

Abstract
This study aimed to investigate the inhibitory effect of EM-2, a natural active monomer purified from Elephantopusmollis H.B.K., on the proliferation of human hepatocellular carcinoma cells and the molecular mechanism involved. The results from the MTT assay revealed that EM-2 significantly inhibited the proliferation of human hepatocellular carcinoma (HCC) cells in a dose-dependent manner but exhibited less cytotoxicity to the normal liver epithelial cell line LO2. EdU staining and colony formation assays further confirmed the inhibitory effect of EM-2 on the proliferation of Huh-7 hepatocellular carcinoma cells. According to the RNA sequencing and KEGG enrichment analysis results, EM-2 markedly activated the MAPK pathway in Huh-7 cells, and the results of Western blotting further indicated that EM-2 could activate the ERK and JNK pathways. Meanwhile, EM-2 induced apoptosis in a dose-dependent manner and G2/M phase arrest in Huh-7 cells, which could be partially reversed when treated with SP600125, a JNK inhibitor. Further study indicated that EM-2 induced endoplasmic reticulum stress and blocked autophagic flux in Huh-7 cells by inhibiting autophagy-induced lysosome maturation. Inhibition of autophagy by bafilomycin A1 could reduce cell viability and increase the sensitivity of Huh-7 cells to EM-2. In conclusion, our findings revealed that EM-2 not only promoted G2/M phase arrest and activated ER stress but also induced apoptosis by activating the JNK pathway and blocked autophagic flux by inhibiting autolysosome maturation in Huh-7 hepatocellular carcinoma cells. Therefore, EM-2 is a potential therapeutic drug with promising antitumor effects against hepatocellular carcinoma and fewer side effects.
Keywords: Elephantopusmollis H.B.K., JNK, hepatocarcinoma cells, apoptosis, autophagy
Introduction
Primary liver disease is one of the major health problems in the world, and liver cancer is the most debilitating liver disease and the second leading cause of cancer-related mortality [1]. Most patients suffer from hepatocellular carcinoma (HCC), which accounts for more than 90% of all patients with liver cancer [2]. Chemotherapy is the conventional treatment for cancer, which is beset with serious side effects. There are numerous chemotherapy drugs available for liver cancer treatment, such as oxaliplatin, capecitabine, gemcitabine, and 5-fluorouracil. However, most patients with HCC have a poor prognosis after chemotherapy, with a 5-year survival rate between 17% and 53% and a recurrence rate as high as 50% within 2 years [3, 4], mostly because HCC cells develop strong resistance to clinical chemotherapy drugs [5]. Therefore, it is urgent to develop efficacious treatments for HCC that have few side effects.
Accumulating studies have reported that monomers purified from some Chinese herbal medicines exhibit better antitumor effects with lower toxicity [6, 7]. Elephantopusmollis H.B.K. is a traditional Chinese herbal medicine with a long history in the treatment of epistaxis, jaundice, edema, bruises, dysentery, etc [8, 9]. Increasing evidence suggests that Elephantopusmollis H.B.K. has noteworthy pharmacological effects, including antimelanogenic [10], antiproliferative [11], and proapoptotic activities [12]. Recently, Shao et al. reported that ROS accumulation and ASK1/JNK signaling pathway activation by EM23, a natural sesquiterpene lactone family member isolated from Elephantopusmollis, were critical for the induction of apoptosis in human cervical cancer (CaSki and SiHa) cell lines [13]. However, the antitumor effectors and underlying molecular mechanism of EM-2, a novel monomer purified from Elephantopusmollis H.B.K., on HCC remain poorly understood.
The mitogen-activated protein kinase (MAPK) family is a kind of serine/threonine protein kinase superfamily that is activated in response to numerous cellular stressors. To date, the known MAPKs mainly consist of p38 MAPK, extracellular signal-regulated kinase (ERK), and c-Jun NH2-terminal kinase (JNK). MAPKs play critical roles in controlling cellular responses to environmental stressors and in regulating numerous cellular activities including gene expression, cell growth, migration, differentiation, and apoptosis, which have made these proteins a priority for research related to many human diseases [14, 15]. Tumorigenesis and development are complicated processes affected by multiple signaling pathways, including MAPK signaling pathways. Xu et al. found that apoptosis and S-phase arrest were induced via ROS-dependent JNK and ERK activation in the human pancreatic cancer cell line Miapaca-2 upon treatment with sophoridine [16]. Similarly, Mhaidat et al. revealed that ER stress is involved in docetaxel-induced JNK-dependent apoptosis of human melanoma [17]. In our previous studies, PP-22, a Paris polyphylla monomer, was shown to induce autophagy and apoptosis in CNE-2 nasopharyngeal carcinoma cells by activating p38 signaling and endoplasmic reticulum stress [18]. However, the effect of EM-2 on MAPK family activity in HCC is unknown.
In our previous work, we isolated 25 monomer compounds (named EM-1 to EM-25) from Elephantopusmollis H.B.K. The MTT assay results showed that among the 25 monomers, EM-2, EM-3, EM-10, and EM-12 had strong antitumor effects. In this study, we demonstrated that EM-2 exhibited excellent antitumor effects in human HCC cells with few adverse effects on normal cells. The compound affected Huh-7 HCC cells by activating the JNK pathway and promoting G2/M arrest and apoptosis. Moreover, we showed that EM-2 induced endoplasmic reticulum stress and inhibited autophagy in Huh-7 cells by blocking autolysosome maturation and thus increasing the sensitivity of Huh-7 cells to EM-2. These data suggested that EM-2 is a potential drug with promising antitumor effects for the treatment of HCC.
Materials and methods
Materials
EM-2 a monomeric sesquiterpene lactone called 2β-methoxy-2-deethoxyphantomolin, was kindly provided by Professor Guo-cai Wang (College of Pharmacy, Jinan University). The degree of purity of EM-2 used in the present study was ≥98% as determined by high-performance liquid chromatography. EM-2 was dissolved in dimethyl sulfoxide (DMSO) and diluted DMEM/F12 medium (Gibco, CA) without fetal bovine serum (Gibco, CA). DMSO and trypsin-EDTA were purchased from Sigma. MTT was purchased from Beyotime Biotechnology (Shanghai, China). Polyvinylidene fluoride (PVDF) was purchased from Millipore (Burlington, MA). Antibodies against p38, p-p38, ERK1/2, JNK1/2, p-JNK1/2, c-Jun, p-c-Jun, Bcl-2, Bax, Caspase-9, Caspase-3, cleaved-Caspase-3, PARP, Cyclin A2, Cyclin B1, Cyclin D1, CDK4, Myt1, cdc2, p-cdc2 (Tyr15), p53, IR1α, p-IR1α, eIF2α, p-eIF2α, Bip, ATF4, p62, and LC3 were purchased from Cell Signaling Technology (Boston, MA). In addition, anti-Wee1, anti-p-ERK1/2, anti-p21, goat anti-rabbit IgG-HRP, and goat anti-mouse IgG-HRP antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and anti-Cathepsin L was purchased from BD Biosciences.
Cells and cell culture
Human hepatoma cell lines Hep3B, CRL-8024, SMMC-7721, QGY-7703, and Huh-7; LO2 cells (normal liver epithelial cells) and MIHA cells (immortalized normal liver cells) were obtained from the Cancer Center of Sun Yat-Sen University (Guangdong, China). Cells were maintained in DMEM/F12 medium with 10% FBS and cultured at 37 °C in an atmosphere containing 5% CO2.
Cell viability assay
Cells were seeded into 96-well plates at a density of 5000 cells per well and allowed to attach overnight in DMEM/F12 medium containing 10% FBS. The cells were then incubated with different concentrations of EM-2 (0, 1.25, 2.5, 5, 10, and 20 μM) for 48 h before the MTT assay. Then, 20 μL of MTT solution (5 mg/mL) was added to each well, and the plates were incubated for another 4 h at 37 °C before the optical densities were measured at 570 nm by a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA).
EdU staining assay
Huh-7 cells were treated with different concentrations of EM-2 for 24 h after they were seeded in 96-well plates at a density of 5000 cells per well. Then, EdU staining assays were performed to detect cell proliferation activity using a Cell-LightTM EdU Apollo488 Imaging kit (RiboBio, Guangzhou, China) according to the manufacturer’s instructions.
Colony formation assay
Huh-7 cells were seeded in 6-well plates at a density of 500 cells per well and then exposed to different concentrations of EM-2 (0, 0.25, 0.5 μM) for 7 days. Then, cell colonies were fixed with 4% paraformaldehyde for 20 min and stained with 0.5% crystal violet solution (Sigma, USA) for 30 min. After three gentle washes and air drying, the stained colonies were counted, and photographed.
RT-qPCR and RNA sequencing
Total RNA was isolated from cultured cells using TRIzol reagent (Invitrogen, CA, USA) and reverse-transcribed using a HiScript II Q Select RT SuperMix reverse transcription kit for qPCR (Vazyme Biotech, Nanjing, China). qPCR assays were performed with ChamQ SYBR Color qPCR Master Mix (Vazyme Biotech, Nanjing, China), and the primers used in the present study are listed in Table 1. After extraction and treatment with DNase, total RNA was sent to BGI (Shenzhen, China) for RNA sequencing on a BGISEQ-500 sequencer.
Table 1.
Primer sequences for qRT-PCR.
| Gene | sequence | |
|---|---|---|
| ATF4 | Forward | 5′-CTTTCTCTCCTCCTGCTTCT-3′ |
| Reverse | 5′-GAGTCACACGACCCAATCA-3′ | |
| ATF6 | Forward | 5′-CTGTGGTGAAACCTCCACCT-3′ |
| Reverse | 5′-CATGGTGACCACAGGAGATG-3′ | |
| EIF2AK3 | Forward | 5′-TTTTGCTGCAGAACGGACATG-3′ |
| Reverse | 5′-ATCTTCACCGGGACACTTCAC-3′ | |
| DDIT3 | Forward | 5′-GAGTTGGAGGCGTGGTATGA-3′ |
| Reverse | 5′-CCTTGGTGGCGATTGGTGAA-3′ | |
| HSPA5 | Forward | 5′-ACGAGAACACAGAAGACGGG-3′ |
| Reverse | 5′-ATCCCAATCACTGTCCCAAC-3′ | |
| ERN1 | Forward | 5′-CGGCCTCGGGATTTTTGGAA-3′ |
| Reverse | 5′-CCTGCAGGACTGGATCTTCT-3′ | |
| SERP1 | Forward | 5′-GTCGCCAAGACCTCGAGAAA-3′ |
| Reverse | 5′-GCAAGGCACTGTGGTATGGA-3′ | |
| GAPDH | Forward | 5′-GTAACTCCGCAGAAAAGCCAGAC-3′ |
| Reverse | 5′-CAAAAGAAACTAACACACACACA-3′ |
Cell cycle distribution and apoptosis analysis
Propidium iodide (PI) staining was performed to analyze the cell cycle distribution. Cells were seeded in 6-well plates and exposed to different concentrations of EM-2 for 48 h before they were harvested and fixed with 70% ethanol for 12 h at 4 °C. After resuspension in phosphate-buffered solution (PBS) and centrifugation (1500 rpm, 5 min), the cells were stained with PI according to the instructions of the Cell Cycle Analysis Kit (4A Biotech, Beijing, China). Apoptosis was assessed according to the instructions of the Annexin V-FITC/PI apoptosis detection kit (4A Biotech, Beijing, China). Both cell cycle distribution and apoptosis were analyzed using flow cytometry (Becton, Dickinson and Company, VT).
Immunofluorescence assay
Huh-7 cells were seeded at a density of 1 × 105 cells per well on 20 mm glass coverslips that were placed at the bottom of a 12-well plate and allowed to adhere overnight, followed by exposure to EM-2 (0, 8 μM) for 24 h. After washing twice with 1× phosphate-buffered saline, cells were treated with 4% paraformaldehyde for 10 min and 0.1% Triton X-100 for another 10 min, followed by incubation with 10% goat serum (Boster, Wuhan, China) for 1.5 h at 37 °C. Cells were incubated with LC3B/MAP1LC3B (Novus, USA) and p62 primary antibody overnight at 4 °C. Then, the cells were incubated with FITC-conjugated goat anti-rabbit IgG (H + L) (ABclonal, Wuhan, China) or DyLight 649-conjugated goat anti-mouse IgG (H + L) (Abbkine, CA, USA) for 1.5 h at room temperature in the dark. Cell slides were then sealed with Antifade Mounting Medium (with DAPI) (Solarbio, Beijing, China). The fluorescence intensities of LC3 and p62 staining were observed under a confocal laser scanning microscope (Lecia TCS SP8, Germany) or an inverted fluorescence microscope (Lecia DMi8, Germany).
Western blot assay
Cells were homogenized in 1× RIPA buffer (Cell Signaling Technology, MA, USA), and the protein supernatant was separated by centrifugation at 12,000 × g for 15 min at 4 °C. The protein concentrations were determined by using a BCA Protein Assay Kit (Thermo Scientific, Shanghai, China). Protein samples were loaded into SDS-PAGE gels and transferred to 0.45 μm PVDF membranes (Millipore, Billerica, MA, USA). After they were blocked with 5% skimmed milk in 1× TBST for 1 h at room temperature, the membranes were incubated with primary antibody overnight at 4 °C followed by secondary antibody for 1 h at room temperature. The immunoreactive bands were visualized with a Gel Imaging system (Bio-Rad, CA, USA) using an ECL kit (Bio-Rad, CA, USA) and analyzed using ImageJ software.
Statistical analysis
All the data are presented as the mean ± standard error of the mean (SEM). Student’s t test and one-way ANOVA were performed to analyze the significance of the difference between two groups or among multiple groups, respectively. Statistical analysis was performed using SPSS 16.0 (SPSS software, IBM, USA) and GraphPad Prism 7.0 (GraphPad software, San Diego, CA, USA). P values < 0.05 were considered statistically significant.
Results
EM-2 inhibited cell proliferation in human hepatocellular carcinoma cells
EM-2 is a monomeric sesquiterpene lactone extracted from Elephantopusmollis H.B.K., and its chemical structure is depicted in Fig. 1a. To investigate the cytotoxic effects of EM-2 on human liver cancer cell lines (Huh-7, Hep3B, QGY-7703, CRL-8024, and SMMC-772), normal liver epithelial cells (LO2) and normal immortal liver cells (MIHA), were exposed to different concentrations of EM-2 for 48 h. The results from the MTT assay showed that EM-2 significantly inhibited the proliferation of human HCC cell lines in a dose-dependent manner; furthermore, EM-2 showed less cytotoxicity in normal human liver epithelial LO2 cells (Fig. 1b) and exhibited better antitumor effects than other liver cancer chemotherapy drugs, such as oxaliplatin, fluorouracil, sorafenib, or cisplatin (Fig. 1c). The IC50 values of EM-2 against different cell lines after 48 h of treatment are presented in Table 2.
Fig. 1. Cytotoxic effect of EM-2 on human hepatocellular carcinoma cell lines.

a The chemical structure of EM-2. b Cell viability analysis of various human hepatoma cell lines and normal liver epithelial cell lines cultured in the presence of EM-2 as determined by the MTT assay. c Cell viability analysis of Huh-7 cells treated with EM-2 and various liver cancer chemotherapy drugs as determined by the MTT assay. d Huh-7 cells were seeded in 6-well plates at a density of 500 cells per well and treated with different concentrations of EM-2 (0, 0.25, and 0.5 μM) for 7 days. The results represent three independent experiments. e Statistical analysis of colony formation (*** compared with the control group, P ≤ 0.001). f The effects of EM-2 on Huh-7 cell proliferation were determined by EdU staining; the red dots represent the population of daughter cells. After exposure to EM-2 (2 μM) for 24 h followed by EdU staining, the cells were examined under a multifunctional fluorescence microscope. Scale bars, 50 μm. g Statistical analysis of the EdU staining assay (** compared with the control group, P < 0.01).
Table 2.
IC50 of EM-2 against various human hepatocellular carcinoma cell lines and normal liver epithelial cell lines for 48 h.
| Cell lines | IC50 (μM) |
|---|---|
| Hep3B | 8.81 ± 0.11 |
| CRL-8024 | >20 |
| SMMC-7721 | 8.77 ± 0.26 |
| QGY-7703 | 8.55 ± 0.21 |
| Huh-7 | 5.89 ± 0.08 |
| MIHA | 14.45 ± 0.29 |
| LO2 | >20 |
(Means ± standard deviation).
Colony formation and EdU assays were performed to further investigate the inhibitory effect of EM-2 on Huh-7 cell proliferation. The results showed that the numbers of colonies formed and EdU-positive cells significantly decreased with increasing EM-2 concentrations in Huh-7 cells (Fig. 1d–g). Overall, these findings suggested that EM-2 inhibited the proliferation of human HCC cells in a dose-dependent manner.
EM-2 activated the MAPK signaling pathway
To determine which pathway was associated with EM-2 mediated inhibition of HCC cell proliferation, we performed RNA-seq analysis of total RNA extracted from Huh-7 cells with or without EM-2 treatment. The sequencing results were analyzed by GO function and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis, which showed that EM-2 treatment significantly activated the MAPK signaling pathway and tumor necrosis factor (TNF) signaling pathway in Huh-7 cells (Fig. 2a). Furthermore, the Western blotting results showed that EM-2 treatment increased the expression levels of p-ERK1/2, p-JNK, and p-c-Jun in a dose-dependent manner but failed to alter the expression of total ERK1/2, JNK, and c-Jun as well as the levels of p-p38 (Fig. 2b). To further verify whether EM-2 induced increases in p-ERK1/2 levels, we treated Huh-7 cells with 4 μM EM-2 for 0, 3, 6, 12, 24, and 36 h and examined p-ERK1/2 levels by Western blotting. Our results demonstrated that the level of p-ERK1/2 increased in a time-dependent manner upon EM-2 treatment (Fig. 2c). All these data suggested that EM-2 activated the JNK and ERK1/2 proteins of the MAPK signaling pathway in Huh-7 cells.
Fig. 2. EM-2 activated the MAPK signaling pathway.

a KEGG pathway enrichment analysis was performed in Huh-7 cells treated with EM-2 (4 μM). b Huh-7 cells were exposed to EM-2 (0, 2, 4, and 8 μM) for 24 h, and the expression levels of proteins associated with the MAPK pathway were analyzed by Western blotting. c Huh-7 cells were exposed to 4 μM EM-2 for 0 to 36 h, and the protein levels of ERK and p-ERK were analyzed by Western blotting. Representative immunoblot of an experiment repeated three times is shown.
EM-2 induced G2/M arrest and apoptosis in Huh-7 hepatocellular carcinoma cells
To further investigate the effect of EM-2 on the physiological functions of Huh-7 cells, an Annexin V/PI double staining assay was performed to examine the apoptosis activity of Huh-7 cells upon EM-2 treatment. The results showed that apoptosis was induced by EM-2 in a concentration-dependent manner in Huh-7 cells after 48 h of treatment, and the apoptosis rate was up to 83.94% when treated with 8 μM EM-2 (Fig. 3a, c). Cell cycle progression was also analyzed by flow cytometry. The results showed that treatment with EM-2 for 24 h significantly induced G2/M arrest in Huh-7 cells based on the significant increase in the G2/M fraction in a concentration-dependent manner, but the S fraction had little change, and the G0/G1 fraction decreased (Fig. 3b, d). In addition, the expression levels of proteins associated with apoptosis and the cell cycle were detected by Western blot assay. We found that after treatment with EM-2 for 24 h, the expression levels of Bcl-2, Caspase-9, and Caspase-3 decreased but those of cl-Caspase-3, cl-Caspase-9 and cl-PARP increased. The expression levels of Cyclin A2 and Cyclin B1 increased while those of CDK4 and Cyclin D1 decreased (Fig. 3e, f). In addition, we also observed an obvious increase in the levels of p-cdc2, Wee1, and Myt1, while there was no change in the level of total cdc2. Interestingly, our results suggested that although the expression of p21 and p53 were both elevated as the concentration of EM-2 increased, the increase in p21 expression was much more significant than that of p53 (Fig. 3f). Taken together, these results suggested that EM-2 induces G2/M arrest and apoptosis in Huh-7 cells in a concentration-dependent manner.
Fig. 3. EM-2 induced G2/M arrest and apoptosis in Huh-7 hepatoma cells.

a Huh-7 cells were exposed to EM-2 (0, 2, 4, and 8 μM) for 48 h, and Annexin V-FITC/PI staining analysis was conducted to evaluate the percentage of apoptotic cells using flow cytometry. b Huh-7 cells were exposed to EM-2 (0, 2, 4, and 8 μM) for 24 h, and PI staining analysis was conducted to analyze the cell cycle distribution using flow cytometry. c, d The percentage of apoptotic cells and cell cycle distribution fractions of Huh-7 cells are displayed as the mean ± standard deviation of triplicate determinations (** compared with control group, P < 0.01; *** compared with control group, P < 0.001). e, f Huh-7 cells were exposed to 0, 2, 4, or 8 μM EM-2 for 24 h. The protein expression levels of genes associated with apoptosis and the cell cycle were analyzed by Western blot analysis. Representative immunoblot of an experiment repeated three times is shown.
EM-2 induced apoptosis and G2/M arrest in Huh-7 cells by activating JNK
Previous studies reported that the MAPK signaling pathway is involved in regulating numerous cellular processes, such as cell growth and proliferation, cycle, migration, differentiation, and cell death, in eukaryotes [19]. To investigate the relationship among MAPK family proteins, apoptosis, and G2/M phase arrest in Huh-7 cells after EM-2 treatment, we further examined Huh-7 cell apoptosis levels and the cell cycle distribution in the presence of the MEK inhibitor trametinib and/or the JNK inhibitor SP600125.
The flow cytometry and MTT assay results showed that Huh-7 cell apoptosis levels increased and cell viability decreased in the (Trametinib+, EM-2+) group compared with those of the (Trametinib−, EM-2+) group (Fig. 4a, c, d). Additionally, the results from the Western blot analysis showed that the protein levels of cl-PARP increased in the (Trametinib+, EM-2+) group compared with the (Trametinib−, EM-2+) group (Fig. 4g). This result suggested that ERK activation induced by EM-2 in Huh-7 cells was cytoprotective, and suppression of ERK activity could render Huh-7 cells sensitive to EM-2. However, when Huh-7 cells were pretreated with the JNK inhibitor SP600125 before exposure to EM-2, opposing results were obtained. Compared with the (SP600125−, EM-2+) group, the (SP600125+, EM-2+) group showed decreased apoptosis levels and increased cell viability, which revealed that EM-2 induced apoptosis by activating JNK (Fig. 4b, e, f). Furthermore, consistent with the flow cytometry and MTT results, the protein levels of p21 and cl-PARP were decreased upon SP600125 treatment (Fig. 4h). The above results indicated that EM-2 induced Huh-7 cell apoptosis by activating JNK.
Fig. 4. EM-2 induced apoptosis and G2/M arrest in Huh-7 cells by activating JNK.

a, b Apoptosis levels were analyzed by the Annexin V-FITC/PI staining assay in Huh-7 cells with or without trametinib (a) and SP600125 (b) pretreatment prior to EM-2 (4 μM) exposure for 48 h. c, e The percentage of apoptotic Huh-7 cells with or without trametinib (c) and SP600125 (e) pretreatment are displayed as the mean ± standard deviation of triplicate determinations (**P < 0.01; ***P < 0.001). d, f Cell viability was detected by the MTT assay in Huh-7 cells with or without trametinib (d) and SP600125 (f) pretreatment prior to EM-2 (4 μM) exposure for 48hr. g, h Protein expression levels were examined by Western blot in Huh-7 cells with or without trametinib (g) and SP600125 (h) pretreatment prior to EM-2 (4 μM) exposure for 24 h. All experiments were repeated three times.
EM-2 induced endoplasmic reticulum stress in Huh-7 hepatocellular carcinoma cells
There are many stimulating factors that can activate the JNK/SPAK pathway, such as TNF, ROS, and endoplasmic reticulum (ER) stress. As one of the most important cellular organelles, the ER plays a critical role in maintaining cell homeostasis. Triggered by numerous intracellular and extracellular factors, ER stress usually causes disruption of cellular homeostasis, which can be salvaged by an adaptive mechanism called the unfolded protein response (UPR) in order to maintain cell survival [20]. However, the UPR may also promote apoptosis, and increasing evidence has reported that ER stress causes dimerization and autophosphorylation of inositol requiring-enzyme 1 alpha (IRE1α), which triggers the activation of ASK1 (apoptotic signaling kinase 1) and thereby activates JNK [21]. Therefore, to demonstrate whether ER stress was activated in Huh-7 cells upon EM-2 treatment, the protein expression levels of ER stress-related proteins were examined by Western blotting. The increased levels of p-IRE1α, p-eIF2α, ATF4, and Bip indicated that EM-2 could induce ER stress (Fig. 5a). Meanwhile, the mRNA levels of these proteins were detected by RT-qPCR in Huh-7 cells treated with different concentrations of EM-2. Consistent with the Western blotting results, the mRNA levels of ER stress-related genes, such as EIF2AK3(PERK), ERN1(IRE1α), DDIT3(CHOP), HSPA5(Bip), SERP1, ATF6, and ATF4, significantly increased with increasing EM-2 concentrations (Fig. 5b). These findings demonstrated that EM-2 induced ER stress in Huh-7 HCC cells.
Fig. 5. EM-2 induced endoplasmic reticulum stress in Huh-7 hepatoma cells.

a Huh-7 cells were exposed to 0, 2, 4, or 8 μM EM-2 for 24 h. The protein expression levels of proteins associated with endoplasmic reticulum stress were detected by Western blotting. b Huh-7 cells were exposed to 0, 2, 4, or 8 μM EM-2 for 24 h. The mRNA expression levels of ER stress-related pathway genes were analyzed by RT-qPCR. All experiments were performed in three independent replicates (*P < 0.05; **P < 0.01; ***P < 0.001).
EM-2 blocked autophagy flux in Huh-7 cells by inhibiting autolysosome maturation and cell proliferation
Accumulating evidence suggests that blocking autophagic flux activates ER stress [22, 23]. We also observed that EM-2 treatment could induce ER stress in Huh-7 cells (Fig. 5), so we speculated that EM-2 treatment also has a similar effect on autophagy flux to activate ER stress. To determine whether autophagy is involved in apoptosis and G2/M arrest in Huh-7 cells treated with EM-2, the expression of proteins associated with autophagy was detected by Western blotting. The results showed that the protein expression levels of LC3-II increased with increasing EM-2 concentrations (Fig. 6a). Similarly, an enormous increase in LC3 fluorescence intensity was observed in Huh-7 cells treated with 8 μM EM-2 compared to cells in the control group (Fig. 6c). Interestingly, we also found that the fluorescence intensity and protein expression levels of p62 increased significantly in a dose-dependent manner upon EM-2 treatment (Fig. 6a, c). Meanwhile, the expression levels of precursor-Cathepsin L increased, while the expression levels of mature-Cathepsin L decreased in EM-2-treated Huh-7 cells, which indicated that EM-2 inhibited autophagy-mediated lysosome maturation (Fig. 6b). Moreover, pretreatment with bafilomycin A1, an autophagy inhibitor, significantly aggravated the damage to Huh-7 cell viability caused by EM-2 (Fig. 6d), which means autophagy promoted cell survival, and blocking autophagy promoted cell death. These results revealed that EM-2 blocked autophagy by inhibiting autophagy-mediated lysosome maturation.
Fig. 6. EM-2 inhibited autophagy in Huh-7 cells by blocking the fusion of autophagosomes with lysosomes.

a, b Huh-7 cells were exposed to 0, 2, 4, or 8 μM EM-2 for 24 h. The protein expression levels of genes associated with autophagy were analyzed by Western blotting. c Immunostaining of LC3 and p62 in Huh-7 cells treated with different concentrations of EM-2 (0 and 8 μM). LC3 staining was imaged under a confocal laser scanning microscope, and p62 staining was imaged by an inverted fluorescence microscope. d Huh-7 cells with or without 25 μM bafilomycin A1 pretreatment were exposed to 4 μM EM-2 for 48 h, and cell viability was detected by the MTT assay. All experiments were performed in three independent replicates (*P < 0.05).
Discussion
Liver cancer is the third most common cancer in China, and every year, 50% of global new cases are in China [1]. Although there are various chemotherapeutic drugs available for the clinical treatment of liver cancer, poor prognosis and high recurrence rate have always been impending problems due to the acquired resistance of HCC cells to chemotherapeutic drugs [5]. In this study, we first demonstrated that EM-2, a novel natural monomer extracted from Elephantopusmollis H.B.K., had a strong inhibitory effect on the proliferation of human hepatoma cell lines in a dose-dependent manner but exerted little cytotoxicity in LO2 normal liver epithelial cells (Fig. 1b). We also observed that EM-2 had a better antitumor effect than some liver cancer chemotherapeutic drugs, such as oxaliplatin, fluorouracil, sorafenib, or cisplatin (Fig. 1c). The lowest IC50 value (5.89 ± 0.08 μM) was observed in Huh-7 cells upon treatment with EM-2 for 48 h (Table 1). Moreover, the numbers of EdU-positive cells and cell colonies in EM-2-treated Huh-7 cells were significantly reduced compared with those in the control cells, which further confirmed the inhibitory effect of EM-2 on hepatoma cell proliferation (Fig. 1d–g).
To determine the underlying mechanisms involved in the inhibition of hepatoma cell proliferation induced by EM-2, total RNA from Huh-7 cells with or without EM-2 treatment was extracted and sequenced. The sequencing results were analyzed by GO function and KEGG enrichment analysis, and the results showed that EM-2 treatment significantly activated the MAPK signaling pathway in Huh-7 cells (Fig. 2a). Meanwhile, we investigated the effects of EM-2 on the physiological functions of Huh-7 cells, including apoptosis activity and cell cycle arrest. As the EM-2 concentration increased, the number of Annexin V-positive cells and the levels of cl-Caspase-3, cl-Caspase-9 and cl-PARP were increased in addition to the Bax/Bcl-2 ratio (Fig. 3a, e). Moreover, we also found that the G2/M fraction in EM-2-treated Huh-7 cells was significantly increased while the G0/G1 fraction was decreased as the EM-2 concentration increased. Nevertheless, EM-2 treatment had little effect on the S fraction (Fig. 3b, d). Moreover, the protein expression levels of Cyclin A2 and Cyclin B1 were upregulated, but Cyclin D1 and CDK4 expression levels were downregulated upon EM-2 treatment. Cdc2 expression showed no obvious changes, but the levels of p-cdc2 (Tyr15), Wee1 and Myt1 increased in an EM-2 concentration-dependent manner (Fig. 3f). Interestingly, we found that the expression levels of p21 increased significantly, but the change in the level of its upstream protein p53 was modest, suggesting that the increase in p21 expression was independent of the expression of p53 in Huh-7 cells, which was probably because Huh-7 is a p53-mutant cell line [24]. Previous studies showed that activation of the TNF-α signaling pathway is an important factor in determining p21 expression and stabilization [25]. In our findings, EM-2 treatment significantly activated the TNF signaling pathway in Huh-7 cells (Fig. 2a) and thus may be indicative of p21 expression via activation of the TNF-α pathway in a p53-independent manner.
C-Jun N-terminal kinases (JNKs) and ERKs are members of the MAPK family, which can be activated by multiple cellular stressors and cytokines. As serine–threonine kinases, both play pivotal roles in signaling cascades to perform diverse biological functions and exert physiological effects under both normal and pathological conditions. ERK hyperactivation plays an important role in cancer development and progression, including tumor proliferation, invasion, metastasis, and angiogenesis [26]. JNK is generally activated by cellular stress and closely associated with cell death [16, 17, 27]. In this study, we found that EM-2 treatment could activate JNK and ERK but not p38 in Huh-7 hepatoma cells (Fig. 2b, c). Pretreatment with trametinib, an ERK inhibitor, decreased cell viability and increased the number of Annexin V-positive Huh-7 cells and the level of cl-PARP, indicating that ERK activation is a prosurvival mechanism in EM-2-treated Huh-7 cells. When JNK was inhibited by SP600125, the apoptosis level decreased and cell viability increased in Huh-7 cells (Fig. 4). In addition, the protein levels of p21 and cl-PARP decreased in the presence of SP600125 (Fig. 4h). These results suggested that apoptosis is mediated by JNK activation in EM-2-treated Huh-7 hepatoma cells.
Previous studies reported that ER stress caused dimerization and autophosphorylation of IRE1α, which triggers the activation of ASK1 and thereby activates JNK [21]. Thus, to determine whether the ER stress response in Huh-7 cells was activated by EM-2 treatment, the mRNA and protein expression of ER stress-related genes was detected. With increasing concentrations of EM-2 in Huh-7 hepatoma cells, the protein levels of p-IRE1α, p-eIF2α, ATF4, and Bip increased, and the mRNA levels of EIF2AK3(PERK), ERN1(IRE1α), HSPA5(Bip), DDIT3(CHOP), ATF6, ATF4, and SERP1 also increased significantly (Fig. 5). These results suggested that EM-2 treatment could induce ER stress in hepatoma Huh-7 cells.
Autophagy is a basic physiological process in cells and is usually activated when cells are exposed to environmental stresses such as hypoxia, malnutrition, chemicals, and radiation [28, 29]. Hoo et al. found that impaired autophagy could promote ER stress, which in turn activates proinflammatory signaling pathways, increases the production of inflammatory cytokines and eventually contributes to the development of obesity-related inflammatory diseases [30]. In addition, diminished autophagy activity with aging contributes to increased ER stress and inflammation in adipose tissue [23]. In this study, we demonstrated that the protein level of LC3-II and the fluorescence intensity of LC3 increased significantly, indicating that autophagy occurred in Huh-7 cells upon EM-2 treatment. Interestingly, the fluorescence intensity and protein expression levels of p62 also drastically increased, suggesting that EM-2 blocks autophagic flux (Fig. 6a, c). Furthermore, the expression of mature-cathepsin L decreased with increasing EM-2 concentrations (Fig. 6b). We also observed that cells cotreated with EM-2 and bafilomycin A1 showed lower cell viability than those treated with EM-2 or bafilomycin A1 alone (Fig. 6d). These results indicated that EM-2 blocked autophagy flux in Huh-7 cells by inhibiting autophagy-mediated lysosome maturation.
Conclusion
We discovered that EM-2 significantly inhibited the proliferation of HCC cells with few adverse effects on normal liver epithelial cells. In addition, we also indicated that EM-2 could block autophagic flux, induce ER stress, and promote apoptosis by activating JNK in Huh-7 hepatoma cells. Meanwhile, EM-2 treatment upregulated the expression of p21 and caused G2/M phase arrest (Fig. 7). Overall, this study illustrated that EM-2 had a promising antitumor effect on HCC cells, which provided an experimental basis for the future application of EM-2 in the clinical treatment of HCC.
Fig. 7. Schematic demonstrating the molecular mechanism of the antitumor effects of EM-2 on hepatocellular carcinoma cells.
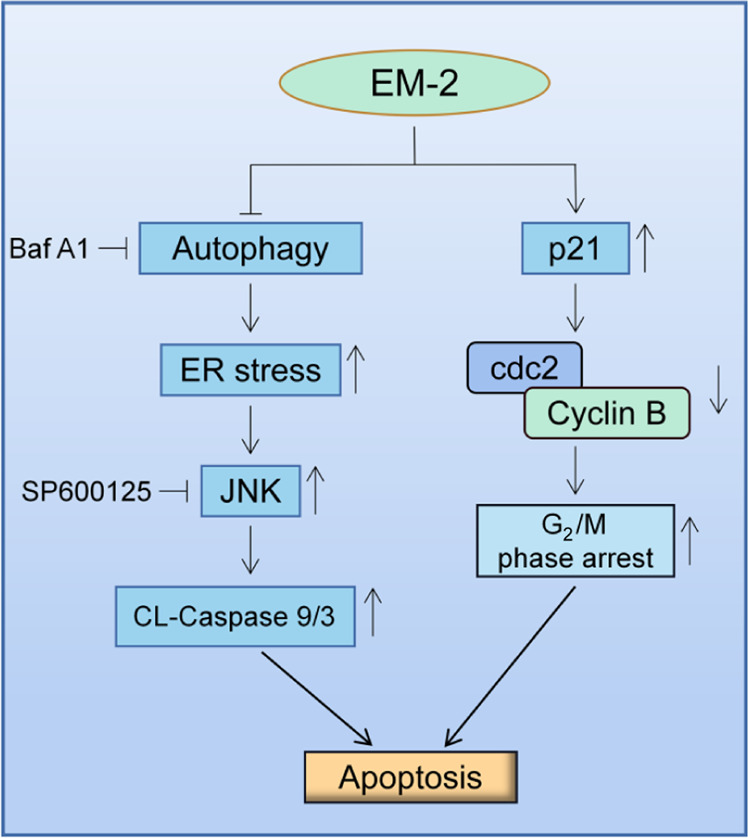
EM-2 activated the JNK and Caspase-9/3 signaling pathways to trigger apoptosis by blocking autophagic flux and inducing endoplasmic reticulum stress. Meanwhile, EM-2 promoted p21 expression and then inhibited cdc2/Cyclin B complex activity, causing G2/M phase arrest. JNK C-Jun N-terminal kinase, ER endoplasmic reticulum, Baf A1 Bafilomycin A1.
Acknowledgements
This research was supported by the Science and Technology Project of Guangdong Province (No. 2016ZC0044), the Science and Technology Planning Project of Guangdong Province (No. 2018B030320007), the Clinical research projects of the first affiliated hospital of Jinan university (No. 2018008, 2019315). Meanwhile, this research was also supported by the Guangzhou Science and Technology Program (No. 202002030087) and the National Natural Science Foundation of China (No. 82074064).
Author contributions
JWJ and XFZ contributed to the conception of the study. JY and ZDL contributed to the design, performance, and data analysis of the experiments. CYH and ZYL contributed to the writing and submitting of the paper. CYH contributed to the revise of this paper. XYZ and QL contributed to the collection and assembly of data. SYM and QZ contributed to the typesetting and correction of the paper. XFZ and JWJ contributed to the critical revision and final approval of the article.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Jie Yang, Zhen-dong Li.
Contributor Information
Xiao-feng Zhu, Email: zhuxfeng@mail.sysu.edu.cn.
Jian-wei Jiang, Email: jjw703@jnu.edu.cn.
References
- 1.Wu J. The changing epidemiology of hepatocellular carcinoma in Asia versus United States and Europe. Adv Mod Oncol Res. 2017;3:51–8. doi: 10.18282/amor.v3.is1.182. [DOI] [Google Scholar]
- 2.Jean-Luc R, Marine G. Hepatocellular carcinoma: slow progress in a booming epidemic. J Oncol Pract. 2017;13:365–6. doi: 10.1200/JOP.2017.024364. [DOI] [PubMed] [Google Scholar]
- 3.Shiffman ML. The next wave of hepatitis C virus: the epidemic of intravenous drug use. Liver Int. 2018;38:34–9. doi: 10.1111/liv.13647. [DOI] [PubMed] [Google Scholar]
- 4.Niu J, Lin Y, Guo Z, Niu M, Su C. The epidemiological investigation on the risk factors of hepatocellular carcinoma: a case-control study in Southeast China. Medicine. 2016;95:e2758. doi: 10.1097/MD.0000000000002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeff H, Ahmed K, Economides MP, Manal H, Harrys T. Effect of sorafenib on hepatitis C viremia in cirrhotic patients with hepatocellular carcinoma. OFID. 2016;3:459. doi: 10.1093/ofid/ofw172.323. [DOI] [Google Scholar]
- 6.Francine G, Sylvain G, Stamatiki R, Annelise L, Barbara F, Nikolaus S, et al. Chemopreventive properties of apple procyanidins on human colon cancer-derived metastatic SW620 cells and in a rat model of colon carcinogenesis. Carcinogenesis. 2005;26:1291–5. doi: 10.1093/carcin/bgi074. [DOI] [PubMed] [Google Scholar]
- 7.Dai X, Yin C, Zhang Y, Guo G, Zhao C. Osthole inhibits triple negative breast cancer cells by suppressing STAT3. J Exp Clin Cancer Res. 2018;7:322. doi: 10.1186/s13046-018-0992-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabopda TK, Ngoupayo J, Liu J, Shaiq Ali M, Khan SN, Ngadjui BT, et al. Further cytotoxic sesquiterpene lactones from Elephantopus mollis KUNTH. Chem Pharm Bull. 2008;56:231–3. doi: 10.1248/cpb.56.231. [DOI] [PubMed] [Google Scholar]
- 9.Gibert-Tisseuil F. Reflections on traditional Chinese medicine and its pharmacopoeia. Ann Pharm Fr. 1998;56:282. [PubMed] [Google Scholar]
- 10.Hasegawa K, Furuya R, Mizuno H, Umishio K, Sato K. Inhibitory effect of Elephantopus mollis HB and K. Extract on melanogenesis in B16 murine melanoma cells by downregulating microphthalmia-associated transcription factor expression. Biosci Biotechnol Biochem. 2010;74:1908–12.. doi: 10.1271/bbb.100318. [DOI] [PubMed] [Google Scholar]
- 11.Ooi KL, Muhammad TST, Mei LT, Sulaiman SF. Cytotoxic, apoptotic and anti-α-glucosidase activities of 3,4-di-O-caffeoyl quinic acid, an antioxidant isolated from the polyphenolic-rich extract of Elephantopus mollis Kunth. J Ethnopharmacol. 2011;135:685–95.. doi: 10.1016/j.jep.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Ooi KL, Muhammad TST, Lam LY, Sulaiman SF. Cytotoxic and apoptotic effects of ethyl acetate extract of Elephantopus mollis Kunth. In human liver carcinoma HepG2 cells through caspase-3 activation. Integr Cancer Ther. 2014;13:NP1–9. doi: 10.1177/1534735411433203. [DOI] [PubMed] [Google Scholar]
- 13.Shao F, Wang S, Li H, Chen W, Wang G, Ma D, et al. EM23, a natural sesquiterpene lactone, targets thioredoxin reductase to activate JNK and cell death pathways in human cervical cancer cells. Oncotarget. 2016;7:6790–808.. doi: 10.18632/oncotarget.6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Low HB, Zhang Y. Regulatory roles of MAPK phosphatases in cancer. Immune Netw. 2016;16:85–98. doi: 10.4110/in.2016.16.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways medicated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 16.Xu Z, Zhang F, Bai C, Yao C, Zhong H, Zou C, et al. Sophoridine induces apoptosis and S phase arrest via ROS-dependent JNK and ERK activation in human pancreatic cancer cells. J Exp Clin Cancer Res. 2017;36:124. doi: 10.1186/s13046-017-0590-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mhaidat NM, Thorne R, Zhang XD, Hersey P. Involvement of endoplasmic reticulum stress in docetaxel-induced JNK-dependent apoptosis of human melanoma. Apoptosis. 2008;13:1505–12.. doi: 10.1007/s10495-008-0276-8. [DOI] [PubMed] [Google Scholar]
- 18.Tan G, Wang X, Tang Y, Cen W, Li Z, Wang G, et al. PP-22 promotes autophagy and apoptosis in the nasopharyngeal carcinoma cell line CNE-2 by inducing endoplasmic reticulum stress, downregulating STAT3 signaling, and modulating the MAPK pathway. J Cell Physiol. 2019;234:2618–30.. doi: 10.1002/jcp.27076. [DOI] [PubMed] [Google Scholar]
- 19.Zhang W, Liu H. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 20.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Wu Q, Ding Y, Zhou W, Liu R, Chen H, et al. YD277 suppresses triple-negative breast cancer partially through activating the endoplasmic reticulum stress pathway. Theranostics. 2017;7:2339–49.. doi: 10.7150/thno.17555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh AK, Mau T, O’Brien M, Garg S, Yung R. Impaired autophagy activity is linked to elevated ER-stress and inflammation in aging adipose tissue. Aging. 2016;8:2525–37.. doi: 10.18632/aging.101083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biczo G, Vegh ET, Shalbueva N, Mareninova OA, Elperin J, Lotshaw E, et al. Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models. Gastroenterology. 2018;154:689–703. doi: 10.1053/j.gastro.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanno SI, Kurauchi K, Tomizawa A, Yomogida S, Ishikawa M. Pifithrin-alpha has a p53-independent cytoprotective effect on docosahexaenoic acid-induced cytotoxicity in human hepatocellular carcinoma HepG2 cells. Toxicol Lett. 2015;232:393–402. doi: 10.1016/j.toxlet.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi N, Takada Y, Hachiya M, Ando K, Nakajima N, Akashi M. TNF-alpha induced p21(WAF1) but not Bax in colon cancer cells WiDr with mutated p53: important role of protein stabilization. Cytokine. 2000;12:1745–54.. doi: 10.1006/cyto.2000.0782. [DOI] [PubMed] [Google Scholar]
- 26.Guo Y, Pan W, Liu S, Shen Z, Hu L. ERK/MAPK signalling pathway and tumorigenesis (Review) Exp Ther Med. 2020;19:1997–2007. doi: 10.3892/etm.2020.8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ballif BA, Blenis J. Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ. 2001;12:397–408. [PubMed] [Google Scholar]
- 28.Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol. 2010;21:683–90.. doi: 10.1016/j.semcdb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–30.. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoo RLC, Shu L, Cheng KKY, Wu X, Liao B, Wu D, et al. Adipocyte fatty acid binding protein potentiates toxic lipids-induced endoplasmic reticulum stress in macrophages via inhibition of janus kinase 2-dependent autophagy. Sci Rep. 2017;7:40657. doi: 10.1038/srep40657. [DOI] [PMC free article] [PubMed] [Google Scholar]