

Abstract
The PARK7 gene (encode DJ-1 protein) was first discovered as an oncogene and later found to be a causative gene for autosomal recessive early onset Parkinson’s disease. DJ-1 has been proposed as a potential therapeutic anticancer target due to its pivotal role in tumorigenesis and cancer progression. Based on the homodimer structure of DJ-1, a series of bis-isatin derivatives with different length linkers were designed, synthesized, and evaluated as dimeric inhibitors targeting DJ-1 homodimer. Among them, DM10 with alkylene chain of C10 displayed the most potent inhibitory activity against DJ-1 deglycase. We further demonstrated that DM10 bound covalently to the homodimer of DJ-1. In human cancer cell lines H1299, MDA-MB-231, BEL7402, and 786-O, DM10 (2.5–20 μM) inhibited the cell growth in a concentration-dependent manner showing better anticancer effects compared with the positive control drug STK793590. In nude mice bearing H1299 cell xenograft, intratumor injection of DM10 (15 mg/kg) produced significantly potent tumor growth inhibition when compared with that caused by STK793590 (30 mg/kg). Moreover, we found that DM10 could significantly enhance N-(4-hydroxyphenyl)retinamide-based apoptosis and erastin-based ferroptosis in H1299 cells. In conclusion, DM10 is identified as a potent inhibitor targeting DJ-1 homodimer with the potential as sensitizing agent for other anticancer drugs, which might provide synergistical therapeutic option for cancer treatment.
Keywords: DJ-1 inhibitor, DM10, isatin derivatives, homodimer, anticancer therapy, apoptosis, ferroptosis, STK793590
Introduction
The PARK7 gene (encodes the DJ-1 protein) was first discovered as an oncogene and later found to be a causative gene for autosomal recessive early onset Parkinson’s disease (PD) [1, 2]. Increasing evidence suggests that the dysfunction of DJ-1 leads to the onset of oxidative stress-related diseases, including neurodegenerative diseases [3–6], type 2 diabetes [7], male infertility [8, 9], and cancer [10, 11]. The DJ-1 protein has multiple functions through various mechanisms. To protect cells from death, DJ-1 preferentially translocates to mitochondria, subsequently regulating mitochondrial function [12, 13]. Notably, DJ-1 plays a role in antioxidative stress through the direct elimination of reactive oxygen species (ROS) by oxidizing itself on the Cys106 residue to –SO2 or –SO3 [14–16]. In addition, DJ-1 stabilizes the antioxidant transcriptional master regulator NRF2 [17], which orchestrates the expression of genes encoding stress response and antioxidant proteins. Furthermore, we recently identified the critical role of DJ-1 in cancer development and treatment response with distinct mechanisms [18, 19]. On the one hand, the different oxidation states of DJ-1 function as a cellular redox sensor caused by chemotherapy, thereby determining the cell fate of autophagy or apoptosis by regulating the activity of ASK1 [18]. On the other hand, DJ-1 determines the response of cancer cells to ferroptosis, maintaining cysteine synthesized from the transsulfuration pathway by preserving the activity of S-adenosyl homocysteine hydrolase [19]. Importantly, DJ-1 knockdown potently strengthens the sensitivity to cytotoxic antitumor drugs both in vitro and in vivo, suggesting that DJ-1 serves as a therapeutic target in cancer.
To intervene DJ-1, researchers have used RNA interference to block DJ-1 protein expression in the past decade. Although DJ-1 knockdown leads to enhanced sensitivity of tumor cells to chemotherapeutic drugs [20–23], initial difficulties remain to be solved in achieving efficacious results with RNAi-based DJ-1 intervention therapeutics without toxic side effects but with advances in delivery and improved physical and chemical properties [24]. Thus, there is a long-standing question of how to manipulate the function of DJ-1 through small molecules. The answer was not clear until DJ-1 was recently identified as a deglycase that repairs methylglyoxal (MGO)- and glyoxal-glycated amino acids and proteins by acting on early glycation intermediates and releases repaired proteins and lactate or glycolate, respectively [25, 26]. It has been suggested that the deglycase activity of DJ-1 may constitute a major enzymatic basis in its biological function. Based on this, the Shinya Tashiro group recently identified and optimized a series of isatin (1H-indole-2,3-dione) derivatives as DJ-1 inhibitors [27]. Among them, STK793590 (STK) demonstrated the highest affinity and most robust inhibitory activity against DJ-1 in vitro; however, its anticancer effect has not been evaluated. Though there is no experimental evidence to show the selectivity of STK in targeting DJ-1 in cells, it may be of the main concern due to the small size of the inhibitor (200 Da). Thus, the efficacy and selectivity of DJ-1 inhibitors can be optimized for potential cancer therapy.
An X-ray crystal structure study showed that DJ-1 functions as a homodimer [28, 29], and the dimer-deficient Lys166Pro mutant was confirmed to be a disease-causing mutation associated with autosomal early onset PD [30–32]. In addition, our group also found that the homodimeric form of DJ-1 is required for the response to oxidative stress triggered by N-(4-hydroxyphenyl)retinamide (4HPR), and the integrity of DJ-1 is related to its ferroptosis-blocking effect [18]. Based on this information, we hypothesized that compounds that can target the DJ-1 dimer simultaneously will exert more powerful efficacy on DJ-1 inhibition. Therefore, the aim of this study was to design an STK-derived inhibitor that specifically binds to the DJ-1 homodimer and to evaluate its efficacy in cancer therapy. Our results suggest that DM10 targets dimeric DJ-1 and is effective in various human cancer cells in either a single treatment or combination treatment with other cell death inducers.
Materials and methods
Antibodies and reagents
Antibodies against PARP (ET1608-56), cleaved-poly (ADP-ribose) polymerase (c-PARP) (ET1608-10), and c-caspase-3 (ET1602-47) were obtained from HuaAn Biotechnology (Hangzhou, China). Antibodies against DJ-1 (db1214), GAPDH (db106), and FLAG (db7002) were obtained from Diagnostic Biosystems (Hangzhou, China). NAC (A7250) and MGO (W296902) were obtained from Sigma (St. Louis, MO, USA). Erastin (S7242) and ferrostatin-1 (Fer-1) (S7243) were obtained from Selleck Chemicals (Houston, TX, USA). STK793590 was obtained from Vitas-M Laboratory (Apeldoorn, The Netherlands).
Synthesis of compounds
General procedure for the synthesis of DM2, DM4, DM6, DM8, DM10, and DM12
1,1′-(Ethane-1,2-diyl)bis(5-fluoroindoline-2,3-dione) (DM2)
![]()
A mixture of 5-fluoroindoline-2,3-dione (1.0 mM), 1,3-dibromopropane (0.5 mM), and K2CO3 (1.2 mM) was stirred in 4 mL of DMSO at room temperature for 14 h. Then, the reaction was extracted with dichloromethane (25 mL, ×3), the combined organic layers were dried over anhydrous sodium sulfate, and the solvent was evaporated in vacuum to give the crude product, which was purified carefully by column chromatography using dichloromethane and petroleum ether as eluents to afford the product DM2 as a red solid. DM4, DM6, DM8, DM10, and DM12 were prepared in a similar manner.
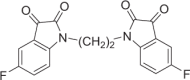
Red solid, 45% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.58 (td, J = 9.0, 3.0 Hz, 2H), 7.49 (dd, J = 7.0, 2.5 Hz, 2H), 7.31 (dd, J = 8.5, 4.0 Hz, 2H), and 4.01–3.98 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 182.71, 160.20 (d, J = 242.3 Hz), 158.93, 146.86, 124.80 (d, J = 24.3 Hz), 118.78 (d, J = 7.4 Hz), 112.70 (d, J = 7.3 Hz), 112.17 (d, J = 24.5 Hz), and 37.52. 19F NMR (376 MHz, DMSO-d6) δ −119.77 to −119.83 (m). HRMS (ESI): m/z calculated for C18H11F2N2O4 [M + H]+ 357.0681; experimental: 357.0682.
1,1′-(Butane-1,4-diyl)bis(5-fluoroindoline-2,3-dione) (DM4)
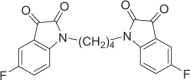
Red solid, 57% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.53 (td, J = 9.0, 3.0 Hz, 2H), 7.45 (dd, J = 7.0, 2.5 Hz, 2H), 7.24 (dd, J = 8.5, 3.5 Hz, 2H), 3.70–3.68 (m, 4H), and 1.68–1.66 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 183.27, 160.12 (d, J = 242.0 Hz), 158.67, 147.32, 124.56 (d, J = 24.0 Hz), 118.90 (d, J = 7.2 Hz), 112.67 (d, J = 7.6 Hz), 112.00 (d, J = 24.3 Hz), and 24.44. 19F NMR (376 MHz, DMSO-d6) δ −120.28 to −120.33 (m). HRMS (ESI): m/z calculated for C20H15F2N2O4 [M + H]+ 385.0994: experimental: 385.0996.
1,1′-(Hexane-1,6-diyl)bis(5-fluoroindoline-2,3-dione) (DM6)
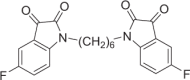
Red solid, 68% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.52 (td, J = 9.0, 2.5 Hz, 2H), 7.45 (dd, J = 7.0, 3.0 Hz, 2H), 7.20 (dd, J = 8.5, 3.5 Hz, 2H), 3.64 (t, J = 7.0 Hz, 4H), 1.60–1.51 (m, 4H), and 1.38–1.30 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 183.37, 160.08 (d, J = 241.9 Hz), 158.53, 147.43, 124.57 (d, J = 24.0 Hz), 118.89 (d, J = 7.2 Hz), 112.57 (d, J = 7.4 Hz), 112.02 (d, J = 24.4 Hz), 27.02, and 26.32. 19F NMR (376 MHz, DMSO-d6) δ −120.35 to −120.40 (m). HRMS (ESI): m/z calculated for C22H19F2N2O4 [M + H]+ 413.1307, experimental: 413.1316.
1,1′-(Octane-1,8-diyl)bis(5-fluoroindoline-2,3-dione) (DM8)
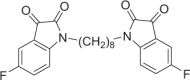
Red solid, 55% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.54 (td, J = 9.0, 3.0 Hz, 2H), 7.46 (dd, J = 7.0, 3.0 Hz, 1H), 7.22 (dd, J = 8.5, 4.0 Hz, 2H), 3.65 (t, J = 7.5 Hz, 4H), 1.58–1.54 (m, 4H), and 1.32–1.28 (m, 8H). 19F NMR (376 MHz, DMSO-d6) δ −120.36 to −120.42 (m). HRMS (ESI): m/z calculated for C24H23F2N2O4 [M + H]+ 441.1620, experimental: 441.1622.
1,1′-(Decane-1,10-diyl)bis(5-fluoroindoline-2,3-dione) (DM10)
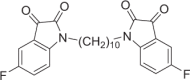
Red solid, 67% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.67–7.49 (m, 2H), 7.48–7.34 (m, 2H), 7.30–7.08 (m, 2H), 3.80–3.50 (m, 2H), 1.69–1.47 (m, 4H), and 1.38–1.06 (m, 12H). 13C NMR (101 MHz, DMSO-d6) δ 183.42, 160.09 (d, J = 242.0 Hz), 158.53, 147.46, 124.63 (d, J = 24.1 Hz), 118.88 (d, J = 7.2 Hz), 112.61 (d, J = 7.4 Hz), 112.04 (d, J = 24.5 Hz), 29.31, 29.11, 27.12, and 26.64. 19F NMR (376 MHz, DMSO-d6) δ −120.35 to −120.40 (m) HRMS (ESI): m/z calculated for C26H27F2N2O4 [M + H]+ 469.1933, experimental: 469.1931.
1,1′-(Dodecane-1,12-diyl)bis(5-fluoroindoline-2,3-dione) (DM12)
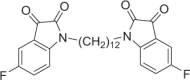
Red solid, 56% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.55 (td, J = 9.0, 2.5 Hz, 2H), 7.47 (dd, J = 7.0, 3.0 Hz, 2H), 7.23 (dd, J = 8.5, 3.5 Hz, 1H), 3.64 (t, J = 7.0 Hz, 4H), 1.59–1.54 (m, 4H), 1.31–1.24 (m, 8H), and 1.23–1.19 (m, 8H). 13C NMR (101 MHz, DMSO-d6) δ 183.40, 160.08 (d, J = 241.8 Hz), 158.51, 147.46, 124.62 (d, J = 24.0 Hz), 118.87 (d, J = 7.2 Hz), 112.61 (d, J = 7.1 Hz), 112.03 (d, J = 24.5 Hz), 29.36, 29.13, 27.11, and 26.64. 19F NMR (376 MHz, DMSO-d6) δ −120.35 to −120.40 (m). HRMS (ESI): m/z calculated for C28H31F2N2O4 [M + H]+ 497.2246, experimental: 497.2240.
Procedure for the synthesis of compound A
![]()
A mixture of 5-fluoroindoline-2,3-dione (1.0 mM), 1-bromodecane (1.1 mM), K2CO3 (2.0 mM), and 3.0 mL of DMSO was stirred at room temperature for 10 h. Then, the mixture was poured into a beaker containing 30 mL of H2O and extracted with ethyl acetate (25 mL, ×3). The combined organic layers were washed with brine and dried over anhydrous sodium sulfate, and the solvent was evaporated in vacuum to give the crude product, which was purified by column chromatography using ethyl acetate and petroleum ether as eluents to afford compound A as a red solid. 67% yield. 1H NMR (500 MHz, DMSO-d6) δ 7.52 (td, J = 9.0, 3.0 Hz, 1H), 7.45 (dd, J = 7.5, 2.5 Hz, 1H), 7.21 (dd, J = 9.0, 3.5 Hz, 1H), 3.64 (t, J = 7.0 Hz, 2H), 1.57 (m, 2H), 1.36–1.14 (m, 14H), and 0.84 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 183.43, 158.91 (d, J = 242.0 Hz), 158.53, 147.46, 124.52 (d, J = 24.1 Hz), 118.84 (d, J = 7.2 Hz), 112.60 (d, J = 7.3 Hz), 111.00 (d, J = 24.5 Hz), 31.78, 29.43, 29.19, 27.15, 26.68, 22.60, and 14.43. 19F NMR (376 MHz, DMSO-d6) δ −120.39 to −120.344 (m). HRMS (ESI): m/z calculated for C18H25FNO2 [M + H]+ 306.1869, experimental: 306.1873.
DJ-1 expression and purification
The cDNA of DJ-1 was cloned into the pET28a vector (#69864-3, Madison, WI, USA) containing an N-terminal His6-tag. pET28a-DJ-1-WT, pET28a-DJ-1-ΔC2, and pET28a-DJ-1-ΔC3 were constructed using the primers listed in Supplementary Table S1. Briefly, Escherichia coli BL21(DE3) cells transformed with a plasmid containing DJ-1 were grown in LB medium at 37 °C. Protein expression was induced with isopropyl-beta-D-thiogalactoside (IPTG, 0.5 mM) (V900917, Sigma-Aldrich, St Louis, MO, USA) when the OD600 reached 0.4. Cells were grown for an additional 16 h at 25 °C. After centrifugation, the cell pellets were resuspended in lysis buffer (20 mM phosphate buffer, 500 mM NaCl, and 50 mM imidazole (pH 8.0)) and homogenized by a high-pressure cell lyser followed by homogenization. The soluble fraction was separated by centrifugation and loaded into a Ni2+-NTA column (His-Trap HP, GE Healthcare). The protein was eluted with 500 mM imidazole and then subjected to dialysis buffer (50 mM Tris-HCl, 150 mM NaCl, 0.5 mM EDTA, and 10% glycerol (pH 8.0)). The purified DJ-1 protein was stored at −80 °C.
N-acetylcysteine deglycation assay
Experiments were performed in 50 mM sodium phosphate buffer (pH 7.0) at room temperature. Compounds were incubated with r-DJ-1 for 30 min and then added to a mixture of MGO and N-acetylcysteine. The final concentrations were 2 μM DJ-1, 20 mM N-acetylcysteine, and 20 mM MGO. An identical volume of sodium phosphate buffer was used as a control. The deglycase activity was determined by monitoring the absorbance of hemithioacetal at 288 nm for 60 min using a Molecular Devices SpectraMax M5 plate reader and SoftMax Pro 6.3 software. Enzyme activity under test conditions was reported as a ratio relative to 0 min.
Cell culture
The H1299, PC9, MDA-MB-231, HCC1937, BEL7402, HepG2, 786-O, RCC4, and HEK293T cell lines were purchased from the Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). H1299, PC9, MDA-MB-231, HCC1937, BEL7402, 786-O, and RCC4 cells were cultured in RPMI-1640 medium (HyClone, SH30809). HepG2 and HEK293T cells were cultured in DMEM (HyClone, SH30022). All media were supplemented with 10% fetal bovine serum (HyClone, SV30160) and maintained at 37 °C in a humidified atmosphere of 5% CO2. The cell line was authenticated by STR profiling. Cell lines were monitored for mycoplasma contamination every 6 months.
Lentivirus transduction
The lentiviral shRNA vector pCDH-EF1-puro (CD710B-1) was obtained from System Biosciences (Palo Alto, CA, USA). pCDH-DJ-1-FLAG was constructed using the primers listed in Supplementary Table S1. Lentivirus was produced by transfecting HEK293T cells with the pCMV-R8.91 (packaging vector), pMD2-VSVG (envelope vector), and shRNA plasmids or pCDH plasmid by Lipofectamine 2000 (#11668019, Invitrogen, Carlsbad, CA, USA). Virus-containing medium was harvested 48 h after transfection. For infection, cells were grown in six-well plates at 30%–40% confluency, and virus (multiplicity of infection of 10) was added with 1 μL of polybrene (6 mg/mL).
Growth inhibition assay
Cell growth inhibition was typically assessed using the sulforhodamine B (SRB, #S1402, Sigma-Aldrich, St. Louis, MO, USA) assay based on the measurement of the cellular protein content. Briefly, cell monolayers were fixed with 10% (wt/vol) trichloroacetic acid and stained for 30 min, after which the excess dye was removed by washing repeatedly with 1% (vol/vol) acetic acid. The protein-bound dye was dissolved in 10 mM Tris base solution for the OD determination at 510 nm using a microplate reader (Multiskan Spectrum, Thermo). The cell viability under test conditions is reported as a percentage relative to the negative control treatment.
Immunoblotting analysis
Cells were lysed with 1% NP-40 buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.1 mM sodium vanadate, 5 μg/mL leupeptin, and 0.1 mM phenylmethylsulfonyl fluoride (pH 7.4)) and incubated at 4 °C for 30 min. The lysate was then centrifuged at 14,000 × g for 20 min at 4 °C to remove insoluble proteins. Protein concentrations of whole-cell lysates were determined using the DC Protein Assay Kit (#5000111, Bio-Rad, Hercules, CA, USA). The proteins were then electrophoresed in SDS-PAGE gels and transferred to polyvinylidene difluoride membranes (#00010, Millipore, Bedford, MA, USA). After blocking with 5% nonfat milk, the membranes were incubated with various primary antibodies overnight at 4 °C, and the proteins were visualized by enhanced chemiluminescence detection (NEL103E001EA, PerkinElmer, Waltham, MA, USA) by AI600 (GE Healthcare, Chicago, IL, USA) after incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies.
Analysis of lipid ROS production
The day before the experiment, 200,000 cells/well were seeded in six-well dishes. On the day of the experiment, cells were incubated with DM10 for 12 h, followed by erastin for another 24 h, harvested by trypsinization, resuspended in 500 μL of PBS containing 2 μM C11-BODIPY (581/591) (#D3861, Invitrogen, Carlsbad, CA, USA), and incubated for 30 min at 37 °C. Cells were then resuspended in 500 μL of fresh PBS, filtered through a 40 μm cell strainer, and analyzed using a flow cytometer (FACSuite, BD Biosciences, Bedford, MA, USA) equipped with a 488 nm laser for excitation. Data were collected from the FL1 channel (527 nm). A minimum of 10,000 cells were analyzed per condition. Data analysis was performed by using FlowJo Version 7.6 software.
In vivo xenograft mouse study
Tumors were established by injecting 200 μL of H1299 cell suspensions (2 × 106) into BALB/c female athymic nude mice (5 weeks, National Rodent Laboratory Animal Resource, Shanghai, China). When the tumor volume reached an average size of 80 mm3, tumor-bearing mice were subsequently randomly divided into three groups (n = 7 for each group). STK or DM10 was intratumorally administered as a single agent in mouse tumors at a dose of 30 or 15 mg/kg only once. The weight and tumor size were measured every 3 days, and volumes were calculated using the formula: (length × width2)/2. At the end of the experiment, the tumor weights were calculated. Maintenance and experimental procedures for the mouse studies were approved by Zhejiang University’s IACUC (IACUC-s20-030).
Statistical analysis
All statistical analyses were performed by using Prism 5.0c (GraphPad Software). All of the experiments were performed three times. Data are presented as the mean ± standard deviation (SD). Differences between means were determined using unpaired Student’s t-tests and were considered significant at P < 0.05.
Results
Design and synthesis of inhibitors targeting the DJ-1 homodimer
DJ-1 is a homodimer mediated by helices at the C-terminus (PDB ID: 1UCF) (Fig. 1a), and these helices play important roles in the biological function of DJ-1. Near the dimer interface, there is a pocket formed by amino acid residues [29], including Cys106, His126, and Glu18, with favorable properties for binding small molecules (Fig. 1b). Recently, the Shinya Tashiro laboratory identified a series of isatin derivatives as potent DJ-1 deglycase inhibitors [27]. The X-ray crystal structure of DJ-1 and the inhibitor complex reveals that the sulfur atom of Cys106 can form a covalent bond with the 3-carbonyl moiety of isatin. The crystal structure also showed that the 7-fluorine on the isatin scaffold can interact with Arg28 and Pro184, which is beneficial for the stabilization of this covalent binding, and the substituent at position N1 can also affect the binding affinity (Fig. 1c). Thus, there are strategic opportunities to optimize the efficacy and the selectivity of DJ-1 inhibitors. Considering the homodimer structure of DJ-1, we designed dimeric DJ-1 inhibitors. The scaffold of STK, 5-fluoroindoline-2,3-dione, was chosen as the pharmacophore binding to Cys106 of DJ-1, and alkylene chains of different lengths were used to connect the N1 position of the two moieties to obtain a novel series of bis-isatin derivatives, namely, DM2, DM4, DM6, DM8, DM10, and DM12. These compounds were synthesized from 5-fluoroindoline-2,3-dione and α,ω-dibromo alkanes in the presence of potassium carbonate in DMSO and purified carefully by column chromatography (Fig. 1d, e).
Fig. 1. Design and synthesis of inhibitors targeting the DJ-1 homodimer.

a, b The X-ray crystal structure of the DJ-1 dimer (Protein Data Bank [PDB] ID: 1UCF) shows as a putative active site near the dimer interface, which includes residues Cys106, His126, and Glu18. c The chemical structure of STK: the isatin scaffold is the main functional group that can covalently bind to DJ-1, and the substituent at position N1 can result in a variety of affinities (red sticks). d Synthesis of DMs from 5-fluoroindoline-2,3-dione and α,ω-dibromo. Conditions: reagents were stirred with K2CO3 in 4 mL of DMSO at room temperature for 14 h. e Chemical structures of DM2, DM4, DM6, DM10, and DM12.
Inhibitory potency of DM10 on DJ-1 deglycase activity by targeting the homodimer of DJ-1
To elucidate the biological properties of DMs, we first performed an N-acetylcysteine deglycation assay based on the absorbance at 288 nm, which indicated the degradation of hemithioacetal formed between N-acetylcysteine and MGO into N-acetylcysteine and lactate in the presence of DJ-1 (Fig. 2a). Because the compounds have a double functional structure compared with the structure of STK, it was expected that they would exert a strong inhibitory effect on the function of DJ-1. As shown in Fig. 2b, STK and all DMs inhibited the deglycase activity of DJ-1 (compared with the black line). Among these DMs, the inhibitory potency of DM4–DM12 was stronger than that of STK at the same concentration, while DM2 showed an inferior inhibitory property than STK alone. The inhibitory potency of DM6–DM12 was similar to or even stronger than that of STK at two concentrations (Fig. 2b). Furthermore, DM10 showed the best inhibitory property and was significantly higher than two concentrations of STK (Fig. 2c). It is conceivable that the compound could not target the DJ-1 homodimer well when the length of the linker was too short, and therefore, the remaining half of the functional group could not covalently bind to the second Cys106 of the DJ-1 homodimer, which resulted in its weak inhibitory potency. Collectively, these data further suggest that the combination of functional groups linked by the C chain is not a linear response, but there is a preference for the length of the C chain, which may be related to the size of the DJ-1 dimerization interface.
Fig. 2. Inhibitory potency of DM10 on DJ-1 deglycase activity by targeting the DJ-1 homodimer.

a The schematic representation of the N-acetylcysteine deglycation assay. The N-acetylcysteine deglycation assay based on the absorbance at 288 nm indicates the degradation of hemithioacetal formed between N-acetylcysteine and methylglyoxal (MGO) into N-acetylcysteine and lactate in the presence of DJ-1. b Human recombinant wild-type DJ-1 protein was purified and incubated with STK or DMs for 30 min, and then the deglycase activity was assayed. The data are reported as a ratio relative to the initial OD value under test conditions. “1STK” represents the molar ratio of DJ-1:STK = 1:1; “2STK” represents the molar ratio of DJ-1:STK = 1:2; and “1DMs” represents the molar ratio of DJ-1:DMs = 1:1. Independent experiments were repeated three times, and representative data are shown. c The statistical analysis in (b). Data under the test conditions were reported as a ratio relative to the 2STK treatment. Data shown represent the mean ± SD, n = 3; comparisons were made using the two-tailed, unpaired Student’s t test. *P < 0.05 (versus 2STK group). d–f DM10 binds to the DJ-1 homodimer. d The human recombinant wild-type DJ-1 protein was purified and incubated with STK, DM2, or DM10 for 30 min and then analyzed by Western blotting. “2STK” represents the molar ratio of DJ-1:STK = 1:2; “1DM2” represents the molar ratio of DJ-1:DM2 = 1:1; and “1DM10” represents the molar ratio of DJ-1:DM10 = 1:1. Independent experiments were repeated three times, and representative data are shown. e H1299 cells were infected with lentivirus (DJ-1-FLAG) for 72 h. The indicated H1299 cells with DJ-1 re-overexpression were incubated with DM10 (2.5–20 μM) for 48 h and then analyzed by Western blotting. Independent experiments were repeated three times, and representative data are shown. f Recombinant proteins DJ-1-WT (wild type), DJ-1-ΔC2 (deletion of two amino acids from the C-terminus), and DJ-1-ΔC3 (deletion of three amino acids from the C-terminus) were purified and incubated with DM10 (DJ-1:DM10 = 1:1) for 30 min and then analyzed by Western blotting. Independent experiments were repeated three times, and representative data are shown. g A competitive assay between DM10 and STK or DM2. The human recombinant wild-type DJ-1 protein was purified and incubated with STK or DM2 for 30 min, followed by incubation with DM10 for another 30 min, and then analyzed by Western blotting. “2STK” represents the molar ratio of DJ-1:STK = 1:2; “1DM2” represents the molar ratio of DJ-1:DM2 = 1:1; and “1DM10” represents the molar ratio of DJ-1:DM10 = 1:1. Independent experiments were repeated three times, and representative data are shown.
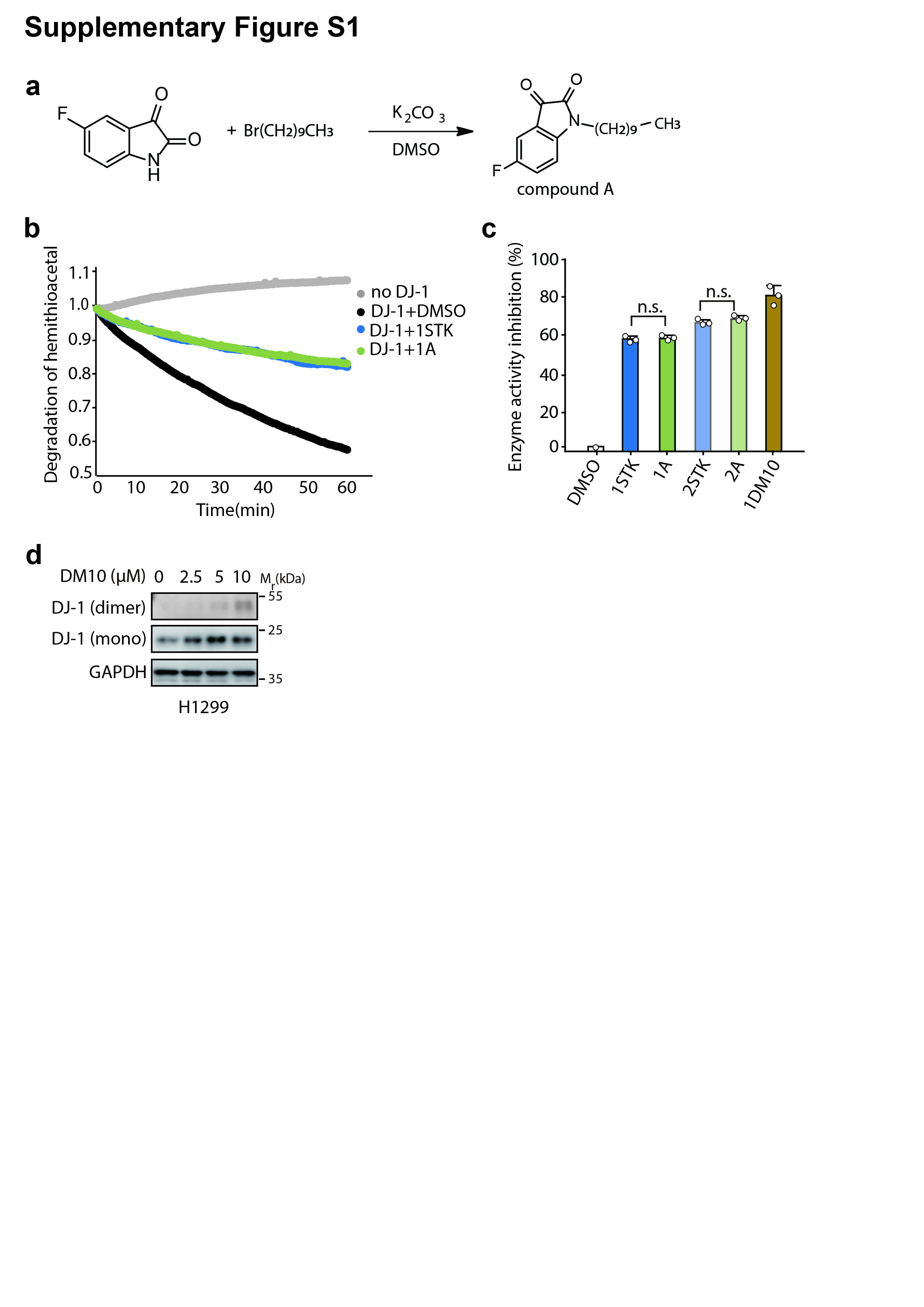
In our model, the C chain in DMs functions as a linker to adapt to the DJ-1 dimerization interface. To further verify that the C linker would not change the warhead chemical properties, we synthesized N-decane-(5-fluoroindoline-2,3-dione) as one piece of isatin combined with a chain of C10 (named compound A) (Supplementary Fig. S1a) and carried out an acetylcysteine deglycation assay. As shown in Supplementary Fig. S1b, c, the inhibitory potency of compound A was the same as that of STK, as expected. Moreover, to determine whether DM10 can target the DJ-1 homodimer, STK and DM10 were incubated with the human recombinant DJ-1 protein (r-DJ-1) and subsequently analyzed in SDS-PAGE gels. Since immunoblotting was performed under reducing conditions, the dimeric form could not be detected unless the protein was covalently crosslinked. As shown in Fig. 2d, compared to STK and DM2, the dimeric form of DJ-1 only appeared in the presence of DM10, suggesting that DM10 acts on the DJ-1 homodimer. Similar results were observed at the cellular level, where DM10 triggered DJ-1 dimer formation in a concentration-dependent manner in H1299 cells with or without the exogenous overexpression of FLAG-tagged DJ-1 (Fig. 2e and Supplementary Fig. S1d).
Next, we evaluated the role of DM10 in dimer-deficient DJ-1. Given that the last three amino acids of the C-terminus determine dimeric DJ-1 formation [33], the recombinant proteins DJ-1-ΔC2 (deletion of two amino acids from the C-terminus), and DJ-1-ΔC3 (deletion of three amino acids from the C-terminus) were purified and incubated with DM10, followed by immunoblotting. As shown in Fig. 2f, compared to the dimer formation of wild-type DJ-1, DM10 triggered the formation of the DJ-1 dimer in the dimer-preserved DJ-1 mutant (DJ-1-ΔC2) but not in the dimer-deficient mutant DJ-1 (DJ-1-ΔC3). Additionally, a competitive assay was performed among DM10 and STK or DM2, where DM10 could not trigger DJ-1 dimer formation with STK or DM2 pretreatment (Fig. 2g), which indicated that DM10 binds to Cys106 of DJ-1, the same amino acid that binds to STK.
Taken together, these results suggest that DM10 may selectively bind to the DJ-1 dimerization interface and inhibit the functional form of DJ-1, thereby serving as a dimeric DJ-1 inhibitor and exerting a potential inhibitory potency on DJ-1 deglycase activity.
The antitumor activity of DM10
Some studies have shown that modulating DJ-1 protein levels by biological methods affects the outcome of chemotherapy; however, there are no studies on the effect of DJ-1 inhibitors in cancer. Hence, we next investigated the biological activities of DM10 in human cancer cells. Growth inhibition was measured in H1299 cells, a human non-small cell lung carcinoma (NSCLC) cell line, to investigate DJ-1 overexpression compared with growth inhibition in normal adjacent control tissues [34]. As the most potent inhibitor of dimeric DJ-1, DM10 significantly inhibited the growth of H1299 cells in a concentration- and time-dependent manner (Supplementary Fig. S2a).
Thus, we conducted a further evaluation of the anticancer effect of DM10 and STK, and growth inhibition was measured after 72 h of treatment. DM10 showed a better anticancer effect than STK, whose inhibition ratio at 10–20 μM was 85%–93%, whereas the inhibition ratio of STK was only 61% at 20 μM (Fig. 3a). Similar sensitization was observed in PC9 cells, another NSCLC cell line, with an inhibition ratio of 85%–93% in the DM10 treatment group at 10–20 μM and 6% in the STK treatment group at 10–20 μM (Supplementary Fig. S2b). We also investigated the antitumor activity of DM10 in other cancer cells. As shown in Fig. 3b and Supplementary Fig. S2c, more intense suppression was found in the DM10 treatment group than in the STK treatment group at the same concentration in MDA-MB-231 and HCC1937 breast cancer cells. Similar results were observed in BEL7402 hepatocellular carcinoma cells (Fig. 3c), HepG2 cells (Supplementary Fig. S2d), 786-O renal cell carcinoma cells (Fig. 3d), and RCC4 cells (Supplementary Fig. S2e). These data demonstrate that DM10 significantly affects the viability of cancer cells after structural modification from STK.
Fig. 3. The antitumor activity of DM10.

a–d The indicated cells were incubated with STK (2.5–20 μM) or DM10 (2.5–20 μM) for 72 h, followed by the SRB assay. The growth inhibition of H1299 (a), MDA-MB-231 (b), BEL7402 (c), and 786-O (d) cells is shown. Data shown represent mean ± SD, n = 3.
DM10 enhances 4HPR-induced apoptosis
Our group and others have reported that the cancer chemopreventive and therapeutic retinoid 4HPR mainly suppresses cancer cells by inducing apoptosis, and DJ-1 overexpression weakens the therapeutic effect of chemotherapy. Thus, we next wanted to test whether the dimeric DJ-1 inhibitor DM10 could enhance 4HPR-triggered apoptotic cell death. H1299 cells were exposed to DM10 before 4HPR treatment, and then cell survival was examined using the SRB assay. As illustrated in Fig. 4a, b, combination treatment with DM10 and 4HPR resulted in significantly increased cell death (14% in the DM10 treatment group, 11% in the 4HPR treatment group, and 54% in the combination treatment group). Of note, the c-PARP protein and the cleaved-caspase-3 protein (two classic apoptosis markers) were observed only under combination treatment with DM10 and 4HPR (Fig. 4c). Similar sensitization was also observed in three other cancer cells, MDA-MB-231 cells (8% in the DM10 treatment group, 6% in the 4HPR treatment group, and 85% in the combination treatment group) (Supplementary Fig. S3a), BEL7402 cells (41% in the DM10 treatment group, 18% in the 4HPR treatment group, and 81% in the combination treatment group) (Supplementary Fig. S3b), and 786-O cells (3% in the DM10 treatment group, 15% in 4HPR treatment group, and 80% in the combination treatment group) (Supplementary Fig. S3c). Taken together, these results suggest that the DJ-1 inhibitor DM10 activates apoptosis triggered by 4HPR and then enhances the anticancer activity.
Fig. 4. DM10 enhanced 4HPR-induced apoptosis.

a–c H1299 cells were preincubated with DM10 (10 μM) for 12 h, incubated with or without 4HPR (10 μM) for another 36 h, and evaluated to determine apoptosis. a Growth inhibition was detected using the SRB assay. Data shown represent the mean ± SD, n = 3; comparisons were made using the two-tailed, unpaired Student’s t test. **P < 0.01. b The phase-contrast images of H1299 cells are shown. Independent experiments were repeated three times, and representative data are shown. c Total cell extracts were probed with antibodies against PARP, cleaved PARP (c-PARP), cleaved caspase-3 (c-caspase-3), and GAPDH. Independent experiments were repeated three times, and representative data are shown.
DM10 enhances erastin-induced ferroptosis
Ferroptosis is a process driven by accumulated iron-dependent lipid ROS that leads to cell death [35], and increasing evidence suggests that ferroptosis plays a crucial role in the suppression of tumorigenesis and may be a beneficial strategy for anticancer treatment [36–40]. We recently found that DJ-1 determines the response of cancer cells to ferroptosis and that DJ-1 depletion potently enhances the sensitivity of ferroptosis inducers both in vitro and in vivo [19]. Therefore, we were also interested in the effect of the DJ-1 inhibitor DM10 on ferroptosis-based cell death. We first used erastin to establish a model of ferroptosis-based cell death. H1299 cells were exposed to DM10 before treatment with erastin, a specific inducer of ferroptosis, and then cell survival was examined using the SRB assay. Interestingly, we found that DM10 dramatically increased the effect of erastin-triggered ferroptotic cell death induction, with a growth inhibition rate of 20% in the erastin treatment group and 70% in the combination treatment group (Fig. 5a). Similar results were observed in three other cancer cell lines, MDA-MB-231 cells (8% in the erastin treatment group and 57% in the combination treatment group) (Supplementary Fig. S4a, b), BEL7402 cells (7% in the erastin treatment group and 80% in the combination treatment group) (Supplementary Fig. S4c, d), and 786-O cells (5% in the erastin treatment group and 62% in the combination treatment group) (Supplementary Fig. S4e, f). To validate that DM10 specifically enhances the ferroptotic process, we used Fer-1, a specific inhibitor of ferroptosis [41]. Notably, Fer-1 treatment completely reversed cell death in the combination treatment with DM10 and erastin (Fig. 5b, c). We next monitored the accumulation of lipid ROS, a classic biomarker of ferroptosis [35], with flow cytometry using the fluorescent probe C11-BODIPY. Similar sensitization was observed in the combination treatment group (Fig. 5d). Thus, under ferroptotic stimuli, the DJ-1 inhibitor DM10 markedly amplified the ferroptosis-inducing effect.
Fig. 5. DM10 enhanced erastin-induced ferroptosis.

a H1299 cells were preincubated with DM10 (10 μM) for 12 h and then incubated with or without erastin (1 μM) for another 36 h, and growth inhibition was detected using the SRB assay. b, c Fer-1 inhibited the effect of DM10. H1299 cells were preincubated with DM10 (10 μM) for 12 h and then incubated with or without erastin (1 μM) or Fer-1 (1.5 μM) for another 36 h. Growth inhibition was detected using the SRB assay (b), and phase-contrast images of H1299 cells are shown (c). d H1299 cells were preincubated with DM10 (10 μM) for 12 h and then incubated with or without erastin (1 μM) or Fer-1 (1.5 μM) for another 24 h, and lipid ROS production was assayed by flow cytometry using C11-BODIPY. Representative data (left view) and the statistical analysis (right view) are shown. Data shown represent the mean ± SD, n = 3; comparisons were made using the two-tailed, unpaired Student’s t test. **P < 0.01; ***P < 0.001.
The in vivo study of DM10 antitumor activity
To evaluate whether treatment with DM10 could inhibit tumor growth in vivo, we established a xenograft nude mouse model generated with H1299 cells. Beginning at day 12, the xenograft nude mice were divided into three groups (n = 7 for each group) and incubated with vehicle, 30 mg/kg STK or 15 mg/kg DM10 through one intratumor injection. The tumor volume and body weight were measured every 3 days. After 3 weeks of treatment, compared with the tumor growth in the vehicle control group, tumor growth in the 30 mg/kg STK and 15 mg/kg DM10 groups was inhibited (Fig. 6a, b). Of note, DM10 had a stronger antitumor activity than STK. The tumor masses were ~1.5- and 8-fold higher in control mice than in those incubated with STK and DM10, respectively, without affecting the mouse body weight (Fig. 6c, d). Thus, our data suggest that DM10 has high antitumor activity and could reduce tumor growth in vivo.
Fig. 6. DM10 inhibited the tumor growth in vivo.

The mice were injected subcutaneously with the indicated H1299 cells and incubated with 15 mg/kg DM10, 30 mg/kg STK or vehicle control. a Tumor volume was monitored every 3 days. b Representative images of tumors in each treatment group. c Weight of excised tumors. d Changes in body weight of mice pretreatment and posttreatment. n = 7. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Discussion
The involvement of DJ-1 in a plethora of oncogenic pathways has positioned DJ-1 as a prominent therapeutic target [42]. Given that DJ-1 is frequently overexpressed in many cancers, cancer cells are particularly sensitive to chemotherapeutic drugs when DJ-1 is suppressed. Over the last decade, DJ-1 interventions, from RNA interference to small-molecule inhibitors, have been developed. Accordingly, the isatin family defined by the Shinya Tashiro group showed potent inhibition of the purified DJ-1 protein with a defined mechanism of action [27]; however, these inhibitors have not been assessed at the cellular level, especially the anticancer effect, but have been used only as a tool to analyze the functions of DJ-1. The DJ-1 targeting selectivity in cancer cells is a major concern with these inhibitors. None of these inhibitors have been reported to target the functional dimeric form of DJ-1. Here, we developed DM10, a compound with a symmetric structure linked by the C chain that blocks DJ-1 deglycase activity by specifically binding to the dimer interface of DJ-1 and then inhibits the biological function of DJ-1 in cancer. In addition, DM10 facilitates the cell death triggered by 4HPR and erastin, showing vulnerability to apoptosis and ferroptosis, which are both effective cancer therapy strategies.
It has been reported that the isatin family inhibits DJ-1 deglycase activity by covalently binding to DJ-1 [27]. Thus, our designed compounds (DM2–DM12) are a combination of two identical functional groups via a carbon chain, are symmetric structures and have stronger inhibitory effects than STK. It is interesting that the effect of DM2–DM12 on the DJ-1 deglycase activity is not a linear phenomenon, as DM10 appears to have the most potent effect since it has the best fit between the length of the C chain of DM10 and the size of the DJ-1 dimerization interface among these DMs. These results indicate that a proper length of this symmetric structure is important and depends on the dimer interface of DJ-1. However, the anticancer effects in H1299 cells seemed not exactly consistent with the results from the in vitro deglycase assay; DM2 showed less inhibitory deglycase activity but had the best anticancer effect. We think this is probably because of the small size of DM2. DM2 is active with two functional groups but shows weak selectivity for DJ-1; thus, its inhibitory effect on DJ-1 is mild in the purified protein system, but in cells, DM2 could bind to other proteins with a cysteine and lead to severe cell death. These results highlight the importance of the size of DJ-1 inhibitors, and proper size may improve the selectivity and achieve a potent anticancer effect. Moreover, it is reasonable to aim for better selectivity and anticancer properties by applying additional optimization schemes, especially by exploiting a larger affinity at the dimer interface of DJ-1.
However, DJ-1 also plays a pivotal role in PD [2, 43], which is a progressive neurodegenerative disorder associated with the degeneration of dopamine neurons of the substantia nigra pars compacta and the presence of intraneuronal aggregates of α-synuclein and its posttranslational products [44–46]. Indeed, α-synuclein acts as a substrate for the deglycase activity of DJ-1, and reduced DJ-1 activity may lead to the accumulation of glycated α-synuclein and its aggregates in dopaminergic cells and then cause PD [47]. It is not clear whether DM10 influences DJ-1 functions in dopaminergic cells. Therefore, although DJ-1-based cancer therapy is promising, it will be meaningful to test the permeation of blood–brain barrier of DM10.
Cancer treatment is becoming increasingly personalized and driven by effective therapies in biomarker-defined populations. An increasing number of studies have found that the expression of DJ-1 is enhanced in NSCLC compared to normal lung tissue, and DJ-1 can serve as a predictor of recurrence [48, 49]. Regarding the DJ-1 protein expression level and cancer progression, patients with NSCLC, breast cancer, hepatocellular carcinoma, and renal cell carcinoma with high DJ-1 protein frequently showed lymph node metastases [34], shorter overall survival time [10, 50, 51], and drug resistance [52]. These findings highlight the potential target of suppressing DJ-1. Here, we showed the anticancer activity of DM10 both in vitro and in vivo. Additionally, DM10 enhanced the susceptibility of several kinds of cancer cells, including H1299, MDA-MB-231, BEL7402, and 786-O cells, to apoptosis and ferroptosis inducers. All these results supported our previous discovery about the role of DJ-1 in apoptosis and ferroptosis. Thus, in the future, DM10 or its analogs could also be applied to other cancer cells that are characterized by DJ-1 overexpression, such as pancreatic cancer [11] and acute leukemia [22], which have an intolerably poor prognosis.
In summary, we report a new inhibitor of DJ-1 deglycase activity that targets the DJ-1 homodimer and enhances the effect of anticancer therapies based on apoptosis and ferroptosis, suggesting a synergistic therapeutic option for cancer.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81773757 to MDY; No. 81402951 to JC), the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program,” China (2018ZX09711002-003-008 to HZ), and the Zhejiang Provincial Natural Science Foundation (No. Y18H310005 to JC).
Author contributions
JC, RS, and XBC designed the research; JC, RS, and XBC wrote the paper; KB and RS performed the chemical synthesis studies; XBC, HYZ, and LJ performed the biochemical and cellular studies; XBC, HYZ, JC, MDY, and HZ analyzed the results; and JC, RS, QJH, and BY supervised the study.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Xiao-bing Chen, Hai-ying Zhu
Contributor Information
Rong Sheng, Email: shengr@zju.edu.cn.
Ji Cao, Email: caoji88@zju.edu.cn.
Supplementary information
The online version of this article (10.1038/s41401-020-00600-5) contains supplementary material, which is available to authorized users.
References
- 1.Nagakubo D, Taira T, Kitaura H, Ikeda M, Tamai K, Iguchi-Ariga SM, et al. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun. 1997;231:509–13. doi: 10.1006/bbrc.1997.6132. [DOI] [PubMed] [Google Scholar]
- 2.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 3.Aleyasin H, Rousseaux MW, Phillips M, Kim RH, Bland RJ, Callaghan S, et al. The Parkinson’s disease gene DJ-1 is also a key regulator of stroke-induced damage. Proc Natl Acad Sci USA. 2007;104:18748–53. doi: 10.1073/pnas.0709379104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi J, Sullards MC, Olzmann JA, Rees HD, Weintraub ST, Bostwick DE, et al. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem. 2006;281:10816–24. doi: 10.1074/jbc.M509079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sajjad MU, Green EW, Miller-Fleming L, Hands S, Herrera F, Campesan S, et al. DJ-1 modulates aggregation and pathogenesis in models of Huntington’s disease. Hum Mol Genet. 2014;23:755–66. doi: 10.1093/hmg/ddt466. [DOI] [PubMed] [Google Scholar]
- 6.Yanagisawa D, Kitamura Y, Inden M, Takata K, Taniguchi T, Morikawa S, et al. DJ-1 protects against neurodegeneration caused by focal cerebral ischemia and reperfusion in rats. J Cereb Blood Flow Metab. 2008;28:563–78. doi: 10.1038/sj.jcbfm.9600553. [DOI] [PubMed] [Google Scholar]
- 7.Jain D, Jain R, Eberhard D, Eglinger J, Bugliani M, Piemonti L, et al. Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J Mol Cell Biol. 2012;4:221–30. doi: 10.1093/jmcb/mjs025. [DOI] [PubMed] [Google Scholar]
- 8.An CN, Jiang H, Wang Q, Yuan RP, Liu JM, Shi WL, et al. Down-regulation of DJ-1 protein in the ejaculated spermatozoa from Chinese asthenozoospermia patients. Fertil Steril. 2011;96:19–23.e2. doi: 10.1016/j.fertnstert.2011.04.048. [DOI] [PubMed] [Google Scholar]
- 9.Okada M, Matsumoto K, Niki T, Taira T, Iguchi-Ariga SM, Ariga H. DJ-1, a target protein for an endocrine disrupter, participates in the fertilization in mice. Biol Pharm Bull. 2002;25:853–6. doi: 10.1248/bpb.25.853. [DOI] [PubMed] [Google Scholar]
- 10.Zeng HZ, Qu YQ, Zhang WJ, Xiu B, Deng AM, Liang AB. Proteomic analysis identified DJ-1 as a cisplatin resistant marker in non-small cell lung cancer. Int J Mol Sci. 2011;12:3489–99. doi: 10.3390/ijms12063489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Kang M, Lu W, Guo Q, Zhang B, Xie Q, et al. DJ-1, a novel biomarker and a selected target gene for overcoming chemoresistance in pancreatic cancer. J Cancer Res Clin Oncol. 2012;138:1463–74. doi: 10.1007/s00432-012-1205-3. [DOI] [PubMed] [Google Scholar]
- 12.Blackinton J, Lakshminarasimhan M, Thomas KJ, Ahmad R, Greggio E, Raza AS, et al. Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J Biol Chem. 2009;284:6476–85. doi: 10.1074/jbc.M806599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, et al. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci USA. 2004;101:9103–8. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinumi T, Kimata J, Taira T, Ariga H, Niki E. Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;317:722–8. doi: 10.1016/j.bbrc.2004.03.110. [DOI] [PubMed] [Google Scholar]
- 15.Zhou W, Zhu M, Wilson MA, Petsko GA, Fink AL. The oxidation state of DJ-1 regulates its chaperone activity toward alpha-synuclein. J Mol Biol. 2006;356:1036–48. doi: 10.1016/j.jmb.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 16.Wilson MA. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid Redox Signal. 2011;15:111–22. doi: 10.1089/ars.2010.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JPY. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci USA. 2006;103:15091–6. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao J, Ying M, Xie N, Lin G, Dong R, Zhang J, et al. The oxidation states of DJ-1 dictate the cell fate in response to oxidative stress triggered by 4-hpr: autophagy or apoptosis? Antioxid Redox Signal. 2014;21:1443–59. doi: 10.1089/ars.2013.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao J, Chen X, Jiang L, Lu B, Yuan M, Zhu D, et al. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat Commun. 2020;11:1251. doi: 10.1038/s41467-020-15109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacKeigan JP, Clements CM, Lich JD, Pope RM, Hod Y, Ting JP. Proteomic profiling drug-induced apoptosis in non-small cell lung carcinoma: identification of RS/DJ-1 and RhoGDIalpha. Cancer Res. 2003;63:6928–34. [PubMed] [Google Scholar]
- 21.Hod Y. Differential control of apoptosis by DJ-1 in prostate benign and cancer cells. J Cell Biochem. 2004;92:1221–33. doi: 10.1002/jcb.20159. [DOI] [PubMed] [Google Scholar]
- 22.Liu H, Wang M, Li M, Wang D, Rao Q, Wang Y, et al. Expression and role of DJ-1 in leukemia. Biochem Biophys Res Commun. 2008;375:477–83. doi: 10.1016/j.bbrc.2008.08.046. [DOI] [PubMed] [Google Scholar]
- 23.Zhu H, Liao SD, Shi JJ, Chang LL, Tong YG, Cao J, et al. DJ-1 mediates the resistance of cancer cells to dihydroartemisinin through reactive oxygen species removal. Free Radic Biol Med. 2014;71:121–32. doi: 10.1016/j.freeradbiomed.2014.03.026. [DOI] [PubMed] [Google Scholar]
- 24.Chen X, Mangala LS, Rodriguez-Aguayo C, Kong X, Lopez-Berestein G, Sood AK. RNA interference-based therapy and its delivery systems. Cancer Metastasis Rev. 2018;37:107–24. doi: 10.1007/s10555-017-9717-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richarme G, Mihoub M, Dairou J, Bui LC, Leger T, Lamouri A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J Biol Chem. 2015;290:1885–97. doi: 10.1074/jbc.M114.597815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richarme G, Liu C, Mihoub M, Abdallah J, Leger T, Joly N, et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science. 2017;357:208–11. doi: 10.1126/science.aag1095. [DOI] [PubMed] [Google Scholar]
- 27.Tashiro S, Caaveiro JMM, Nakakido M, Tanabe A, Nagatoishi S, Tamura Y, et al. Discovery and optimization of inhibitors of the Parkinson’s disease associated protein DJ-1. ACS Chem Biol. 2018;13:2783–93. doi: 10.1021/acschembio.8b00701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Honbou K, Suzuki NN, Horiuchi M, Niki T, Taira T, Ariga H, et al. The crystal structure of DJ-1, a protein related to male fertility and Parkinson’s disease. J Biol Chem. 2003;278:31380–4. doi: 10.1074/jbc.M305878200. [DOI] [PubMed] [Google Scholar]
- 29.Tao X, Tong L. Crystal structure of human DJ-1, a protein associated with early onset Parkinson’s disease. J Biol Chem. 2003;278:31372–9. doi: 10.1074/jbc.M304221200. [DOI] [PubMed] [Google Scholar]
- 30.Olzmann JA, Brown K, Wilkinson KD, Rees HD, Huai Q, Ke H, et al. Familial Parkinson’s disease-associated L166P mutation disrupts DJ-1 protein folding and function. J Biol Chem. 2004;279:8506–15. doi: 10.1074/jbc.M311017200. [DOI] [PubMed] [Google Scholar]
- 31.Baulac S, LaVoie MJ, Strahle J, Schlossmacher MG, Xia W. Dimerization of Parkinson’s disease-causing DJ-1 and formation of high molecular weight complexes in human brain. Mol Cell Neurosci. 2004;27:236–46. doi: 10.1016/j.mcn.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Moore DJ, Zhang L, Dawson TM, Dawson VL. A missense mutation (L166P) in DJ-1, linked to familial Parkinson’s disease, confers reduced protein stability and impairs homo-oligomerization. J Neurochem. 2003;87:1558–67. doi: 10.1111/j.1471-4159.2003.02265.x. [DOI] [PubMed] [Google Scholar]
- 33.Jiang L, Chen X, Wu Q, Zhu H, Du C, Ying M, et al. C terminus of DJ-1 determines its homodimerization, deglycation activity and suppression of ferroptosis. Acta Pharmacol Sin. 2020. 10.1038/s41401-020-00531-1. [DOI] [PMC free article] [PubMed]
- 34.Bai J, Guo C, Sun W, Li M, Meng X, Yu Y, et al. DJ-1 may contribute to metastasis of non-small cell lung cancer. Mol Biol Rep. 2012;39:2697–703. doi: 10.1007/s11033-011-1024-7. [DOI] [PubMed] [Google Scholar]
- 35.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2:e1054549. doi: 10.1080/23723556.2015.1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–84. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4. doi: 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–6. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao J, Chen X, Ying M, He Q, Yang B. DJ-1 as a therapeutic target against cancer. Adv Exp Med Biol. 2017;1037:203–22. doi: 10.1007/978-981-10-6583-5_13. [DOI] [PubMed] [Google Scholar]
- 43.Dolgacheva LP, Berezhnov AV, Fedotova EI, Zinchenko VP, Abramov AY. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J Bioenerg Biomembr. 2019;51:175–88. doi: 10.1007/s10863-019-09798-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forno LS. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol. 1996;55:259–72. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 45.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 46.Dickson DW. Neuropathology of Parkinson disease. Parkinsonism Relat Disord. 2018;46 Suppl 1:S30–3. [DOI] [PMC free article] [PubMed]
- 47.Sharma N, Rao SP, Kalivendi SV. The deglycase activity of DJ-1 mitigates alpha-synuclein glycation and aggregation in dopaminergic cells: role of oxidative stress mediated downregulation of DJ-1 in Parkinson’s disease. Free Radic Biol Med. 2019;135:28–37. doi: 10.1016/j.freeradbiomed.2019.02.014. [DOI] [PubMed] [Google Scholar]
- 48.Fan J, Yu H, Lv Y, Yin L. Diagnostic and prognostic value of serum thioredoxin and DJ-1 in non-small cell lung carcinoma patients. Tumour Biol. 2016;37:1949–58. [DOI] [PubMed]
- 49.Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell. 2005;7:263–73. doi: 10.1016/j.ccr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 50.Kawate T, Iwaya K, Kikuchi R, Kaise H, Oda M, Sato E, et al. DJ-1 protein expression as a predictor of pathological complete remission after neoadjuvant chemotherapy in breast cancer patients. Breast Cancer Res Treat. 2013;139:51–9. doi: 10.1007/s10549-013-2523-0. [DOI] [PubMed] [Google Scholar]
- 51.Liu S, Yang Z, Wei H, Shen W, Liu J, Yin Q, et al. Increased DJ-1 and its prognostic significance in hepatocellular carcinoma. Hepatogastroenterology. 2010;57:1247–56. [PubMed] [Google Scholar]
- 52.Trivedi R, Dihazi GH, Eltoweissy M, Mishra DP, Mueller GA, Dihazi H. The antioxidant protein PARK7 plays an important role in cell resistance to cisplatin-induced apoptosis in case of clear cell renal cell carcinoma. Eur J Pharmacol. 2016;784:99–110. doi: 10.1016/j.ejphar.2016.04.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.