

Summary
Limited therapeutic options are available for advanced colorectal cancer (CRC). Herein, we report that exposure to a neo-synthetic bis(indolyl)thiazole alkaloid analog, nortopsentin 234 (NORA234), leads to an initial reduction of proliferative and clonogenic potential of CRC sphere cells (CR-CSphCs), followed by an adaptive response selecting the CR-CSphC-resistant compartment. Cells spared by the treatment with NORA234 express high levels of CD44v6, associated with a constitutive activation of Wnt pathway. In CR-CSphC-based organoids, NORA234 causes a genotoxic stress paralleled by G2-M cell cycle arrest and activation of CHK1, driving the DNA damage repair of CR-CSphCs, regardless of the mutational background, microsatellite stability, and consensus molecular subtype. Synergistic combination of NORA234 and CHK1 (rabusertib) targeting is synthetic lethal inducing death of both CD44v6-negative and CD44v6-positive CRC stem cell fractions, aside from Wnt pathway activity. These data could provide a rational basis to develop an effective strategy for the treatment of patients with CRC.
Subject areas: Cancer, Cell Biology, Drugs, Molecular Physiology
Graphical abstract
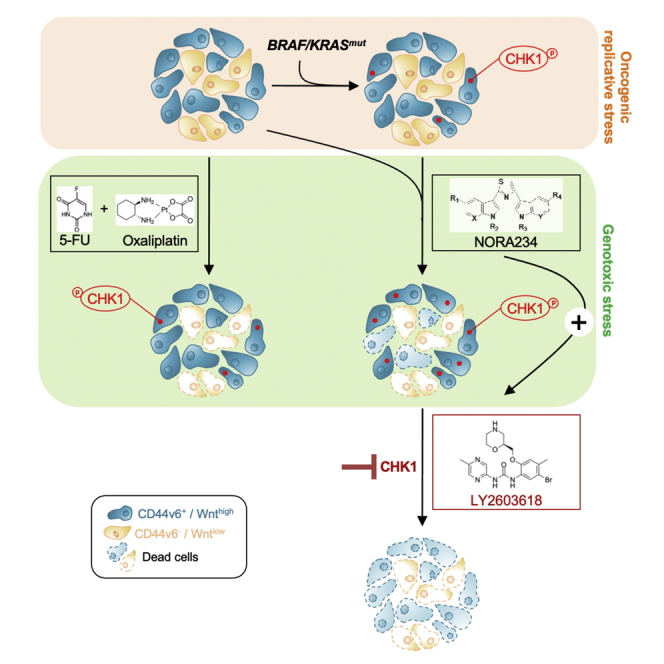
Highlights
-
•
CR-CSCs acquire a long-term resistance to the NORA234 treatment
-
•
Replicative and genotoxic stress induces the activation of CHK1
-
•
Adaptive response to NORA234 is associated with high expression levels of CHK1
-
•
NORA234 together with targeting of CHK1 leads to depletion of CR-CSC compartment
Cancer; Cell biology; Drugs; Molecular physiology
Introduction
Notwithstanding the recent advances in cancer research and therapy, in terms of early diagnosis and treatment options, colorectal cancer (CRC) represents the second-leading cause of cancer-related death, owing to the phenomena of primary and acquired resistance to antitumor therapy, as well as the onset of recurrence and metastatic disease (Dillekas et al., 2019; Siegel et al., 2020). All these biological mechanisms have been recently demonstrated to be driven by a specific cancer cell subset, called cancer stem cells (CSCs), which is endowed with peculiar features shared with healthy stem cells responsible for tumor initiation, promotion, and progression (Puglisi et al., 2009; Todaro et al., 2014; Turdo et al., 2019). CSCs are characterized by several hallmarks that render them resistant to conventional therapy and able to disseminate to distant organs, including slow cycling, stemness/differentiative capacity, high expression of adenosine-triphosphate-binding cassette transporters, antiapoptotic proteins, and DNA damage repair machinery (Di Franco et al., 2014; Saigusa et al., 2009). Despite the advent of targeted therapies for the treatment of cancer, the resistance phenomenon and, in most cases, the low specificity are the key challenges. Thus, there is an urgent need to design innovative antitumor therapies that can efficiently target CRC stem cells (CR-CSCs) and, at the same time, reduce conventional treatment side effects (Veschi et al., 2020). We showed that activated CR-CSCs express CD44v6 and depend on the PI3K/AKT and Wnt pathway for their survival and spreading (Todaro et al., 2014; Vermeulen et al., 2010). Importantly from a clinical perspective, we have recently demonstrated that the expression of CD44v6, as well as the regulation of Wnt pathway, is a highly dynamic process during tumor progression, which is finely modulated by the tumor microenvironment (Lenos et al., 2018).
Recent findings have shown the promising use, as potential adjuvant, of natural compounds of different sources (Nobili et al., 2009). Although natural compounds need high doses for the completion of their biological activity, the low toxicity and capability of inhibiting multiple pathways represent a resourceful long-term strategy to boost standard anticancer therapeutic regimens (Lodi et al., 2017). In this context, alkaloids, being characterized by a nitrogen atom within a heterocyclic ring, interact with a wide spectrum of molecules and have been recently recognized as an important source for cancer treatment (Carbone et al., 2020; Cascioferro et al., 2020a, 2020b; Habli et al., 2017; Jung et al., 2019; Kumar and Agnihotri, 2019; Lu et al., 2012). Among alkaloids, particular attention was given to nortopsentin, whose analogs showed good antiproliferative activity against several human tumor cell lines (Cascioferro et al., 2019; Kumar et al., 2011). We have reported that nortopsentin analogs effectively inhibit the in vitro activity of CDK1, causing a block in the G2/M cell cycle phase increasing the apoptotic events of pancreatic cancer cells (Carbone et al., 2015). Based on the significant functional effects of natural derivative compounds toward cancer (Habli et al., 2017; Moudi et al., 2013; Nobili et al., 2009), it is reasonable to hypothesize that CSCs could represent an elective target.
Importantly, in addition to the targeting of multiple intracellular pathways (Millimouno et al., 2014), alkaloids have been demonstrated to trigger DNA damage response (DDR) in cancer cells (Ehrhardt et al., 2013; Habli et al., 2017). After DNA damage, specific kinases such as ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3 related (ATR) are recruited and activated, thus leading to activation of downstream effectors, including CHK1 (Walker et al., 2009). The activation of the ATR/CHK1 axis, which was highly detected in CRC, is pivotal for the cell cycle arrest in G2/M phases and activation of the DDR pathway (Gralewska et al., 2020; Zhang and Hunter, 2014). This hallmark enhances the capability of CSCs to activate DDR and induce resistance to anticancer therapy. CHK1 inhibitors are currently under clinical trial aiming to sensitize cancer cells to genotoxic agents (Hong et al., 2018; Rogers et al., 2020). In particular, the CHK1 inhibitor LY2603618 (rabusertib) displays a potent effect and a highly selective CHK1 inhibitor activity, without targeting CHK2 (King et al., 2014; Klaeger et al., 2017; van Harten et al., 2019).
Considering the potent antitumor activity of bis-indole neo-synthetic alkaloid compounds (Gul and Hamann, 2005), herein, we evaluate the biological activity of the bis-indole neo-synthetic alkaloid NORA234, as an anticancer agent that efficiently targets CR-CSCs.
Results
CRC sphere cells possess intrinsic resistance to conventional chemotherapy
Taking advantage of our broad collection of primary cell lines, which recapitulate the main characteristics of parental cancer cells including the genomic and transcriptomic survey, we sought to explore the molecular mechanisms underlying the resistance of CRC sphere cells (CR-CSphCs) to standard chemotherapeutic regimen. Regardless from mutational status, CR-CSphCs displayed a stable proliferation after treatment with 5-fluorouracil (5-FU) in combination with oxaliplatin (Figure 1A). Of note, treated CR-CSphCs underwent a block on S phase, likely owing to the occurrence of DNA damage in the replication phase at early time point (Figure 1B).
Figure 1.

CR-CSphCs are endowed with innate chemoresistance
(A) Cell growth kinetics of CR-CSphCs treated with a vehicle or 5-FU in combination with oxaliplatin, up to 4 days. Data represent the mean ± SD (n = 3) using 2 different CR-CSphC lines for each subgroup (wt, #7 and #21; Braf, #3 and #5; Kras #8 and #9; Res #R3 and #R4). Statistical significance between 2 groups was determined by unpaired Student's t-test (2-tailed). ns, nonsignificant.
(B) Cell cycle analysis in CR-CSphCs treated as in (A) for 24 h. Data show percentage of cell number in G0–G1, S, and G2–M phases. Data are expressed as mean ± SD of three independent experiments performed in CR-CSphCs isolated from patients with wt (#7 and #21), Braf (#3 and #5), Kras (#8 and #9) and chemoresistant (#R3 and #R4) CRC. (Right panels) Representative cell cycle analysis of CR-CSphCs treated with a vehicle or 5-FU in combination with oxaliplatin, for 24 h (blue color = G0-G1; yellow color = S; green color = G2-M).
(C) Percentage of viability in flow-cytometry-sorted TOP-GFP CR-CSphCs, treated as in (A) up to 120 h. Data are expressed as mean ± SD of three independent experiments performed using two different CR-CSphC lines (#8, #9). (Right panel) Representative flow cytometry analysis of TOP-GFP expression in spared CR-CSphCs, at 5 days. Statistical significance between 2 groups was determined by unpaired Student's t-test (2-tailed). ∗∗p ≤ 0.01.
(D) Cell viability percentage of CR-CSphCs enriched for CD44v6 expression and treated with vehicle or 5-FU in combination with oxaliplatin up to 120 h. Data are expressed as mean ± SD of three independent experiments performed using four different CR-CSphCs lines (#3, #9, #21, #R4). (Right panel) Representative flow cytometry analysis of the percentage of CD44v6 positivity in spared CR-CSphCs, at 5 days. Statistical significance between 2 groups was determined by unpaired Student's t-test (2-tailed). ∗p ≤ 0.05.
(E) Tumor size of CR-CSphCs subcutaneously injected into immunocompromized mice, treated for 4 weeks (from sixth to ninth week) with vehicle or 5-FU in combination with oxaliplatin. Data represent the mean ± SD of tumor size measured in six mice per group, using 2 different CR-CSphC lines (#8, #21). Black arrowheads indicate weeks of treatment. Statistical significance between 2 groups was determined by unpaired Student's t-test (2-tailed). ns, nonsignificant.
(F) Cell viability analysis of healthy cells (IMEC and AD-MSCs) treated as in (D), for 3 days. Data are expressed as mean ± SD of three independent experiments. Statistical significance between 2 groups was determined by unpaired Student's t-test (2-tailed).∗∗p ≤ 0.01; ∗∗∗∗p ≤ 0.0001. See also Figure S1.
Given that CR-CSphCs represent a heterogeneous cell compartment, characterized by the presence of cells at different stage of differentiation, such as progenitor and terminally differentiated cells, we investigated which cell fraction is spared by the chemotherapy treatment taking over and gaining a proliferative advantage. Among CR-CSphCs transduced with a GFP Wnt reporter gene, we observed that the GFP-negative fraction was sensitive to chemotherapy-induced cell death, whereas exposure to 5-FU in combination with oxaliplatin for 5 days led to a significant increase of survival of GFP+ CR-CSphCs, paralleled by a β-catenin activation (Figure 1C) (Vermeulen et al., 2010). We formerly provided evidence that while CD44v6 is a functional marker that pinpoints CRC cells endowed with the ability to in vivo generate and recapitulate the parental tumor, CD44v6-negative compartment is constituted by the transit-amplifying and differentiated cells (Todaro et al., 2014). As observed for Wnt pathway activity, chemotherapy did not efficiently target CD44v6-positive fraction (Figure 1D), thus confirming that standard antitumor therapy is able to only target differentiated CRC cells. Mouse avatars generated by subcutaneous injection of CR-CSphCs, treated with 5-FU in combination with oxaliplatin, showed a growth similar to treatment with a vehicle (Figure 1E). This poor effect on cancer cells was also associated with a significant decrease of the cell viability of healthy cells, already at a short term of exposure (Figure 1F). These data indicate the possibility that resistant tumorigenic cells can emerge and expand after the chemotherapy treatment.
NORA234 reduces CR-CSphCs proliferation and clonogenic potential
Because CR-CSphCs become resistant to the chemotherapy, we evaluated whether neo-synthetic alkaloid compounds could be exploited as innovative antitumor agents. Of six alkaloid derivatives tested, NORA234 showed a great antiproliferative effect on both established CRC cell lines and primary CR-CSphCs, in a time- and concentration-dependent manner (Figures S1A–S1C). Given the importance of mutational profile (Domingo et al., 2018), microsatellite instability (MSI) (Fornasarig et al., 2000; Ward et al., 2001), consensus molecular subtype (CMS) (Guinney et al., 2015), and CD44v6 expression (Todaro et al., 2014) in the response to standard and targeted therapies, we investigated the ability of NORA234 to target CR-CSphCs on each of these subgroups.
Treatment with NORA234 reduced the cell viability of CR-CSphCs, regardless of their mutational/MSI status, CMS, and basal expression levels of CD44v6 (Figures S1D–S1G). Although NORA234 treatment alone led to delayed proliferation rate of CR-CSphCs unrelated to the mutational status, after 48 h, cells acquired resistance and started regrowing (Figure 2A and Table S1).
Figure 2.

CD44v6+ CR-CSCs acquire resistance to NORA234 treatment
(A) Kinetics of cell growth of CR-CSphCs treated with vehicle or NORA234, up to 4 days. Data represent the mean ± SD (n = 3) using CR-CSphCs isolated from wt (#7 and #21), Braf (#3 and #5), Kras (#8 and #9) and chemoresistant(#R3 and #R4) CRC patients. Statistical significance between 2 groups was determined by unpaired Student's t-test (2-tailed). ∗p ≤ 0.05; ∗∗p ≤ 0.01.
(B) Viability percentage of low and high TOP-GFP cell fraction treated as in (A) up to 120 h. Data are expressed as mean ± SD of three independent experiments performed using two different CR-CSphC lines (#8, #9).
(C) Viability percentage of CR-CSphCs enriched for CD44v6 expression and treated as in (A) up to 120 h. Data are expressed as mean ± SD of three independent experiments performed using four different CR-CSphC lines (#3, #9, #21, #R4).
(D) Colony-forming assay of CR-CSphCs treated with a vehicle or NORA234, at 21 days. The number of colonies represents mean ± SD of 3 independent experiments performed with cells isolated from 4 different patients with CRC (CR-CSphCs #3, #9, #21, #R4). Statistical significance between 2 groups was determined by unpaired Student's t-test (2-tailed). ns, nonsignificant; ∗∗p ≤ 0.01. See also Figure S1.
In addition, while NORA234 mainly targeted the Wntlow cell compartment, only the 30% of CR-CSCs endowed with high Wnt pathway activity, highlighted by β-catenin-driven GFP expression, resulted affected (Figures 2B and S1H). In line with this phenomenon, 65% of CD44v6-positive CR-CSCs survived, whereas the differentiated fraction exhibited a pronounced sensitivity to NORA234 treatment (Figures 2C and S1I). These data indicate that this alkaloid derivative selectively target CD44v6-negative cells, sparing most CD44v6-positive CR-CSCs.
Of note, while long-term exposure to NORA234, by selecting the treatment-resistant cell clones, showed a stability of clonogenic potential, healthy cells did not show any significant toxicity (Figures 2D and S1J–S1L).
Acquired resistance to NORA234 is mediated by activation of DDR pathways driven by CHK1
To explore the molecular mechanisms involved in the adaptive response of CR-CSphCs, we evaluated whether NORA234 could induce a genotoxic defect that is repaired during the DNA replication. CR-CSphCs in response to NORA234 showed an increase G2-M phase, thus indicating that cell cycle arrest induced by DNA damage is required for the triggering of DDR machinery (Figure 3A). Proteomic analysis of DNA damage biomarkers exhibited increased expression levels of γ-H2AX and cleaved PARP, paralleled by upregulation of the homologous recombination repair protein RAD51 in CR-CSphCs after exposure to NORA234 (Figure 3B). Such treatment led to an induction of DNA damage and consequent cell death, which is more pronounced in wt CR-CSphCs (Figures S2A–S2C). Moreover, transcriptomic analysis revealed that CR-CSphCs respond to NORA234 treatment, regardless the mutational background, by increasing the expression of CHK1-related proteins, which are restored to basal levels on drug washout (Figure 3C). This phenomenon could be driven by genotoxic stress (Figures 3D, 3E, S2D, and S2E). Of note, CR-CSphCs bearing Braf or Kras oncogenic mutation displayed higher basal activation levels of CHK1, likely owing to the replicative stress induced by a dysregulated proliferative pathway (Figures 3F, 3G, and S2F) (Colomer et al., 2019; Manic et al., 2018). Accordingly, analysis of a large cohort of patients with cancer showed a significant upregulation of CHK1 at both RNA and protein levels in CRC compared with healthy mucosa (Figures S2F–S2H) (Madoz-Gurpide et al., 2007). Altogether, these data suggest that CR-CSphCs are highly dependent on CHK1-mediated DDR in the resistance to genotoxic stress agents including NORA234.
Figure 3.

CR-CSphCs resistance to NORA234 is mediated by increased activity of CHK1-mediated DDR
(A) Cell cycle analysis in CR-CSphCs treated with a vehicle or NORA234 (red bars), for 24 h. The data show percentage of cell number in G0–G1, S, and G2–M phases. Data are expressed as mean ± SD of three independent experiments performed with cells isolated from 4 different patients with CRC (CR-CSphCs #3, #9, #21, #R4). (Lower panels) Representative cell cycle analysis of CR-CSphCs (#R3) treated with vehicle or NORA234, for 24 h.
(B) Representative immunoblot analysis and quantification of DNA damage (y-H2AX), apoptosis (cleaved PARP), and DDR (RAD51) markers expression in CR-CSphCs treated with vehicle or NORA234 at the indicated time points. β-actin was used as loading control. Data are presented as mean ± S.D. of 2 independent experiments performed with cells isolated from 4 different patients with CRC (CR-CSphCs #3, #9, #21, #R4).
(C) Heatmap of DNA damage – ATM/ATR regulation of G2-M-checkpoint-related genes (2−ΔΔCt expression values) in CR-CSphCs treated as in (B). Data are presented as normalized expression values of cells isolated from 4 different CRC patients (CR-CSphCs #3, #9, #21, #R4). Black arrow heads indicate genes upregulated upon 24 h of treatment with NORA234, which values are restored to basal levels after 48 h of drug washout.
(D) Representative immunocytochemistry analysis of nuclear p-CHK1, in CR-CSphCs (#21) treated with a vehicle, 5-FU in combination with oxaliplatin, or NORA234, at 24 h. Scale bars, 100 μm.
(E) Immunoblot analysis of p-CHK1 and CHK1 in wt CR-CSphCs (CSphC #21, #24, and #33) treated as indicated for 24 h. β-actin was used as a loading control.
(F) Immunoblot and its relative OD ratio analysis of p-CHK1 and CHK1 in wt, Braf, Kras, or chemoresistant CR-CSphCs. β-actin was used as a loading control. Data are presented as mean ± S.D. of 2 independent experiments performed with cells isolated from 4 different patients with CRC (CR-CSphCs # 21, # 3, # 9, #R4).
(G) Box and violin plot of p-CHK1 positivity, evaluated by flow cytometry, in wt (CR-CSphCs #7, #14, #24, #35, #37, #58), Braf (CR-CSphCs #1, #2, #3, #4), Kras (CR-CSphCs #8, #9, #10, #11, #12, #15, #16, #20, #22, #57, #59), or chemoresistant CR-CSphCs (CR-CSphCs #R2, #R3, #R4). Data are expressed as mean ± SD of three independent experiments. Statistical significance between 2 groups was determined by unpaired Student's t-test (2-tailed). ∗p ≤ 0.05; ∗∗p ≤ 0.01.
See also Figure S2.
NORA234 in combination with CHK1 inhibitor abrogates the CR-CSC compartment
We next investigated the role of CHK1 in the acquired resistance to NORA234 treatment of CR-CSCs. In accordance with literature, inducible silencing of CHK1 led to a block in S phase of the cell cycle and stabilization of CR-CSphCs proliferation (Figures S3A–S3D). After CHK1 downregulation, while the expression γ-H2AX highly increased, a reduction of DDR response, highlighted by RAD51 expression, was observed in CR-CSphCs. As consequence, CR-CSphCs, harboring silenced CHK1, slightly enhanced apoptotic events (Figures S3E and S3F). Interestingly, after NORA234 treatment, downregulation of CHK1 sensitized CR-CSphCs to induced cell death, associated with a significant decrease of cell proliferation and clonogenicity regardless the mutational background (Figures 4A–4C). Notwithstanding blockade of CHK1, by pharmacological inhibition with rabusertib (LY2603618), in combination with NORA234, had a limited toxic effect on healthy cells, CR-CSCs-expressing CD44v6, endowed with high activity of Wnt pathway and marked by nuclear β-catenin, were efficiently targeted (Todaro et al., 2014; Vermeulen et al., 2010) (Figures 4D–4F and S4A–S4F).
Figure 4.

Inhibition of CHK1 activity sensitized CR-CSphCs to NORA234 by synthetic lethality
(A) Fold variation of cell number in CR-CSphCs transduced with shCHK1 or ns shRNA and treated with vehicle, 5-FU in combination with oxaliplatin, or NORA234, up to 72 h. Data are mean ± S.D. of 3 independent experiments performed with cells isolated from patients with wt (#21), Braf (#3), Kras (#9), or chemoresistant (#R4) CRC.
(B) Cell viability analysis of cells treated as in (A) for 72 h. Data are mean ± S.D. of 3 independent experiments performed with cells isolated from 4 different CRC patients (CR-CSphCs #3, #9, #21, #R4). Statistical significance between 2 groups was determined by unpaired Student's t-test (2-tailed). ns, nonsignificant; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001.
(C) Representative colony forming assay of CR-CSphCs (R#4) transduced with shCHK1 or ns shRNA and treated with vehicle or NORA234, at 21 days. n represents the number of colonies. Scale bars, 1000 μm.
(D) Percentage of Annexin V positivity in CR-CSphCs treated with vehicle or rabusertib (LY2603618), for 24 h. Data represent mean ± S.D. of 3 independent experiments performed with cells isolated from patients with wt (#21), Braf (#3), Kras (#9) or chemoresistant (#R4) CRC. Statistical significance between 2 groups was determined by unpaired Student's t-test (2-tailed). ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001.
(E) Cell cycle analysis in CR-CSphCs treated with vehicle or NORA234 in combination with Rabusertib (LY2603618), for 48 h. Data show percentage of cell number in subG0 (red color), G0–G1 (blue color), S (brow color), and G2–M (green color) cell cycle phase. Data are expressed as mean ± SD of three independent experiments using cells isolated from 4 different patients with CRC (CR-CSphCs #3, #9, #21, #R4).
(F) Flow cytometry analysis of GFP and CD44v6 positivity percentage in CR-CSphCs transduced with TOP-GFP and treated with a vehicle, NORA234, alone or in combination with rabusertib (LY2603618), for 48 hr. Data are expressed as mean ± SD of three independent experiments using cells isolated from 2 different patients with CRC (CR-CSphCs #8, #9).
(G) 3D synergy map of viability in CR-CSphCs treated alone or in combination with NORA234 and rabusertib (LY2603618) at the indicated doses, for 48 h. Data are mean of 3 independent experiments using cells isolated from patients with wt (#21), Braf (#3), Kras (#9) or chemoresistant (#R4) CRC. See also Figures S3 and S4.
Notably, the combinatorial treatment based on the use of NORA234 and CHK1 inhibitor showed a synergistic effect possibly driven by the concomitant induction of DNA damage followed by the inhibition of DDR molecular machinery (Figure S4G).
These data indicate that inhibition of CHK1 together with a genotoxic stress may be considered as promising synthetic lethal antitumor regimen for patients with CRC regardless the mutational status.
Discussion
Treatment of CRC is currently based on chemotherapy regimen and EGFR- or VEGF-based targeted therapies. These treatments are unable to target CR-CSCs expressing CD44v6, which acquire an adaptive therapy resistance owing to the constitutive activation of PI3K and Wnt pathway (Mangiapane et al., 2021; Todaro et al., 2014). CR-CSphCs exposed to NORA234 led to an initial decrease of proliferative capacity and clonogenicity, which was followed by the regrowth of resistant subclones, characterized by high expression levels of CD44v6 and β-catenin activity. The block in G2-M of CR-CSphCs induced by NORA234-driven genotoxic stress is likely paralleled with the inhibition of ATM-regulated DNA damage checkpoint. However, the adaptive response to NORA234 led to a restore of genome integrity mediated by RAD51 and highlighted by the reduction of y-H2AX, thus giving evidence of an alternative DDR mechanism. Transcriptomic and proteomic analysis showed that the transient effect induced by NORA234 was followed by accumulation of a cell fraction endowed with highly activation of CHK1-mediated DDR machinery. In presence of replication stress (RS), CHK1 directly interacts with ATR (Chen and Poon, 2008; Madoz-Gurpide et al., 2007), by playing a crucial role in DNA replication, response to RS, and cell cycle progression (Zeman and Cimprich, 2014; Zhang and Hunter, 2014).
Here, we show that CR-CSphCs are highly dependent on CHK1-mediated DDR in the resistance to genotoxic stress induced by alkaloid derivate, NORA234, probably reducing the genomic instability, sustained by RS (Aguilera and Garcia-Muse, 2013; Cancer Genome Atlas, 2012; Gorgoulis et al., 2005; Pearl et al., 2015). Proliferative cancer cells are highly dependent on cell cycle checkpoints controlled by CHK1 posing this mechanism as the “Achilles' heel” (Dietlein et al., 2015). Blockade of CHK1 activity inhibits cancer cell replication and cell cycle checkpoints, thus leading CR-CSphCs to apoptosis regardless of their mutational status (Manic et al., 2018, 2021). On these premises, CHK1 inhibitors have been recently exploited in clinical studies to block RS-induced pathways in cancer cells endowed with DDR defects (NCT01341457, NCT01296568, NCT00415636) (Reilly et al., 2019). Treatment with CHK1 inhibitor (LY2603618) in combination with gemcitabine and pemetrexed reported adverse effects (NCT01139775) (Wehler et al., 2017). Although the results obtained using CHK1-based double combinations showed acceptable safety and pharmacokinetics, the response of patients with cancer was below expectation (Calvo et al., 2016; Gorecki et al., 2021; Weiss et al., 2013). Thus, all together these findings point out the urgent need to improve patient selection criteria (i.e., mutational profile) and design a new combinatorial treatment strategy.
Addition of CHK1 inhibitor to the NORA234 treatment was able to synergistically target CR-CSphCs, regardless of their mutational background, having a limited toxic effect on healthy cells. Of note, the double combination led to a marked apoptotic induction in the CSC compartment, as highlighted by the reduction of CD44v6+/Wnthigh cells, and the concurrent abrogation of CR-CSphCs proliferation and clonogenicity. Such treatment overcame the acquired resistance mechanisms dictated by chemotherapy and targeted therapies, which mostly affect the more differentiated CD44v6-negative cell compartment (Mangiapane et al., 2021; Siravegna et al., 2015).
Our results provide evidence that CR-CSphCs circumvent the genotoxic stress and induction of apoptosis driven by NORA234 by hijacking toward the activation of CHK1. Accordingly, the inhibition of CHK1, by both genetic and pharmacological approach, sensitizes CR-CSphCs to NORA234, thus suggesting that the pharmacological inhibition of two DDR effectors, such as ATM and CHK1, might be exploited in preclinical models (Mauri et al., 2020; Rundle et al., 2017).
DDR is a key mechanism whose molecular components are finely regulated during CRC progression, thus conferring a selective advantage in the acquisition of genetic alterations and chemoresistance. A major limitation in the use of genotoxic antitumor drugs is represented by the presence of CSCs, which are endowed with innate chemoresistance. Here, we demonstrate that nortopsentin analog, NORA234, targets CR-CSphCs, which acquire an adaptive resistance by upregulating CHK1-driven DDR pathways. The combinatorial treatment based on the use of NORA234 and CHK1 inhibitor was sufficient to abrogate CR-CSCs proliferative and clonogenic potential, regardless the mutational status.
Although further advances are needed to elucidate the clinical relevance of presented findings, our results demonstrate that NORA234 displays a synthetic lethality effect with the deficiency of CHK1-mediated DDR mechanisms, thus suggesting its putative exploitation in clinical setting as adjuvant therapy to sensitize CR-CSCs to targeted therapy.
Limitations of the study
Although our in vitro analyses clearly demonstrated that CR-CSCs are targeted by the combination treatment based on the use of nortopsentin analog and CHK1 inhibitor, its exploitation in adjuvant clinical setting should be previously validated in a metastatic preclinical study.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Giorgio Stassi (giorgio.stassi@unipa.it). This study led to the generation of new unique reagents.
Materials availability
All the reagents and data generated in this study will be made available on request. A payment and/or a completed Materials Transfer Agreement could be required if there is potential for commercial application.
Data and code availability
This study did not generate/analyze data sets/codes.
STAR★Methods
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human CD44 v6 APC-conjugated Antibody | R&D system | Cat#FAB3660A; AB_621925 |
| Mouse IgG1 APC-conjugated Antibody | R&D system | Cat#IC002A; AB_357239 |
| Phospho-Histone H2A.X (Ser139) (20E3) | Cell Signaling Technology | Cat# 9718; AB_2118009 |
| Rabbit (DA1E) mAb IgG XP® Isotype Control | Cell Signaling Technology | Cat# 3900; AB_1550038 |
| Rad51 (D4B10) | Cell Signaling Technology | Cat# 8875; AB_272110 |
| Phospho-Chk1 (Ser345) (133D3) | Cell Signaling Technology | Cat# 2348; AB_331212 |
| Chk1 (2G1D5) | Cell Signaling Technology | Cat# 2360; AB_2080320 |
| β-actin (8H10D10) | Cell Signaling Technology | Cat# 3700; AB_2242334 |
| Chk2 (1C12) | Cell Signaling Technology | Cat# 3440; AB_2229490 |
| p53 Antibody | Cell Signaling Technology | Cat# 9282; AB_331476 |
| Histone H2A.X | Cell Signaling Technology | Cat# 2595; AB_10694556 |
| Cleaved PARP (Asp214) | Cell Signaling Technology | Cat# 9541; AB_331426 |
| Phospho-Chk1 (Ser317) (D12H3) | Cell Signaling Technology | Cat# 12302; AB_2783865 |
| Cdk1 (POH1) | Cell Signaling Technology | Cat# 9116; AB_2074795 |
| Phospho-cdk1 (Tyr15) (10A11) | Cell SignalingTechnology | Cat# 4539; AB_560953 |
| Alexa Fluor 488 goat anti-rabbit | ThermoFisher Scientific | Cat# A11008; AB_143165 |
| Goat anti-rabbit HRP-linked | ThermoFisher Scientific | Cat# 31460; AB_228341 |
| Biological samples | ||
| Subcutaneous CRC xenografts | This paper | N/A |
| CR-CSphC lines | Mangiapane et al. (2021) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase, Type II | ThermoFisher Scientific | Cat# 17101015 |
| Hyaluronidase | Sigma-Aldrich | Cat# H4272 |
| StemPro Accutase Cell Dissociation Reagent | ThermoFisher Scientific | Cat# A1110501 |
| Rabusertib (LY2603618) | Selleckchem | Cat# S2626 |
| NORA234 | ||
| 5-FU | Selleckchem | Cat# S1209 |
| Oxaliplatin | Sigma | Cat# O9512 |
| Polybrene | Sigma-Aldrich | Cat# H9268 |
| Puromycin | Sigma-Aldrich | Cat# A1113803 |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| TOTO-3 Iodide | ThermoFisher Scientific | Cat# T3604 |
| DAPI | ThermoFisher Scientific | Cat# D1306 |
| 7-AAD | BD Pharmingen | Cat# 559925 |
| SeaPlaque Agarose | Lonza | Cat# 50101 |
| Propidium Iodide | Sigma-Aldrich | Cat# P4170 |
| TRIzol™ Reagent | Thermo Fisher | Cat# 15596026 |
| Critical commercial assays | ||
| iScript gDNA Clear cDNA Synthesis Kit | BIO-RAD | Cat# 1725034 |
| SsoAdvanced Universal SYBR Green Supermix | BIO-RAD | Cat# 1725271 |
| DNA damage-ATM ATR regulation of G2 M checkpoint | BIO-RAD | Cat# 10030616 |
| CaspGLOW™ Fluorescein Active Caspase-3 Staining Kit | Biovision | Cat# K183 |
| CaspGLOW™ Red Active Caspase-3 Staining Kit | Biovision | Cat# K193 |
| FITC Annexin V Apoptosis Detection Kit I | BD | Cat#556547 |
| Experimental models: cell lines | ||
| Adipose-Derived Mesenchymal Stem Cells | ATCC | PCS-500-011 |
| hTERT immortalized IMECs | Alessio Zippo | N/A |
| Experimental models: organisms/strains | ||
| NOD/SCID mice | Charles River Laboratories | Cat# 634 |
| Recombinant DNA | ||
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| TRIPZ inducible Lentiviral Non-silencing shRNA control | Dharmacon | Cat# RHS4743 |
| TRIPZ Inducible Lentiviral Human CHEK1 shRNA | Dharmacon | Cat# RHS4696-200700465 |
| TOP-GFP | Addgene | Cat# 35489 |
| Software and algorithms | ||
| Graphpad Prism 8 | GraphPad Software | http://www.graphpad.com/scientificsoftware/prism/ |
| R version 3.6.1 | R software | https://www.r-project.org/ |
| FlowJo_v10.7.1 | BD | https://www.flowjo.com/solutions/flowjo |
| Fiji | ImageJ software | https://imagej.net/Fiji/Downloads |
Experimental models and subject details
Colorectal cancer cells isolation and culture
CRC specimens were provided by the University Hospital “P. Giaccone,” in accordance with the ethical policy of the Institutional Committee for Human Experimentation (authorization CE9/2015, Policlinico Paolo Giaccone, Palermo). Human samples were digested with collagenase (0.6 mg/mL) and hyaluronidase (10 μg/mL), and cell suspension was cultured in ultralow adhesion using serum-free stem cell medium (SCM) supplemented with EGF and b-FGF (Todaro et al., 2014). Chemoresistant CR-CSphCs line was obtained from metastatic liver lesions of patients receiving chemotherapy who have undergone hepatectomy at the University Polyclinic A. Gemelli, Rome (Table S1). CR-CSphCs lines and the related tumor tissues were routinely authenticated by short tandem repeat (STR) analysis using a multiplex PCR assay (GlobalFiler™ STR kit, Applied Biosystem) and analyzed by ABIPRISM 3130 genetic analyzer (Applied Biosystems) (Mangiapane et al., 2021). The presence of mycoplasma contamination was checked by using the MycoAlertTM Plus Mycoplasma Detection Kit (Lonza) as per the manufacturer's instructions every 3 months. All the experiments were performed with early passage cultures and the expression of stem-like markers was regularly assessed. Adipose-derived mesenchymal stem cells (PCS-500-011) and hTERT immortalized mammary epithelial cells (kindly provided by Prof. Alessio Zippo) were cultured as per the manufactures’ instructions and used for cell viability assay as healthy control cells.
Animals and tumor models
Six- to 8-week-old male NOD/SCID mice were purchased by Charles River Laboratories, and in vivo experiments were performed as per the ARRIVE and Animal Care Committee Guidelines of the University of Palermo (Italian Ministry of Health authorization n. 154/2017-PR). 2.5x105 CR-CSphCs were subcutaneously injected in the flank of NOD/SCID mice, in 150 μl of 1:1 SCM/Matrigel (BD) solution. Mice were treated for 4 weeks (weeks 6–9) by intraperitoneal (i.p.) injection with phosphate-buffered saline (PBS) (vehicle) with 5-FU (15 mg/kg, 2 days/week) in combination with oxaliplatin (0.25 mg/kg, once a week). Tumor volume was calculated using the formula: largest diameter x (smallest diameter)2 x π/6. To evaluate possible toxic effects of NORA234, mice were treated for 3 weeks with a vehicle (PBS) or NORA234 (8 mg/kg, 2 days/week) by i.p. injection. At the end of treatment, animals were sacrificed accordingly to Directive 2010/63/EU guidelines (D.lgs 26/2016), and the liver, colon, kidney, spleen, lungs and pancreas were collected for histopathological examination.
Methods details
In vitro treatment of CR-CSphCs
CR-CSphCs were treated with 5-fluorouracil 10 μM (Selleckchem) and oxaliplatin 10 μM (Sigma-Aldrich), NORA234 (0,3 μM), and LY2603618 0,1μM (rabusertib, Selleckchem) alone or in combination. All the compounds were replenished in culture media every 48 h. To determine the IC50 or drug-combination efficiency of LY2603618 and NORA234, 6×103 CR-CSphCs were seeded in 96-well plates and, after 24 h, treated with different concentrations (vehicle, 0.01 μM, 0.1 μM, 1 μM, 10 μM) up to 96 h. For drug screening, CR-CSphCs were treated with vehicle or different neo-synthetic alkaloid compounds at the indicated concentrations (Vehicle, 0.03 μM, 0.3 μM, 3 μM).
Synthesis of nortopsentin analog compounds
To carry out the synthesis of nortopsentin analog compounds, a suspension of the appropriate carbothioamide (5.0 mmol) and holo-acetyl compounds (5.0 mmol) in ethanol (3.0 ml) was heated under reflux for 30 min–3 h. The resulting precipitate was filtered off, dried, and recrystallized from ethanol to afford the pure compounds (Cascioferro et al., 2019).
Cell viability, proliferation, and clonogenic assay
CR-CSphCs viability was assessed using the CellTiter 96® AQueous One Solution Cell Proliferation 369 Assay (MTS, Promega) as per the manufacturer’s instructions and analyzed by the GDV MPT 370 reader (DV 990 BV6). To evaluate cell proliferation, Cell Titer-Glo Luminescent Cell Viability Assay Kit (Promega) was used as per the manufacturer's instruction, and luminescence was measured by using Infinite F500 (Tecan). Bliss synergistic score was calculated by using SynergyFinder (Ianevski et al., 2017). For colony-formation assay, CR-CSphCs were seeded at a clonal density on 0.3% Agarose SeaPlague Agar (Invitrogen) and cultured up to 21 days. Colonies were stained with 0.01% crystal violet and counted using ImageJ software.
Flow cytometry, cell sorting, and cell cycle analysis
CR-CSphCs were harvested, washed with PBS, and stained with CD44v6 (2F10 APC, mouse IgG1, R&D systems), p-CHK1 (Ser345, 133D3, Rabbit IgG, Cell Signaling Technology), or corresponding IMC for 1 h at 4°C. The dead cells were excluded with 7-AAD (0.25 μg/1x106cells, BD Biosciences).
For intracellular staining, cells were fixed in 4% paraformaldehyde, permeabilized with 100% ice-cold methanol and stained with Phospho-Histone H2A.X (γ-H2AX,Ser139) (20E3, rabbit IgG, Cell signaling technology) and isotype-matched control (DA1E) (rabbit mAb IgG, Cell signaling technology) for 1 h at room temperature. After incubation with secondary antibody goat-anti-rabbit IgG (H+L), AlexaFluor-488 (Thermofisher), CR-CSphCs were washed with PBS and incubated with RNAse A (10 μg/mL, Qiagen Cat# 19101) and propidium iodide (5 μg/mL, Sigma-Aldrich) for 20 min at 4°C. Samples were analyzed by flow cytometer. Apoptotic cells were identified by using the CaspGLOW Fluorescein or Red Active Caspase 3 Staining Kit (Biovision)or the FITC Annexin V Apoptosis Staining Kit (BD Bioscience) as per the manufacturer’s protocol. The caspase 3 activity and the percentage of early and late apoptotic cells were analyzed by flow cytometry.
To enrich Wnt- and CD44v6-expressing cells, CR-CSphCs were resuspended in PBS supplemented with 2% bovine serum albumin and 2mM ethylenediaminetetraacetate and filtered with a 70-μm mesh to avoid cell sorter (FACSMelody, BD Bioscience) plugging. To verify the purity of sorted cells, a postsorting acquisition was performed.
For cell cycle analysis, CR-CSphCs were centrifuged, and cell pellet was incubated with 1 ml of Nicoletti Buffer (0.1% of sodium citrate, 0.1% of Triton x-100, 50 μg/ml of propidium iodide, 10 μg/ml of RNAse solution) in the dark at 4°C overnight. DNA content was evaluated by BD FACSLyric flow cytometer (BD Clinical system, BD Bioscience).
The overall obtained data were analyzed by FlowJo software.
Cell transfection and lentiviral transduction
To generate lentiviral particles, packaging cell line HEK-293T was transfected with TOP-dGFP-reporter (Addgene, 35489), TRIPZ inducible lentiviral nonsilencing shRNA control (ns shRNA, Dharmacon), or human CHK1 shRNA (shCHK1, Dharmacon) plasmids in association with psPAX2 (Addgene, 12260) and pMD2.G (Addgene, 12259) in OPTIMEM (Gibco) supplemented with XtremeGENE HP DNA transfection reagent (Roche).
Lentiviral supernatants were concentrated using the Lenti-X Concentrator reagent (Clontech) and CR-CSphCs were transduced in presence of 8 μg/mL of polybrene (Sigma-Aldrich). Transduced clones were selected by using puromycin (1 μg/ml, Sigma-Aldrich). shCHK1 was induced treating transduced cells with doxycycline (1 μg/ml, Sigma-Aldrich) for 5–10 days.
Immunohistochemistry and immunofluorescence
Immunohistochemical analysis was performed on cytospins, using phospho-CHK1 (S317, D12H3; Cell Signaling Technology). Single staining was revealed using biotin-streptavidin system (Dako) and detected with 3-amino-9-ethylcarbanzole (AEC, Dako). Double staining was performed using antibodies against CD44v6 (2F10 APC, mouse IgG1, R&D systems) and p-CHK1 (Ser345, 133D3, Rabbit IgG, Cell Signaling Technology), revealed by the MACH 2 double stain 2 kit conjugated goat antimouse polymer horseradish peroxidase (HRP) and the conjugated goat antirabbit polymer alkaline phosphatase (Biocare Medical), and detected by DAB and Vulcan Fast Red chromogen. Nuclei were counterstained with aqueous hematoxylin (Sigma-Aldrich). Hematoxylin and eosin stainings were performed using standard protocols.
Cytospun of CR-CSphCs untreated or treated with NORA234 were fixed, permeabilized, and incubated overnight with RAD51 (D4B10, cell signaling technology). To reveal, primary antibody cells were stained with Alexa Fluor-488 Goat antirabbit IgG (Life Technologies) secondary antibody. Nuclei were counterstained using Toto-3 iodide (Life Technologies) or DAPI (33258, Thermofisher).
Real-time polymerase chain reaction
Total RNA was obtained using the TRIzol™ Reagent (Thermo Fisher) protocol, and 1 μg of total RNA, after the removal of genomic DNA, was retrotranscribed as per the manufacturer’s instructions. The expression analysis of genes involved in DNA damage signaling pathway was evaluated using a PrimePCR designed panel (Bio-Rad). Relative mRNA expression levels were normalized with the endogenous control (GAPDH) and calculated using the comparative Ct method (2−ΔΔCt).
Western blot
CR-CSphCs were lyzed in ice-cold lysis buffer (Tris-HCL 10 mM, NaCl 50 mM, sodium pyruvate 30 mM, NaF 50 nM, ZnCl2 5 μM, triton 1, sodium orthovanadate 0.1 nM, sodium butyrate 10 mM and PMSF 1 mM) supplemented with protease and phosphatase inhibitors (Sigma-Aldrich). Whole-cell lysates were loaded in sodium dodecyl sulfate-polyacrylamide-gel electrophoresis gels and blotted on nitrocellulose membranes. Membranes were blocked with a 5% nonfat dry milk and 0.1% Tween 20 PBS solution for 1 h at room temperature and then incubated with specific antibodies against γ-H2AX (Ser139, 20E3, Rabbit IgG, Cell Signaling Technology), cleaved PARP (Asp214, D64E10, Rabbit IgG, Cell Signaling Technology), RAD51 (D4B10, rabbit IgG, Cell Signaling Technology), CHK1 (2G1D5, Mouse IgG1, Cell Signaling Technology), CHK2 (1C12, Mouse IgG2b, Cell Signaling Technology), P53 (Rabbit, Cell Signaling Technology), H2AX (Rabbit, Cell Signaling Technology), pCHK1 (Ser345, 133D3, Rabbit IgG, cell Signaling Technology), p-CDK1 (Tyr15, 10A11, Rabbit, Cell Signaling Technology), CDK1 (POH1, Mouse IgG2a, Cell Signaling Technology), and β-actin (8H10D10, mouse IgG2b, Cell Signaling Technology). Primary antibodies were revealed using antimouse or antirabbit HRP-conjugated (goat IgG; Thermo Fisher Scientific) and detected by Amersham imager 600 (GE Healthcare). Protein levels were normalized with β-actin and calculated by densitometric analysis using ImageJ software.
Quantification and statistical analysis
Database and statistical analysis
Transcriptomic data of healthy and tumor tissues have been collected by using the Gene Expression Profile Interactive Analysis (GEPIA) database (http://gepia.cancer-pku.cn/), by matching TCGA normal and GTEx data. Data were shown as mean ± standard deviation. Statistical significance was estimated by unpaired t-test. Results were referred to statistically significant as p<0.05. ∗ indicates p< 0.05, ∗∗ indicate p < 0.01, ∗∗∗ indicate p < 0.001, and ∗∗∗∗ indicate p < 0.0001.
Acknowledgments
AT and VV are research fellows funded by European Union FESR FSE, PON Ricerca e Innovazione 2014–2020 (AIM line 1). This work was supported by Italian Association of Cancer Research Investigator Grant (21445) and PRIN (2017WNKSLR) to G.S., RF2018-12367044 to R.D.M. and M.T, and PON ARS01_00432 to R.D.M., I.S., P.D., and G.S.
Author contributions
Conceptualization and visualization: S.D.F., B.P., M.G., and G.S.; Methodology, S.D.F., B.P., M.G., V.D.P., P.B., A.N., L.R.M., M.L.I., G.G., V.V., A.G., A.T., I.P., and M.T.; Software and formal analysis: S.D.F., B.P., M.G., and A.T.; Writing – original draft: S.D.F., B.P., M.G., P.D., and G.S.; Writing – review & editing: S.D.F., M.G., M.T., R.D.M., P.D., and G.S.; Resources: B.P., L.C., S.C., D.C., C.P., M.E.F., G.C., P.D., and G.S.; Supervision: M.T., R.D.M., I.S., P.D., and G.S.; Funding acquisition: R.D.M., M.T., I.S., P.D., and G.S.
Declaration of interests
The authors have no conflicts of interests to declare.
Inclusion and diversity
The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: June 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102664.
Supplemental information
References
- Aguilera A., Garcia-Muse T. Causes of genome instability. Annu. Rev. Genet. 2013;47:1–32. doi: 10.1146/annurev-genet-111212-133232. [DOI] [PubMed] [Google Scholar]
- Calvo E., Braiteh F., Von Hoff D., McWilliams R., Becerra C., Galsky M.D., Jameson G., Lin J., McKane S., Wickremsinhe E.R. Phase I study of CHK1 inhibitor LY2603618 in combination with gemcitabine in patients with solid tumors. Oncology. 2016;91:251–260. doi: 10.1159/000448621. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone A., Parrino B., Di Vita G., Attanzio A., Spano V., Montalbano A., Barraja P., Tesoriere L., Livrea M.A., Diana P. Synthesis and antiproliferative activity of thiazolyl-bis-pyrrolo[2,3-b]pyridines and indolyl-thiazolyl-pyrrolo[2,3-c]pyridines, nortopsentin analogues. Mar. Drugs. 2015;13:460–492. doi: 10.3390/md13010460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone D., Parrino B., Cascioferro S., Pecoraro C., Giovannetti E., Di Sarno V., Musella S., Auriemma G., Cirrincione G., Diana P. 1,2,4-Oxadiazole topsentin analogs with antiproliferative activity against pancreatic cancer cells, targeting GSK3beta kinase. ChemMedChem. 2020;16:537–554. doi: 10.1002/cmdc.202000752. [DOI] [PubMed] [Google Scholar]
- Cascioferro S., Attanzio A., Di Sarno V., Musella S., Tesoriere L., Cirrincione G., Diana P., Parrino B. New 1,2,4-oxadiazole nortopsentin derivatives with cytotoxic activity. Mar. Drugs. 2019;17:35. doi: 10.3390/md17010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascioferro S., Li Petri G., Parrino B., El Hassouni B., Carbone D., Arizza V., Perricone U., Padova A., Funel N., Peters G.J. 3-(6-Phenylimidazo [2,1-b][1,3,4]thiadiazol-2-yl)-1H-indole derivatives as new anticancer agents in the treatment of pancreatic ductal adenocarcinoma. Molecules. 2020;25:329. doi: 10.3390/molecules25020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascioferro S., Petri G.L., Parrino B., Carbone D., Funel N., Bergonzini C., Mantini G., Dekker H., Geerke D., Peters G.J. Imidazo[2,1-b] [1,3,4]thiadiazoles with antiproliferative activity against primary and gemcitabine-resistant pancreatic cancer cells. Eur. J. Med. Chem. 2020;189:112088. doi: 10.1016/j.ejmech.2020.112088. [DOI] [PubMed] [Google Scholar]
- Chen Y., Poon R.Y. The multiple checkpoint functions of CHK1 and CHK2 in maintenance of genome stability. Front Biosci. 2008;13:5016–5029. doi: 10.2741/3060. [DOI] [PubMed] [Google Scholar]
- Colomer C., Margalef P., Villanueva A., Vert A., Pecharroman I., Sole L., Gonzalez-Farre M., Alonso J., Montagut C., Martinez-Iniesta M. IKKalpha kinase regulates the DNA damage response and drives chemo-resistance in cancer. Mol. Cell. 2019;75:669–682.e65. doi: 10.1016/j.molcel.2019.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Franco S., Todaro M., Dieli F., Stassi G. Colorectal cancer defeating? Challenge accepted! Mol. Aspects Med. 2014;39:61–81. doi: 10.1016/j.mam.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Dietlein F., Kalb B., Jokic M., Noll E.M., Strong A., Tharun L., Ozretic L., Kunstlinger H., Kambartel K., Randerath W.J. A synergistic interaction between Chk1- and MK2 inhibitors in KRAS-mutant cancer. Cell. 2015;162:146–159. doi: 10.1016/j.cell.2015.05.053. [DOI] [PubMed] [Google Scholar]
- Dillekas H., Rogers M.S., Straume O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019;8:5574–5576. doi: 10.1002/cam4.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E., Camps C., Kaisaki P.J., Parsons M.J., Mouradov D., Pentony M.M., Makino S., Palmieri M., Ward R.L., Hawkins N.J. Mutation burden and other molecular markers of prognosis in colorectal cancer treated with curative intent: results from the QUASAR 2 clinical trial and an Australian community-based series. Lancet Gastroenterol. Hepatol. 2018;3:635–643. doi: 10.1016/S2468-1253(18)30117-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt H., Pfeiffer S., Schrembs D., Wachter F., Grunert M., Jeremias I. Activation of DNA damage response by antitumor therapy counteracts the activity of vinca alkaloids. Anticancer Res. 2013;33:5273–5287. [PubMed] [Google Scholar]
- Fornasarig M., Viel A., Valentini M., Capozzi E., Sigon R., De Paoli A., Della Puppa L., Boiocchi M. Microsatellite instability and MLH1 and MSH2 germline defects are related to clinicopathological features in sporadic colorectal cancer. Oncol. Rep. 2000;7:39–43. doi: 10.3892/or.7.1.39. [DOI] [PubMed] [Google Scholar]
- Gorecki L., Andrs M., Korabecny J. Clinical candidates targeting the ATR-CHK1-WEE1 Axis in cancer. Cancers (Basel) 2021;13:795. doi: 10.3390/cancers13040795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V.G., Vassiliou L.V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., Ditullio R.A., Jr., Kastrinakis N.G., Levy B. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Gralewska P., Gajek A., Marczak A., Rogalska A. Participation of the ATR/CHK1 pathway in replicative stress targeted therapy of high-grade ovarian cancer. J. Hematol. Oncol. 2020;13:39. doi: 10.1186/s13045-020-00874-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinney J., Dienstmann R., Wang X., de Reynies A., Schlicker A., Soneson C., Marisa L., Roepman P., Nyamundanda G., Angelino P. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gul W., Hamann M.T. Indole alkaloid marine natural products: an established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005;78:442–453. doi: 10.1016/j.lfs.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habli Z., Toumieh G., Fatfat M., Rahal O.N., Gali-Muhtasib H. Emerging cytotoxic alkaloids in the battle against cancer: overview of molecular mechanisms. Molecules. 2017;22:250. doi: 10.3390/molecules22020250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D.S., Moore K., Patel M., Grant S.C., Burris H.A., 3rd, William W.N., Jr., Jones S., Meric-Bernstam F., Infante J., Golden L. Evaluation of Prexasertib, a checkpoint kinase 1 inhibitor, in a phase Ib study of patients with squamous cell carcinoma. Clin. Cancer Res. 2018;24:3263–3272. doi: 10.1158/1078-0432.CCR-17-3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianevski A., He L., Aittokallio T., Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics. 2017;33:2413–2415. doi: 10.1093/bioinformatics/btx162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y.Y., Shanmugam M.K., Narula A.S., Kim C., Lee J.H., Namjoshi O.A., Blough B.E., Sethi G., Ahn K.S. Oxymatrine attenuates tumor growth and deactivates STAT5 signaling in a lung cancer xenograft model. Cancers (Basel) 2019;11:49. doi: 10.3390/cancers11010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C., Diaz H., Barnard D., Barda D., Clawson D., Blosser W., Cox K., Guo S., Marshall M. Characterization and preclinical development of LY2603618: a selective and potent Chk1 inhibitor. Invest New Drugs. 2014;32:213–226. doi: 10.1007/s10637-013-0036-7. [DOI] [PubMed] [Google Scholar]
- Klaeger S., Heinzlmeir S., Wilhelm M., Polzer H., Vick B., Koenig P.A., Reinecke M., Ruprecht B., Petzoldt S., Meng C. The target landscape of clinical kinase drugs. Science. 2017;358:eaan4368. doi: 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D., Kumar N.M., Chang K.H., Gupta R., Shah K. Synthesis and in-vitro anticancer activity of 3,5-bis(indolyl)-1,2,4-thiadiazoles. Bioorg. Med. Chem. Lett. 2011;21:5897–5900. doi: 10.1016/j.bmcl.2011.07.089. [DOI] [PubMed] [Google Scholar]
- Kumar S., Agnihotri N. Piperlongumine, a piper alkaloid targets Ras/PI3K/Akt/mTOR signaling axis to inhibit tumor cell growth and proliferation in DMH/DSS induced experimental colon cancer. Biomed. Pharmacother. 2019;109:1462–1477. doi: 10.1016/j.biopha.2018.10.182. [DOI] [PubMed] [Google Scholar]
- Lenos K.J., Miedema D.M., Lodestijn S.C., Nijman L.E., van den Bosch T., Romero Ros X., Lourenco F.C., Lecca M.C., van der Heijden M., van Neerven S.M. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat. Cell Biol. 2018;20:1193–1202. doi: 10.1038/s41556-018-0179-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodi A., Saha A., Lu X., Wang B., Sentandreu E., Collins M., Kolonin M.G., DiGiovanni J., Tiziani S. Combinatorial treatment with natural compounds in prostate cancer inhibits prostate tumor growth and leads to key modulations of cancer cell metabolism. NPJ Precis Oncol. 2017;1:18. doi: 10.1038/s41698-017-0024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.J., Bao J.L., Chen X.P., Huang M., Wang Y.T. Alkaloids isolated from natural herbs as the anticancer agents. Evid. Based Complement Alternat Med. 2012;2012:485042. doi: 10.1155/2012/485042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madoz-Gurpide J., Canamero M., Sanchez L., Solano J., Alfonso P., Casal J.I. A proteomics analysis of cell signaling alterations in colorectal cancer. Mol. Cell Proteomics. 2007;6:2150–2164. doi: 10.1074/mcp.M700006-MCP200. [DOI] [PubMed] [Google Scholar]
- Mangiapane L.R., Nicotra A., Turdo A., Gaggianesi M., Bianca P., Di Franco S., Sardina D.S., Veschi V., Signore M., Beyes S. PI3K-driven HER2 expression is a potential therapeutic target in colorectal cancer stem cells. Gut. 2021 doi: 10.1136/gutjnl-2020-323553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manic G., Musella M., Corradi F., Sistigu A., Vitale S., Soliman Abdel Rehim S., Mattiello L., Malacaria E., Galassi C., Signore M. Control of replication stress and mitosis in colorectal cancer stem cells through the interplay of PARP1, MRE11 and RAD51. Cell Death Differ. 2021 doi: 10.1038/s41418-020-00733-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manic G., Signore M., Sistigu A., Russo G., Corradi F., Siteni S., Musella M., Vitale S., De Angelis M.L., Pallocca M. CHK1-targeted therapy to deplete DNA replication-stressed, p53-deficient, hyperdiploid colorectal cancer stem cells. Gut. 2018;67:903–917. doi: 10.1136/gutjnl-2016-312623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri G., Arena S., Siena S., Bardelli A., Sartore-Bianchi A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann. Oncol. 2020;31:1135–1147. doi: 10.1016/j.annonc.2020.05.027. [DOI] [PubMed] [Google Scholar]
- Millimouno F.M., Dong J., Yang L., Li J., Li X. Targeting apoptosis pathways in cancer and perspectives with natural compounds from mother nature. Cancer Prev. Res. (Phila) 2014;7:1081–1107. doi: 10.1158/1940-6207.CAPR-14-0136. [DOI] [PubMed] [Google Scholar]
- Moudi M., Go R., Yien C.Y., Nazre M. Vinca alkaloids. Int. J. Prev. Med. 2013;4:1231–1235. [PMC free article] [PubMed] [Google Scholar]
- Nobili S., Lippi D., Witort E., Donnini M., Bausi L., Mini E., Capaccioli S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009;59:365–378. doi: 10.1016/j.phrs.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Pearl L.H., Schierz A.C., Ward S.E., Al-Lazikani B., Pearl F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer. 2015;15:166–180. doi: 10.1038/nrc3891. [DOI] [PubMed] [Google Scholar]
- Puglisi M.A., Sgambato A., Saulnier N., Rafanelli F., Barba M., Boninsegna A., Piscaglia A.C., Lauritano C., Novi M.L., Barbaro F. Isolation and characterization of CD133+ cell population within human primary and metastatic colon cancer. Eur. Rev. Med. Pharmacol. Sci. 2009;13(Suppl 1):55–62. [PubMed] [Google Scholar]
- Reilly N.M., Novara L., Di Nicolantonio F., Bardelli A. Exploiting DNA repair defects in colorectal cancer. Mol. Oncol. 2019;13:681–700. doi: 10.1002/1878-0261.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers R.F., Walton M.I., Cherry D.L., Collins I., Clarke P.A., Garrett M.D., Workman P. CHK1 inhibition is synthetically lethal with loss of B-family DNA polymerase function in human lung and colorectal cancer cells. Cancer Res. 2020;80:1735–1747. doi: 10.1158/0008-5472.CAN-19-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundle S., Bradbury A., Drew Y., Curtin N.J. Targeting the ATR-CHK1 Axis in cancer therapy. Cancers (Basel) 2017;9:41. doi: 10.3390/cancers9050041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saigusa S., Tanaka K., Toiyama Y., Yokoe T., Okugawa Y., Ioue Y., Miki C., Kusunoki M. Correlation of CD133, OCT4, and SOX2 in rectal cancer and their association with distant recurrence after chemoradiotherapy. Ann. Surg. Oncol. 2009;16:3488–3498. doi: 10.1245/s10434-009-0617-z. [DOI] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Siravegna G., Mussolin B., Buscarino M., Corti G., Cassingena A., Crisafulli G., Ponzetti A., Cremolini C., Amatu A., Lauricella C. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015;21:827. doi: 10.1038/nm0715-827b. [DOI] [PubMed] [Google Scholar]
- Todaro M., Gaggianesi M., Catalano V., Benfante A., Iovino F., Biffoni M., Apuzzo T., Sperduti I., Volpe S., Cocorullo G. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. 2014;14:342–356. doi: 10.1016/j.stem.2014.01.009. [DOI] [PubMed] [Google Scholar]
- Turdo A., Veschi V., Gaggianesi M., Chinnici A., Bianca P., Todaro M., Stassi G. Meeting the challenge of targeting cancer stem cells. Front Cell Dev Biol. 2019;7:16. doi: 10.3389/fcell.2019.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Harten A.M., Buijze M., van der Mast R., Rooimans M.A., Martens-de Kemp S.R., Bachas C., Brink A., Stigter-van Walsum M., Wolthuis R.M.F., Brakenhoff R.H. Targeting the cell cycle in head and neck cancer by Chk1 inhibition: a novel concept of bimodal cell death. Oncogenesis. 2019;8:38. doi: 10.1038/s41389-019-0147-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen L., De Sousa E.M.F., van der Heijden M., Cameron K., de Jong J.H., Borovski T., Tuynman J.B., Todaro M., Merz C., Rodermond H. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- Veschi V., Mangiapane L.R., Nicotra A., Di Franco S., Scavo E., Apuzzo T., Sardina D.S., Fiori M., Benfante A., Colorito M.L. Targeting chemoresistant colorectal cancer via systemic administration of a BMP7 variant. Oncogene. 2020;39:987–1003. doi: 10.1038/s41388-019-1047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M., Black E.J., Oehler V., Gillespie D.A., Scott M.T. Chk1 C-terminal regulatory phosphorylation mediates checkpoint activation by de-repression of Chk1 catalytic activity. Oncogene. 2009;28:2314–2323. doi: 10.1038/onc.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward R., Meagher A., Tomlinson I., O'Connor T., Norrie M., Wu R., Hawkins N. Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut. 2001;48:821–829. doi: 10.1136/gut.48.6.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehler T., Thomas M., Schumann C., Bosch-Barrera J., Vinolas Segarra N., Dickgreber N.J., Dalhoff K., Sebastian M., Corral Jaime J., Alonso M. A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non-small cell lung cancer. Lung Cancer. 2017;108:212–216. doi: 10.1016/j.lungcan.2017.03.001. [DOI] [PubMed] [Google Scholar]
- Weiss G.J., Donehower R.C., Iyengar T., Ramanathan R.K., Lewandowski K., Westin E., Hurt K., Hynes S.M., Anthony S.P., McKane S. Phase I dose-escalation study to examine the safety and tolerability of LY2603618, a checkpoint 1 kinase inhibitor, administered 1 day after pemetrexed 500 mg/m(2) every 21 days in patients with cancer. Invest New Drugs. 2013;31:136–144. doi: 10.1007/s10637-012-9815-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman M.K., Cimprich K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer. 2014;134:1013–1023. doi: 10.1002/ijc.28226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze data sets/codes.