R, Rich KM, Chicoine MR, Dowling JL, Leuthardt EC, Zipfel GJ, Kim AH, Huang J, Radiation therapy dose escalation for glioblastoma multiforme in the era of temozolomide. Int. J. Radiat. Oncol. Biol. Phys. 90, 877–885 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Prados MD, Wara WM, Sneed PK, McDermott M, Chang SM, Rabbitt J, Page M, Malec M, Davis RL, Gutin PH, Lamborn K, Wilson CB, Phillips TL, Larson DA, Phase III trial of accelerated hyperfractionation with or without difluromethylornithine (DFMO) versus standard fractionated radiotherapy with or without DFMO for newly diagnosed patients with glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 49, 71–77 (2001). [DOI] [PubMed] [Google Scholar]
- 66.Bruna A, Darken RS, Rojo F, Ocaña A, Peñuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, Seoane J, High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11, 147–160 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Fulton B, Short SC, James A, Nowicki S, McBain C, Jefferies S, Kelly C, Stobo J, Morris A, Williamson A, Chalmers AJ, PARADIGM-2: Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin. Transl. Radiat. Oncol. 8, 12–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Teicher BA, Maehara Y, Kakeji Y, Ara G, Keyes SR, Wong J, Herbst R, Reversal of in vivo drug resistance by the transforming growth factor-β inhibitor decorin. Int. J. Cancer 71, 49–58 (1997). [DOI] [PubMed] [Google Scholar]
- 69.Zhang M, Herion TW, Timke C, Han N, Hauser K, Weber KJ, Peschke P, Wirkner U, Lahn M, Huber PE, Trimodal glioblastoma treatment consisting of concurrent radiotherapy, temozolomide, and the novel TGF-β receptor I kinase inhibitor LY2109761. Neoplasia 13, 537–549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang M, Kleber S, Rohrich M, Timke C, Han N, Tuettenberg J, Martin-Villalba A, Debus J, Peschke P, Wirkner U, Lahn M, Huber PE, Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 71, 7155–7167 (2011). [DOI] [PubMed] [Google Scholar]
- 71.Liu P, Menon K, Alvarez E, Lu K, Teicher BA, Transforming growth factor-β and response to anticancer therapies in human liver and gastric tumors in vitro and in vivo. Int. J. Oncol. 16, 599–610 (2000). [DOI] [PubMed] [Google Scholar]
- 72.DiBiase SJ, Zeng ZC, Chen R, Hyslop T, Curran WJ Jr., Iliakis G, DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer Res. 60, 1245–1253 (2000). [PubMed] [Google Scholar]
- 73.Muraki K, Han L, Miller D, Murnane JP, The role of ATM in the deficiency in nonhomologous end-joining near telomeres in a human cancer cell line. PLoS Genet. 9, e1003386 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pierce AJ, Johnson RD, Thompson LH, Jasin M, XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 13, 2633–2638 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bindra RS, Goglia AG, Jasin M, Powell SN, Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 41, e115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Singh SK, Wang M, Staudt C, Iliakis G, Post-irradiation chemical processing of DNA damage generates double-strand breaks in cells already engaged in repair. Nucleic Acids Res. 39, 8416–8429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andarawewa KL, Erickson AC, Chou WS, Costes SV, Gascard P, Mott JD, Bissell MJ, Barcellos-Hoff MH, Ionizing radiation predisposes nonmalignant human mammary epithelial cells to undergo transforming growth factor b induced epithelial to mesenchymal transition. Cancer Res. 67, 8662–8670 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Hanzelmann S, Castelo R, Guinney J, GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gu Z, Eils R, Schlesner M, Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Wilkerson DM, Hayes ND, ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Whiteaker JR, Zhao L, Lin C, Yan P, Wang P, Paulovich AG, Sequential multiplexed analyte quantification using peptide immunoaffinity enrichment coupled to mass spectrometry. Mol. Cell Proteomics 11, M111.015347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maclean B, Tomazela DM, Abbatiello SE, Zhang S, Whiteaker JR, Paulovich AG, Carr SA, Maccoss MJ, Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal. Chem. 82, 10116–10124 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ, Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.