

Key Points
Increased EPO sensitivity or activity improves erythropoiesis in β-thalassemia.
Combinatorial use of erythroid-stimulating agents and erythroid iron restriction agents correct anemia in β-thalassemia.
Abstract
β-Thalassemia intermedia is a disorder characterized by ineffective erythropoiesis (IE), anemia, splenomegaly, and systemic iron overload. Novel approaches are being explored based on the modulation of pathways that reduce iron absorption (ie, using hepcidin activators like Tmprss6-antisense oligonucleotides [ASOs]) or increase erythropoiesis (by erythropoietin [EPO] administration or modulating the ability of transferrin receptor 2 [Tfr2] to control red blood cell [RBC] synthesis). Targeting Tmprss6 messenger RNA by Tmprss6-ASO was proven to be effective in improving IE and splenomegaly by inducing iron restriction. However, we postulated that combinatorial strategies might be superior to single therapies. Here, we combined Tmprss6-ASO with EPO administration or removal of a single Tfr2 allele in the bone marrow of animals affected by β-thalassemia intermedia (Hbbth3/+). EPO administration alone or removal of a single Tfr2 allele increased hemoglobin levels and RBCs. However, EPO or Tfr2 single-allele deletion alone, respectively, exacerbated or did not improve splenomegaly in β-thalassemic mice. To overcome this issue, we postulated that some level of iron restriction (by targeting Tmprss6) would improve splenomegaly while preserving the beneficial effects on RBC production mediated by EPO or Tfr2 deletion. While administration of Tmprss6-ASO alone improved the anemia, the combination of Tmprss6-ASO + EPO or Tmprss6-ASO + Tfr2 single-allele deletion produced significantly higher hemoglobin levels and reduced splenomegaly. In conclusion, our results clearly indicate that these combinatorial approaches are superior to single treatments in ameliorating IE and anemia in β-thalassemia and could provide guidance to translate some of these approaches into viable therapies.
Visual Abstract

Introduction
β-Thalassemias are a heterogeneous group of hereditary disorders yielded by several hundred mutations that strike in the β-globin gene or its regulatory elements.1-3 Depending on the genetic background and consequent levels of functional hemoglobin (Hb) produced, the severity of the disease can range from the asymptomatic thalassemia minor to the most severe β-thalassemia major.1-3
β-Thalassemia intermedia (non–transfusion-dependent thalassemia [NTDT]) is characterized by mild to severe ineffective erythropoiesis (IE), exhibiting anemia and increased iron absorption, eventually leading to iron overload.1,3,4 NTDT does not require blood transfusion for survival.4 However, the anemia in patients often worsens over time, resulting in the development of transfusion dependence, a feature of the most severe form (β-thalassemia major or transfusion-dependent thalassemia).1,3,5 Impaired synthesis of β-globin chains leads to the excess of α-globin chains and consequent production of insoluble hemichromes.6,7 These accumulate in erythroid progenitors, triggering IE, which is characterized by increased number of erythroid progenitors, reduced differentiation, and increased apoptosis of late erythroblasts.7-9 Some of the major consequences of IE are anemia, extramedullary hematopoiesis, splenomegaly, and systemic iron overload mediated by downregulation of hepcidin (the master regulator of iron homeostasis) due to the increased synthesis of erythroferrone (ERFE).10,11 In addition, the anemia leads to hypoxia, which triggers upregulation of iron-related genes such as DMT1 and ferroportin (FPN) in the duodenum, further exacerbating iron absorption and iron overload.12 Therefore, improvement of anemia could slow down or even reverse the deteriorating effects observed in aging patients, such as organ iron overload and the comorbidities associated with it.13,14
In our mouse model of NTDT (Hbbth3/+), we have shown that increasing hepcidin levels causes iron restriction, thus improving anemia and reducing splenomegaly. This effect is associated with a reduction in the formation of hemichromes and an improved lifespan of red blood cells (RBCs).15-18 However, the exact mechanism through which iron restriction ameliorates erythropoiesis has not yet been elucidated. These findings emphasize the crosstalk among erythropoiesis, RBC synthesis, and iron metabolism. In theory, agents that can enhance erythropoiesis (like erythropoietin [EPO]) could improve anemia by increasing the production of RBC and concurrently consume iron stored in organs, mitigating the iron overload. However, in this scenario, we would also predict that EPO alone may have some negative effect on hepcidin, with potential increased iron absorption and transferrin saturation. This may neutralize the beneficial effect of “erythroid-mediated consumption of stored iron." Therefore, we postulated that upon increasing erythropoiesis by EPO administration or by modulating the activity of transferrin receptor 2 (Tfr2) in the bone marrow (BM), some level of iron restriction would be required to achieve the beneficial effects of improving anemia and, concurrently, decreasing iron overload in a mouse model of β-thalassemia intermedia.
Over the last decade, several strategies have been investigated in order to enhance hepcidin activity (such as hepcidin inducers, mimetic, or FPN inhibitors) in diseases associated with iron overload due to low hepcidin expression.15-21 It has been already established by us and other groups that increasing hepcidin expression by reducing the transmembrane serine protease Tmprss6 is an effective approach for the treatment of iron overload–associated diseases, such as hereditary hemochromatosis and β-thalassemia.15,18,19,22,23
In the present study, we enhanced erythropoiesis in our β-thalassemia intermedia mouse model by 2 approaches. On one hand, we used a new ex vivo technology called TARGT (transduced autologous restorative gene therapy) for prolonged production and secretion of therapeutic agents (EPO). The TARGTEpo approach uses mouse dermal fibroblasts transduced with adenovirus vector expressing the Epo gene under the EF1α promoter.24 The transduced fibroblasts, subcutaneously implanted in our mouse model (Hbbth3/+), started producing EPO immediately and continuously for the length of the experiment. On the other hand, we used Hbbth3/+ mice lacking one of the Tfr2 alleles in the BM (Tfr2BM hetero/Hbbth3/+). TFR2 in the liver contributes to hepcidin regulation, but in erythroid cells, it is known to be a component of the EPO receptor complex, with an important but still not completely elucidated role in efficient erythropoiesis.25-28 We have shown that mice lacking Tfr2 in the BM have higher Hb levels and RBC number most likely due to increased erythroblast sensitivity to EPO.25
In both animal models, enhanced erythropoiesis leads to improved Hb levels and RBC number but no changes, or even worsening, of splenomegaly.25 We then induced iron restriction by targeting the Tmprss6 messenger RNA (mRNA) using a Tmprss6-ASO. Combining enhanced erythropoiesis with iron restriction allowed us to achieve a significant improvement in anemia. The remarkable improvement in Hb levels and RBC number not only significantly improved anemia but also significantly reduced splenomegaly.
Finally, in search of an approach potentially translatable to the clinic, we proved that a pretreatment of Hbbth3/+ mice with Tmprss6-ASO followed by a simultaneous administration of constrained doses of recombinant human EPO (rhEPO) improves anemia more effectively than Tmprss6-ASO alone.
Overall, our results show that combinatorial treatments are superior to approaches targeting single pathophysiological cues and may represent a future option for the management of β-thalassemia.
Methods
Animal models
Hbbth3/+ mice (with heterozygous deletion of β1 and β2 genes)29 were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice receiving fibroblasts were bred in the animal facility at the Children’s Hospital of Philadelphia and kept in a pure C57BL/6J background. Hbbth3/+ mice (on a pure C57BL/6N background) were crossed with Tfr2+/− mice (on a pure 129S2 background) in the animal facility at San Raffaele Scientific Institute. The phenotype of mice has been evaluated as described in the supplemental Material, available on the Blood Web site. The sequences of the oligos utilized for gene expression analysis are reported in Table 1. Hbbth3/+ and Tfr2+/−/Hbbth3/+ progenies are on a mixed C57/129S2 background.30
Table 1.
Oligonucleotide primers used for quantitative reverse transcription polymerase chain reaction by TaqMan
| Transcript | Assay ID |
|---|---|
| Hprt1 | Mm01318743_m1 |
| Hamp | Mm00519025_m1 |
| Tmprss6 | Mm00551119_m1 |
| Epo | Mm01202755_m1 |
Bone marrow transplantation (BMT)
BM cells isolated from 10-week-old C57/129S2 Tfr2+/−/Hbbth3/+ and Hbbth3/+ male mice were used for BM transplantation (BMT), as described previoulsy.26 In brief, 5 × 106 BM cells recovered from the femur of Tfr2+/−/Hbbth3/+ double mutants and Hbbth3/+ mice (expressing the CD45.2 surface antigen) were transplanted into lethally irradiated C57BL/6-Ly-5.1 male mice (expressing the CD45.1 surface antigen), generating thalassemic mice with both (tHbbth3/+) or a single Tfr2 allele (Tfr2BM hetero/Hbbth3/+) in the BM. Nine weeks after BMT, mice were treated with triantennary N-acetyl galactosamine (GalNAc)-conjugated antisense oligonucleotide (ASO)t against Tmprss6 mRNA or control (Table 2).
Table 2.
Sequences of the ASOs used
| ASOs | Sequence (5′-3′) |
|---|---|
| GalNAc Tmprss6 | GCTTAGAGTACAGCCCACTT |
| GalNAc control | CCTTCCCTGAAGGTTCCTCC |
Fibroblasts expansion and transduction
Cryopreserved C57BL/6N mouse primary dermal fibroblasts (catalog number C57-6067, Cell Biologics) and/or fibroblasts extracted in-house from C57BL/6N mice were expanded in full media (Dulbecco’s modified Eagle medium supplemented with 10% serum, 2.5 µg/mL amphotericin B, and 50 µg/mL gentamycin sulfate) until generation of a sufficient number of plates with 90% of confluency. Transduction of the fibroblasts was performed as follows: per one 100-mm plate, the viral vector (HDAd-EF1α-mEpo, 5.79 × 1012 viral particles/mL) was diluted in 3 mL “full media” to a final amount of 1 × 1010 viral particles in 3 mL (1.72 µL vector + 3 mL media). Some plates were not transduced and kept as control. Plates were kept at 37°C, 5% CO2 for 16 to 24 hours. After 16 to 24 hours, the cells were washed, and the transduction media was replaced with 10 mL of full media and then kept in incubator until the implantation day (≤48 hours after transduction). Transduced and nontransduced cells were embedded in Matrigel (Corning) and then injected subcutaneously in the dorsal area of 2- to 5-month-old Hbbth3/+ female. The following day, the mice receiving fibroblasts overexpressing Epo were injected with GalNAc-conjugated ASO against Tmprss6 mRNA, while animals receiving empty (nontransduced) fibroblasts were treated with a control ASO (CTRL-ASO).
Hemichrome analysis
To visualize membrane-bound α-globins (hemichromes) in control and treated animals, we used Triton/acetic acid/urea (TAU) gel as previously described.17,18
Animal studies
All animal studies were conducted under protocols approved by the institutional animal care committees of the Children’s Hospital of Philadelphia and San Raffaele Scientific Institute and in accordance with European Union and United States guidelines.
Statistical analysis
Data are presented as mean ± standard deviation (SD). One-way analysis of variance (with multiple comparisons tests) or unpaired Student t test analysis was performed using Prism 5.0 or 8.0 (GraphPad). Only comparisons with P < .05 were considered statistically significant and included in the figures.
Results
A helper-dependent adenoviral vector system allows sustained production of high levels of mouse EPO in vivo
In order to express sustained and high levels of EPO and increase erythropoiesis, we used an ex vivo approach called TARGT for prolonged production and secretion of therapeutic agents.24 We implanted wild-type (WT) and β-thalassemic (Hbbth3/+) animals with murine primary dermal fibroblasts transduced with a helper-dependent adenoviral vector carrying the Epo gene (TARGTEpo). Three groups were generated for both WT and Hbbth3/+ mice, using different amounts of genetically modified fibroblasts (1 × 105, 5 × 105, and 1 × 106 transduced cells). One additional group received 1 × 106 untransduced fibroblasts as control. Complete blood count (CBC) and serum harvest for EPO measurement were performed 1 week after implantation and then every 10 days until the end of the experiment at 7 weeks. This pharmacokinetic study indicated that 1 × 106 cells were sufficient to significantly increase circulating EPO levels in WT and Hbbth3/+ mice after 7 weeks (supplemental Figure 1A-B). This led to an increase in Hb levels of ∼3 g/dL in both WT and Hbbth3/+ mice as early as 1 week after fibroblasts implantation (not shown). At the end of the treatment, WT animals showed significantly increased Hb levels (18.5 ± 1.7 g/dL, 19.4 ± 0.5 g/dL and 19.8 ± 0.8 g/dL in the groups implanted with 1 × 105, 5 × 105, and 1 × 106 fibroblasts, respectively, compared with 13.5 ± 0.8 g/dL in controls; Figure 1A), high hematocrit levels, and erythrocytosis (supplemental Figure 1C-D).
Figure 1.
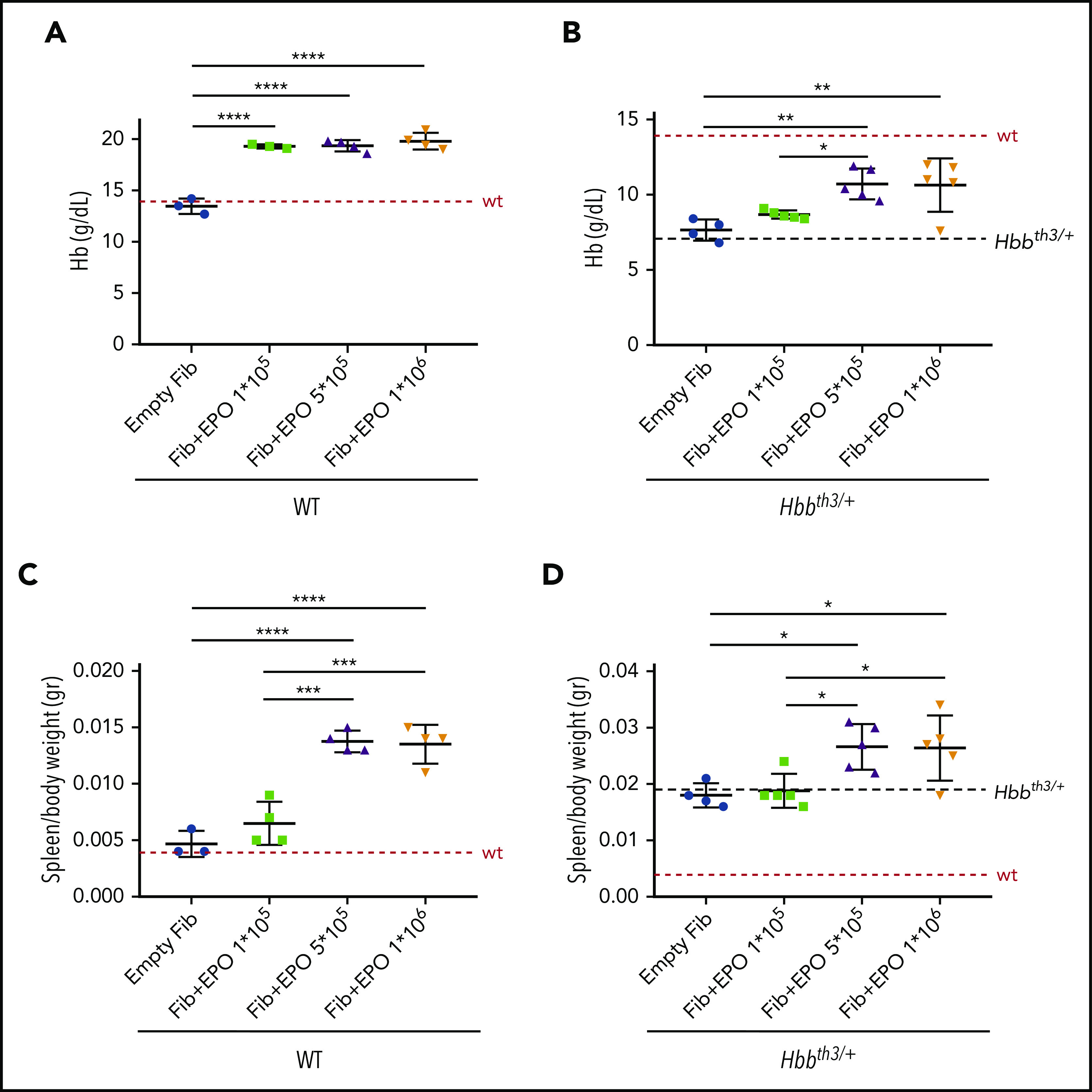
Pharmacokinetic studies in WT and Hbbth3/+ mouse models. Wild type (WT) and β-thalassemic (Hbbth3/+) animals were implanted with murine primary dermal fibroblasts (Fib) overexpressing Epo. Three groups were generated for each genotype (1 × 105, 5 × 105, and 1 × 106 transduced cells). Increased expression of Epo leads to significant increased Hb levels in both WT (A) and Hbbth3/+ (B) mice. As expected, enhanced erythropoiesis was also associated with splenomegaly in WT (C) and worsening of it in β-thalassemic animals (D). Dotted red line indicates mean values for untreated WT mice. Dashed black lines indicates mean values for untreated Hbbth3/+ mice. Bars represent SD. Asterisks refer to statistically significant differences (****P ≤ .001; ***P ≤ .005; **P ≤ .01; *P ≤ .05).
In Hbbth3/+ mice at the end of the treatment, Hb levels reached 8.7 ± 0.3 g/dL, 10.7 ± 1.0 g/dL, and 10.6 ± 1.8 g/dL in the groups implanted with 1 × 105, 5 × 105, and 1 × 106 fibroblasts respectively, compared with 7.7 ± 0.7 g/dL in controls (Figure 1B). Hematocrit levels and RBC count followed the same trend as in WT mice, showing significant increases in the groups implanted with transduced fibroblasts (supplemental Figure 1E-F).
However, as expected, stimulation of erythropoiesis led to increased spleen weight in WT mice (Figure 1C) and exacerbation of splenomegaly in Hbbth3/+ animals (Figure 1D).
This was associated with increased reticulocyte numbers and suppression of hepcidin in WT mice (supplemental Figure 1G-H, respectively), while we only observed a trend for these 2 parameters in Hbbth3/+ animals (supplemental Figure 1I,L). This likely prevented the beneficial effect of erythroid-mediated consumption of stored iron in Hbbth3/+ animals, as indicated by unchanged LIC (263 µg/g in controls and 265 µg/g in animals receiving 1 × 106 fibroblasts producing EPO). We then postulated that, in this setting, some level of iron restriction is required to improve anemia without exacerbation of splenomegaly.
Combination of EPO administration and Tmprss6-ASO corrects anemia and reduces splenomegaly of β-thalassemia animals
We then combined the fibroblasts overexpressing Epo with an ASO targeting the Tmprss6 mRNA (Tmprss6-ASO). Previously, we have shown that deletion or inhibition of Tmprss6 increased expression of endogenous hepcidin, which decreased iron absorption and limited erythroid iron intake. This improved IE, splenomegaly, and iron overload in Hbbth3/+ mice.15,18,23 For this study, we used a GaINAc-conjugated Tmprss6-ASO, which enables selective delivery of the ASO to hepatocytes where Tmprss6 is expressed.31,32
In agreement with published data, GalNAc-conjugated Tmprss6-ASO treatment significantly increased Hb levels (Figure 2A) and RBC count (Figure 2B) of Hbbth3/+ mice. Animals receiving Tmprss6-ASO also showed reduced mean corpuscular volume (MCV) (Figure 2C), while minimal effects were observed on mean corpuscular Hb (MCH) (Figure 2D). Improvement in anemia was also associated with significant reduction in reticulocyte number (Figure 2E) and spleen size (Figure 2F). At the end of the 6-week treatment, high levels of EPO (mediated by TARGTEpo) ameliorated anemia (Figure 2A-B) but also significantly increased reticulocyte number (Figure 2E) and spleen size (Figure 2F) as expected.
Figure 2.
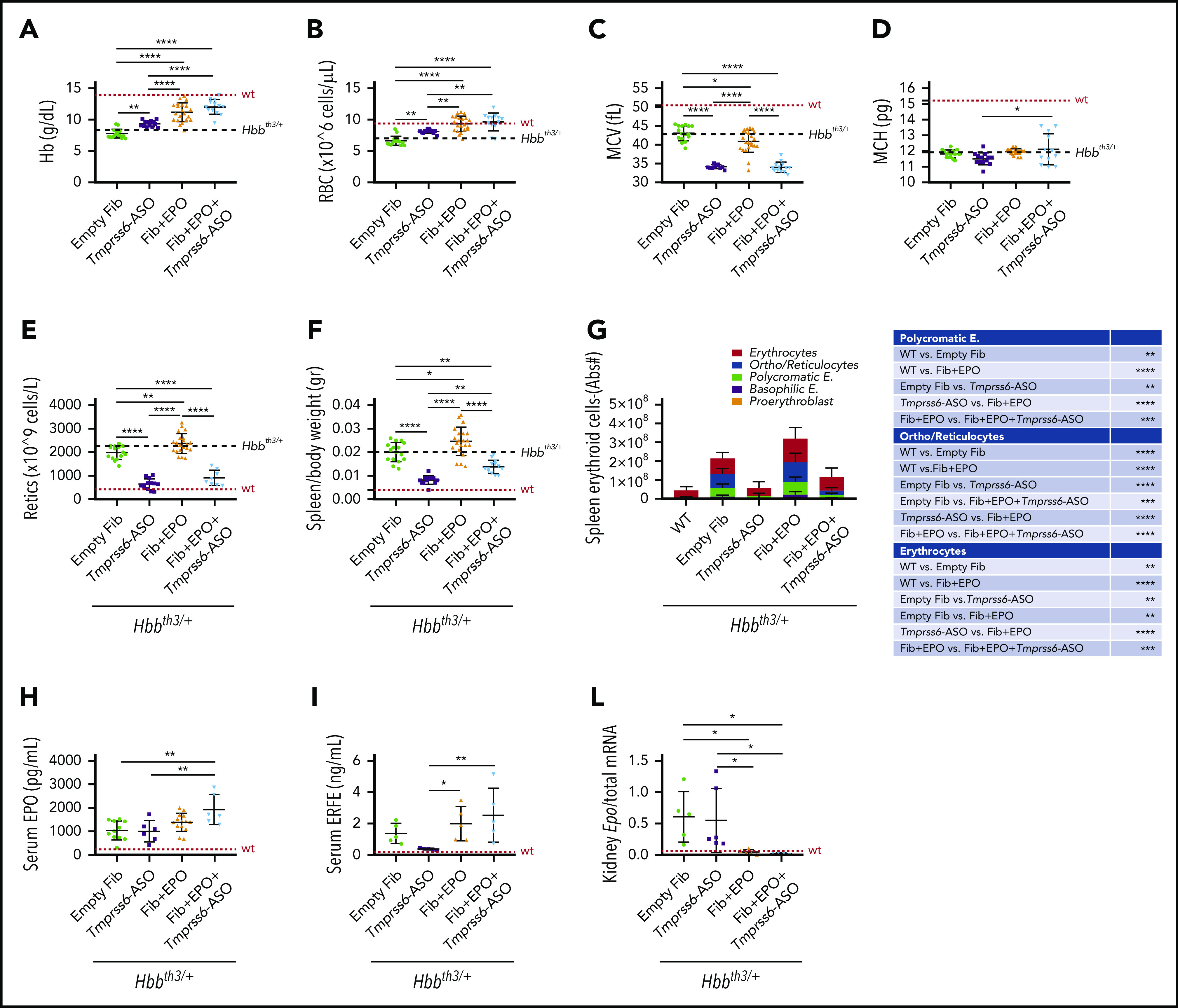
Tmprss6-ASO treatment, together with overexpression of Epo, significantly improves anemia and reduces splenomegaly in Hbbth3/+ animals. Treatment with Tmprss6-ASO induced increased Hb levels (A) and RBC count (B) in Hbbth3/+ mice. Tmprss6-ASO also induced a reduction of MCV (C) and had a minimal effect on MCH (D). Improvement in anemia was also associated with a significant reduction in reticulocyte (Retics) number (E) and splenomegaly (F) when compared with animals receiving empty fibroblasts. When EPO was coadministered with Tmprss6-ASO, animals displayed normalization of Hb levels and RBC number when compared with controls (A-B), as well as normalization of reticulocyte number and splenomegaly, when compared with animals receiving fibroblasts overexpressing Epo (E-F). This was also confirmed by flow cytometry analysis of the erythroid compartment in the spleen (expressed in Abs#). (G). Coadministration of EPO with Tmprss6-ASO led to higher levels of serum EPO (H) as well as serum ERFE (I). Suppression of endogenous Epo expression was observed in the kidney of animals receiving fibroblasts overexpressing Epo (L). Dotted red line indicates mean values for untreated WT mice. Dashed black lines indicates mean values for untreated Hbbth3/+ mice. Bars represent SD. Asterisks refer to statistically significant differences (****P ≤ .001; ***P ≤ .005; **P ≤ .01; *P ≤ .05). E, erythroblasts.
The combination of Tmprss6-ASO and TARGTEpo did not further increase RBC count and Hb levels (Figure 2A-B), but it significantly reduced reticulocyte number and spleen size, reaching values comparable to mice receiving Tmprss6-ASO alone (Figure 2E-F). Reduction of splenomegaly was also confirmed by flow cytometry analysis of the erythroid compartment in the spleen (Figure 2G).
Interestingly, we observed higher levels of EPO in circulation in animals treated with Tmprss6-ASO and TARGTEpo than in those treated with TARGTEpo alone (Figure 2H). Since splenomegaly and the number of erythroid cells was superior in animals treated with TARGTEpo alone (Figure 2F-G), we interpreted this effect as the result of the different amounts of EPO used per mass of erythroid cells. In other words, if we assume the production of EPO was the same in each animal, then the consumption of EPO was higher in animals treated with TARGTEpo than in those with TARGTEpo + Tmprss6-ASO, which showed better erythroid differentiation, longer lifespan, less apoptosis, and an overall lower number of erythroid cells.
ERFE levels in the serum were increased by EPO administration in Tmprss6-ASO–treated mice (Figure 2I), in agreement with high circulating EPO and erythropoiesis expansion. Similar results comparing Tmprss6-ASO vs Tmprss6-ASO + EPO were observed with Erfe transcript levels in the BM and spleen (supplemental Figure 2A-B). Administration of exogenous EPO, achieved by engineered fibroblasts, led to the suppression of endogenous Epo expression in the kidney (Figure 2L).
Iron restriction induced by Tmprss6-ASO in association with EPO significantly reduces iron parameters and improves RBC survival in Hbbth3/+ animals
As previously reported, GalNAc-conjugated Tmprss6-ASO administration suppressed Tmprss6 mRNA in livers (≤96%) (supplemental Figure 3A) of Hbbth3/+ mice, even in the presence of high EPO levels.15,18 This corresponded to a 2.5-fold increase in hepcidin mRNA expression in the liver (supplemental Figure 3B) and protein level in the serum (Figure 3A). The iron-restrictive effect induced by Tmprss6-ASO led to a significant reduction in serum iron (Figure 3B) and transferrin saturation (Figure 3C). While liver iron concentration was significantly reduced in animals treated with Tmprss6-ASO alone, we only observed a trend toward reduction in mice treated with the combination of the 2 drugs (Figure 3D). In particular, we observed that mice that had the lowest liver iron concentration (LIC) showed less iron in parenchymal cells but some accumulation (as expected) in Kupffer cells. In contrast, those that showed higher LIC still presented iron not only in Kupffer cells but also in parenchymal cells. This suggests that for some animals, longer treatment with Tmprss6-ASO should have been required to significantly reduce total LIC. This was due to iron retention in macrophages. However, iron retention in macrophages was not observed in animals with very low levels of iron in the liver (supplemental Figure 3C-D), suggesting that in some animals, longer treatment is required to significantly decrease the LIC.
Figure 3.
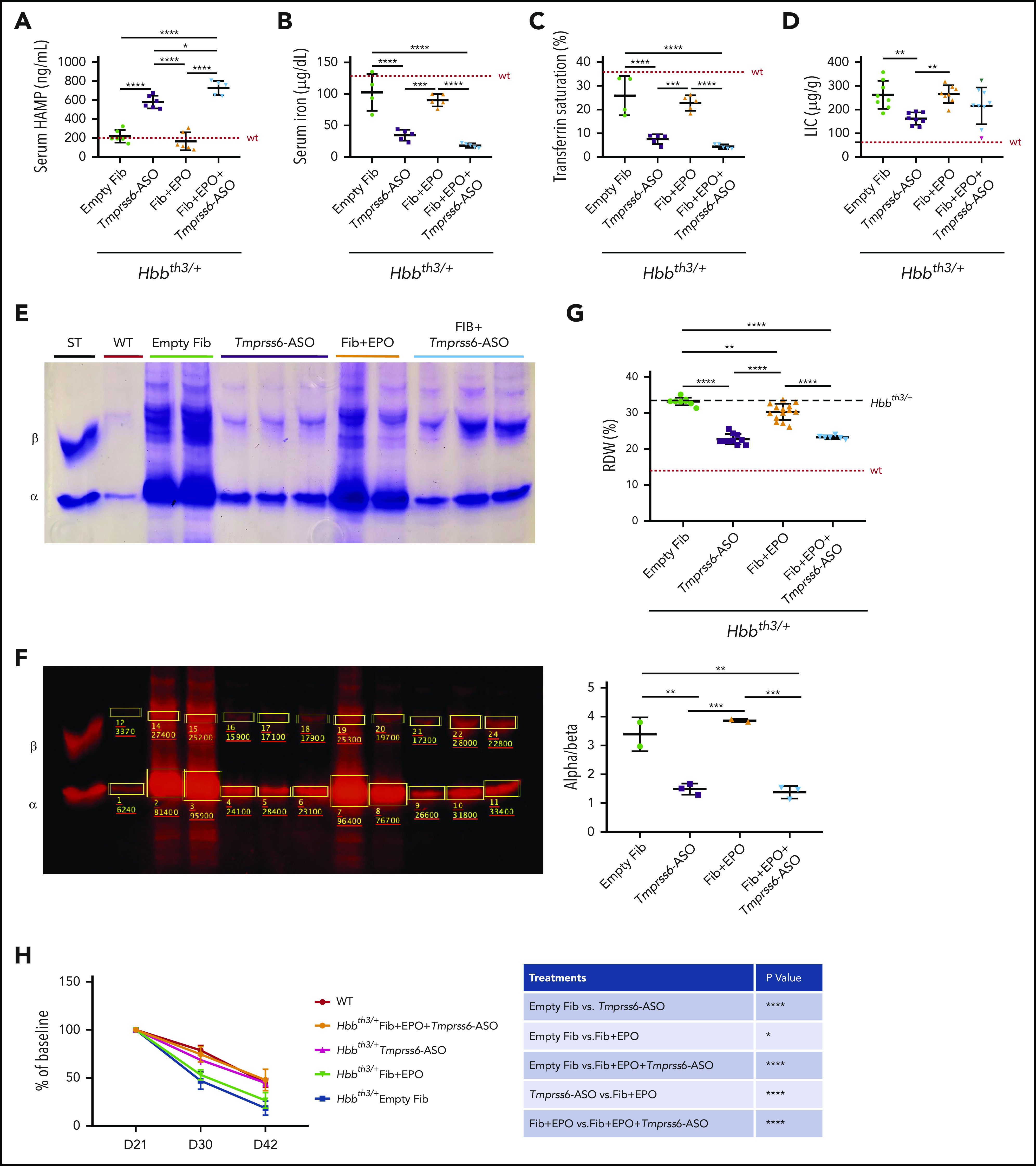
Improved iron parameters due to the iron-restrictive effect of Tmprss6-ASO. Administration of Tmprss6-ASO (with or without overexpression of Epo) to thalassemic animals resulted in significant increase in serum HAMP concentration (A). This was associated with reduced serum iron (B) and transferrin saturation (C) when compared with control animals and animals receiving only fibroblasts overexpressing Epo. Also, liver iron was reduced in a subset of animals (D). Reduction in erythroid iron intake also resulted in a remarkable decrease in ⍺ precipitates on the RBC membrane (E), as corroborated by quantification of the relative abundance of the ⍺ and β globin chains (ImageStudio Lite software) (F). Reduction in ⍺ precipitates induced by Tmprss6-ASO resulted in significant improvement of RBC distribution width (RDW) (G) and normalization of the lifespan of peripheral erythroid cells of treated animals compared with controls (H). Dotted red line indicates mean values for untreated WT mice. Dashed black lines indicates mean values for untreated Hbbth3/+ mice. Bars represent SD. Asterisks refer to statistically significant differences (****P ≤ .001; ***P ≤ .005; **P ≤ .01; *P ≤ .05). ST, standard.
Furthermore, reduced erythroid iron intake due to Tmprss6-ASO administration significantly reduced the formation of α precipitates (Figure 3E). Quantification of the globin proteins (Figure 3F) confirmed that combined treatment did not worsen the ⍺/β-globin chains balance compared with Tmprss6-ASO alone. This was also associated with improved RBC distribution width (RDW) (Figure 3G) and normalized RBCs lifespan (Figure 3H).
The combination of Tfr2-haploinsufficiency and Tmprss6-ASO strongly ameliorates anemia in Hbbth3/+ mice
In order to validate our results, we decided to investigate whether Tmprss6-ASO treatment synergizes with Tfr2 deletion in the BM, which increases EPO sensitivity of erythroid cells, for the correction of β-thalassemia. In view of a potential pharmacologic approach able to inhibit TFR2 only in erythroid cells, we decided to verify whether BM Tfr2 haploinsufficiency (which does not impact systemic iron homeostasis) is also able to improve the thalassemic phenotype. To this aim, we generated Hbbth3/+ mice with either (tHbbth3/+[t, transplanted]) or a single Tfr2 allele (Tfr2BM hetero/Hbbth3/+) in the BM through BMT.
Nine weeks after BMT, mice were treated with GalNAc-conjugated Tmprss6-ASO or CTRL-ASO twice a week for 6 weeks. Deletion of a single Tfr2 allele in the BM of Hbbth3/+ mice increased RBC count (Figure 4A) and Hb levels (Figure 4B) while reducing the percentage of reticulocytes (Figure 4C) and the erythrocyte indexes MCV (Figure 4D) and MCH (Figure 4E). A comparable beneficial effect was reached at the end of Tmprss6-ASO treatment in tHbbth3/+ mice.
Figure 4.

Tfr2 haploinsufficiency in combination with Tmprss6-ASO significantly ameliorates anemia of β-thalassemic mice. Tfr2BM hetero/Hbbth3/+ and tHbbth3/+ mice were treated with Tmprss6-ASO or CTRL-ASO (5mg/kg, intraperitoneally) twice a week for 6 weeks starting 9 weeks after BMT. Complete blood count was determined at the beginning, middle, and end of treatment. The combinatorial treatment showed improvement of RBC count (A), Hb levels (B), reticulocyte (Retics) percentage (C), MCV (D), and MCH (E). Dotted red line indicates mean values for untreated WT mice. Bars represent SD. Color-coded asterisks refer to statistically significant differences between each group and CTRL-ASO–treated Hbbth3/+ mice. ***P < .005; **P < .01; *P < .05.
Treatment of Tfr2BM hetero/Hbbth3/+ animals with Tmprss6-ASO induced a significant increase in the number of RBCs, reaching the normal range after 3 weeks of treatment. Surprisingly, at the end of treatment RBC count was even greater than that of WT mice (Figure 4A).
Hb levels of Tfr2BM-hetero/Hbbth3/+ were increased by ∼2 g/L compared with tHbbth3/+ after 3 weeks of Tmprss6-ASO administration, and only a modest further improvement was achieved at the end of treatment (Figure 4B). After 6 weeks of treatment, the reticulocyte count normalized, while MCV and MCH were strongly reduced, indicating a disproportionate erythropoietic expansion relative to the available iron (Figure 4C-E).
The combination of Tfr2 haploinsufficiency and Tmprss6-ASO induces iron restriction, improves erythropoiesis, and reduces splenomegaly in Hbbth3/+ mice
We confirmed that the 6-week-long treatment with GalNAc-conjugated Tmprss6-ASO efficiently inhibited Tmprss6 expression in the liver of both Tfr2BM-hetero/Hbbth3/+ and tHbbth3/+ mice in a similar fashion (∼94%; supplemental Figure 4A), with a consequent fourfold increase of hepatic hepcidin (Hamp) expression (supplemental Figure 4B). Transferrin saturation (supplemental Figure 4C) and serum iron (supplemental Figure 4D) were reduced in CTRL-ASO–treated Tfr2BM hetero/Hbbth3/+ mice compared with tHbbth3/+ controls, likely because of the enhanced erythropoiesis induced by Tfr2 deletion. As expected, treatment with Tmprss6-ASO resulted in a strong reduction of transferrin saturation and serum iron, reaching comparable values in both genotypes. Following Tmprss6-ASO treatment, spleen iron content was significantly augmented in Tfr2BM hetero/Hbbth3/+ mice, compatible with high hepcidin levels, whereas it was only mildly increased in tHbbth3/+ animals (supplemental Figure 4E). LIC was comparable among all groups (supplemental Figure 4F), likely due to iron sequestration into macrophages, as previously shown (supplemental Figure 3D).
Epo transcript levels in the kidney (Figure 5A) and protein levels in the serum (Figure 5B) inversely correlated to Hb levels. EPO levels were reduced in CTRL-ASO–treated Tfr2BM-hetero/Hbbth3/+ and Tmprss6-ASO–treated tHbbth3/+ mice when compared with tHbbth3/+ controls. Furthermore, they were even lower (within the normal range) in Tmprss6-ASO–treated Tfr2BM-hetero/Hbbth3/+ animals. This was also confirmed by similar alterations in the expression levels of the EPO-target gene Erfe both in the BM (Figure 5C) and in the spleen (Figure 5D).
Figure 5.
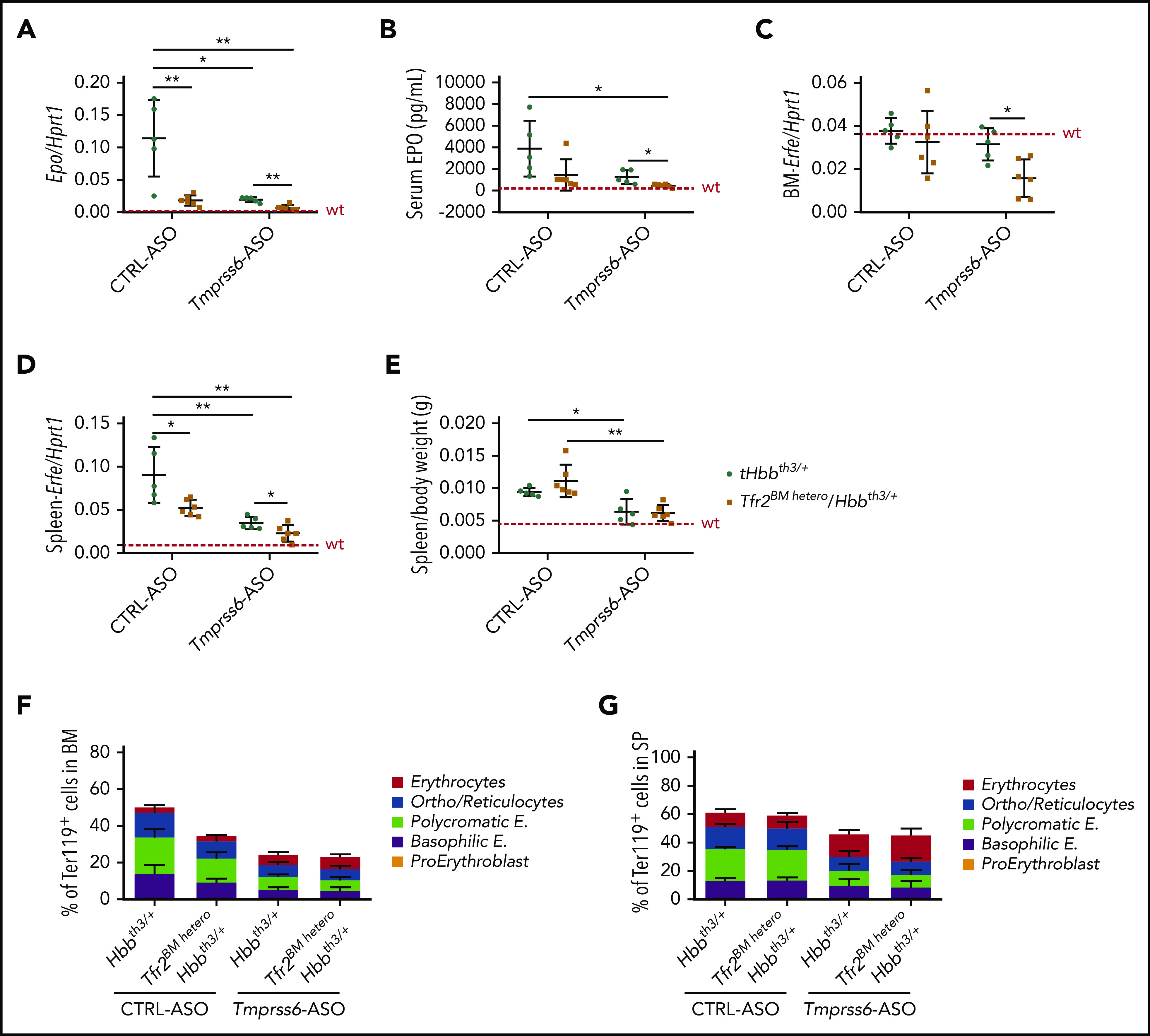
Tfr2 haploinsufficiency in combination with Tmprss6-ASO significantly ameliorates erythropoiesis and splenomegaly of β-thalassemic mice. At the end of the treatment with Tmprss6-ASO or CTRL-ASO, Tfr2BM hetero/Hbbth3/+ and tHbbth3/+ mice were euthanized, and we evaluated kidney mRNA expression of EPO (Epo) relative to hypoxanthine phosphoribosyltransferase 1 (Hprt1) (A), serum EPO levels (B), Erfe mRNA levels in the BM (C) and spleen (D), spleen size relative to body weight (E), percentage of Ter119+ cells on alive cells, and subpopulation composition (gated clusters: proerythroblasts [I], basophilic erythroblasts [II], polychromatic erythroblasts [III], orthochromatic erythroblasts and immature reticulocytes [IV], and mature red cells [V]) based on Ter119/CD44 expression and forward scatter (reflecting cell size) both in the BM (F) and in the spleen (SP) (G). Dotted red line indicates mean values for untreated WT mice. Bars represent SD. Asterisks refer to statistically significant differences between age-matched tHbbth3/+ and Tfr2BMKO/Hbbth3/+ mice (***P < .005; **P < .01; *P < .05). Complete statistical analysis relative to panels F and G (1-way analysis of variance with Bonferroni’s multiple comparison test) is reported in supplemental Tables 1 and 2.
As expected from our previous findings, Tfr2 haploinsufficiency did not alter spleen size in thalassemic mice, while treatment with Tmprss6-ASO significantly reduced splenomegaly in both Tfr2BM-hetero/Hbbth3/+ and tHbbth3/+ animals in a comparable manner (Figure 5E).26 Tfr2 deletion modestly improved IE in the BM (Figure 5F), but not in the spleen (Figure 5G). Tmprss6-ASO administration, on the other hand, strongly ameliorated BM and spleen erythropoiesis of both Tfr2BM-hetero/Hbbth3/+ and tHbbth3/+ animals in a comparable manner. The RDW SD was significantly reduced in Tmprss6-ASO–treated Tfr2BM-hetero/Hbbth3/+ animals relative to Tmprss6-ASO–treated tHbbth3/+ (32.9 ± 2.0 fL and 37.3 ± 3.9 fL, respectively; P = .045), suggesting better fitness of Tfr2-haploinsufficient β-thalassemic erythrocytes.
Iron restriction mediated by Tmprss6-ASO mitigates the effect of EPO on Akt and Jak2 activation
We next inquired how the combination of iron restriction and enhanced EPO stimulation improved erythropoiesis. We quantified the nucleated erythroid cells positive for the phosphorylated forms of Akt and Jak2, proteins activated by EPO but whose activity is potentially limited by iron restriction.33 For instance, the total number of pAkt+ and pJak2+ proerythroblasts in the spleen, normalized to circulating EPO levels, was reduced in Hbbth3/+ mice treated with GalNAc-conjugated Tmprss6-ASO or Tmprss6-ASO + TARGTEpo (respectively −96% and −84% for pAkt+ and −91% and −84% for Jak2+ when compared with control Hbbth3/+ mice; Figure 6A-B). The total number of progenitor erythroid cells (erythroid colony-forming unit [CFU-E]) was also decreased with Tmprss6-ASO treatment, and that of erythroid burst-forming units followed a similar trend (Figure 6C; supplemental Figure 5).
Figure 6.
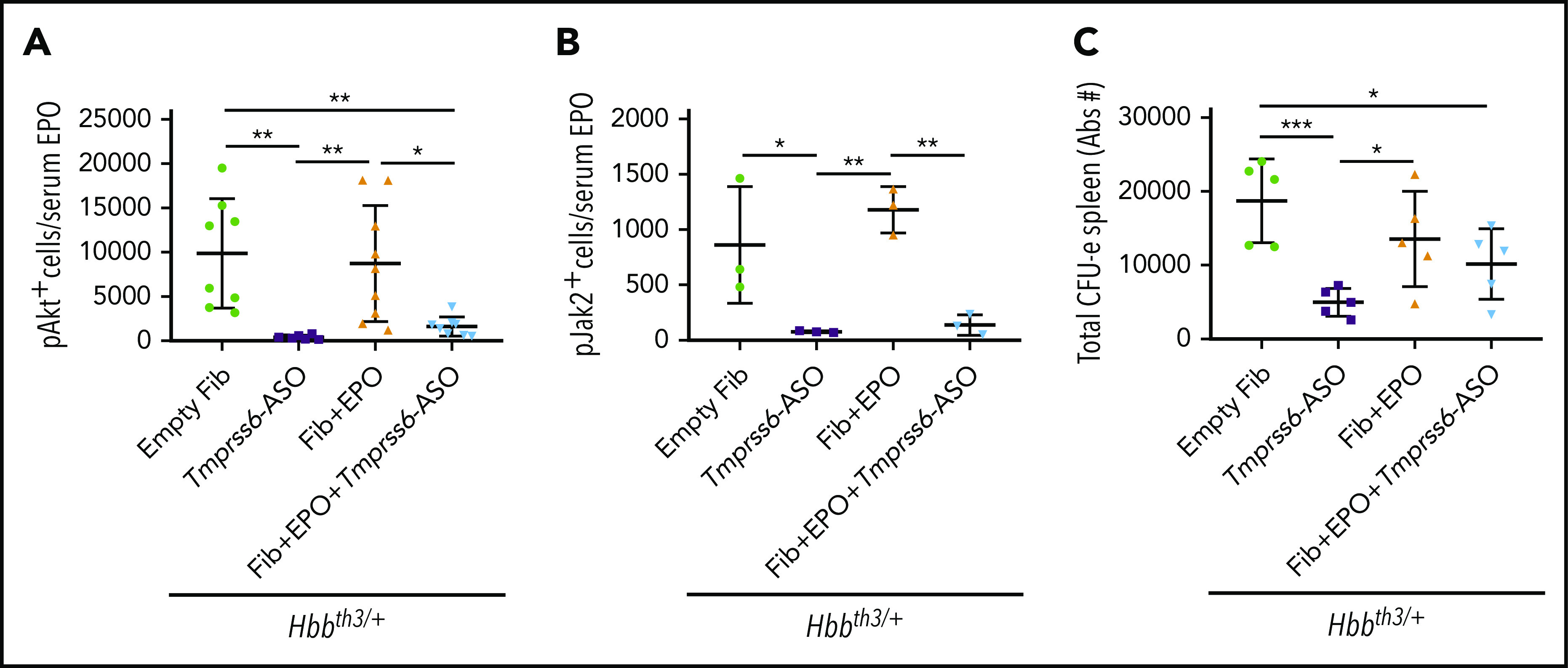
The effect of EPO is modulated by the iron-restrictive properties of Tmprss6-ASO. In Hbbth3/+ animals, administration of Tmprss6-ASO induced reduction of the total amount of splenic proerythroblasts positive for the phosphorylated form of Akt (A) and Jak2 (B) when normalized to the total amount of EPO in circulation. The total number of splenic CFU-Es (expressed in Abs#). followed a similar profile (C). Bars represent SD. Asterisks refer to statistically significant differences (***P ≤ .005, **P ≤ .01; *P ≤ .05).
Altogether, these data suggest that under conditions of IE, the combination of erythroid iron restriction with strategies aimed to increase RBC synthesis improves the quality of RBC and does not reduce the overall production of erythrocytes, despite lower levels of erythroid progenitors being produced.
Iron restriction induced by Tmprss6-ASO in association with lower doses of EPO significantly improves anemia and reduces splenomegaly in Hbbth3/+ animals
To overcome the possible issues associated with the use of very high levels of EPO in a clinical setting, we evaluated additional treatment regimens. Hbbth3/+ animals were first pretreated with the regular doses of Tmprss6-ASO for 3 weeks to abate IE, followed by the coadministration of lower doses of EPO (administered as rhEPO or fibroblasts overexpressing Epo) in the last 2 weeks of the experiment. This strategy was based on the hypothesis that less EPO would be needed once IE was controlled. At the end of the study, animals receiving the lower doses of rhEPO (Figure 7A-C) or a lower dose of fibroblasts overexpressing Epo (Figure 7D-F) displayed a significant improvement in anemia (as shown by increased RBC number and Hb levels) and reduction of splenomegaly when compared with animals receiving Tmprss6-ASO alone (Figure 7A-F). These data provide the proof of principle that a tailored combination therapy could be extremely beneficial in the clinical setting, allowing for a further improvement of the thalassemic phenotype without detrimental effects on the reduced splenomegaly induced by Tmprss6-ASO alone.
Figure 7.
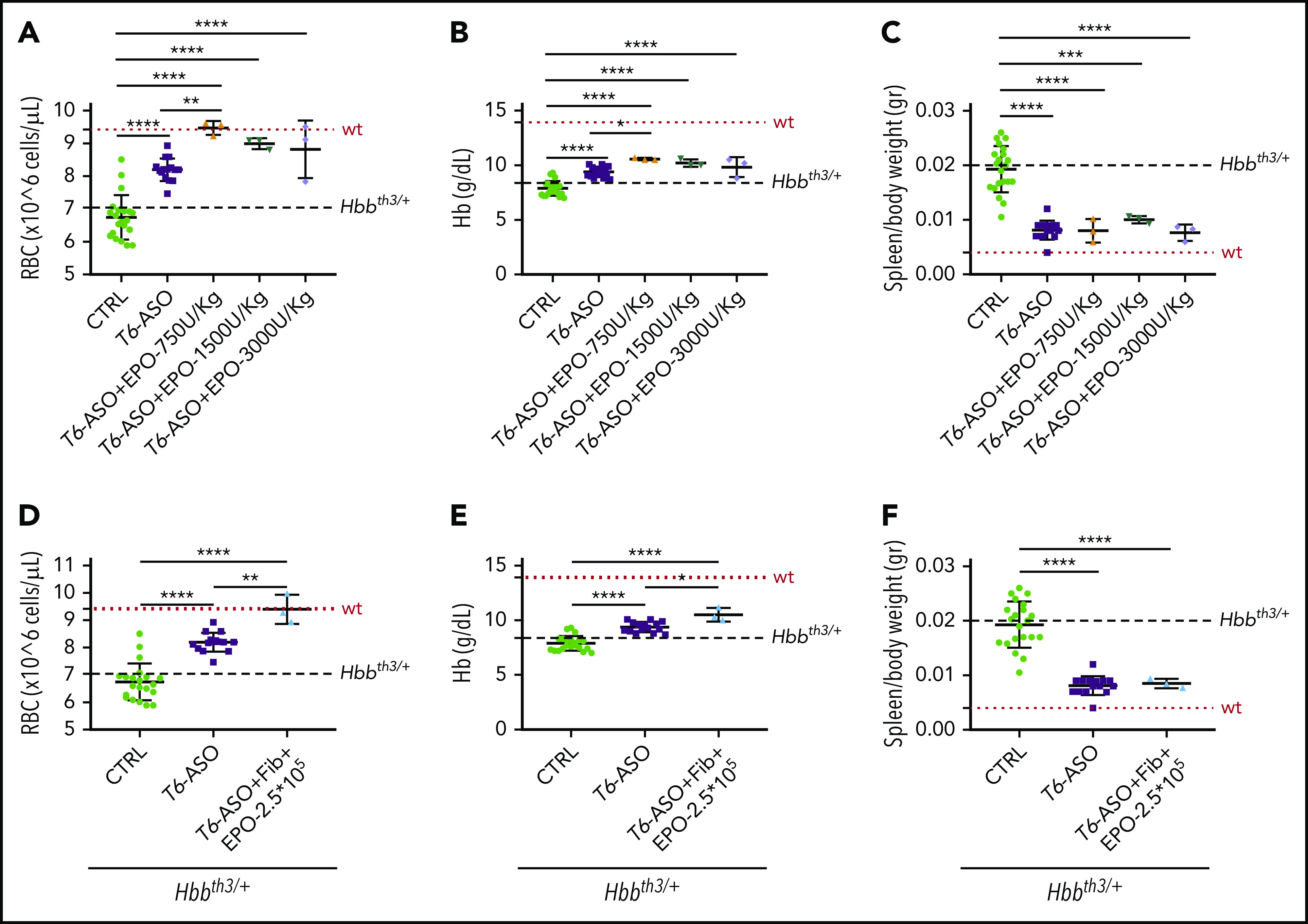
Pretreatment with Tmprss6-ASO followed by a concomitant lower dose of EPO is more effective than Tmprss6-ASO alone in improving anemia in Hbbth3/+ mice. Thalassemic mice receiving cotherapy with Tmprss6-ASO (T6-ASO) and lower dose of EPO (in the form of rhEPO or fibroblasts overexpressing Epo) following a pretreatment with Tmprss6-ASO showed significantly improved anemia when compared with animals receiving Tmprss6-ASO alone. At the end of the treatment we observed increased RBC number, Hb levels and reduced splenomegaly when both a low dose of rhEPO (A-C) or lower dose of fibroblasts overexpressing Epo (D-F) were coadministered with Tmprss6-ASO. Dotted red line indicates mean values for untreated WT mice. Dashed black lines indicates mean values for untreated Hbbth3/+ mice. Bars represent SD. Asterisks refer to statistically significant differences (****P ≤ .001; ***P ≤ .005; **P ≤ .01; *P ≤ .05).
Discussion
Traditional treatment of β-thalassemia is still far from being optimal, and all of the ongoing experimental approaches are based on the administration of single agents. We decided to investigate the possibility of increasing the effectiveness of β-thalassemia treatment through a “combinatorial” approach, in analogy with current therapies for hematological malignancies. Past studies indicated that despite high EPO levels in β-thalassemia, additional EPO administration was effective in improving anemia in patients.34-40 However, this approach was abandoned in light of potential cardiovascular complications associated with the high dose of rhEPO required, the transient effect, costs, and inconvenient route of administration of the drug.41
Furthermore, we have shown that iron restriction improves anemia in β-thalassemia mice and reduces oxidative stress, hemichrome formation, and splenomegaly.15,17,18 Therefore, we decided to evaluate whether iron restriction could prevent the side effects of EPO administration (ie, splenomegaly) and its inability to improve hemichromes formation without impairing RBC production. Here, we demonstrated that the induction of iron restriction through Tmprss6-ASO administration and the stimulation of EPO activity (through either EPO administration or reducing Tfr2 in BM cells) synergize to improve the thalassemic phenotype and ameliorate or correct hematological parameters, erythropoiesis, and splenomegaly.
However, the results obtained with the 2 therapeutic approaches did not overlap completely. With EPO administration, the increase in RBC count was exclusively EPO mediated (as no additive effect with Tmprss6-ASO was observed) and reached levels observed in WT mice. However, the contribution of Tmprss6-ASO was clear when we characterized the quality of the red cells and, in the spleen, the total number of erythroid cells, the portion of these that were pAkt+ and pJak2+, and the total number of CFU-Es. Administration of Tmprss6-ASO not only improved red cell quality and lifespan but also reduced the number of erythroid progenitors that exacerbated the splenomegaly by EPO administration, as indicated by the number of pAkt+ and pJak2+ erythroid cells normalized by serum EPO levels. In other words, while EPO was required to increase RBC synthesis, administration of Tmprss6-ASO induced some level of iron restriction that limited overproduction of erythroid cells and, simultaneously, corrected IE. In contrast, in Tfr2-haploinsufficient β-thalassemic mice, Tmprss6-ASO administration further increased RBC count, to levels superior to those of WT mice. Following implantation of fibroblasts overexpressing Epo, RBC production might reach plateau levels, which do not further increase with additional Tmprss6-ASO administration. If this is the case, then why did Tfr2-deficient β-thalassemic mice have higher levels of RBCs in circulation? One possibility is that lack of Tfr2 may reset the EPO sensitivity of the erythroid cells so that lower or similar levels of EPO may lead to higher production of RBCs. Alternatively, since the diet administered to the Tfr2-deficient β-thalassemic mice was more iron rich, this could positively impact RBC synthesis. Future studies will address these questions.
EPO administration alone is not an option for β-thalassemic patients because of the risk of side effects associated with the high doses required.37,42 However, our results prove that a pretreatment with Tmprss6-ASO, aimed at limiting IE, strongly reduces the amount of EPO required to achieve a beneficial effect on the thalassemic phenotype in mice, making this approach a potential therapeutic option for β-thalassemic patients. Selective targeting of Tfr2 on erythroid cells may represent an alternative strategy for the future. Once the TFR2-EPO receptor–interacting site is identified, small molecules/peptides that interfere with binding may be designed and tested, or an approach based on specific ASOs or small interfering RNAs could be envisioned. As we have shown here and previously, Tfr2 haploinsufficiency is enough to improve the β-thalassemic phenotype, without impairing hepcidin expression in the liver and causing iron overload.25
Although our results suggest that the combination of EPO administration or Tfr2-targeting and anti-Tmprss6 therapy may be clinically effective, the induction of excessive iron restriction has to be avoided. Indeed, we have shown that at the end of the 6-week-long protocol, iron restriction induced by Tmprss6 inhibition in Tfr2 haploinsufficient mice is disproportionate relative to the strong erythropoietic stimulus, leading to exaggerated erythrocytosis, microcytosis, and hypochromia. However, the time-course analysis of hematological parameters revealed that earlier in the course of treatment, RBC and reticulocyte counts are within the normal range, Hb levels are already high, and only a small effect on MCV and MCH is observed. These findings suggest that in view of a potential therapeutic approach, an optimal Tmprss6-ASO treatment regimen can be identified. Nevertheless, here, we showed that in non–transfusion-dependent β-thalassemia, the combination of iron restriction and strategies aimed at stimulating erythropoiesis maximizes the correction of anemia while improving splenomegaly and, to some extent, iron overload. Whether this approach might also benefit transfusion-dependent β-thalassemia is still not known and worthwhile to be tested in future studies.
In addition, our findings pave the way for and strongly support the use of additional drugs that target iron metabolism (ie, hepcidin mimetics, FPN inhibitors, or TMPRSS6 inhibitors) in combination with erythroid enhancer agents (ie, EPO, TFR2 inhibitors, and luspatercept) to improve the treatment of β-thalassemia. In conclusion, we suggest that combinatorial therapies targeting different pathophysiological pathways of this disorder could represent a valid and superior approach for the management of β-thalassemia.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors are indebted to Clara Camaschella (San Raffaele Scientific Institute, Milan, Italy) for criticism and suggestions.
This work was supported by Ionis Pharmaceuticals and Aevi Genomic Medicine (S.R.); the Cooley’s Anemia Foundation, the Cariplo Foundation (“Young Investigator” grant 2017-0916), and the American Society of Hematology (ASH Global Research Award 2018) (A.N.); the Telethon Foundation (SR-TIGET core grant) (G.F.); the US Department of Defense (W81XWH-16-1-0598) (D.S.); and the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases Institute (R01 DK090554 and R01 DK095112) (S.R.).
Footnotes
For original data, please e-mail the corresponding author.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.C. and M.P. performed research, analyzed results, and contributed to paper writing; A.L., M.A., V.L.P., M.R.L., K.A.M., E.O., V.O., S.M.D.M., S.B., and H.D. performed experiments; S.G. and M.A. generated and characterized the ASOs and edited the paper; G.N., R.M., N.S., I.Z.-L., G.F., and D.S. contributed to critical data analysis; A.N. and S.R. conceived the experiments, analyzed results, and wrote and reviewed the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: S.R. is a member of scientific advisory board of Ionis Pharmaceuticals, Meira GTx, and Disc Medicine and owns stock options in Disc Medicine and Meira GTx. S.R. has been or is consultant for Cambridge Healthcare Res, Celgene Corporation, Ghost Tree Capital, Noble insight, Protagonist Therapeutics, Sanofi Aventis U.S., Slingshot Insight, Techspert.io, venBio Select, and Disc Medicine. A.N. has filed a patent application on the results presented in the paper. S.R. and C.C. have filed a patent application on the results presented in the paper. S.G., M.A., and S.B. are employees and shareholders of Ionis Pharmaceuticals. G.N. is chief scientific officer of Aevi Genomic Medicine. The remaining authors declare no competing financial interests.
Correspondence: Stefano Rivella, Children’s Hospital of Philadelphia, Abramson Research Center, 3615 Civic Center Blvd, Room 316B, Philadelphia, PA 19104; e-mail: rivellas@e-mail.chop.edu; and Antonella Nai, San Raffaele Scientific Institute, and Vita-Salute San Raffaele University, Via Olgettina, 58, 20132 Milan, Italy; e-mail: nai.antonella@hsr.it.
REFERENCES
- 1.Oikonomidou PR, Rivella S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. 2018;32(2):130-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2017;391(10116):155-167. [DOI] [PubMed] [Google Scholar]
- 3.Rivella S. β-thalassemias: paradigmatic diseases for scientific discoveries and development of innovative therapies. Haematologica. 2015;100(4):418-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rivella S. Iron metabolism under conditions of ineffective erythropoiesis in β-thalassemia. Blood. 2019;133(1):51-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta R, Musallam KM, Taher AT, Rivella S. Ineffective erythropoiesis: anemia and iron overload. Hematol Oncol Clin North Am. 2018;32(2):213-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan J, Kannan R, Shinar E, Rachmilewitz EA, Low PS. Isolation, characterization, and immunoprecipitation studies of immune complexes from membranes of beta-thalassemic erythrocytes. Blood. 1992;79(11):3007-3013. [PubMed] [Google Scholar]
- 7.Rachmilewitz EA. Formation of hemichromes from oxidized hemoglobin subunits. Ann N Y Acad Sci. 1969;165(1):171-184. [DOI] [PubMed] [Google Scholar]
- 8.Melchiori L, Gardenghi S, Rivella S. Beta-thalassemia: HiJAKing ineffective erythropoiesis and iron overload. Adv Hematol. 2010;2010:938640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forster L, Cornwall S, Finlayson J, Ghassemifar R. Cell cycle, proliferation and apoptosis in erythroblasts cultured from patients with β-thalassaemia major. Br J Haematol. 2016;175(3):539-542. [DOI] [PubMed] [Google Scholar]
- 10.Kautz L, Jung G, Du X, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood. 2015;126(17):2031-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism [published correction appears in Nat Genet. 2020;52(4):463]. Nat Genet. 2014;46(7):678-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson ER, Taylor M, Xue X, et al. Intestinal HIF2α promotes tissue-iron accumulation in disorders of iron overload with anemia. Proc Natl Acad Sci USA. 2013;110(50):E4922-E4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bou-Fakhredin R, Bazarbachi AH, Chaya B, Sleiman J, Cappellini MD, Taher AT. Iron overload and chelation therapy in non-transfusion dependent thalassemia. Int J Mol Sci. 2017;18(12):E2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sleiman J, Tarhini A, Bou-Fakhredin R, Saliba AN, Cappellini MD, Taher AT. Non-transfusion-dependent thalassemia: an update on complications and management. Int J Mol Sci. 2018;19(1):E182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casu C, Aghajan M, Oikonomidou PR, Guo S, Monia BP, Rivella S. Combination of Tmprss6- ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of beta-thalassemia intermedia. Haematologica. 2016;101(1):e8-e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casu C, Chessa R, Liu A, et al. Minihepcidins improve ineffective erythropoiesis and splenomegaly in a new mouse model of adult beta-thalassemia major. Haematologica. 2019;haematol.2018.212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casu C, Oikonomidou PR, Chen H, et al. Minihepcidin peptides as disease modifiers in mice affected by β-thalassemia and polycythemia vera. Blood. 2016;128(2):265-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo S, Casu C, Gardenghi S, et al. Reducing TMPRSS6 ameliorates hemochromatosis and β-thalassemia in mice. J Clin Invest. 2013;123(4):1531-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt PJ, Toudjarska I, Sendamarai AK, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine β-thalassemia intermedia. Blood. 2013;121(7):1200-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Liu W, Liu Y, et al. New thiazolidinones reduce iron overload in mouse models of hereditary hemochromatosis and β-thalassemia. Haematologica. 2019;104(9):1768-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manolova V, Nyffenegger N, Flace A, et al. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of β-thalassemia. J Clin Invest. 2019;130(1):491-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt PJ, Racie T, Westerman M, Fitzgerald K, Butler JS, Fleming MD. Combination therapy with a Tmprss6 RNAi-therapeutic and the oral iron chelator deferiprone additively diminishes secondary iron overload in a mouse model of β-thalassemia intermedia. Am J Hematol. 2015;90(4):310-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nai A, Pagani A, Mandelli G, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of β-thalassemia. Blood. 2012;119(21):5021-5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shapir N, Miari R, Blum S, et al. Preclinical and preliminary clinical evaluation of genetically transduced dermal tissue implants for the sustained secretion of erythropoietin and interferon α. Hum Gene Ther Clin Dev. 2015;26(4):216-227. [DOI] [PubMed] [Google Scholar]
- 25.Artuso I, Lidonnici MR, Altamura S, et al. Transferrin receptor 2 is a potential novel therapeutic target for β-thalassemia: evidence from a murine model [published correction appears in Blood. 2019;134(1):94]. Blood. 2018;132(21):2286-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nai A, Lidonnici MR, Rausa M, et al. The second transferrin receptor regulates red blood cell production in mice. Blood. 2015;125(7):1170-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nai A, Pellegrino RM, Rausa M, et al. The erythroid function of transferrin receptor 2 revealed by Tmprss6 inactivation in different models of transferrin receptor 2 knockout mice. Haematologica. 2014;99(6):1016-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silvestri L, Nai A, Pagani A, Camaschella C. The extrahepatic role of TFR2 in iron homeostasis. Front Pharmacol. 2014;5:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang B, Kirby S, Lewis J, Detloff PJ, Maeda N, Smithies O. A mouse model for beta 0-thalassemia. Proc Natl Acad Sci USA. 1995;92(25):11608-11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roetto A, Di Cunto F, Pellegrino RM, et al. Comparison of 3 Tfr2-deficient murine models suggests distinct functions for Tfr2-alpha and Tfr2-beta isoforms in different tissues. Blood. 2010;115(16):3382-3389. [DOI] [PubMed] [Google Scholar]
- 31.Crooke ST, Baker BF, Xia S, et al. integrated assessment of the clinical performance of galnac3-conjugated 2′-o-methoxyethyl chimeric antisense oligonucleotides: I. Human volunteer experience. Nucleic Acid Ther. 2019;29(1):16-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prakash TP, Graham MJ, Yu J, et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014;42(13):8796-8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khalil S, Delehanty L, Grado S, et al. Iron modulation of erythropoiesis is associated with Scribble-mediated control of the erythropoietin receptor. J Exp Med. 2018;215(2):661-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amer J, Dana M, Fibach E. The antioxidant effect of erythropoietin on thalassemic blood cells. Anemia. 2010;2010:978710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asadov C, Alimirzoyeva Z, Hasanova M, Mammadova T, Shirinova A. Clinical application of recombinant erythropoietin in beta-thalassaemia intermedia. Georgian Med News. 2016;(255):86-92. [PubMed] [Google Scholar]
- 36.Bourantas K, Economou G, Georgiou J. Administration of high doses of recombinant human erythropoietin to patients with beta-thalassemia intermedia: a preliminary trial. Eur J Haematol. 1997;58(1):22-25. [DOI] [PubMed] [Google Scholar]
- 37.Chaidos A, Makis A, Hatzimichael E, et al. Treatment of beta-thalassemia patients with recombinant human erythropoietin: effect on transfusion requirements and soluble adhesion molecules. Acta Haematol. 2004;111(4):189-195. [DOI] [PubMed] [Google Scholar]
- 38.Nişli G, Kavakli K, Vergin C, Oztop S, Cetingül N. Recombinant human erythropoietin trial in thalassemia intermedia. J Trop Pediatr. 1996;42(6):330-334. [DOI] [PubMed] [Google Scholar]
- 39.Olivieri NF, Freedman MH, Perrine SP, et al. Trial of recombinant human erythropoietin: three patients with thalassemia intermedia. Blood. 1992;80(12):3258-3260. [PubMed] [Google Scholar]
- 40.Rachmilewitz EA, Aker M. The role of recombinant human erythropoietin in the treatment of thalassemia. Ann N Y Acad Sci. 1998;850(1 COOLEY’S ANEM):129-138. [DOI] [PubMed] [Google Scholar]
- 41.Borgna-Pignatti C. Modern treatment of thalassaemia intermedia. Br J Haematol. 2007;138(3):291-304. [DOI] [PubMed] [Google Scholar]
- 42.Singbartl G. Adverse events of erythropoietin in long-term and in acute/short-term treatment. Clin Investig. 1994;72(suppl 6):S36-S43. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.