

Abstract
Sepsis is life-threatening organ dysfunction due to dysregulated systemic inflammatory and immune response to infection, often leading to cognitive impairments. Growing evidence shows that artemisinin, an antimalarial drug, possesses potent anti-inflammatory and immunoregulatory activities. In this study we investigated whether artemisinin exerted protective effect against neurocognitive deficits associated with sepsis and explored the underlying mechanisms. Mice were injected with LPS (750 μg · kg−1 · d−1, ip, for 7 days) to establish an animal model of sepsis. Artemisinin (30 mg · kg−1 · d−1, ip) was administered starting 4 days prior LPS injection and lasting to the end of LPS injection. We showed that artemisinin administration significantly improved LPS-induced cognitive impairments assessed in Morris water maze and Y maze tests, attenuated neuronal damage and microglial activation in the hippocampus. In BV2 microglial cells treated with LPS (100 ng/mL), pre-application of artemisinin (40 μΜ) significantly reduced the production of proinflammatory cytokines (i.e., TNF-α, IL-6) and suppressed microglial migration. Furthermore, we revealed that artemisinin significantly suppressed the nuclear translocation of NF-κB and the expression of proinflammatory cytokines by activating the AMPKα1 pathway; knockdown of AMPKα1 markedly abolished the anti-inflammatory effects of artemisinin in BV2 microglial cells. In conclusion, atemisinin is a potential therapeutic agent for sepsis-associated neuroinflammation and cognitive impairment, and its effect is probably mediated by activation of the AMPKα1 signaling pathway in microglia.
Keywords: artemisinin, sepsis, cognitive dysfunction, neuroinflammation, microglia, AMP-activated protein kinases
Introduction
Sepsis is defined as life-threatening organ dysfunction due to dysregulated systemic inflammation and immune responses to infection [1]. Cognitive impairments are common in patients with sepsis and are associated with poor prognosis [2, 3]. Neuroinflammation plays a key role in sepsis-induced cognitive impairments. In the early stage of sepsis, inflammatory mediators and neurotoxic factors enter the brain through the injured blood–brain barrier and lead to microglial activation [1]. Severe microglial activation induces the release of proinflammatory cytokines (TNF-α, IL-6, etc.) and causes brain damage, specifically in the hippocampus [4]. Research suggests that neurocognitive dysfunction persists even after the patient has recovered from sepsis [5]. Unfortunately, there are currently no effective preventive or therapeutic measures for sepsis-associated cognitive deficits [6]. Since the discovery of artemisinin in the 1970s, artemisinin and its derivatives (artemisinins) have been used as first-line antimalarial drugs and have saved millions of malaria patients worldwide [7, 8]. In recent years, considerable efforts have been made to explore the unique chemical and pharmacological properties of this remarkable phytochemical. Accumulating evidence reveals that artemisinin also possesses potent anti-inflammatory, antitumor, and antifibrotic properties [9–12]. We recently reported that artemisinin protects neuronal HT-22 cells from oxidative injury by activating the Akt pathway [13]. This finding suggests that artemisinin may have a potential therapeutic effect on nervous system diseases. However, the role of artemisinin in neuroinflammation and cognitive dysfunction associated with sepsis remains unknown. In the present study, we evaluated the in vivo and in vitro therapeutic effects of artemisinin on neuroinflammation and cognitive dysfunction associated with sepsis. We also characterized the possible protective mechanism of artemisinin.
Materials and methods
Experimental animals
Male C57BL/6 mice (4 weeks old, 15–20 g, certificate No. SCXK2018-0002) were purchased from the Guangdong Laboratory Animal Central (Guangzhou, China) and housed under standard conditions. These mice were fed a standard laboratory diet. One week later, the animals were randomly divided into the following three groups: the sham group, LPS group, and LPS + ART group. There were forty mice in each group. The mice received artemisinin (Sigma Aldrich, MO, USA) and LPS (Sigma Aldrich, MO, USA) by intraperitoneal (ip) injection, as shown in Fig. 1a. From day 1 to day 7, artemisinin was injected first, and LPS was injected 3 h later. Artemisinin was dissolved in DMSO. The doses of artemisinin (30 mg · kg−1 · d−1) and LPS (750 μg · kg−1 · d−1) were chosen based on previous studies [14–16]. The study protocol was approved by the Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University (permit No. 2016-102), and all procedures were carried out according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (USA) [17].
Fig. 1. Artemisinin improved cognitive impairment in an LPS-induced sepsis model.

a Timeline of the in vivo experimental treatments. b Murine sepsis score (MSS) at different time points. c Neurobehavioral score at different time point. d The expression levels of IL-6 and TNF-α in the serum and brain were measured by ELISA. e Analysis of the data obtained in the Morris water maze test. The escape latency was recorded in the positioning navigation experiment. The average swimming speed, swimming distance, and percentage of total time in the target quadrant were recorded in the space exploration experiment. f The percentage of alternations was observed in the Y maze spontaneous alternation test. *P < 0.05 versus sham group mice. #P < 0.05 versus LPS-treated mice. n = 6.
Murine sepsis score
The murine sepsis score (MSS) was calculated to assess sepsis severity in the animal model. The MSS consists of seven indices: appearance, consciousness, activity, response to stimulus, eyes, respiration rate, and respiration quality. Each index has a score from “0” to “4”. A higher score is associated with more severe sepsis [18]. Two independent and blinded researchers determined the MSS.
Neurobehavioral score
Neurobehavioral scoring was used to assess neural reflexes to confirm the development of septic encephalopathy in the mice. The point scoring consists of the pinna reflex, righting reflex, corneal reflex, tail flexion, and escape response. A score of “0” means that the mouse has no reflex, “1” means the mouse has a reflex longer than 1 s, and “2” means the mouse has a reflex in 1 s. The lower the score, the more severe the neurological damage to the brain [19]. Two independent and blinded researchers determined the neurobehavioral score.
Morris water maze test
To evaluate cognitive disorders and the therapeutic effects of artemisinin in mice, the Morris water maze (MWM) test was conducted as described previously [16, 20]. First, 4 days of training trials were conducted for all groups of animals prior to ip LPS injection. The mice were trained twice to find a hidden platform in the maze within 60 s. If a mouse failed to find the platform within 60 s, it was guided to the platform and allowed to stay on it for 10 s. The positioning navigation experiment was conducted 5 days after LPS injection, and the escape latency was recorded. On the 6th day after LPS injection, a space exploration experiment was performed in which the platform was removed from the pool. All mice were monitored for 60 s to observe the average swimming speed, distance of swimming, and percentage of total time in the targeted quadrant. The data were analyzed using SMART 3.0 software (Harvard Apparatus Technology Co., Ltd., MA, USA), which was designed for the MWM test.
Y maze test
The Y maze was used to test short-term spatial memory. The Y maze consisted of a chamber with three arms (A, B, and C) at a 120° angle to each other. In our experiment, Y maze testing was conducted 4 days after LPS injection. During the experiments, each mouse was allowed to explore the Y maze for 8 min. All exploratory behaviors of the mouse were recorded. Alternation behavior was defined as entry into all three arms without repeated entries. Percentage of alteration (%) = number of alteration behaviors/(total entries − 2) × 100.
Tissue preparation and histological analysis
The mice were euthanized and transcardially perfused with phosphate-buffered saline (PBS), followed by a solution of 4% paraformaldehyde. Sections of hippocampal tissue from the brain were prepared and fixed in a 4% paraformaldehyde solution overnight. After being dehydrated in ethanol, the tissues were embedded in paraffin and cut into 4-µm sections. Paraffin-embedded sections of brain tissue were deparaffinized in xylene and rehydrated through descending grades of ethanol (Tianjin Sheng Winton Chemical Co., Ltd., Tianjin, China). The sections were then stained with hematoxylin and eosin to visualize tissue structures. The sections were examined under a light microscope (Nikon Technology Co., Ltd., Japan). The number of hippocampal Cornu Ammonis 1 (CA1) pyramidal neurons per mm2 was used to calculate the neuronal density.
Immunofluorescence and histochemistry
After deparaffinization and rehydration, the slides were immersed in EDTA antigen retrieval buffer and heated for antigen retrieval. The sections were blocked with 3% BSA for 30 min at room temperature and then incubated with primary antibodies against cleaved caspase 3 (Wuhan Servicebio Biotechnology, Co., Ltd., China, GB11532, 1:500) or ionized calcium-binding adaptor molecule-1 (Iba-1, Abcam, Cambridge, UK. ab5076, 1:200), followed by incubation with fluorescent secondary antibodies. The sections were also incubated with DAPI solution at room temperature for 10 min to visualize the nuclei. The sections were imaged with a fluorescence microscope (Axio Observer Z1; Carl Zeiss AG, Germany).
Automated microglial morphological analysis was performed using Fiji Image J software as previously described [21, 22]. Measurements of microglial morphology were performed in a blinded manner. Three consecutive sections of the CA1 areas from each mouse were analyzed. In each section, at least five microglial cells were evaluated. Briefly, microglial cells were randomly selected using a region of interest. The extra signal was eliminated using the brush tool, and binary images of microglial cells were obtained. For fractal analysis, the outlined cell in the binary image was scanned to obtain the hull and circle results, including lacunarity, perimeter, and radius data. For skeleton analysis, the binary image was converted into a skeletonized format. The plugin “skeleton analysis” was applied, and the numbers of branches, endpoints, and junctions were obtained.
Cell culture
BV2 microglial cells were cultured in DMEM (Thermo Fisher Scientific, MA, USA) supplemented with 10% FBS (Thermo Fisher Scientific, MA, USA), 0.5% penicillin, and 0.5% streptomycin (Thermo Fisher Scientific, MA, USA) at 37 °C with a humidified atmosphere of 5% CO2 and 95% air. BV2 cells at passages 10–20 were then divided into three groups: the control group, LPS group (treated with 100 ng/mL LPS for 12 h), and LPS + ART group (pretreated with 40 μΜ artemisinin for 2 h and then treated with 100 ng/mL LPS for another 12 h). Our preliminary experiment showed no cytotoxic effect in response to artemisinin treatment at concentrations ≤80 µM in BV2 cells (Supplementary Fig. S1). Dose-dependent suppressive effects of artemisinin on inflammation were indicated. It was found that 40 μM artemisinin was the minimum effective concentration (Supplementary Fig. S2). Therefore, 40 μΜ artemisinin was chosen as the optimum dose for reversing neuroinflammation. The concentration of LPS was chosen based on previous studies [23, 24].
ELISA
The expression levels of IL-6, TNF-α, IL-1α, IL-1β, MCP-1, and MIP-2 in the cell supernatants, serum, or hippocampal homogenates were measured using ELISA kits (Dakewe Bioengineering Co., Ltd., Shenzhen, China) according to the manufacturer’s instructions. The optical density values were measured at 450 nm by a microplate reader within 5 min, and standard curves were plotted [25]. Both standards and samples were measured in triplicate.
Quantitative RT-PCR
The mRNA expression of IL-6, TNF-α, and TGF-β was determined by qRT-PCR as described previously [26]. Briefly, total RNA was isolated using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocol. Total RNA was reverse-transcribed to synthesize cDNA using a SuperScript III Reverse Transcription Kit (Invitrogen, USA). qRT-PCR was conducted using SYBR Green qPCR SuperMix (Invitrogen, USA) in the ViiA™ 7 Real-time PCR System (Applied Biosystems, USA). The primers used in this protocol were listed in Table 1. The results were analyzed using the 2−ΔΔCt method.
Table 1.
Primers used in this study.
| Primers | Sequence (5′-3′) | |
|---|---|---|
| IL-6 | Forward | CACATGTTCTCTGGGAAATCG |
| Reverse | TTGTATCTCTGGAAGTTTCAGATTGTT | |
| TNF-α | Forward | GCCACCACGCTCTTCTGTCTAC |
| Reverse | GGGTCTGGGCCATAGAACTGAT | |
| TGF-β | Forward | CCAGATCCTGTCCAAACTAAGG |
| Reverse | CTCTTTAGCATAGTAGTCCGCT | |
| β-actin | Forward | GTACCACCATGTACCCAGGC |
| Reverse | AACGCAGCTCAGTAACAGTCC |
Western blot analysis
Western blot analysis was performed on cell lysates according to standard protocols, as previously described [13, 27]. Briefly, total protein was extracted from cells with the radioimmunoprecipitation assay lysis buffer (Kaiji Company, Shenzhen, China) containing a complete protease inhibitor mixture and protein phosphatase inhibitor (Kaiji Company, Shenzhen, China). Nuclear fractions of the cells were prepared using a Nuclear Extraction Kit (Mei5 Biotechnology, Co., Ltd. Beijing, China). Protein samples were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. We then incubated the membranes with diluted primary antibodies against AMPKα1 (Cell Signaling Technology Inc., Danvers, USA; cst 2795s, 1:1000), p-AMPKα1 (Cell Signaling Technology Inc., Danvers, USA; cst 4185s, 1:1000), inducible nitric oxide synthase (iNOS; Cell Signaling Technology Inc., Danvers, USA; cst 13120s, 1:1000), IL-6 (Cell Signaling Technology Inc., Danvers, USA; cst 12912, 1:1000), TNF-α (Abcam, Cambridge, UK; ab1793, 1:1000), TGF-β (Cell Signaling Technology Inc., Danvers, USA; cst 3711, 1:1000), β-actin (Cell Signaling Technology Inc., Danvers, USA; cst 4970s, 1:1000), NF-κB (Cell Signaling Technology Inc., Danvers, USA; cst 8242s, 1:1000), or PCNA (Cell Signaling Technology Inc., Danvers, USA; cst 13100s, 1:1000) overnight at 4 °C. On the following day, diluted secondary antibodies were used to detect the corresponding primary antibodies. The relative expression levels of the target proteins were analyzed using Image-Pro Plus (IPP) 6.0 (Media Cybernetics Inc., Bethesda, MD, USA).
Wound healing assay
For the migration assay, BV2 microglial cells were seeded in six-well plates and cultured until the cells formed a 90% confluent monolayer. A linear wound was made by scratching the cell monolayer with a sterile 200-μL pipette tip. After the wells were washed with PBS to remove cell fragments, the cells were incubated in FBS-free medium. The wound area and cell migration were photographed by a phase-contrast microscope at 0 and 12 h after wounding. All images were analyzed by IPP and the number of migrating cells was counted.
Transwell assay
A total of 100,000 BV2 cells were seeded in FBS-free DMEM in the upper chamber of a Transwell insert (Corning Incorporated, NY, USA). The lower chamber was filled with DMEM supplemented with 10% FBS. After 12 h of incubation, the cells in the upper chamber were removed, and the cells that had invaded the membrane were fixed with 4% paraformaldehyde and stained with crystal violet for 20 min. Five randomly selected fields were captured with an optical microscope, and the number of invading cells was counted.
Small interfering RNA (siRNA) transfection
AMPKα1-targeted siRNA was synthesized by Gene Pharma Company (Gene Pharma, China) and used to transfect BV2 cells. The sequence of the specific AMPKα1 siRNA was 5′-GCAUAUGCUGCAGGUAGAUTT-3′. The sequence of a scrambled control siRNA was 5′-UUCUCCGAACGUGUCACGUTT-3′. Cells were transfected with AMPKα1 siRNA (si-AMPKα1) or scrambled control siRNA (si-NC) 8 h prior to further experimentation using Lipofectamine iMAX (Invitrogen, USA) according to the manufacturer’s instructions.
Immunocytochemical staining
Cells were fixed in BD Cytofix/Cytoperm solution (BD Biosciences, NJ, USA) and permeabilized with 0.1% Triton-X. The cells were incubated overnight with primary antibodies against nuclear factor kappa B (NF-κB; Cell Signaling Technology Inc., Danvers, USA; cst 8242s, 1:500) followed by labeling with FITC-conjugated goat anti-rabbit IgG (Wuhan Servicebio Technology Co., Ltd, Wuhan, China; 1:400). Later, the cells were incubated with 0.5 mg/mL DAPI to stain the nuclei. Images were obtained using a fluorescence microscope. To analyze the ratio of nuclear and cytoplasmic NF-κB fluorescence, the percentage of cells exhibiting high fluorescence in the different areas (either nucleus or cytoplasm) was counted. A total of 200 cells were counted per group. The nuclear translocation of NF-κB was expressed as a percentage of the total cells.
Statistical analysis
The experimental results are expressed as the mean ± SD. Statistical analysis was performed with GraphPad Prism 7.0 software (San Diego, CA, USA) using one-way ANOVA followed by the Tukey’s post hoc analysis. A P value < 0.05 was considered statistically significant.
Materials
BV2 microglial cells were purchased from Guangzhou Jennio Biotech Co., Ltd. (Guangzhou, China). Artemisinin (purity > 98%), LPS, and DMSO were purchased from Sigma Aldrich (MO, USA). Anti-Iba-1 (ab5076) and anti-TNF-α (ab1793) antibodies were purchased from Abcam (Cambridge, UK). Anti-AMPKα1 (cst 2795s), anti-p-AMPKα1 (cst 4185s), anti-iNOS (cst 13120s), anti-IL-6 (cst 12912), anti-NF-κB (cst 8242s), anti-PCNA (cst 13100s), anti-TGF-β (cst 3711), and anti-β-actin (cst 4970s) antibodies were purchased from Cell Signaling Technology Inc. (Danvers, USA). Anti-cleaved caspase 3 (GB11532) antibodies were purchased from Wuhan Servicebio Biotechnology Co., Ltd. (Wuhan, China). TNF-α, IL-6, IL-1α, IL-1β, MCP-1, and MIP-2 ELISA kits were purchased from Dakewe Bioengineering Co., Ltd. (Shenzhen, China).
Results
Artemisinin ameliorated neuronal cell death and improved cognitive impairment in an LPS-induced murine sepsis model
The MSS was determined for all mice (baseline and the 1st, 3rd, 5th, and 7th days after sepsis induction) to assess disease progression. The MSS increased significantly in the LPS group compared with the sham group on the 3rd, 5th, and 7th days. The ART group showed a significant reduction in the MSS on the 3rd, 5th, and 7th days after sepsis induction compared with the LPS group (Fig. 1b). Neurobehavioral scoring was used to further investigate the effect of LPS-induced sepsis on the brain. The neurobehavioral score was significantly lower in the LPS group than in the sham group on the 1st, 3rd, 5th, and 7th days. Treatment with artemisinin increased the neurobehavioral score compared with that of the LPS group on the 5th and 7th days after sepsis induction (Fig. 1c). The inflammatory response in the serum and hippocampus was also evaluated by measuring the expression of IL-6 and TNF-α. The ELISA results showed that exposure to LPS resulted in significant IL-6 and TNF-α secretion and that artemisinin reversed this inflammatory response (at 12 h and on the 7th day after sepsis induction) (Fig. 1d).
The Morris water maze test was used to assess the efficacy of artemisinin treatment on LPS-induced cognitive impairment. Five days after systemic injection of LPS, a positioning navigation experiment was performed to observe the cognitive function of the mice. As shown in Fig. 1e, compared with sham group mice, mice exposed to LPS exhibited significantly prolonged escape latencies to find the platform, while those that were pretreated with artemisinin exhibited shorter escape latencies (P < 0.05). On the 6th day, the space exploration experiment was performed in the absence of the platform. Our results demonstrated that the percentage of the total time in the target quadrant was significantly reduced in the LPS-challenged group compared with the sham group, while those in the artemisinin-pretreated group spent significantly more time in the target quadrant than those in the LPS-challenged group (P < 0.05). There was no significant difference in the average speed or the distance traveled in the space exploration test among the three groups of mice (P > 0.05). The Y maze was used to monitor spatial cognition and exploratory behavior in the animals. The percentage of alterations was significantly decreased in the LPS group compared with the sham group. However, the percentage of alterations was increased in artemisinin-treated mice (Fig. 1f). These findings suggest that artemisinin can improve LPS-induced memory and cognitive impairments.
The hippocampus is closely associated with cognitive and memory functions. To determine whether artemisinin could affect neuronal survival in the hippocampal region and improve cognition, we performed hematoxylin and eosin staining of the hippocampus. Our data showed that LPS induced a significant reduction in the number of hippocampal CA1 neurons. Notably, artemisinin pretreatment ameliorated neuronal cell death in the CA1 region (Fig. 2a, b). Cleaved caspase 3 staining also demonstrated that artemisinin pretreatment protected CA1 neurons from LPS-induced injury (Fig. 2c, d). Moreover, Iba-1-immunoreactive microglia were abundant in the hippocampus in the LPS group. Artemisinin administration induced a dramatic decrease in Iba-1-positive cells in the hippocampal region (Fig. 3a, b). Microglial morphology is associated with microglial activation [21]. Fractal analysis was used to evaluate changes in microglial shape. The lacunarity, perimeter, and radius were significantly decreased in the LPS group compared with the control group. Furthermore, the decreases in lacunarity, perimeter, and radius were reversed in the artemisinin pretreatment group. Skeleton analysis was used to evaluate differences in the ramified structure of microglial cells by measuring the branches, junctions, and endpoints. As expected, LPS induced significant decreases in branch length and the numbers of endpoints and junctions, and these effects were reversed by artemisinin pretreatment (Fig. 3c, d). These data demonstrated that artemisinin could ameliorate hippocampal neuronal cell death and improve cognitive impairment caused by LPS by suppressing microglial activation.
Fig. 2. Artemisinin ameliorated neuronal cell death in an LPS-induced murine sepsis model.

a Immunohistochemical staining was used to examine neuronal survival in the CA1 area of the hippocampus. Magnification: ×200. b Quantitative analysis of the data in a. c Representative images of cleaved caspase 3 staining. Magnification: ×200. d Statistical analysis of the relative fluorescence intensity of cleaved caspase 3. *P < 0.05 versus sham group mice. #P < 0.05 versus LPS-treated mice. n = 4–6.
Fig. 3. Artemisinin attenuated LPS-induced microglial activation in the hippocampus.

a Immunofluorescence staining showing Iba-1-positive cells. Magnification: ×200. b Quantitative analysis of the data in a. c Iba-1 staining, binary, outline, and skeleton images of microglia. d Statistical analysis of microglial lacunarity, perimeter, radius, branches, endpoints, and junctions. *P < 0.05 versus sham group mice. #P < 0.05 versus LPS-treated mice. n = 6.
Artemisinin regulated proinflammatory cytokines and the migratory ability of BV2 microglia
Microglial activation and proliferation precede the onset of CNS injury [28]. Activated microglia produce a wide range of proinflammatory mediators. To investigate the effects of artemisinin on LPS-induced BV2 microglia, ELISA was used to measure the levels of inflammatory cytokines in the supernatant of BV2 cells. We observed a marked increase in proinflammatory cytokines (TNF-α, IL-6, IL-1α, and IL-1β) after 12 h of LPS stimulation, which was abolished by artemisinin treatment (Fig. 4). Furthermore, changes in TNF-α and IL-6 mRNA and protein expression were confirmed by qRT-PCR and Western blotting, respectively. Our data showed significant increases in the mRNA expression levels of IL-6 and TNF-α in the LPS-activated group, which were markedly inhibited by artemisinin (Fig. 5a, b). Similarly, Western blot analysis confirmed that artemisinin treatment significantly inhibited the protein expression of IL-6 and TNF-α after LPS stimulation (Fig. 5c, d).
Fig. 4. The effects of artemisinin on proinflammatory cytokines in BV2 microglia.

The levels of the proinflammatory factors IL-6 (a), TNF-α (b), IL-1α (c), and IL-1β (d) were measured by ELISA. *P < 0.05 versus the control group. #P < 0.05 versus the LPS-treated group. n = 6.
Fig. 5. Artemisinin preconditioning reduced LPS-induced upregulation of the mRNA and protein expression of IL-6 and TNF-α.

a The fold changes in IL-6 expression were estimated by qRT-PCR. b The fold changes in TNF-α expression were estimated by qRT-PCR. n = 6. c The level of IL-6 protein was measured by Western blotting. d The level of TNF-α protein was measured by Western blotting. *P < 0.05 versus the control group. #P < 0.05 versus the LPS-treated group. n = 5.
To observe the effect of artemisinin on microglial motility, wound healing assays and Transwell chambers were used. Compared with the control treatment, LPS stimulation efficiently enhanced the migratory ability of BV2 cells. However, this effect was diminished in the presence of artemisinin (Fig. 6a, b). The ELISA results showed that artemisinin reversed the LPS-induced increases in the chemokines MCP-1 and MIP-2 (Fig. 6c, d). These findings indicate that artemisinin can reduce the production of proinflammatory cytokines and suppress microglial migration. Artemisinin also significantly increased the mRNA and protein expression of TGF-β (Fig. 6e, f).
Fig. 6. Artemisinin inhibited the migratory ability of BV2 microglia.

a Wound healing assay and the relative migrated cells 12 h after wounding. b Transwell chamber migration of BV2 microglial cells. Magnification: ×100 or ×200. c The levels of the chemokine MCP-1 were measured by ELISA. d The levels of the chemokine MIP-2 were measured by ELISA. e The mRNA expression of TGF-β. f The level of TGF-β protein was measured by Western blotting. *P < 0.05 versus the control group. #P < 0.05 versus the LPS-treated group. n = 3–6.
The AMPKα1 pathway is involved in the anti-inflammatory effect of artemisinin
AMPK was reported to be an important therapeutic target of artemisinin in some disease models, such as mouse models of atherosclerosis and Alzheimer’s disease [29, 30]. However, whether AMPK is a therapeutic target of artemisinin in sepsis-induced neuroinflammation remains unknown. Therefore, we next investigated whether the AMPKα1 pathway was involved in the anti-inflammatory effect of artemisinin on neuroinflammation. Preliminary Western blot results demonstrated that artemisinin could activate AMPKα1 in BV2 cells in a dose-dependent manner (Fig. 7a). Accordingly, artemisinin treatment significantly upregulated the expression of AMPKα1 in comparison with that in the LPS group (Fig. 7b). In addition, reduced expression levels of iNOS, IL-6, and TNF-α (Fig. 7b) and the cytoplasmic translocation of NF-κB (Fig. 7c, d) were also observed in the artemisinin group. These results suggest that artemisinin can inhibit inflammation in BV2 cells by increasing AMPKα1 protein levels.
Fig. 7. Artemisinin activates AMPKα1 and suppresses the inflammatory response in BV2 cells.

a Western blots showing that treatment with artemisinin increased the phosphorylation of AMPKα1. b Protein levels of AMPKα1, iNOS, IL-6, and TNF-α were measured by Western blotting. c NF-κB cellular localization analysis by immunocytochemical staining. Scale bars = 50 μm. d Nuclear levels of NF-κB were analyzed by Western blotting. *P < 0.05 versus the control group. #P < 0.05 versus the LPS-treated group. n = 5.
The essential role of AMPKα1 activation in the anti-inflammatory effect of artemisinin was confirmed using AMPKα1-specific siRNA. As shown in Fig. 8a, the immunoblot results showed excellent knockdown efficiency of the AMPKα1-specific siRNA. We observed that artemisinin inhibited the LPS-induced increase in TNF-α protein production, but this effect was significantly reduced after transfection with AMPKα1-specific siRNA (Fig. 8b). The ELISA results demonstrated that the anti-inflammatory effect of artemisinin was partially abolished by AMPKα1-specific siRNA (Fig. 8c, d). Therefore, these results suggest that artemisinin attenuates LPS-induced inflammation by activating AMPKα1.
Fig. 8. The anti-inflammatory effect of artemisinin on LPS-stimulated BV2 microglia was blocked by AMPKα1-specific siRNA.

a Efficiency of siRNA-mediated AMPKα1 knockdown in BV2 cells. *P < 0.05 versus the si-NC group. b TNF-α protein levels in BV2 cells after siRNA treatment. n = 5. c TNF-α production in BV2 cells after siRNA treatment. d IL-6 production in BV2 cells after siRNA treatment. *P < 0.05 the versus the control group. #P < 0.05 versus the LPS-treated group. &P < 0.05 versus the LPS + ART group. n = 6.
Discussion
In our study, artemisinin exhibited powerful anti-inflammatory and neuroprotective effects on the sepsis model. This protection was mediated by the activation of AMPKα1 signaling in microglia (Fig. 9).
Fig. 9. The possible neuroprotective mechanism of artemisinin in sepsis-associated neuroinflammation.
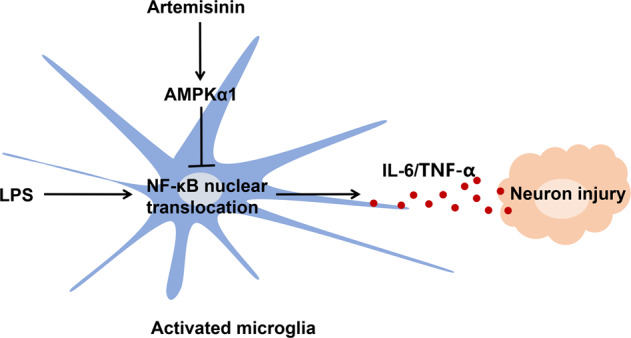
Artemisinin inhibits the LPS-induced activation of microglia by regulating AMPKα1/NF-κB axis, which attenuates the inflammatory injury of neuron.
Cognitive impairment is an essential manifestation of sepsis [31]. Unfortunately, the treatment outcomes remain unsatisfactory. Many patients continue to have cognitive dysfunction after recovering from sepsis [5]. The cognitive impairment associated with sepsis is caused by the immune response to bacterial LPS or other endotoxic components of bacteria in the absence of direct central nervous system infection [32]. Therefore, we chose systemic LPS injection to induce the experimental sepsis model, and relevant experiments were conducted using this model. The Morris water maze test results showed that artemisinin could improve cognitive function in septic mice. Artemisinin-preconditioned mice exhibited better spatial learning-memory than LPS-challenged mice. The Y maze test also demonstrated that artemisinin could improve spatial cognition in sepsis models. It has been reported that artemisinin B, one of the compounds isolated from Artemisia annua L., improved spatial memory in the water maze test in a mouse model of dementia [33]. This study provides preliminary evidence for the neuroprotective effect of artemisinins against neurodegenerative diseases. Our results showed that artemisinin could improve cognitive dysfunction in a sepsis mouse model and further illustrated the effects of artemisinin on neurological disorders.
A previous clinical study of brain tissue showed an association between the activation of microglia and sepsis [34]. Excessive microglial activation has been demonstrated to be one of the main pathogenic mechanisms of neurocognitive deficits associated with sepsis [6, 35]. Activated microglia release multiple inflammatory factors, resulting in hippocampal neuronal damage, which ultimately leads to cognitive dysfunction [33]. Therefore, suppressing microglial activation has therapeutic potential in the treatment of sepsis-induced neurocognitive deficits [36, 37]. Pathological changes in the hippocampus were measured to detect neuronal survival and microglial activation in our study. Our results showed that artemisinin inhibited LPS-induced activation of microglia and neuronal death in the hippocampal CA1 region. Overall, this study demonstrates that artemisinin can restore hippocampus-mediated cognition by inhibiting microglial activation and neuronal death in the CA1 region.
To further evaluate the potential pharmacological mechanism of artemisinin in microglia, ELISA was used to measure the expression levels of cytokines in the BV2 cell supernatants. We observed marked production of proinflammatory cytokines (TNF-α, IL-6, IL-1α, and IL-1β) following LPS stimulation for 12 h, which was abolished by artemisinin treatment. Previous in vitro experiments have also shown that artemisinins can inhibit TNF-α and IL-6 release from LPS-stimulated BV2 cells [38], which is in accordance with our results. Our study further demonstrated the inhibitory effect of artemisinin on inflammation in the brain tissue of the sepsis model. The ELISA results showed that artemisinin decreased the expression levels of IL-6 and TNF-α in the mouse hippocampus. Neuroinflammation, which is mainly characterized by the activation of microglial cells and the massive production of proinflammatory cytokines, is involved in cognitive impairment associated with sepsis [39, 40]. Excessive neuroinflammation causes neuronal damage and cognitive impairment by inducing the overproduction of proinflammatory cytokines [41–43]. Inhibiting overactivated microglia appears to be a potential therapeutic strategy for treating cognitive impairment associated with sepsis [35]. Based on our findings, it can be concluded that artemisinins have appreciable anti-inflammatory effects and thus decrease the toxic effects of proinflammatory cytokines on neurons.
The ELISA results also revealed that artemisinin inhibited LPS-induced chemokine release (MCP-1 and MIP-2). Chemokines are crucial factors in the early inflammatory response that stimulate the migration of microglial cells [44]. Microglial cells migrate to the site of injury or lesion and play a pivotal role in the occurrence and development of inflammation [45]. It was reported that microglial migration is associated with proinflammatory responses, which cause chronic inflammation and neuronal damage [46]. In this study, Iba-1 immunofluorescence, wound healing, and Transwell migration assays were performed to examine whether artemisinin could reduce the migratory capacity of microglial cells. Immunofluorescence analysis of Iba-1 showed that artemisinin significantly inhibited LPS-induced migration of microglia in the brain. Wound healing and Transwell migration assays also revealed that artemisinin inhibited the migratory ability of BV2 microglia. Our data suggest that the anti-inflammatory effect of artemisinin can be exerted by suppressing microglial cell motility via the inhibition of chemokine activity.
TGF-β is an M2 polarization marker of microglia [47]. TGF-β released by microglia plays a role in inhibiting inflammation and promoting phagocytosis, thereby promoting neuronal repair[48, 49]. We found that treatment with artemisinin significantly increased the expression of TGF-β in BV2 cells. This result suggested that artemisinin regulated microglial activation and inhibited neuronal damage by modulating TGF-β.
AMPK has been reported to be involved in the pathogenesis of cognitive impairment in sepsis, and activation of the AMPK pathway could attenuate cognitive impairment in septic mice [50, 51]. Recent studies also showed that AMPK was an important therapeutic target of artemisinin in some disease models, such as atherosclerosis and Alzheimer’s disease [29, 30]. However, whether AMPK is a therapeutic target of artemisinin in sepsis-induced neuroinflammation remains unknown. Therefore, we explored whether the effect of artemisinin on sepsis-induced neuroinflammation occurs through the regulation of the AMPK pathway. AMPK is a highly conserved serine/threonine protein kinase that is present in eukaryotic cells [52, 53]. AMPK participates in multiple biological processes by modulating cellular energy homeostasis. AMPK has also become an attractive drug target for diabetes, stroke, and tumors [54]. Recent studies have shown that AMPK is also involved in the inflammatory response through the regulation of the NF-κB signaling pathway [55]. It has been reported that phosphorylated AMPK inhibits the activation of NF-κB and decreases the production of IL-6, TNF-α, and IL-1β [56–59]. These results suggest that AMPK phosphorylation may also serve as an important therapeutic target for inflammation [52, 60]. However, whether artemisinin inhibits neuroinflammation through the AMPK/NF-κB pathway remains unexplored. First, our study demonstrated that artemisinin could activate AMPK in a dose-dependent manner in microglial cells. Furthermore, pretreatment with artemisinin markedly decreased the nuclear translocation of NF-κB in BV2 cells. PCR, Western blotting, and ELISA showed that LPS induced the upregulation of IL-6 and TNF-α, and this effect was reversed by artemisinin pretreatment. Therefore, the findings of the present study suggest that artemisinin increases the expression of AMPK and blocks the nuclear translocation of NF-κB, which can initiate transcription by binding to the gene promoter elements of IL-6 and TNF-α. These results suggest that the AMPK/NF-κB pathway is involved in the anti-inflammatory effect of artemisinin on neuroinflammation. Previous studies have shown that artemisinins can regulate the AMPK pathway, which is an attractive drug target for cancer and oxidative stress injury [61, 62]. The current study demonstrates for the first time that the AMPK pathway can be a therapeutic target of artemisinin for the treatment of neuroinflammation.
Conclusion
In summary, our data have suggested for the first time that artemisinin inhibits hippocampal neuronal death and cognitive impairment by inhibiting microglial activation in a sepsis mouse model. In addition, artemisinin significantly increased AMPKα1 levels to inhibit the LPS-induced neuroinflammatory response in BV2 microglial cells. Our findings indicate that artemisinin may be a potential preventive and therapeutic agent in neurocognitive deficits associated with sepsis.
Supplementary Information
Acknowledgements
This work was partly supported by the National Natural Science Foundation of China (Grant No. 81641088), the Natural Science Foundation of Guangdong Province (Grant No. 2017B030311019), the Science and Technology Planning Project of Guangdong Province (Grant No. 2015A030302091), the Science and Technology Planning Project of Guangzhou (Grant No. 201607010160), and the Key Medical Disciplines and Specialties Program of Guangzhou (2017-2019). Guangzhou Institute of Cardiovascular Disease, the Second Affiliated Hospital of Guangzhou Medical University, provided us with some experimental instruments.
Author contributions
XHC, PYL, and SPL designed the experiments. JXW, JSH, JYB, LDZ, QL, HJL, and SQC conducted the experiments. SPL and SY wrote the manuscript. All the authors analyzed the data, revised the manuscript, and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00634-3.
References
- 1.Annane D, Sharshar T. Cognitive decline after sepsis. Lancet Respir Med. 2015;3:61–9. doi: 10.1016/S2213-2600(14)70246-2. [DOI] [PubMed] [Google Scholar]
- 2.Zhang LN, Wang XT, Ai YH, Guo QL, Huang L, Liu ZY, et al. Epidemiological features and risk factors of sepsis-associated encephalopathy in intensive care unit patients: 2008-11. Chin Med J. 2012;125:828–31. [PubMed] [Google Scholar]
- 3.Silva AYO, Amorim EA, Barbosa-Silva MC, Lima MN, Oliveira HA, Granja MG, et al. Mesenchymal stromal cells protect the blood-brain barrier, reduce astrogliosis, and prevent cognitive and behavioral alterations in surviving septic mice. Crit Care Med. 2020;48:e290–e98. doi: 10.1097/CCM.0000000000004219. [DOI] [PubMed] [Google Scholar]
- 4.Zong MM, Zhou ZQ, Ji MH, Jia M, Tang H, Yang JJ. Activation of β2-adrenoceptor attenuates sepsis-induced hippocampus-dependent cognitive impairments by reversing neuroinflammation and synaptic abnormalities. Front Cell Neurosci. 2019;13:293. doi: 10.3389/fncel.2019.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Widmann CN, Heneka MT. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014;13:630–6. doi: 10.1016/S1474-4422(14)70017-1. [DOI] [PubMed] [Google Scholar]
- 6.Sonneville R, de Montmollin E, Poujade J, Garrouste-Orgeas M, Souweine B, Darmon M, et al. Potentially modifiable factors contributing to sepsis-associated encephalopathy. Intensive Care Med. 2017;43:1075–84. doi: 10.1007/s00134-017-4807-z. [DOI] [PubMed] [Google Scholar]
- 7.Tu Y. Artemisinin—a gift from traditional chinese medicine to the world (Nobel Lecture) Angew Chem Int Ed Engl. 2016;55:10210–26. doi: 10.1002/anie.201601967. [DOI] [PubMed] [Google Scholar]
- 8.Miller LH, Su X. Artemisinin: discovery from the Chinese herbal garden. Cell. 2011;146:855–8. doi: 10.1016/j.cell.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das AK. Anticancer effect of antimalarial artemisinin compounds. Ann Med Health Sci Res. 2015;5:93–102. doi: 10.4103/2141-9248.153609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen W, Li FF, Li C, Sui JK, Meng QF, Li XL, et al. Artemisinin ameliorates the symptoms of experimental autoimmune myasthenia gravis by regulating the balance of TH1 cells, TH17 cells and Treg cells. J Biol Regul Homeost Agents. 2018;32:1217–23. [PubMed] [Google Scholar]
- 11.Tilaoui M, Mouse HA, Jaafari A, Zyad A. Differential effect of artemisinin against cancer cell lines. Nat Prod Bioprospect. 2014;4:189–96. doi: 10.1007/s13659-014-0024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, Lu J, Liao Y, Liu S, Chen Y, He R, et al. Dihydroartemisinin attenuates lipopolysaccharide-induced acute kidney injury by inhibiting inflammation and oxidative stress. Biomed Pharmacother. 2019;117:109070. doi: 10.1016/j.biopha.2019.109070. [DOI] [PubMed] [Google Scholar]
- 13.Lin SP, Li W, Winters A, Liu R, Yang SH. Artemisinin prevents glutamate-induced neuronal cell death via Akt pathway activation. Front Cell Neurosci. 2018;12:108. doi: 10.3389/fncel.2018.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao Y, Guo Q, Cao Y, Qiu Y, Tan R, Yu Z, et al. Artemisinin derivatives inactivate cancer-associated fibroblasts through suppressing TGF-beta signaling in breast cancer. J Exp Clin Cancer Res. 2018;37:282. doi: 10.1186/s13046-018-0960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie LH, Li Q, Zhang J, Weina PJ. Pharmacokinetics, tissue distribution and mass balance of radiolabeled dihydroartemisinin in male rats. Malar J. 2009;8:112. doi: 10.1186/1475-2875-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang BN, Wu CB, Chen ZM, Zheng PP, Liu YQ, Xiong J, et al. DL-3-n-butylphthalide ameliorates diabetes-associated cognitive decline by enhancing PI3K/Akt signaling and suppressing oxidative stress. Acta Pharmacol Sin. 2021;42:347–60. doi: 10.1038/s41401-020-00583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.National Research Council (U.S.). Committee for the update of the Guide for the Care and Use of Laboratory Animals., Institute for Laboratory Animal Research (U.S.), National Academies Press (U.S.). Guide for the care and use of laboratory animals. 8th ed. Washington, D.C.: National Academies Press; 2011.
- 18.Shrum B, Anantha RV, Xu SX, Donnelly M, Haeryfar SM, McCormick JK, et al. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res Notes. 2014;7:233. doi: 10.1186/1756-0500-7-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou R, Yang X, Li X, Qu Y, Huang Q, Sun X, et al. Recombinant CC16 inhibits NLRP3/caspase-1-induced pyroptosis through p38 MAPK and ERK signaling pathways in the brain of a neonatal rat model with sepsis. J Neuroinflammation. 2019;16:239. doi: 10.1186/s12974-019-1651-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao J, Bi W, Xiao S, Lan X, Cheng X, Zhang J, et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci Rep. 2019;9:5790. doi: 10.1038/s41598-019-42286-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vargas-Caraveo A, Sayd A, Robledo-Montana J, Caso JR, Madrigal JLM, Garcia-Bueno B, et al. Toll-like receptor 4 agonist and antagonist lipopolysaccharides modify innate immune response in rat brain circumventricular organs. J Neuroinflammation. 2020;17:6. doi: 10.1186/s12974-019-1690-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young K, Morrison H. Quantifying microglia morphology from photomicrographs of immunohistochemistry prepared tissue using ImageJ. J Vis Exp. 2018;136:57648. doi: 10.3791/57648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung EH, Hwang JS, Kwon MY, Kim KH, Cho H, Lyoo IK, et al. A tryptamine-paeonol hybridization compound inhibits LPS-mediated inflammation in BV2 cells. Neurochem Int. 2016;100:35–43. doi: 10.1016/j.neuint.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Park J, Ha SH, Abekura F, Lim H, Chang YC, Lee MJ, et al. 4-O-carboxymethylascochlorin protected against microglial-mediated neurotoxicity in SH-SY5Y and BV2 cocultured cells from LPS-induced neuroinflammation and death by inhibiting MAPK, NF-kappaB, and Akt pathways. J Cell Biochem. 2018;120:1742–53. doi: 10.1002/jcb.27464. [DOI] [PubMed] [Google Scholar]
- 25.Lin SP, Ye S, Chen XH, Jiang HL, Mao HF, Chen MT, et al. Increased expression of microRNA-21 in peripheral blood mediates the down-regulation of IFN-gamma and increases the prevalence of stroke-associated infection. J Neurol Sci. 2016;366:235–9. doi: 10.1016/j.jns.2016.03.034. [DOI] [PubMed] [Google Scholar]
- 26.Lin SP, Ye S, Long Y, Fan Y, Mao HF, Chen MT, et al. Circular RNA expression alterations are involved in OGD/R-induced neuron injury. Biochem Biophys Res Commun. 2016;471:52–6. doi: 10.1016/j.bbrc.2016.01.183. [DOI] [PubMed] [Google Scholar]
- 27.Peng X, Wang Y, Li H, Fan J, Shen J, Yu X, et al. ATG5-mediated autophagy suppresses NF-kappaB signaling to limit epithelial inflammatory response to kidney injury. Cell Death Dis. 2019;10:253. doi: 10.1038/s41419-019-1483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81:374–89. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- 29.Cao Q, Du H, Fu X, Duan N, Liu C, Li X. Artemisinin attenuated atherosclerosis in high-fat diet-Fed ApoE−/− mice by promoting macrophage autophagy through the AMPK/mTOR/ULK1 pathway. J Cardiovasc Pharmacol. 2020;75:321–32. doi: 10.1097/FJC.0000000000000794. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Zhao X, Lazarovici P, Zheng W. Artemether activation of AMPK/GSK3beta(ser9)/Nrf2 signaling confers neuroprotection towards beta-amyloid-induced neurotoxicity in 3xTg Alzheimer’s mouse model. Oxid Med Cell Longev. 2019;2019:1862437. doi: 10.1155/2019/1862437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stubbs DJ, Yamamoto AK, Menon DK. Imaging in sepsis-associated encephalopathy-insights and opportunities. Nat Rev Neurol. 2013;9:551–61. doi: 10.1038/nrneurol.2013.177. [DOI] [PubMed] [Google Scholar]
- 32.Schedlowski M, Engler H, Grigoleit JS. Endotoxin-induced experimental systemic inflammation in humans: a model to disentangle immune-to-brain communication. Brain Behav Immun. 2014;35:1–8. doi: 10.1016/j.bbi.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Qiang W, Cai W, Yang Q, Yang L, Dai Y, Zhao Z, et al. Artemisinin B improves learning and memory impairment in AD dementia mice by suppressing neuroinflammation. Neuroscience. 2018;395:1–12. doi: 10.1016/j.neuroscience.2018.10.041. [DOI] [PubMed] [Google Scholar]
- 34.Lemstra AW, Groen in’tWoudJC, Hoozemans JJ, van Haastert ES, Rozemuller AJ, Eikelenboom P, et al. Microglia activation in sepsis: a case-control study. J Neuroinflammation. 2007;4:4. doi: 10.1186/1742-2094-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan C, Si Y, Meng Q, Jing L, Chen L, Zhang Y, et al. Suppression of the RAC1/MLK3/p38 signaling pathway by beta-elemene alleviates sepsis-associated encephalopathy in mice. Front Neurosci. 2019;13:358. doi: 10.3389/fnins.2019.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beggs S, Salter MW. SnapShot: microglia in disease. Cell. 2016;165:1294–94.e1. doi: 10.1016/j.cell.2016.05.036. [DOI] [PubMed] [Google Scholar]
- 37.Sui DM, Xie Q, Yi WJ, Gupta S, Yu XY, Li JB, et al. Resveratrol protects against sepsis-associated encephalopathy and inhibits the NLRP3/IL-1beta axis in microglia. Mediators Inflamm. 2016;2016:1045657. doi: 10.1155/2016/1045657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu C, Xiong Z, Chen X, Peng F, Hu X, Chen Y, et al. Artemisinin attenuates lipopolysaccharide-stimulated proinflammatory responses by inhibiting NF-kappaB pathway in microglia cells. PLoS ONE. 2012;7:e35125. doi: 10.1371/journal.pone.0035125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Block ML, Calderon-Garciduenas L. Air pollution: mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 2009;32:506–16. doi: 10.1016/j.tins.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- 42.Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53:1181–94. doi: 10.1007/s12035-014-9070-5. [DOI] [PubMed] [Google Scholar]
- 43.Wang YW, Zhou Q, Zhang X, Qian QQ, Xu JW, Ni PF, et al. Mild endoplasmic reticulum stress ameliorates lipopolysaccharide-induced neuroinflammation and cognitive impairment via regulation of microglial polarization. J Neuroinflammation. 2017;14:233. doi: 10.1186/s12974-017-1002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piao HZ, Choi IY, Park JS, Kim HS, Cheong JH, Son KH, et al. Wogonin inhibits microglial cell migration via suppression of nuclear factor-kappa B activity. Int Immunopharmacol. 2008;8:1658–62. doi: 10.1016/j.intimp.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 45.Fang Y, Wang J, Yao L, Li C, Wang J, Liu Y, et al. The adhesion and migration of microglia to beta-amyloid (Abeta) is decreased with aging and inhibited by Nogo/NgR pathway. J Neuroinflammation. 2018;15:210. doi: 10.1186/s12974-018-1250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheiblich H, Bicker G. Regulation of microglial migration, phagocytosis, and neurite outgrowth by HO-1/CO signaling. Dev Neurobiol. 2015;75:854–76. doi: 10.1002/dneu.22253. [DOI] [PubMed] [Google Scholar]
- 47.Lecca D, Janda E, Mulas G, Diana A, Martino C, Angius F, et al. Boosting phagocytosis and anti-inflammatory phenotype in microglia mediates neuroprotection by PPARgamma agonist MDG548 in Parkinson’s disease models. Br J Pharmacol. 2018;175:3298–314. doi: 10.1111/bph.14214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vinoth Kumar R, Oh TW, Park YK. Anti-inflammatory effects of ginsenoside-Rh2 inhibits LPS-induced activation of microglia and overproduction of inflammatory mediators via modulation of TGF-beta1/Smad pathway. Neurochem Res. 2016;41:951–7. doi: 10.1007/s11064-015-1804-x. [DOI] [PubMed] [Google Scholar]
- 49.Su F, Bai F, Zhang Z. Inflammatory cytokines and Alzheimer’s disease: a review from the perspective of genetic polymorphisms. Neurosci Bull. 2016;32:469–80. doi: 10.1007/s12264-016-0055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo L, Wu J, Qiao L, Lu G, Li J, Li D. Sestrin 2 attenuates sepsis-associated encephalopathy through the promotion of autophagy in hippocampal neurons. J Cell Mol Med. 2020;24:6634–43. doi: 10.1111/jcmm.15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhuang X, Yu Y, Jiang Y, Zhao S, Wang Y, Su L, et al. Molecular hydrogen attenuates sepsis-induced neuroinflammation through regulation of microglia polarization through an mTOR-autophagy-dependent pathway. Int Immunopharmacol. 2020;81:106287. doi: 10.1016/j.intimp.2020.106287. [DOI] [PubMed] [Google Scholar]
- 52.Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 2018;27:299–313. doi: 10.1016/j.cmet.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 53.Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 2017;66:789–800. doi: 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS, Cao L, et al. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. Br J Pharmacol. 2014;171:3146–57. doi: 10.1111/bph.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jian M, Kwan JS, Bunting M, Ng RC, Chan KH. Adiponectin suppresses amyloid-beta oligomer (AbetaO)-induced inflammatory response of microglia via AdipoR1-AMPK-NF-kappaB signaling pathway. J Neuroinflammation. 2019;16:110. doi: 10.1186/s12974-019-1492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cacicedo JM, Yagihashi N, Keaney JF, Jr., Ruderman NB, Ido Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;324:1204–9. doi: 10.1016/j.bbrc.2004.09.177. [DOI] [PubMed] [Google Scholar]
- 57.Thirupathi A, de Souza CT. Multi-regulatory network of ROS: the interconnection of ROS, PGC-1 alpha, and AMPK-SIRT1 during exercise. J Physiol Biochem. 2017;73:487–94. doi: 10.1007/s13105-017-0576-y. [DOI] [PubMed] [Google Scholar]
- 58.Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat Immunol. 2017;18:861–9. doi: 10.1038/ni.3772. [DOI] [PubMed] [Google Scholar]
- 59.Salt IP, Palmer TM. Exploiting the anti-inflammatory effects of AMP-activated protein kinase activation. Expert Opin Investig Drugs. 2012;21:1155–67. doi: 10.1517/13543784.2012.696609. [DOI] [PubMed] [Google Scholar]
- 60.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–55. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 61.Cheng C, Wang T, Song Z, Peng L, Gao M, Hermine O, et al. Induction of autophagy and autophagy-dependent apoptosis in diffuse large B-cell lymphoma by a new antimalarial artemisinin derivative, SM1044. Cancer Med. 2018;7:380–96. doi: 10.1002/cam4.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao X, Fang J, Li S, Gaur U, Xing X, Wang H, et al. Artemisinin attenuated hydrogen peroxide (H2O2)-induced oxidative injury in SH-SY5Y and hippocampal neurons via the activation of AMPK pathway. Int J Mol Sci. 2019;20:2680. doi: 10.3390/ijms20112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.