

Abstract
Hepatocellular carcinoma (HCC) is among the most common cancer types worldwide; yet, patients with HCC have limited treatment options. There is an urgent need to identify new drug targets that specifically inhibit the growth of HCC cells. Here, we used a newly-engineered CRISPR library targeting ~2,000 druggable genes to perform a high throughput screen, and identified adenylosuccinate lyase (ADSL) – a key enzyme involved in the de novo purine synthesis pathway – as a potential drug target for HCC. ADSL has been implicated as a potential oncogenic driver in some cancers, but its role in liver cancer progression remains unknown. CRISPR-mediated knockout of ADSL impaired colony formation of liver cancer cells by affecting adenosine monophosphate (AMP) production. In the absence of ADSL, the growth of liver tumors is retarded in vivo. Mechanistically, we found that ADSL knockout caused S-phase cell cycle arrest, not by inducing DNA damage, but by impairing mitochondrial function. Using HCC patient data, we also revealed that high ADSL expression occurs during tumorigenesis and is linked to poor survival rate. In conclusion, our findings uncover the role of ADSL-mediated de novo purine synthesis in fueling mitochondrial ATP production to promote liver cancer cell growth. Targeting ADSL may be a therapeutic approach for HCC patients.
Keywords: Hepatocellular Carcinoma, CRISPR, Cell Cycle, ATP, Mitochondria
Introduction
Hepatocellular carcinoma (HCC), the most common type of adult primary liver cancer, is caused by uncontrolled proliferation and growth of normally quiescent hepatocytes (1, 2). The 5-year survival rate for HCC patients is only 18%, making HCC the second most lethal cancer in the world (3). Standard treatments for HCC include surgical resection, liver transplantation, chemotherapy, and molecular targeted therapy. However, surgical resection and liver transplantation are only available to patients at the early stage of cancer development. Molecular targeted therapy, on the other hand, could be used to treat patients with advanced liver cancer (4). To date, the only FDA-approved targeted therapies for HCC are tyrosine kinase inhibitors, which, on average, can only extend patient life span by 1 year (5). Molecular aberrations, such as genetic mutations, are often considered as potential therapeutic targets for cancers. Yet, very few “actionable” somatic mutations are associated with HCC (6). Thus, it is imperative to utilize new methods to identify and verify novel drug targets that can effectively inhibit HCC progression.
To support cell proliferation, neoplastic cells have increased metabolic demand for nucleotide biosynthesis (7). Based on the source of substrates, nucleotides are synthesized by two different pathways: salvage pathway and de novo pathway. The De novo pathway provides the majority of nucleotides needed during cell division and growth (8, 9). Dysregulation of the enzymes involved in the de novo pathway are often associated with cancer development (10–12), indicating the critical role of de novo nucleotide synthesis in cancer progression. ADSL encodes the only enzyme that catalyzes two distinct steps in the de novo purine synthesis pathway that produces adenosine monophosphate (AMP). Knocking out ADSL decreases AMP levels in cells (13). Its expression is up-regulated in several cancer types, including prostate cancer (14), triple-negative breast cancers (13), and endometrioid carcinoma (15). The mechanism of how ADSL promotes breast cancer growth was recently revealed – it controls the protein expression level of an oncogene, c-Myc, to promote tumorigenesis(13). However, its role in HCC development remains largely elusive.
Here, we engineered a novel guide RNA library targeting the druggable genome. By using a CRISPR/Cas9-based method, we screened the essentiality of ~2,000 druggable genes in mouse liver cancer growth, and identified ADSL as a top-ranked gene. ADSL is indispensable in all tested liver cancer cells, regardless of genetic background. Knocking out ADSL caused cancer cell cycle arrest at S phase. Mechanistically, ADSL depletion greatly reduced intracellular ATP level in HCC cells via disruption of mitochondrial ATP production. ADSL-depleted cells exhibited impaired mitochondrial function – i.e., mitochondrial elongation, increased expression of mitochondrial stress response protein, and decreased oxygen consumption rate. By analyzing patient data, we found that ADSL is specifically overexpressed in HCC tumor tissues and patients with higher ADSL expression level have lower survival rates. Our findings uncover a novel role of ADSL in maintaining mitochondrial function and cell cycle progression, and nominate a potential drug target for HCC treatment.
Materials and Methods
CRISPR/Cas9-based high throughput screen
p53−/−; H-RASG12V mouse liver cancer cells were infected with lentiviral Cas9 twice, and selected with blasticidin. 2x107 mouse cells with stably-expressed Cas9 were infected with the druggable genome library. Triplicates were performed and the initial infection rate was controlled at around 50% (by determining GFP-positive cell population) for each independent experiment to ensure ~600x presentation of the library. After culturing for 20days, genomic DNA was harvested using the PureLink Genomic DNA mini kit (Invitrogen). Single-guide RNA (sgRNA) was amplified using Phusion Flash High-Fidelity PCR Master Mix (Thermo Fisher Scientific). All sequencing data sets were evaluated using FastQC (version 0.11.2) to ensure high quality. Depleted genes were identified as previously described (16). All sequencing primers are listed in Supporting Table S1.
Colony formation
p53−/−; Myc or p53−/−; H-RASG12V mouse liver cancer cells were seeded at 2,000 cells per well onto 6-well plates. After 7 days, cells were fixed with 4% formalin, and stained with 0.5% crystal violet. Huh7, Hep3B, SK-HEP1, SNU449, and HLE cells were seeded at 3,000 cells per well onto 6-well plates. H2.35 cells were seeded at 1x105 cells per well onto 6-well plate. After 12 days, cells were fixed with 4% formalin and stained with 0.5% crystal violet. To quantify colony formation results, cells were incubated with 100% methanol for 15 mins after staining with crystal violet. Next, 100 μL of supernatant were transferred to transparent 96-well plates (Costar). The absorbance was measured with a microplate reader at a wavelength of 570 nm to determine relative colony formation number.
Immunoblot Analysis
Post-selected cells were lysed with RIPA buffer (Boston bioproducts) supplemented with protease inhibitor (Roche) and phosphatase inhibitor (Thermo Fisher). Protein concentration was measured by BCA assay kit (Thermo Fisher). Equal amounts of protein were loaded onto NuPAGE™ 4-12% Bis-Tris Protein Gels (Invitrogen) and run at 125 V for 90 mins. Protein was transferred to nitrocellulose membrane and incubated with indicated antibodies: anti-ADSL (sc-365623, Santa Cruz), anti-HSP90 (610419, BD Biosciences), anti-cleaved Caspase3 (CC3) (9661, Cell Signaling), anti-phospho-H2AX (9718, Cell Signaling), anti-cyclins (9869, Cell Signaling), or anti-CDKs (9868, Cell Signaling), followed by incubation with distinct fluorophore-conjugated secondary antibodies. Images were captured using the Odyssey system (Li-Cor Biosciences).
RNA extraction and quantitative real-time PCR
RNA was extracted from post-selected cells with the RNeasy min kit (Qiagen). One microgram of RNA was used to synthesize complementary DNA (cDNA) using the high-capacity cDNA reverse-transcription kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Real-time quantitative PCR analysis was performed with SsoFast EvaGreen Supermix (Bio-rad). GAPDH served as an internal control for real-time PCR. All the primers for real-time PCR are listed in Supporting Table S1.
Animals
All animal protocols were approved by the UMass Medical School Institutional Animal Care and Use Committee (IACUC). 10 μg pT3-EF1α-Myc (Addgene 92046); 18 μg sgp53 (17) or 10 μg CTNNB1-N90 (Addgene 31785); and 6 μg CMV-SB10 transposase were delivered with px330.sgNT or px330.sgAdsl to 7-10-week-old C57BL/6 mice by hydrodynamic tail vein injection. Plasmid DNAs were prepared with EndoFree Maxiprep DNA Kit (Qiagen). Five to six weeks after injection, mice were humanely euthanized by CO2 asphyxiation. The number of surface tumors were counted. Mouse livers were fixed in 10% (v/v) formalin overnight, washed in 70% ethanol, and embedded in paraffin. 4 μm liver sections were stained with hematoxylin and eosin (H&E).
Flow cytometry analysis
For cell cycle analysis, post-selected SK-HEP1, Huh7, and Hep3B cells were trypsinized and fixed with 70% ethanol overnight. To analyze the impact of mitochondrial function on the cell cycle, SK-HEP1 cells were pre-treated with 0.3 μM oligomycin for 2 days. To perform Aphidicolin synchronization, Huh7 cells were pre-treated with 3 ng/μL Aphidicolin for 24 h, washed with warm culture medium twice, and incubated in fresh culture medium for 18 h before being stained for cell cycle marker. After fixation, cells were washed with PBS twice, and stained with 50ng/mL propidium iodide (PI) (Thermo Fisher Scientific) and 10 μg/mL RNaseA (Thermo Fisher Scientific). Cells were then incubated at 37°C for 30 mins. For each sample, 50,000 cells were analyzed using the MACSQuant VYB Flow Cytometer. All data were analyzed by FlowJo10.0 software. To compare cell cycle differences mediated by ADSL depletion, we used the same gating setting between different experimental groups to define G1 phase. The cell population in S and G2/M phase was calculated by FlowJo Cell Cycle Analysis program (Dean-Jett-Fox model).
Intracellular ATP level measurement
SK-HEP1, Huh7, and Hep3B were trypsinized and washed with PBS. 2x105 cells were used to measure intracellular ATP level by the ATP Assay Kit (Abcam). To measure the ATP produced by glycolysis or oxidative phosphorylation, SK-HEP1 cells were pre-treated with 1 μM oligomycin or 25 μM 2-Deoxy-D-glucose for 4 hours, followed by measurement of intracellular ATP level using the ATP Assay Kit. ATP produced by glycolysis= total ATP in untreated cells - total ATP in oligomycin-treated cells; ATP produced by mitochondria = total ATP in untreated cells - total ATP in Deoxy-D-glucose-treated cells.
Measurement of mitochondrial function
Oxygen consumption was conducted using a Seahorse Extracellular Flux Analyzer XFe96 as previously described (18). Huh7 and SK-HEP1 cells transduced with non-targeting guide RNA (sgNT) or guide RNAs targeting Adsl (sgAdsl.1 and sgAdsl.2) were seeded on Seahorse cell culture microplates (Agilent) at a density of 8,000 and 12,000, respectively. After more than 24h, the oxygen consumption rate (OCR) was determined in a Seahorse XFe96 extracellular flux analyzer. The plates were subjected to analysis using 1 μM oligomycin, 1.5 μM FCCP and 0.5 μM rotenone/antimycin as indicated. Basal respiration and respiration capacity were determined.
Analysis of online gene expression databases
Two HCC databases, GSE 14520 (19) and The Cancer Genome Atlas (TCGA) HCC datasets, were used to compare gene expression levels between tumor and nontumor tissue. The GSE 14520 data set was analyzed using GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/). Gene expression data of TCGA HCC tumors and matched non-tumors were obtained from the University of California, Santa Cruz Xena (http://xena.ucsc.edu/). This dataset was also used for gene set enrichment analysis (GSEA). Specifically, 50 tumor samples with the highest and lowest expression of ADSL mRNA were analyzed for the indicated gene set with the Broad Institute GSEA webserver (http://software.broadinstitute.org/gsea/msigdb). For survival analysis, fragments per kilobase of exon per million fragments mapped (FKPM) values and survival data were obtained from Human Protein Atlas and cBioPortal (http://www.cbioportal.org/), respectively. P values were calculated using the log-rank Mantel-Cox test with GraphPad Prism8.
Results
CRISPR/Cas9-based high throughput screen identifies Adsl as an essential gene in mouse liver cancer growth.
To explore new drug targets for HCC, we designed an sgRNA library targeting the druggable genome. The library contains ~12,000 sgRNAs targeting 2,007 mouse genes (six sgRNAs per gene; guide RNA sequences are listed in Supporting Table S2) that are known targets of existing drug compounds (“drugged” genes) or that belong to gene categories predicted to be druggable (Fig. 1A). We infected p53−/−; H-RASG12V mouse liver cancer cells (20) with this CRISPR library, and cultured cells for 20 days (7 passages). Next, we compared the representation of guide RNAs in cells at the initial infection stage (3 days post infection) versus cells at the end stage (20 days post infection) (Fig. 1B). Forty-eight sgRNAs, targeting 8 genes, were highly depleted (FDR score <0.05) after 20 days (Supporting Fig. S1A), indicating that these genes are essential for liver cancer cell growth. After excluding the genes that were known to be essential in normal mouse cells (16), six genes were identified as being critical for liver cancer growth, including a known tumor suppressor (Bcl2l1), t-RNA synthetases (Lars and Vars), isomerase (Ppia), cytoskeleton protein (Tuba1b), and nucleotide synthesis-related enzyme (Adsl) (Fig. 1C). It has been widely reported that nucleotide synthesis pathways are rewired in cancer cells (21, 22) and play critical roles in cancer development (9, 13). Among all the tested genes involved in nucleotide synthesis pathways, sgRNAs targeting Adsl were the most significantly depleted (Supporting Fig. S1B). We further assessed genetic dependency in human HCC cells by leveraging a large-scale CRISPR screen profiling study: Depmap (Depmap Public 20Q2, https://depmap.org). Among all the key genes involved in purine synthesis, ADSL is highly indispensable in twenty-three human liver cancer cell lines (median dependency score=−0.8; Supporting Fig. S1C). Therefore, we focused on studying the role of ADSL in liver cancer cell growth in subsequent experiment.
Figure 1: CRISPR/Cas9 screen identifies ADSL as a potential drug target in liver cancer.

A, Left: sgRNA content and coverage of the druggable genome library. Right: Distribution of protein categories within the druggable genome library. B, p53−/−; H-RasG12V mouse liver cancer cells were transduced with lentivirus encoding Cas9 and the druggable genome library. After 20 days (7 passages), we identified depleted sgRNAs. C, Top-ranked genes according to false discovery rate (FDR).
To verify the role of ADSL in mouse liver cancer cell growth, we knocked out ADSL using CRISPR (sgAdsl) in p53−/−; H-RASG12V cells (Supporting Fig. S1D). ADSL-depleted cells showed substantially decreased colony formation capacity compared to cells treated with non-targeting sgRNA (sgNT) (Fig. 2A). By contrast, depletion of ADSL did not affect normal hepatocyte growth (Fig. 2B, Supporting Fig. S1E), indicating the selective essentiality of ADSL for cancer cell growth. Given that ADSL catalyzes de novo AMP synthesis, we engineered a doxycycline-inducible Adsl knockout system, then supplemented the cells with exogenous adenosine to promote AMP synthesis through the salvage pathway (Supporting Fig. S1F). Adenosine supplementation rescued the colony formation capacity of ADSL-depleted cells (Fig. 2C). This result suggests that depletion of ADSL affects liver cancer cell growth by disrupting AMP synthesis.
Figure 2. ADSL is indispensable for the growth of mouse liver cancer.

A, Top: Colony formation of p53−/−; H-RasG12V mouse liver cancer cells transduced with non-targeting guide RNA (sgNT) or guide RNAs targeting Adsl (sgAdsl.1 and sgAdsl.2). Bottom: Quantification of colony formation based on three independent assays. ****, P<0.0001 (Two tailed t-test). B, Top: Colony formation of normal mouse hepatocytes. H2.35 cells were transduced with non-targeting guide RNA (sgNT) or guide RNAs targeting Adsl (sgAdsl.1 and sgAdsl.2). Bottom: Quantification of colony formation based on three independent assays. NS: Not significant (Two tailed t-test). C, Top: Colony formation of p53−/−; H-RasG12V mouse liver cancer cells infected with doxycycline-inducible Cas9 and constitutively-expressed sgAdsl. Cells were treated without/with doxycycline in the presence or absence of 100 μM Adenosine. Bottom: Quantification of colony formation based on three independent assays. **, P<0.01; *, P<0.05 (Two tailed t-test). D, E, Left: Representative image of livers from mice treated with c-Myc, sgRNA targeting p53 (sgp53) (D) or CTNNB1-N90 (E), and either sgNT or sgAdsl. Right: Quantification of surface tumor number in the two liver cancer mouse models (Myc, p53−/−:n=5 sgNT-treated mice, n=7 sgAdsl-treated mice; Myc, CTNNB1-N90: n=3 sgNT-treated mice, n=4 sgAdsl-treated mice).
Previous reports demonstrate that nucleotide biosynthetic enzymes can be regulated by specific oncogenes, such as Myc (23). Therefore, we asked whether the essentiality of ADSL in liver cancer cells is dependent on certain oncogenes. To answer this question, we knocked out ADSL in another cell line (20): p53−/−; Myc mouse liver cancer cells. ADSL depletion still significantly reduced colony numbers compared to sgNT (Supporting Fig. S1G). This finding suggests that the effect of ADSL depletion on cancer cell growth is not specific to a particular oncogene.
Finally, we tested the impact of ADSL depletion on in vivo tumor formation. Overexpression of Myc and knockout of P53 can induce mouse liver carcinoma (20). Therefore, we treated adult mice with plasmids of c-Myc, sgRNA targeting p53 (sgp53), and either sgAdsl or sgNT via hydrodynamic tail vein injection. Mice treated with sgNT developed tumors (average 15 tumors per mouse) by 6 weeks post-injection. By contrast, tumors failed to grow in mice treated with sgAdsl, and the liver morphology and histology were normal (Fig. 2D and Supporting Fig. S2A). CTNNB1 is frequently mutated in HCC patients (24). Therefore, we also investigated the effect of ADSL depletion in a liver cancer model that stably expresses Myc and CTNNB1-N90 (25) to testify the robustness of ADSL depletion in inhibiting liver cancer growth. Consistently, we observed surface tumor in sgNT-treated mice, but not in sgAdsl-treated mice after 5 weeks post-injection (25) (Fig. 2E). However, we did observe small tumor nodules in sgAdsl-treated mouse liver sections by H&E staining (Supporting Fig. S2B). The sizes of these nodules were significantly smaller than those in sgNT-treated mice (Supporting Fig. S2B). It is also possible that ADSL was not fully depleted in these small tumor nodules by transiently expressing sgAdsl and Cas9. Nevertheless, these data suggest that ADSL is important for tumor growth in vivo.
Collectively, our finding suggests that ADSL, a key enzyme involved in de novo AMP synthesis, is indispensable for mouse liver cancer growth and development, and its depletion prevents tumor formation in vivo.
ADSL-mediated AMP de novo synthesis is indispensable for human HCC cell growth
Next, we investigated whether ADSL knockout has the same effect in human HCC cells. Oncogenes and tumor suppressors, such as p53, have been identified as key molecular determinants for de novo nucleotide synthesis pathways to promote cancer cell growth (22, 26). Thus, we first used three cancer cell lines (SK-HEP1, Hep3B, and Huh7) with different oncogenic backgrounds and p53 status to knock out ADSL (Supporting Fig. S2C) and measure colony formation capacity. All three cell lines displayed significantly reduced cell growth without ADSL (Fig. 3A).
Figure 3: De novo AMP synthesis pathway is indispensable for human liver cancer cell growth.

A, Top: Colony formation of human cancer cell lines (SK-HEP1, Hep3B, and Huh7) transduced with non-targeting guide RNA (sgNT) or guide RNAs targeting ADSL (sgADSL.1 and sgADSL.2). Bottom: Quantification of colony formation based on three independent assays. *, P<0.05, ****, P<0.0001 (Two tailed t-test). B, Left: Colony formation of HCC cells transduced with non-targeting shRNA (shNT) or four individual shRNAs targeting ADSL (shADSL.1, shADSL.2, shADSL.3, and shADSL.4). Right: ADSL expression levels in treated HCC cells. C, Left: Colony formation of SK-HEP1 cells transduced with sgNT or sgRNAs targeting APRT (sgAPRT.1 and sgAPRT.2). Right: Quantification of colony formation based on three independent assays. NS: Not significant (Two tailed t-test).
Recent studies on CRISPR/Cas9-mediated gene knockout have raised concerns over false positive results produced by off-target effects or p53-mediated DNA damage-induced toxic response (27). To exclude the possibility that the impact of ADSL knockout is caused by CRISPR/Cas9 side effects, and to further confirm the essentiality of ADSL for human HCC cell growth, we used four individual shRNAs to knockdown ADSL in four human HCC cell lines. Consistent with our CRISPR-mediated knockout results, knockdown of ADSL led to fewer cell colonies in all four cell lines (Fig. 3B). Although all four shRNAs mediated similarly reduced protein levels, shADSL.1- and shADSL.4-treated cells grew into more colonies than the cells treated with shADSL.2 and shADSL.3. This may be due to potential off-target effects of shRNA (28). Finally, we applied pooled small interfering RNA (siRNA) to knock down ADSL in SK-HEP1 cells (Supporting Fig. S2D). Consistent with our CRISPR knockout and shRNA knockdown experiments, colony formation was greatly decreased after siRNA-mediated knockdown of ADSL (Supporting Fig. S2E). Given our findings in mouse liver cells showing that ADSL affects cancer cell growth via AMP synthesis (Fig. 2C), we examined whether disrupting the salvage pathway for AMP synthesis via knockout of adenine phosphoribosyltransferase (APRT) would affect HCC cell growth (Supporting Fig. S2F). Consistent with our CRISPR screen results (Supporting Fig. S1B), we found that colony formation capacity was not affected by APRT depletion (Fig. 3C). Collectively, our findings demonstrate that ADSL-mediated AMP de novo synthesis is indispensable for human HCC cell growth.
ADSL depletion causes liver cancer cell cycle arrest at S phase
Next, we sought to determine the mechanism underlying the impact of ADSL on HCC cell growth. Initially, we thought that depletion of ADSL, a critical enzyme in purine synthesis, might lead to unbalanced or limited nucleotide pools, causing DNA replication stress and error. To investigate this, we knocked out ADSL in three HCC cell lines (SK-HEP1, Hep3B, and Huh7), and measured DNA damage levels by staining for phospho-histone H2AX. However, we did not observe significant differences in DNA damage between sgNT control and ADSL knockout cells (Supporting Fig. S3A). We also did not observe significant differences in cell death, as measured by cleaved caspase-3 (CC3) levels in control and ADSL knockout cells (Supporting Fig. S3A). Therefore, we set out to determine the effect of ADSL depletion on cell cycle progression by staining DNA content with Propidium Iodide (PI). In all three cell lines, depletion of ADSL led to an increase in cells at S phase compared to sgNT-treated cells (Fig. 4A and Supporting Fig. S3B, C). However, we did not observe significant alteration in the expression of some cell cycle-related proteins, including cyclins and CDKs (Supporting Fig. S3D). To confirm that ADSL depletion induces cell cycle arrest at S phase, we synchronized Huh7 cells at G1/early S phase using Aphidicolin, a DNA polymerase inhibitor (Fig. 4B). After a 24h synchronization, Aphidicolin was washed out to enable cell cycle progression. Eighteen hours later, we performed cell cycle analysis (PI staining). As expected, ADSL-depleted cells were halted at S phase without further progression, while ~50% of sgNT-treated cells proceeded to G2/M phase (Fig. 4C). Overall, our findings demonstrate that, instead of causing cell death, disruption of de novo purine synthesis by ADSL knockout delays HCC growth by causing cell cycle arrest at S phase.
Figure 4. ADSL depletion induces cell cycle arrest at S phase.

A, Left: Cell cycle analysis of Huh7 cells treated with non-targeting sgRNA (sgNT) or sgRNAs targeting ADSL (sgADSL.1 and sgADSL.2). Cells were stained with PI and analyzed by FACS. Right: Quantification of cell populations in G1, S, and G2/M phases. **, P<0.01, *, P<0.05 (Two-tailed t-test). B, C, Left: Huh7 cells treated with sgNT, sgADSL.1, or sgADSL.2 were synchronized with Aphidicolin (3 ng/μL) for 24 h (B) and washout for 18 h (C). Cells at endpoint were stained with PI and analyzed by FACS. Right: quantification of cell populations at G1, S, and G2/M phases after washout of Aphidicolin. **, P<0.01, ***, P<0.005 (Two-tailed t-test).
ADSL is critical for mitochondrial ATP production
The major biological event in S phase is DNA replication, during which nucleotides are added to the new DNA strand by consumption of ATP. Knockout of ADSL did not cause DNA damage (as indicated by phosphor-H2AX level in Supporting Fig. S3A). Therefore, we investigated the impact of ADSL knockout on intracellular ATP levels. After knocking out ADSL in HCC cells, we observed significantly lower ATP levels compared to sg-NT treated cells (Fig. 5A and Supporting Fig. S4A), indicating that ADSL-mediated de novo AMP synthesis is essential for maintaining cellular ATP levels. Intracellular ATP is produced by two distinct, but related pathways: glycolysis and oxidative phosphorylation. Glycolysis occurs in the cytoplasm, while oxidative phosphorylation occurs at the inner membrane of mitochondria. To determine which ATP production pathway is affected by ADSL depletion, we treated SK-HEP1 cells with a glycolysis inhibitor (2-Deoxy-D-glucose) or an oxidative phosphorylation inhibitor (oligomycin), and measured intracellular ATP levels. ATP produced by oxidative phosphorylation, not by glycolysis, was reduced by ~80% in ADSL knockout cells (Fig. 5B). Furthermore, the major source of ATP shifted from oxidative phosphorylation to glycolysis in ADSL-depleted cells (Mitochondrial: Glycolytic ATP synthesis ratio was ~4:1 in sgNT-treated cells vs. ~0.3:1 in sgADSL-treated cells; Fig. 5C). In the TCGA dataset, oxidative phosphorylation-related genes are upregulated in HCC patient samples with high ADSL expression (Supporting Fig. 4B), further confirming the role of ADSL in regulating oxidative phosphorylation.
Figure 5. ATP production in mitochondria is disrupted by ADSL depletion.

A, Intracellular ATP level in 2x105 SK-HEP1 cells transduced with non-targeting guide RNA (sgNT) or two guide RNAs targeting ADSL (sgADSL.1 and sgADSL.2). Three independent experiments. **, P<0.01 (Two-tailed t-test). B, ATP produced by oxidative phosphorylation or glycolysis were measured in SK-HEP1 cells treated with sgNT or sgADSL. Three independent experiments. ***, P<0.001 (two-tailed t-test). C, Fraction of ATP produced by oxidative phosphorylation or glycolysis to total ATP production in SK-HEP1 cells treated with sgNT or sgADSL. D, Relative intracellular ATP levels in 2x105 SK-HEP1 cells treated with DMSO or 0.4 μM oligomycin. Relative ATP level was normalized to DMSO-treated cells. Three independent experiments. *, P<0.05 (Two-tailed t-test). E, Cell cycle analysis of SK-HEP1 cells treated with DMSO or 0.4 μM oligomycin. Cells were stained with PI and analyzed by FACS. F, Quantification of cell populations at G1, S, and G2/M as described in (E). Three independent experiments. *, P<0.05, **, P<0.01(Two-tailed t-test).
Given previous evidence that mitochondrial energy metabolism can drive cell cycle progression and cell proliferation (29, 30), we wanted to confirm that the reduction in mitochondrial ATP production by ADSL depletion is the cause of the observed cell cycle arrest at S phase. To this end, we used oligomycin, an ATP synthase inhibitor, to block mitochondrial ATP production in SK-HEP1 cells (Fig. 5D). Oligomycin-treated cell cycle was arrested at S phase (Fig. 5E and F), recapitulating what we observed in ADSL-depleted cells. This result suggests that ADSL promotes cell cycle progression by maintaining mitochondrial ATP production in HCC cells, and reduced mitochondrial ATP synthesis can cause cell cycle arrest at S phase.
Collectively, our findings uncover a role for ADSL in mitochondrial ATP production, and decipher the connection between mitochondrial function and cell cycle progression in HCC.
Depletion of ADSL induces mitochondrial stress
Mitochondria are dynamic organelle with repetitive fission and fusion cycles to adapt to environmental change. For example, mitochondria are fused in G1/S phase to trigger S phase initiation (31), and are fragmented in G2/M phase (32). They also tend to elongate during energy depletion and stress conditions (33). Given our findings that ADSL depletion disrupts ATP synthesis in mitochondria, we investigated how mitochondria may be affected. Using MitoTracker and cytochrome c immunostaining to track mitochondria, we observed hyperfused and elongated mitochondrial network in ADSL-depleted HCC cells (Fig. 6A, B and Supporting Fig. S5A, B). Mitochondria remained intact, as indicated by co-localization of cytochrome c and MitoTracker (Fig. 6A and Supporting Fig. S5A). This finding is consistent with our previous observation of S phase arrest in ADSL-depleted cells, and suggests mitochondrial stress. Indeed, we found that mRNA expression of activating transcription factor 5 (ATF5), a mitochondrial stress response protein, was significantly upregulated in ADSL-depleted HCC cells compared to sgNT-treated cells (Fig. 6C and Supporting Fig. S5C), suggesting ADSL knockout causes mitochondrial stress. To further evaluate the impact of ADSL on mitochondrial function, we compared oxygen consumption rate (OCR) between sgNT- and sgADSL-treated HCC cells. ADSL depletion led to significantly decreased basal respiration rate and impaired maximal respiration capacity in two assayed HCC cell lines (Fig. 6D and Supporting Fig. S5D). Collectively, these data demonstrate that ADSL is essential to maintain mitochondrial function in HCC cells.
Figure 6. Mitochondrial stress induced by ADSL depletion.

A, Immunofluorescent images showing mitochondria morphology in Huh7 cells transduced with non-targeting sgRNA (sgNT) or guide RNAs targeting ADSL (sgADSL.1, sgADSL.2). Huh7 cells were stained for DAPI, Cytochrome C, and MitoTracker. Scale bar=10μm. White arrows indicate representative mitochondrial morphology. B, Quantification of mitochondrial length shown in (A). ****, P<0.0001 (Two tailed t-test). C, ATF5 mRNA level in Huh7 cells treated with sgNT and sgADSL was measured by quantitative real-time PCR and normalized to sgNT-treated cells. *, p<0.05 (Two-tailed t-test). D, Left: Oxygen consumption rate (OCR) in Huh7 cells transduced with non-targeting sgRNA (sgNT) or guide RNAs targeting ADSL (sgADSL.1, sgADSL.2) was measured by seahorse analyzer under basal conditions or in response to indicated inhibitors. Right: basal respiration rate and maximal respiration capacity are summarized. ****, P < 0.0001 (n=14, two tailed t-test).
ADSL is associated with tumorigenesis in HCC patients
To better understand the clinical significance of ADSL in HCC, we sought to correlate our findings in liver cancer cells to HCC patient data. Analysis of two patient datasets (GSE14520 and TCGA) revealed that ADSL expression is significantly higher in tumor samples than in normal liver tissues (Fig. 7A, Supporting Fig. S6A). Furthermore, in most cases (92%), ADSL expression is higher in the tumor section compared to the corresponding peri-tumor liver section in the same patient sample (Fig. 7B), indicating that ADSL is activated during tumorigenesis. Moreover, GSEA data show that liver cancer proliferation-related genes are enriched in HCC patients with higher ADSL expression (Fig. 7C), which is consistent with our colony formation and cell cycle analysis results. Among the three aggressive gastrointestinal cancers – HCC, colorectal and pancreatic – only HCC showed a correlation between ADSL expression and survival rate, with higher ADSL showing poor survival in patients (Fig. 7D). Indeed, compared to other enzymes involved in de novo purine synthesis, the expression of ADSL is the most significantly associated with survival rate of HCC patients (Supporting Fig. S6B). By contrast, HCC patients with higher expression of APRT (the key enzyme in the AMP salvage pathway) do not show any difference in survival rate compared to HCC patients with lower APRT expression (Fig. 7E). Collectively, these data demonstrate the specific role of hepatocellular ADSL level in indicating and predicting HCC progression.
Figure 7. ADSL is upregulated in liver cancer patient samples.
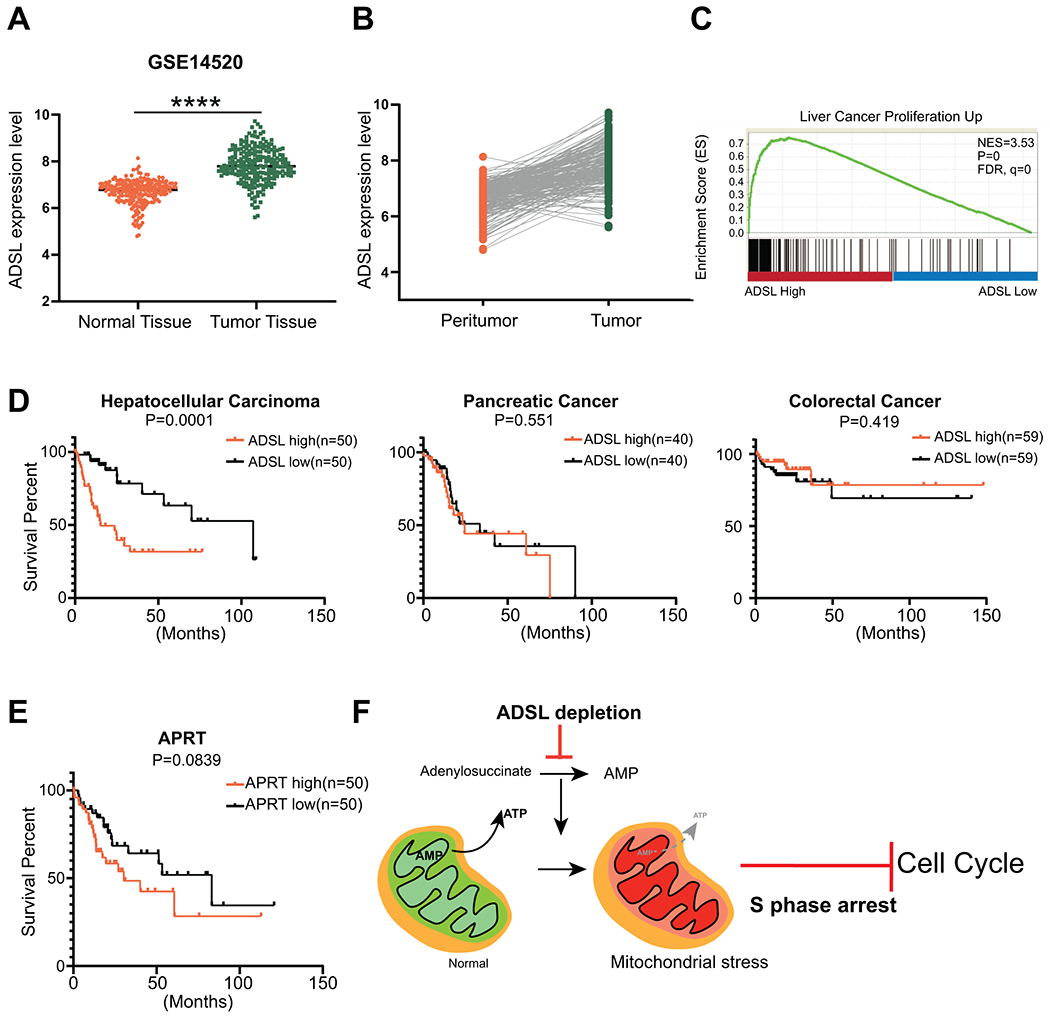
A, ADSL mRNA expression data in non-tumor and tumor samples taken from GSE14520 dataset. B, ADSL mRNA expression level in tumor area and corresponding peri-tumor area. Data are from GSE14520 dataset. C, Expression of genes controlling cell proliferation are significantly upregulated in patients with high ADSL mRNA expression. Gene set enrichment analysis of TCGA dataset (Hepatocellular Cancer). Enrichment Score=0.7514, FDR q-value=0. D, Survival rates in patients with high versus low ADSL mRNA expression in liver (left), pancreatic (middle), and colorectal (right) cancer. P values shown in graph (Log-rank (Mantel-Cox) test). E, Survival rates in HCC patients with high versus low APRT mRNA expression. P values is shown in graph (Log-rank (Mantel-Cox) test). F, Working model: Depletion of ADSL reduces AMP synthesis by disrupting de novo purine synthesis pathway. Lower AMP levels cause less ATP production by oxidative phosphorylation, leading to mitochondrial stress. Lower ATP level induces cell cycle arrest at S phase.
Discussion
Here, by using a novel CRISPR library, we identified ADSL as a potential therapeutic target in HCC, and characterized its role in liver cancer growth. In contrast to a previous report that knockdown of ADSL does not cause decrease in AMP and ATP levels in islet β cells (34), we found that mitochondrial ATP synthesis is substantially perturbed after depletion of ADSL in HCC cells. Mitochondrial dysfunction leads to cell cycle arrest at S phase (potentially due to retarded DNA replication), and delayed cell growth (Fig. 7F). Furthermore, we uncovered the clinical significance of ADSL in HCC tumorigenesis, and correlated its expression with HCC patient survival rates. ADSL expression level might be a specific prognosis marker for HCC.
Our findings are consistent with a previous report that used a genome-wide CRISPR/Cas9-based screen to determine that the ADSL-mediated purine synthesis pathway is essential for nasopharyngeal carcinoma cell growth (35). Moreover, knockout or knockdown of ADSL prevents triple negative breast cancer (13) and endometrial cancer (15) cell growth. Thus, ADSL may be a novel drug target for the treatment of multiple cancer types. Despite ADSL potentially being important for purine metabolism in normal cells, we found that ADSL depletion did not affect the growth of normal hepatocytes. A recent study also reveals that ADSL depletion does not cause growth defects in normal breast epithelial cells (13). However, we cannot exclude the possibility that ADSL might be toxic to other non-cancerous cells. Further work should rigorously compare the effects of ADSL depletion in cancer cells versus various normal cell types. The therapeutic window of ADSL inhibition should also be determined to minimize any potential toxicity. Moreover, the other two enzymatic reaction products of ADSL, fumarate and minoimidazole carboxamide ribotide (AICAR), play important roles in cancer cell growth and invasion (36–38). In this study, we cannot exclude that dysregulation of fumarate/AICAR-mediated pathways might affect liver cancer cell growth in ADSL-depleted cells. Further investigation of the effect of fumarate and AICAR in ADSL-depleted cells should be performed.
Currently, one ADSL-specific inhibitor has been characterized: adenosine phosphonobutyric acid 2’ (3’), 5’-diphosphate (APBADP), a non-cleavable substrate analog that competitively binds to human ADSL to specifically inhibit ADSL-catalyzed reactions (39). However, the effect of APBADP on HCC growth has not been studied. Future studies should investigate its effect on HCC progression to determine the potential of ADSL-specific inhibitors as a therapeutic approach for HCC treatment.
Our finding may also aid current efforts to therapeutically target de novo purine synthesis for cancer treatment. Cancer cells often transcriptionally tune de novo nucleotide synthesis pathway (26), especially the purine synthesis pathway, to promote cell proliferation (11, 40) and to modulate tumor immune response (41, 42), making the de novo purine synthesis pathway an attractive therapeutic target in cancer treatment. Indeed, small molecules resembling nucleotide metabolites, called “antimetabolites”, have been used as chemotherapy drugs to inhibit DNA synthesis in various cancers (7). For example, MIV-818 is an antimetabolite drug in a phase 1b trial that was specifically developed for treating advanced HCC patients. After being taken up into cancer cells, antimetabolites are incorporated into DNA, leading to DNA damage and cell death. However, there are concerns that the impaired DNA damage response and deficiency of targeted protein expression in cancer cells may cause resistance to these compounds (7, 43). In our study, we demonstrate that the effects of ADSL depletion in HCC development are independent of DNA damage. Besides, ADSL is overexpressed in liver tumors. Thus, ADSL-specific inhibitors could serve as an alternative treatment for liver cancers that are resistant to antimetabolites. Given our finding that knockout of ADSL inhibits tumor growth to a different extent in distinct oncogene-induced HCC models, it is possible that some oncogenes could mediate resistance to ADSL depletion. Future work should determine the vulnerability of different HCC genetic backgrounds to ADSL inhibition.
Mitochondrial dysfunction can be reduced by a series of stress response pathways, including the ATF5-mediated unfolded protein response (UPR) pathway (44). Unresolved mitochondrial stress could lead to cancer cell cycle arrest (45, 46) and programmed cell death (47). Growing evidence suggests that there is crosstalk between nucleotide synthesis and mitochondrial function –de novo purine synthesis regulates cell growth by mediating ATP production (48), while mitochondrial ATP fuels nucleotide synthesis in cancer proliferation (49). Mitochondrial stress could also activate the de novo purine synthesis pathway to promote cancer cell growth (50). In this study, we found that disrupting de novo purine synthesis by ADSL depletion decreases mitochondrial ATP production and mediates mitochondria stress, elucidating the connection between nucleotide synthesis and mitochondrial function in cancer cells. Furthermore, impaired mitochondrial function delayed HCC cell cycle progression. Thus, mitochondrial defects might be a therapeutic vulnerability to nucleotide synthesis inhibition in liver cancers.
In summary, using a high throughput CRISPR screen, we identified ADSL – a key enzyme in de novo purine synthesis pathway – as an essential gene for HCC cell growth, and a potential drug target for HCC treatment. Furthermore, we demonstrate that ADSL-mediated nucleotide synthesis promotes mitochondrial function to facilitate HCC cell cycle progression, uncovering a connection between nucleotide metabolism and bioenergetic processes in cancer cells.
Supplementary Material
Acknowledgements
We thank T. Jacks, C. Mello, P. Zamore, S. Wolfe, and E. Sontheimer for discussions and E. Haberlin for editing the manuscript. We thank Y. Liu and E. Kittler in the UMass Morphology and Deep Sequencing Cores for support.
Financial Support
W.X was supported by grants from the National Institutes of Health (DP2HL137167, P01HL131471 and UG3HL147367), American Cancer Society (129056-RSG-16-093), the Lung Cancer Research Foundation, and the Cystic Fibrosis Foundation. C.M.H was supported by grants from the National Institutes of Health (R01AG040061).
Abbreviation
- HCC
hepatocellular carcinoma
- ADSL
adenylosuccinate lyase
- PI
propidium iodide
- sgRNA
single-guide RNA
- TCGA
The Cancer Genome Atlas
- GSEA
gene set enrichment analysis
- ATP
Adenosine triphosphate
- AMP
Adenosine monophosphate
Footnotes
Competing Interests
Authors declare no conflict of interest.
Reference
- 1.Mu X, Espanol-Suner R, Mederacke I, Affo S, Manco R, Sempoux C, Lemaigre FP, et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J Clin Invest 2015;125:3891–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marquardt JU. Deconvolution of the cellular origin in hepatocellular carcinoma: Hepatocytes take the center stage. Hepatology 2016;64:1020–1023. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson B, Mariotto A, et al. Annual Report to the Nation on the Status of Cancer, 1975-2014, Featuring Survival. J Natl Cancer Inst 2017;109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balogh J, Victor D 3rd, Asham EH, Burroughs SG, Boktour M, Saharia A, Li X, et al. Hepatocellular carcinoma: a review. J Hepatocell Carcinoma 2016;3:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 2018;15:599–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luengo A, Gui DY, Vander Heiden MG. Targeting Metabolism for Cancer Therapy. Cell Chem Biol 2017;24:1161–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin J, Ren W, Huang X, Deng J, Li T, Yin Y. Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front Immunol 2018;9:1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson AD, Eich ML, Varambally S. Dysregulation of de novo nucleotide biosynthetic pathway enzymes in cancer and targeting opportunities. Cancer Lett 2020;470:134–140. [DOI] [PubMed] [Google Scholar]
- 11.Li L, Ng SR, Colón CI, Drapkin BJ, Hsu PP, Li Z, Nabel CS, et al. Identification of DHODH as a therapeutic target in small cell lung cancer. Science Translational Medicine 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakravarthi B, Rodriguez Pena MDC, Agarwal S, Chandrashekar DS, Hodigere Balasubramanya SA, Jabboure FJ, Matoso A, et al. A Role for De Novo Purine Metabolic Enzyme PAICS in Bladder Cancer Progression. Neoplasia 2018;20:894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zurlo G, Liu X, Takada M, Fan C, Simon JM, Ptacek TS, Rodriguez J, et al. Prolyl hydroxylase substrate adenylosuccinate lyase is an oncogenic driver in triple negative breast cancer. Nat Commun 2019;10:5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reed VL, Mack DO, Smith LD. Adenylosuccinate lyase as an indicator of breast and prostate malignancies: a preliminary report. Clin Biochem 1987;20:349–351. [DOI] [PubMed] [Google Scholar]
- 15.Park H, Ohshima K, Nojima S, Tahara S, Kurashige M, Hori Y, Okuzaki D, et al. Adenylosuccinate lyase enhances aggressiveness of endometrial cancer by increasing killer cell lectin-like receptor C3 expression by fumarate. Lab Invest 2018;98:449–461. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber J, Ollinger R, Friedrich M, Ehmer U, Barenboim M, Steiger K, Heid I, et al. CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proc Natl Acad Sci U S A 2015;112:13982–13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr Biol 2016;26:2037–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roessler S, Long EL, Budhu A, Chen Y, Zhao X, Ji J, Walker R, et al. Integrative genomic identification of genes on 8p associated with hepatocellular carcinoma progression and patient survival. Gastroenterology 2012;142:957–966 e912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006;125:1253–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halbrook CJ, Wahl DR, Lyssiotis CA. Running the Light: Nucleotide Metabolism Drives Bypass of Senescence in Cancer. Trends Biochem Sci 2019;44:991–993. [DOI] [PubMed] [Google Scholar]
- 22.Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, Carrero ZI, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun 2015;6:7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mannava S, Grachtchouk V, Wheeler LJ, Im M, Zhuang D, Slavina EG, Mathews CK, et al. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008;7:2392–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruiz de Galarreta M, Bresnahan E, Molina-Sanchez P, Lindblad KE, Maier B, Sia D, Puigvehi M, et al. beta-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov 2019;9:1124–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Villa E, Ali ES, Sahu U, Ben-Sahra I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 2018;24:927–930. [DOI] [PubMed] [Google Scholar]
- 28.Singh S, Narang AS, Mahato RI. Subcellular fate and off-target effects of siRNA, shRNA, and miRNA. Pharm Res 2011;28:2996–3015. [DOI] [PubMed] [Google Scholar]
- 29.Xiong W, Jiao Y, Huang W, Ma M, Yu M, Cui Q, Tan D. Regulation of the cell cycle via mitochondrial gene expression and energy metabolism in HeLa cells. Acta Biochim Biophys Sin (Shanghai) 2012;44:347–358. [DOI] [PubMed] [Google Scholar]
- 30.Finkel T, Hwang PM. The Krebs cycle meets the cell cycle: mitochondria and the G1-S transition. Proc Natl Acad Sci U S A 2009;106:11825–11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maycotte P, Marin-Hernandez A, Goyri-Aguirre M, Anaya-Ruiz M, Reyes-Leyva J, Cortes-Hernandez P. Mitochondrial dynamics and cancer. Tumour Biol 2017;39:1010428317698391. [DOI] [PubMed] [Google Scholar]
- 32.Montemurro C, Vadrevu S, Gurlo T, Butler AE, Vongbunyong KE, Petcherski A, Shirihai OS, et al. Cell cycle-related metabolism and mitochondrial dynamics in a replication-competent pancreatic beta-cell line. Cell Cycle 2017;16:2086–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lebeau J, Saunders JM, Moraes VWR, Madhavan A, Madrazo N, Anthony MC, Wiseman RL. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep 2018;22:2827–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gooding JR, Jensen MV, Dai X, Wenner BR, Lu D, Arumugam R, Ferdaoussi M, et al. Adenylosuccinate Is an Insulin Secretagogue Derived from Glucose-Induced Purine Metabolism. Cell Rep 2015;13:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang C, Jiang S, Ke L, Zhang L, Li D, Liang J, Narita Y, et al. Genome-wide CRISPR-based gene knockout screens reveal cellular factors and pathways essential for nasopharyngeal carcinoma. J Biol Chem 2019;294:9734–9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jose C, Hebert-Chatelain E, Bellance N, Larendra A, Su M, Nouette-Gaulain K, Rossignol R. AICAR inhibits cancer cell growth and triggers cell-type distinct effects on OXPHOS biogenesis, oxidative stress and Akt activation. Biochim Biophys Acta 2011;1807:707–718. [DOI] [PubMed] [Google Scholar]
- 37.Su CC, Hsieh KL, Liu PL, Yeh HC, Huang SP, Fang SH, Cheng WC, et al. AICAR Induces Apoptosis and Inhibits Migration and Invasion in Prostate Cancer Cells Through an AMPK/mTOR-Dependent Pathway. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, Drubbel AV, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016;537:544–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sivendran S, Colman RF. Effect of a new non-cleavable substrate analog on wild-type and serine mutants in the signature sequence of adenylosuccinate lyase of Bacillus subtilis and Homo sapiens. Protein Sci 2008;17:1162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Virgilio F Purines, purinergic receptors, and cancer. Cancer Res 2012;72:5441–5447. [DOI] [PubMed] [Google Scholar]
- 41.Antonioli L, Blandizzi C, Pacher P, Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer 2013;13:842–857. [DOI] [PubMed] [Google Scholar]
- 42.Boison D, Yegutkin GG. Adenosine Metabolism: Emerging Concepts for Cancer Therapy. Cancer Cell 2019;36:582–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaaban S, Negm A, Ibrahim EE, Elrazak AA. Chemotherapeutic agents for the treatment of hepatocellular carcinoma: efficacy and mode of action. Oncol Rev 2014;8:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melber A, Haynes CM. UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res 2018;28:281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao JJ, Tan CP, Chen MH, Wu N, Yao DY, Liu XG, Ji LN, et al. Targeting cancer cell metabolism with mitochondria-immobilized phosphorescent cyclometalated iridium(iii) complexes. Chem Sci 2017;8:631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sareen D, van Ginkel PR, Takach JC, Mohiuddin A, Darjatmoko SR, Albert DM, Polans AS. Mitochondria as the primary target of resveratrol-induced apoptosis in human retinoblastoma cells. Invest Ophthalmol Vis Sci 2006;47:3708–3716. [DOI] [PubMed] [Google Scholar]
- 47.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010;11:621–632. [DOI] [PubMed] [Google Scholar]
- 48.Kondo M, Yamaoka T, Honda S, Miwa Y, Katashima R, Moritani M, Yoshimoto K, et al. The rate of cell growth is regulated by purine biosynthesis via ATP production and G(1) to S phase transition. J Biochem 2000;128:57–64. [DOI] [PubMed] [Google Scholar]
- 49.Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab 2015;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez Herrera KN, Zaganjor E, Ishikawa Y, Spinelli JB, Yoon H, Lin JR, Satterstrom FK, et al. Small-Molecule Screen Identifies De Novo Nucleotide Synthesis as a Vulnerability of Cells Lacking SIRT3. Cell Rep 2018;22:1945–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.