

Summary
Here, we describe a rapid and versatile protocol to generate gapped DNA substrates for single-molecule (SM) analysis using optical tweezers via site-specific Cas9 nicking and force-induced melting. We provide examples of single-stranded (ss) DNA gaps of different length and position. We outline protocols to visualize these substrates by replication protein A-enhanced Green Fluorescent Protein (RPA-eGFP) and SYTOX Orange staining using commercially available optical tweezers (C-TRAP). Finally, we demonstrate the utility of these substrates for SM analysis of bidirectional growth of RAD-51-ssDNA filaments.
For complete details on the use and execution of this protocol, please refer to Belan et al. (2021).
Subject areas: Single-molecule Assays, Microscopy, Molecular/Chemical Probes
Graphical abstract
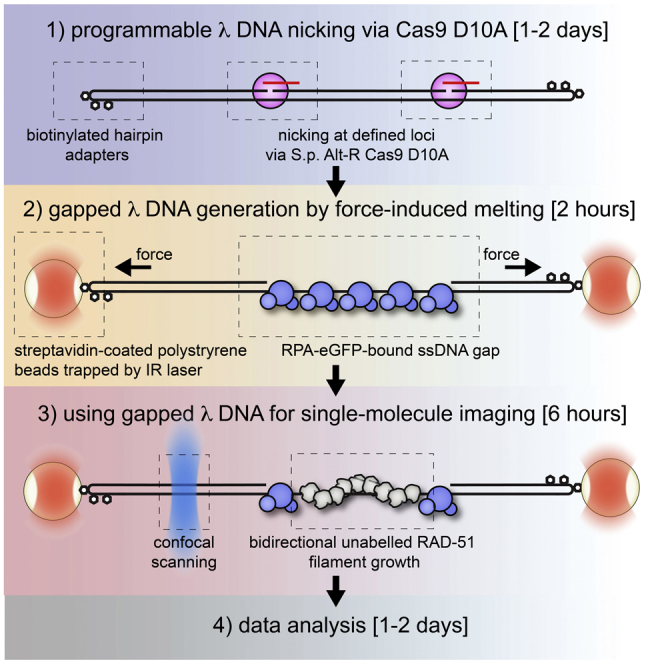
Highlights
-
•
A rapid protocol to generate gapped DNA molecules for single-molecule analysis
-
•
Customizable ssDNA gap length and position
-
•
Suitable for fluorescence imaging of enzymatic reactions
-
•
Includes protein and construct preparation, data acquisition, and analysis
Here, we describe a rapid and versatile protocol to generate gapped DNA substrates for single-molecule (SM) analysis using optical tweezers via site-specific Cas9 nicking and force-induced melting. We provide examples of single-stranded (ss) DNA gaps of different length and position. We outline protocols to visualize these substrates by replication protein A-eGFP and SYTOX Orange staining using commercially available optical tweezers (C-TRAP). Finally, we demonstrate the utility of these substrates for SM analysis of bidirectional growth of RAD-51-ssDNA filaments.
Before you begin
Preparing recombinant RPA-eGFP-his6 protein
Timing: 5 days
-
1.Protein expression in Rosetta(DE3)pLysS.
-
a.The modified polycistronic vector phRPA-eGFP (replication protein A-enhanced Green Fluorescent Protein) containing polyhistidine tag downstream of eGFP fused to C-terminus of RPA70 (Modesti, 2018) is transformed into Rosetta(DE3)pLysS competent cells and plated on LB agar plates supplemented with ampicillin (100 μg/mL) and chloramphenicol (34 μg/mL) and incubated for 16 h at 37°C.
-
b.Next day, three individual colonies are picked up and used to inoculate 50 mL of LB medium supplemented with ampicillin (100 μg/mL) and chloramphenicol (34 μg/mL) and incubated for 16 h at 37°C with shaking.
-
c.Next morning, 3 L of LB media supplemented with ampicillin (100 μg/mL) and chloramphenicol (34 μg/mL) are distributed in three 2 l Erlenmeyer flasks. Each flask is inoculated with 10–15 mL of start culture and incubated at 37°C with shaking (180 rpm) for roughly 2.5–3 h until the bacteria reach OD600 of 0.5. The protein expression is induced by final concentration of 1 mM Isopropyl β- d-1-thiogalactopyranoside (IPTG). Immediately, post induction, the temperature of the incubator is lowered to 15°C and the flasks are incubated at this temperature for 16 h. Next day, cells are harvested by centrifugation at 3500 g for 15 min. The supernatant is removed and cell pellet is transferred to a small plastic bag, which is then spread to form a thin layer inside. The cell paste is weighed and its weight is noted down. The plastic bag containing thinly spread cell pellet is frozen at – 80°C.
-
a.
Pause point: At this stage, frozen pellet can be stored at −80°C over several months.
-
2.Cell lysis and lysate clarification.
-
a.All further steps are performed at 4°C. During the entire procedure, samples should be shielded from light. To resuspend the cell pellet, sufficient (100 mL per 20 g of pellet) RPA lysis buffer (20 mM Tris-HCl pH 7.5, 500 mM NaCl, 2 mM β-mercaptoethanol, 5 mM imidazole pH 8.0 and 10% glycerol) with protease inhibitor tablets (2 tablets per 100 mL) is prepared. The plastic bag containing thin cell paste from −80°C is rapidly crushed using an empty 50 mL falcon tube into small pieces in cold room. Care must be taken to not tear the plastic bag. The crushed cell paste is transferred to appropriate volume of RPA cell lysis buffer and suspension is vigorously stirred until the largest cell paste pieces are fully suspended in lysis buffer.
-
b.The suspension is sonicated using a Branson Sonifier 450 (large flat tip, duty cycle 50%, output control 8) for 3×7 min (200 mL RPA cell lysis buffer) or 2×7 min (100 mL RPA cell lysis buffer). The suspension is stirred using a magnetic stirrer for 5–10 min at 4°C in-between sonication cycles.
-
c.The sonicated sample is clarified by centrifugation at 20 000 g for 1 h at 4°C (Optima LE-80K Ultracentrifuge (Beckman Coulter), Ti45 rotor). The supernatant containing RPA-eGFP is flushed through a filter device with 0.45 μm pores (Millipore).
-
a.
-
3.Protein purification.
-
a.During cell lysate clarification, 2 mL of Ni-NTA slurry per 50 mL of cell suspension from previous steps (typically 4 mL of Ni-NTA slurry is needed per preparation) is washed twice with RPA cell lysis buffer in 50 mL falcon tube (added up to 50 mL volume) using swing-rotor centrifuge (1000 g, 10 min).
-
b.The clarified lysate is divided into 50 mL falcon tube(s) and 2 mL of equilibrated Ni-NTA resin is added equally to all tubes. The resin with clarified lysate in tubes is incubated for 1.5 h at 4°C with gentle rotation.
-
c.Samples are passed through 30 mL Biorad protein purification column (50 mL of sample per column). Each column is washed with 50 mL RPA cell lysis buffer containing 10 mM imidazole. 12 mL RPA cell lysis buffer supplemented with 250 mM imidazole is used for elution per 1 column. 2 mL fractions should be collected and analyzed by SDS-PAGE for RPA-eGFP. Fractions containing most protein should be pooled and dialyzed for 2 h (or for 16 h) against 4 l of RPA buffer R-A (20 mM Tris-HCl pH 7.5, 50 mM KCl, 1 mM Dithiothreitol (DTT), 0.5 mM Ethylenediaminetetraacetic acid (EDTA) and 10% glycerol). Where possible, longer dialysis should be avoided as it results in the precipitation of RPA.
-
d.If precipitation is observed after dialysis, the sample must be spun at maximum g for 10 min to remove precipitated protein.
-
e.The supernatant (soluble protein) is loaded at 1 mL/min speed onto 1 mL HiTrap Heparin column pre-equilibrated with RPA buffer R-A using AKTA FPLC system.
-
f.The HiTrap Heparin column is washed with 10 column volumes of RPA buffer R-A and protein is eluted using linear 0 – 100 % gradient of RPA buffer R-B (20 mM Tris-HCl pH 7.5, 500 mM KCl, 1 mM DTT, 0.5 mM EDTA and 10% glycerol) with 10 column volumes gradient length and 0.2 mL fractions.
-
g.All fractions corresponding to UV peak are analyzed by SDS-PAGE and fractions containing purest RPA (no contamination with other protein bands) are pooled. The protein concentration is determined by Bradford staining and appropriate volume of aliquots are snap frozen in liquid nitrogen. We suggest making 5–10 μL aliquots. This purification procedure yields typically 1–3 mg of protein.
-
a.
Note: Large subunit of trimeric RPA can be fused to other fluorescent protein variants such as RFP, mCherry, mStrawberry or SNAP tag. The purification protocol remains unchanged. This allows for customizable choice of wavelength mainly in combination with other labelled proteins present for multi-colour imaging experiments.
Preparing recombinant untagged RAD-51 protein
-
4.Protein expression in BL21(DE3).
-
a.pET SUMO Champion vector containing nematode rad-51 ORF (Taylor et al., 2015) is transformed into BL21(DE3) competent cells and plated on LB agar plates supplemented with kanamycin (50 μg/mL) and incubated for 16 h at 37°C.
-
b.Next day, a single colony is picked and inoculated in 100 mL of LB medium supplemented with kanamycin (50 μg/mL) and incubated for 16 h at 37°C with shaking.
-
c.Next morning, 6 L of LB media supplemented with kanamycin (50 μg/mL) are distributed in six 2 L Erlenmeyer flasks. Each flask is inoculated with 10–15 mL of start culture and incubated at 37°C with shaking (180 rpm) for roughly 3 h until the bacteria reach OD600 of 0.6. Protein expression is induced by IPTG, which is added to a final concentration of 1 mM. At the same time, temperature of incubator is lowered to 30°C. All flasks are transferred to incubator and further incubated for 4 h. The cells are harvested by centrifugation at 3500 g for 15 min. From this step onwards, cell pellet for RAD-51 is collected and frozen as described for RPA pellet above.
-
a.
-
5.Cell lysis and lysate clarification.
-
a.All further steps are performed at 4°C. Prepare sufficient (100 mL per 20 g) volume of RAD-51 cell lysis buffer (50 mM potassium phosphate pH 7.8, 1 M KCl, 10% glycerol) and add protease inhibitor tablets (2 tablets per 100 mL) to ice-cold RAD-51 cell lysis buffer. The plastic bag containing thin cell paste is taken from –80°C and rapidly crushed using empty 50 mL falcon tube into small pieces. Care is taken not to completely tear the frozen bag. The crushed cell paste is transferred to appropriate volume of RAD-51 cell lysis buffer and suspension is vigorously stirred until the largest cell paste pieces are fully suspended.
-
b.Triton X-100 is added to 0.1% (v/v) final concentration and suspension is kept on the stirrer for further 10 min.
-
c.The suspension is sonicated using a Branson Sonifier 450 (large flat tip, duty cycle 50%, output control 8) for 3×7 min (200 mL RAD-51 cell lysis buffer) or 2×7 min (100 mL RAD-51 cell lysis buffer). The suspension is stirred using a magnetic stirrer for 5–10 min at 4°C in-between sonication cycles.
-
d.The lysate is cleared in an Optima LE-80K Ultracentrifuge (Beckman Coulter) using a Ti45 rotor at 20 000 g for 60 min at 4°C.
-
a.
-
6.Protein purification.
-
a.During cell lysate clarification, 4 mL of Ni-NTA slurry per 50 mL of cell lysate from previous steps (typically 12 mL of Ni-NTA slurry is needed per preparation) is washed twice with RAD-51 cell lysis buffer in 50 mL falcon tube (added up to 50 mL volume) using swing-rotor centrifuge (1000 g, 10 min).
-
b.To reduce non-specific binding of protein to resin, imidazole is added to the supernatant to the final concentration of 25 mM imidazole in the whole supernatant volume. The clarified lysate is then divided equally into 50 mL falcon tubes.
-
c.Next, 4 mL of equilibrated Ni-NTA resin is equally distributed to each falcon tube and incubated with gentle rotation for 1.5 h at 4°C.
-
d.The resin is collected in 30 mL Biorad protein purification column (2 mL of Ni-NTA slurry per 1 column) by flowing through the lysate containing incubated resin. If required, the flowthrough can be collected for future analysis. Each column is washed once with 50 mL RAD-51 cell lysis buffer and once with 50 mL of RAD-51 cell lysis buffer supplemented with 50 mM imidazole. The protein is eluted with 8 mL RAD-51 cell lysis buffer supplemented with 200 mM imidazole by passing through the 8 mL of the buffer twice over each column and once more with 4 mL of the same buffer (12 mL final elution volume per each column). The eluted protein is dialyzed against 4 L RAD-51 Dialysis Buffer (20 mM Tris-HCl pH 8.0, 300 mM KCl, 10% glycerol) for 16 h using 10 kDa MWCO SnakeSkin dialysis tubing (Thermo Scientific).
-
e.Next day, dialyzed sample is spun at maximum g for 10 min to remove precipitated protein.
-
f.The His-SUMO tag is cleaved to yield native RAD-51 by addition of 10 μL His-tagged Ulp1 SUMO protease (10 mg/mL) for 45 min. Immediately after cleavage, sample is spun again at maximum g for 10 min to remove precipitated protein. The sample is incubated with same batch of Ni-NTA agarose after regeneration according to the manufacturer’s instructions for 1.5 h at 4°C. The flowthrough containing untagged RAD-51 is collected and the resin is washed with an additional 3 mL per column of RAD-51 Dialysis Buffer. Fractions are pooled and mixed at 1:1 ratio with RAD-51 Dilution Buffer (20 mM Tris-HCl (pH 8.0), 10% glycerol, 2 mM EDTA, 1 mM DTT).
-
g.The cleaved RAD-51 protein is loaded at 0.5 mL/min speed onto 1 mL MonoQ (5/50 GL) column (GE Healthcare) pre-equilibrated with RAD-51 buffer R-A (20 mM Tris-HCl (pH 8.0), 150 mM KCl, 10% glycerol, 1 mM EDTA, 0.5 mM DTT) using AKTA FPLC system.
-
h.The column is washed with 10 column volumes of RAD-51 buffer R-A.
-
i.The protein is eluted with linear salt gradient (0 – 100 %) in 15 column volume with RAD-51 buffer R-B (20 mM Tris-HCl (pH 8.0), 1000 mM KCl, 10% glycerol, 1 mM EDTA, 0.5 mM DTT). The volume of collected fractions is set to 0.25 mL.
-
j.The fractions corresponding to UV peaks are analyzed by SDS-PAGE. The fractions containing purest protein are pooled and concentrated using a 30 kDa MWCO Amicon Ultra-4 Centrifugal Filter Unit according to manufacturer’s direction. Pre-rinse the unit with water and RAD-51 buffer R-A before use. We suggest making 5× concentrate.
-
k.The protein concentration is determined by Bradford staining and appropriate volumes of aliquots are prepared, and snap frozen in liquid nitrogen. We suggest making 5–10 μL aliquots. This purification procedure yields typically 1–2 mg of protein.
-
a.
Note: After the cleavage with SUMO protease, a significant portion of untagged RAD-51 may still stick to Ni-NTA resin. This is not an issue as RAD-51 yields are generally high enough to obtain sufficient amounts of protein.
Note: RAD-51 forms multimeric species in solution. During concentrating the protein at the final step of purification, may undergo precipitation. To remove the precipitate, protein sample should be spun in a table-top centrifuge at maximum speed for 10 min 4°C.
Preparing oxygen scavenging system
The oxygen scavenging system is important to prevent excessive generation of free oxygen radicals during single-molecule imaging. This increases fluorophore stability and prevents rapid photobleaching of dye-labeled proteins. There are several systems available, we recommend using the PCA/PCD system (Aitken et al., 2008) because it does not affect the solution pH.
-
7.Preparing 124 mM PCA (3,4-Dihydroxybenzoic acid) stock solution.
-
a.Prepare 1 M NaOH.
-
b.Suspend 48.2 mg of PCA in 2 mL of H2O. PCA will not dissolve at this stage.
-
c.Add 0.5 mL of 1 M NaOH to PCA suspension. PCA should dissolve.
-
d.Check pH (using pH paper). It should be in the range of 9–9.5.
-
e.Make 35 μL aliquots and store at −20°C for maximum of 3 months, in a box covered with aluminum foil (to protect from light).
-
a.
-
8.Preparing 10 μM PCD (Protocatechuate 3,4-Dioxygenase) stock solution.
-
a.Determine mass of lyophilized PCD in bottle (usually 8.3 mg).
-
b.Assuming all powder is PCD, determine how many mL of solution will be required to dissolve the PCD to 10 μM or 7 mg/mL (1.186 mL for 8.3 mg). This is the desired total volume of solution.
-
c.Add half of the desired total volume (593 μL) of 2× PCD Buffer (100 mM Tris, pH 7.5, 200 mM NaCl, 200 μM EDTA) to the vial and gently agitate to dissolve the PCD.
-
d.Add an equal volume (593 μL) of anhydrous glycerol to the vial and gently agitate to mix.
-
e.Make 10 μL aliquots of the PCD stock solution. Store at –20°C for maximum of 3 months.
-
a.
CRITICAL: It is important to obtain clear solution of PCA. If pH is out of the proper range, or if PCA is not shielded from light sufficiently, the solution will turn brown. We have observed that not only is PCA prepared in this way inefficient as oxygen scavenger, but it may also interfere with protein binding to DNA. This is indeed the case for RAD-51, where inclusion of degraded PCA inhibits filament assembly and RPA displacement.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli BL21(DE3) | NEB | Cat# C2527I |
| E. coli DH5alpha | NEB | Cat# C2987H |
| E. coli Rosetta(DE3)pLysS | Merck | Cat# 70956-3 |
| Chemicals, peptides, and recombinant proteins | ||
| Ampicillin, Sodium Salt | Merck | Cat# 171254 |
| Kanamycin Sulfate, Streptomyces kanamyceticus | Merck | Cat# 420311 |
| Chloramphenicol | Merck | Cat# 220551 |
| IPTG, Dioxane-Free, High Purity | Merck | Cat# 420322 |
| Sodium hypochlorite | Merck | Cat# XX0637 |
| Sodium thiosulfate | Merck | Cat# 106512 |
| PLURONIC F-127 | Merck | Cat# P2443-250G |
| Albumin from bovine serum | Sigma-Aldrich | Cat# A7030 |
| Mono Q 5/50 GL column | Merck | Cat# GE17-5166-01 |
| HiTrap Heparin HP 1 mL column | Merck | Cat# GE17-0407-01 |
| Ni-NTA Agarose resin | QIAGEN | Cat# 30210 |
| Streptavidin-coated polystyrene particles 0.5% w/v | Spherotech | Cat# SVP-40-5 |
| Lambda DNA | Thermo Fisher | Cat# SD0011 |
| 3,4-Dihydroxybenzoic acid | Merck | Cat# 99-50-3 |
| Protocatechuate 3,4-Dioxygenase | Merck | Cat# P8279-25UN |
| Creatine kinase (CK) | Roche | Cat# 10127566001 |
| Creatine phosphate | Roche | Cat# 10621714001 |
| His6-SUMO protease | Dr. Peter Cherepanov | N/A |
| hRPA-eGFP | This protocol | N/A |
| Untagged RAD-51 | This protocol | N/A |
| Alt-R S. p. Cas9 D10A Nickase | IDT | Cat# 1081062 |
| T4 Polynucleotide Kinase | NEB | Cat# M0201S |
| T4 DNA Ligase | NEB | Cat# M0202S |
| T4 DNA Ligase Reaction Buffer | NEB | Cat# B0202S |
| SYTOX Orange Nucleic Acid Stain | Thermo Fisher | Cat# S11368 |
| Oligonucleotides | ||
| lambda end-cap 1 5’- AGG TCG CCG CCC GGA GTT GAA CG(BT) (BT)T(BT) T(BT)A CGT TCA ACT CC-3’ |
IDT, this study | N/A |
| lambda end-cap 2 5’- GGG CGG CGA CCT CAA GTT GGA CAA (BT)T(BT) T(BT)(BT) TGT CCA ACT TG-3’ |
IDT, this study | N/A |
| tracr RNA (trRNA) 5’-GGACAGCAUAGCAAGU UAAAAUAAGGCUAGUCCGUUAUCAACUUGAAA AAGUGGCACCGAGUCGGUGCUUUUU-3’ |
IDT, this study | N/A |
| crRNA λ2 5’-GUGAUAAGUGGAAUGCCA UGGUUUUAGGAGCUAUGCUGUUUUG-3’ |
IDT, this study | N/A |
| crRNA λ4 5’-CAGATATAGCCTGGTGGTTCG UUUUAGGAGCUAUGCUGUUUUG-3’ |
IDT, this study | N/A |
| crRNA λ5 5’-GGCAAUGCCGAUGGCGAUA GGUUUUAGGAGCUAUGCUGUUUUG-3’ |
IDT, this study | N/A |
| crRNA λ4.2 5’- GCCAUUCUGCUUAUCAGGAA GUUUUAGGAGCUAUGCUGUUUUG -3’ |
IDT, this study | N/A |
| crRNA λ4.1 5’- GGCCAUGUAAGCUGACUUU AGUUUUAGGAGCUAUGCUGUUUUG-3’ |
IDT, this study | N/A |
| crRNA λ4.02 5’- AUUGCCAGGCUUAAAUGAG UGUUUUAGGAGCUAUGCUGUUUUG-3’ |
IDT, this study | N/A |
| Recombinant DNA | ||
| phRPA-eGFP | Dr. Mauro Modesti | N/A |
| Champion pET-SUMO-RAD-51 | This protocol | N/A |
| Software and algorithms | ||
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Fiji | Open source | https://imagej.net/Fiji |
| MATLAB R2018b (9.5.0) | MathWorks | https://uk.mathworks.com |
| Lumicks Pylake | Python package from LUMICKS | https://lumicks-pylake.readthedocs.io/en/latest/index.html# |
| Other | ||
| C-TRAP optical trapping and confocal microscopy setup | Lumicks | N/A |
| ÄKTA pure protein purification system | Cytiva | N/A |
| Optima LE-80K Ultracentrifuge | Beckman Coulter | N/A |
| Branson sonifier 450 | Branson | N/A |
| Econo-Pac Chromatography Columns | Bio-Rad | Cat# 7321010 |
| SnakeSkin Dialysis Tubing, 10 KDa MWCO | Thermo Fisher | Cat# 88243 |
| Amicon Ultra-4 Centrifugal Filter Unit | Merck | Cat# UFC803024 |
Step-by-step method details
Preparing nicked lambda dsDNA with closed biotinylated hairpin handles
See Figure 1A for overview. This part of the protocol describes quick method for generation of site-specific DNA nicks in λ DNA by Cas9 D10A nickase. These nicked substrates are subsequently melted to yield corresponding gapped substrates. Position of the nick is determined by the choice of the guide RNA sequence. Guide RNA consists of universal tracr-RNA scaffold and customizable crRNA component. We utilized previously characterized crRNAs termed crRNA λ2, crRNA λ4 and crRNA λ5 to generate dsDNA constructs containing nicks with 5 kilonucleotides (knt) and 18 knt distance. The distance between the two nicks is dictated by the combination of the guides. These are further referred in the text as λ4/5 (for 5 knt distance) or λ2/5 (for 18 knt distance) gDNA precursors – according to the combination of guide RNAs used to generate them. Further to our previous work, we provide three additional crRNAs (crRNA λ4.02, crRNA λ4.1, crRNA λ4.2) to generate λ gDNA precursors with 0.2 (λ4/4.02 gDNA precursor), 1 (λ4/4.1 gDNA precursor) and 2 (λ4/4.2 gDNA precursor) knt distance between the nicks.
-
1.λ DNA and the end-cap phosphorylation.
-
a.Phosphorylate λ DNA and adapter oligonucleotide end-caps:
-
i.For phosphorylation of 14 nM λ DNA , pipette 41.6 μL of λ DNA (300 μg/mL), 8.4 μL milliQ H2O, 5.6 μL 10×T4 DNA ligase buffer and 0.5 μL polynucleotide kinase (final concentration of 250 U/mL) into 1.5 mL Eppendorf tube.
-
ii.For phosphorylation of 10 μM end-cap 1, pipette 3.2 μL of end-cap 1 (62 μM, PAGE-purified), 14.3 μL milliQ H2O, 2 μL 10×T4 DNA ligase buffer and 0.5 μL polynucleotide kinase (final concentration of 1000 U/mL).
-
iii.For phosphorylation of 10 μM end-cap 2, pipette 2.7 μL of end-cap 2 (73 μM, PAGE-purified), 14.8 μL milliQ H2O, 2 μL 10×T4 DNA ligase buffer and 0.5 μL polynucleotide kinase (final concentration of 1000 U/mL).
-
i.
-
b.Mix very gently with pipette tip. Use cut pipette tips and pipette up-and-down 1–2 times. Avoid excessive up-and-down pipetting. Incubate these three reactions for 1 h at 37°C.
-
a.
-
2.End-cap annealing and ligation (King et al., 2019; Paik and Perkins, 2011).
-
a.Next, incubate the phosphorylated end-cap 1 with phosphorylated λ DNA. Pipette 50 μL of 10× T4 DNA ligase buffer, 400 μL of milliQ H2O, 56 μL of 14 nM phosphorylated λ DNA and 1 μL of 10 μM phosphorylated end-cap 1.
-
b.To ensure that the end-cap annealed in its minimum energy configuration prior to the ligation, heat the above mixture to 80°C for 5 min and subsequently cool rapidly on ice.
-
c.Add T4 DNA ligase (0.5 μL, 400 units) and incubate at 37°C for 1 h (or 25°C for 2 h, or 16°C for 12 h).
-
d.Subsequently, add 10 μL of 10 μM phosphorylated end-cap 2 to the above solution. Heat the mixture to 80°C for 5 min, and cool rapidly on ice. Add T4 DNA ligase enzyme (4 μL, 200 units) and leave to react at 37°C for 1 h, 25°C for 2 h (or 16°C for 12 h).
-
e.Inactivate the ligase at 65°C for 20 min.
-
a.
-
3.Cas9 D10A-gRNA RNP-complex preparation.
-
a.Prepare guide RNA (gRNA) mixes:
-
i.For crRNA λ2:tracr RNA (trRNA) mix pipette 2 μL of 5 μM crRNA λ2, 1 μL of 10 μM trRNA, 4 μL 1× T50 (10 mM Tris-HCl, pH 8.0, 50 mM NaCl), 1 μL milliQ H2O.
-
ii.For crRNA λ5:tracr RNA mix pipette 2 μL of 5 μM crRNA λ5, 1 μL of 10 μM trRNA, 4 μL 1× T50, 1 μL milliQ H2O.
-
iii.For crRNA λ4:tracr RNA mix pipette 2 μL of 5 μM crRNA λ4, 1 μL of 10 μM trRNA, 4 μL 1× T50, 1 μL milliQ H2O.
-
iv.For crRNA λ4.2:tracr RNA mix pipette 2 μL of 5 μM crRNA λ4.2, 1 μL of 10 μM trRNA, 4 μL 1× T50, 1 μL milliQ H2O.
-
v.For crRNA λ4.1:tracr RNA mix pipette 2 μL of 5 μM crRNA λ4.1, 1 μL of 10 μM trRNA, 4 μL 1× T50, 1 μL milliQ H2O.
-
vi.For crRNA λ4.02:tracr RNA mix pipette 2 μL of 5 μM crRNA λ4.02, 1 μL of 10 μM trRNA, 4 μL 1× T50, 1 μL milliQ H2O.
-
i.
-
b.Heat the duplex at 95°C for 2 min and cool it down at 25°C afterward.
-
c.Create the RNP complex:
-
i.Dilute Alt-R S.p. D10A nickase to 5 μM in PBS (0.8 μL of enzyme + 9.2 μL 1×PBS, enzyme stock is 62 μM).
-
ii.Add 2 μL of the diluted enzyme to each of the RNA mixes and incubate at 25°C for 5 min (final Cas9 D10A concentration is 1 μM, final concentration of individual guide RNAs is 1 μM).
-
i.
-
a.
-
4.λ DNA nicking.
-
a.To generate λ2/5 gDNA precursor, add 175 μL of λ DNA from Step 2e, 20 μL 10×Cas9 digestion buffer (200 mM HEPES, pH 7.5, 1 M NaCl, 50 mM MgCl2, 1 mM EDTA), 2 μL of 10 nM Cas9 D10A-crRNA λ2-tracr RNA complex, 2 μL of 10 nM Cas9 D10A-crRNA λ5-tracr RNA complex. Incubate at 37°C for 1 h.
-
b.To generate λ4/5 gDNA precursor, add 175 μL of λ DNA from Step 2e, 20 μL 10×Cas9 digestion buffer, 2 μL of 10 nM Cas9 D10A-crRNA λ4-tracr RNA complex, 2 μL of 10 nM Cas9 D10A-crRNA λ5-tracr RNA complex. Incubate at 37°C for 1 h.
-
c.To generate λ4.2/4 gDNA precursor, add 175 μL of λ DNA from Step 2e, 20 μL 10×Cas9 digestion buffer, 2 μL of 10 nM Cas9 D10A-crRNA λ4.2-tracr RNA complex, 2 μL of 10 nM Cas9 D10A-crRNA λ4-tracr RNA complex. Incubate at 37°C for 1 h.
-
d.To generate λ4.1/4 gDNA precursor, add 175 μL of λ DNA from Step 2e, 20 μL 10×Cas9 digestion buffer, 2 μL of 10 nM Cas9 D10A-crRNA λ4.1-tracr RNA complex, 2 μL of 10 nM Cas9 D10A-crRNA λ4-tracr RNA complex. Incubate at 37°C for 1 h.
-
e.To generate λ4.02/4 gDNA precursor, add 175 μL of λ DNA from Step 2e, 20 μL 10×Cas9 digestion buffer, 2 μL of 10 nM Cas9 D10A-crRNA λ4.02-tracr RNA complex, 2 μL of 10 nM Cas9 D10A-crRNA λ4-tracr RNA complex. Incubate at 37°C for 1 h.
-
f.To terminate the reaction, add 500 mM EDTA (pH 8.0) to a final concentration of 10 mM. Avoid pipetting up-and-down. Store the reaction at 4°C.
-
a.
Note: In λ DNA, GC content is distributed asymmetrically, with one half of λ DNA sequence being more GC-rich than the other. Given ssDNA gaps are generated in the next step by force-induced DNA melting, it should be taken into account that DNA in GC-rich regions of λ DNA, will be more difficult to melt. Although this should not be an issue for short (up to 5 knt) gaps, generation of larger gaps can be significantly more difficult. We suggest choosing position of the gap carefully when designing RNA guides for nicking. If there are no specific underlying sequence requirements, we suggest designing gaps in the GC-poor half of λ DNA first. Troubleshooting 1
Figure 1.

Strategy to generate gapped DNA molecules
(A) A schematic of protocol designed to generate gapped λ DNA (λ gDNA) precursor substrate. Biotinylated end-cap hairpin oligos are annealed and ligated to 48.5 kb long λ DNA. Subsequently, nicking by S.p. Cas9 D10A at desired position is performed. The schematic illustrates two pairs of guide RNAs (λ4 crRNA and λ5 crRNA; λ2 crRNA and λ5 crRNA).
(B) Schematic of the experimental C-trap set-up used for in situ λ gDNA generation by force-induced DNA melting. RPA-eGFP and Sytox Orange staining are employed to image the gap.
In situ DNA melting and generation of gapped lambda DNA substrates
This section describes in situ generation of gapped DNA substrates using a commercially available combination of microfluidics, optical tweezers and fluorescence microscopy setup (C-TRAP, Lumicks). See Figure 1B for overview.
-
5.5-channel flow cell preparation.
-
a.Flush channels with 0.5 mL of 5% bleach solution at 1.5 bar.
-
b.Flush channels with 1.0 mL of milliQ H2O at 1.5 bar.
-
c.Flush channels with 0.5 mL of 10 mM sodium thiosulfate at 1.5 bar.
-
d.Flush channels with 0.5 mL of filtered sterile PBS at 1.5 bar.
-
e.Flush channels with 0.5 mL of 0.5% Pluronics F127 (w/v) in filtered sterile PBS at 1.5 bar.
-
f.Flush channels with 0.5 mL of 0.1% BSA (w/v) in filtered sterile PBS at 1.5 bar.
-
a.
-
6.Setting up single-molecule imaging system.
-
a.Add water on the objective. Raise objective to touch the microfluidics chip. Start trapping laser at 0.31 pN/nm trap stiffness (for 4.5-μm beads). Find appropriate z position for the trapping laser in the middle of the microfluidics channel. Add oil on top of microfluidics chip. Lower the condenser to close position, where reflection image is fully visible on the camera. Inspect the reflection image (‘the moon’) to ensure absence of bubbles in immersion oil and proper condenser position. Lower laser power of the trapping laser to reach 0.14 pN/nm trap stiffness.
-
b.Add 4.5-μm SPHERO Streptavidin Coated polystyrene particles in filtered sterile PBS at 0.005% w/v to channel 1, 0.2% (v/v) λ gDNA precursor constructs prepared in Step 4 of the protocol in filtered sterile PBS to channel 2, filtered sterile PBS to channel 3, 5 nM RPA-eGFP in buffer 0.5×NTM (1×NTM buffer consists of 100 mM NaCl, 50 mM Tris-HCl pH 7.5, 1 mM MgCl2) supplemented with 0.2 mg/mL BSA and the oxygen scavenger system with 5 mM protocatechuic acid (PCA), 100 nM protocatechuic dioxygenase (PCD) to channel 4 and 50 nM SYTOX Orange in filtered sterile PBS to channel 5. Flow enough liquid through the system to observe beads in channel 1 in brightfield.
-
c.Capture a pair of beads in channel 1 and move to channel 2. Perform force calibration according to manufacturer instruction at 0.14 pN/nm trap stiffness.
-
d.Move beads apart to sufficient distance (we suggest 25 μm between the bead edges). Adjust z position in Trap XY settings to get both beads into the focus. Turn on single-photon counter module. Turn on blue laser to 5% laser power. Turn on green laser to 10 % laser power. Set pixel size to 100 nm. Perform a single 2D scan and adjust scanning area to include both beads. Make sure that reflection in the middle of the beads in blue channel is sharp and bright. This indicates proper z position of Trap XY.
-
a.
-
7.λ gDNA precursor capture, force-induced melting and imaging. See Figure 1B for overview.
-
a.Trap two evenly shaped (bead match score > 0.97) streptavidin-coated beads in channel 1.
-
b.Move traps to channel 2, set “ping-pong” option that automatically moves the traps between 5 (‘close’) and 17 μm (‘extended’) distance marks at 6.0 μm/s speed with 1–2 s dwell time at 5 μm position. Set force to zero at 5 μm `close’ distance between the bead edges. Keep pressure of 0.12–0.18 bar to maintain buffer flow. “Ping-pong” regime allows to determine whether dsDNA has been tethered between the beads as evident by the sudden increase of force when the beads are moving apart.
-
c.Monitor force-extension curve. Use built-in worm-like chain models to set an example curve for single λ dsDNA molecule. Small deviations from the model may occur, if the flow is present, but your experimentally obtained force data should roughly follow this model. Once a single molecule of λ gDNA precursor is captured – as judged by force-extension plot, turn off the buffer flow, close all the channels and move the traps to channel 3. It is possible to capture multiple DNA molecules at once. If this occurs, repeat the process by picking up new beads from channel 1 or extend the bead distance beyond 23 μm to break one or more extra molecules captured between the beads.
-
e.In channel 3, move the beads to 'close’ distance, set force to zero and slowly extend the DNA molecule either to 23–25 μm distance between the bead edges (or 65 pN force and above) to generate short gaps as in the case of λ4/5 gDNA and smaller precursors, or 27–29 μm distance (or 100 pN force and above) to generate longer gaps as in the case of λ2/5 gDNA precursor. After 30 – 60 s of holding λ gDNA precursor at high force move the beads to 5 to 10 μm distance.
-
f.Wait for 1 min at low force and then slowly re-extend captured construct to 15 pN force. Monitor force-extension curve (Figure 2A). Repeat the process one more time to ensure that gap melting was complete. Compare recorded force-extension curve to worm-like chain model of λ dsDNA with contour length corresponding to individual λ gDNA constructs (Figure 2B). Contour length of λ gDNA constructs can be used to calculate experimentally obtained gap length and compare it to the expected gap length (Figure 2C). Troubleshooting 2
-
g.Move the traps to channel 4. Then bring in the beads to ‘close’ position. Set force to zero. Activate force clamp and set the value to 5 pN or 15 pN. Turn off the force clamp. Take 2D image of your λ gDNA and beads. The RPA-eGFP signal covering expected segment (ssDNA region) of λ gDNA should be present at the expected genomic position (Figure 3). No additional nicks or gaps should be present.
-
h.To stain for dsDNA content, move the beads to channel 5 containing SYTOX Orange and take another 2D scan.
-
a.
Note: During the re-annealing of gapped DNA constructs various levels of hysteresis in the force-distance curves can be observed (Figure 4). The magnitude of which depends on the following: a) duration of over-stretching: the longer the DNA construct is held at 80 pN, the more hysteresis is observable. Above 65 pN dsDNA gradually melts. The more melted/frayed ssDNA, the longer it takes to re-anneal it to dsDNA upon relaxing the force. b) similarly, the higher the force at which we hold the over-stretched tether, the more hysteresis is observed. c) the speed at which the trap is moved (pulling/relaxation rate). d) the gap size: the longer the size of the gap, the less stable are the corresponding dsDNA segments and the longer the re-annealing might take.
Figure 2.

Gap length validation for individual constructs using force measurement
(A). Force-extension curves of λ dsDNA, λ ssDNA and different λ gDNA constructs. Gap sizes are indicated.
(B) Contour length of λ dsDNA and different λ gDNA constructs calculated from worm-like chain (WLC) fits to individual force-extension curves from 2A. Error bars indicate S.D.
(C) Comparison of expected gap length to gap length calculated from contour lengths in 2B. Error bars indicate S.D.
Figure 3.

Gap length validation for individual constructs using fluorescence
Left. Schematics of the λ gDNA substrates generated in this study. Right. Representative images of individual asymmetrically positioned RPA-eGFP (5 nM RPA-eGFP) coated ssDNA gaps within λ DNA held at 5 pN force. Images display RPA-bound ssDNA (middle panels), and RPA-bound ssDNA with dsDNA counter-strained with 50 nM SYTOX Orange (right panels). Arrow indicates position of λ4 crRNA cut site indicating 5’ end of 0.2, 1, 2 and 5 knt long gaps for reference.
Figure 4.

Examples of forward and reverse force-extension curves of a gapped DNA construct
Different examples of forward and reverse force-extension curves of λ4/5 gDNA constructs showing hysteresis during re-annealing of the DNA. Arrows indicate the direction of the pulling.
Optional: Force-induced DNA melting can be performed in the presence of RPA-eGFP (channel 4) or even in channel 2 containing λ gDNA precursor. In case of DNA melting in the presence of RPA, DNA can be melted faster, but upon returning to the low-force position, longer waiting time is required to fully anneal frayed ssDNA ends on the side of the gap. In case where DNA is melted in the same channel as where λ gDNA precursor is being captured, caution should be taken not to move beads too close (7 μm distance or less) after DNA melting to avoid capturing second DNA molecule. Troubleshooting 2
Using gDNA substrates to monitor molecular dynamics at a single-molecule level
Here we describe an example of λ gDNA construct usage to determine growth polarity of RAD-51 filaments. RAD-51 is a metazoan recombinase that forms helical nucleoprotein filaments on resected RPA-coated ssDNA ends to facilitate DNA double-strand break repair via homologous recombination. Its bacterial homolog, RecA, has been shown to grow on ssDNA bidirectionally with 2-fold kinetic preference in a 5’ to 3’ direction along the ssDNA backbone (Bell et al., 2012). To determine the growth polarity of nematode RAD-51 filaments (Belan et al., 2021), λ2/5 gDNA substrate will be used as described later. It should be noted, however, that gDNA substrates described in this protocol have a wide variety of usage for different enzymatic reactions including helicase unwinding, 3’ end extension by polymerases, nucleolytic processing, etc.
-
8.
The flow cell is washed and the microscope aligned as described in the previous section. However, different buffers are added to individual channels: add 4.5-μm SPHERO Streptavidin coated polystyrene particles in filtered sterile PBS at 0.005% w/v to channel 1, 0.2% (v/v) λ2/5 gDNA precursor construct prepared previously in filtered sterile PBS to channel 2, 5 nM RPA-eGFP in buffer 0.5×NTM (25 mM Tris-HCl pH 7.5, 50 mM NaCl, 0.5 mM MgCl2) supplemented with 0.2 mg/mL BSA and the oxygen scavenger system with 5 mM PCA, 100 nM PCD to channel 3 and 500 nM RAD-51 in buffer 1×NTM (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1.0 mM MgCl2) supplemented with 0.2 mg/mL BSA, 1 mM ATP, ATP regeneration system (20 mM creatine phosphate and 20 μg/mL creatine kinase) and the oxygen scavenger system with 5 mM PCA, 100 nM PCD to channel 4.
-
9.
Perform force calibration, set up confocal imaging, capture beads and tether a single piece of λ gDNA precursor as described previously.
-
10.
Move the traps to channel 3, move the beads to ‘close’ distance, set force to zero and slowly extend the DNA molecule to 27–29 μm distance (or 100 pN force and above) to generate longer gaps as in the case of λ2/5 gDNA precursor. After 30 – 60 s of holding λ gDNA precursor at high force move the beads to 5 to 10 μm distance. Wait for 1–5 min for DNA strands to re-anneal as RPA-eGFP slows it down.
-
11.
Slowly re-extend captured construct to 15 pN force. Monitor force-extension curve. Compare recorded force-extension curve to worm-like chain model of λ2/5 gDNA construct. Take 2D image of your λ2/5 gDNA and beads. The RPA-eGFP signal covering expected length of λ gDNA should be present at the expected genomic position. No additional nicks or gaps should be present. Keep λ gDNA to length corresponding to 15 pN force.
-
12.
Set-up kymograph imaging mode by scanning a single line of pixels through the center of trapped beads and DNA. Set line-time to 5 s and start imaging. Immediately move traps to channel 4 (Figure 5A).
-
13.
Continue building kymograph of RPA-eGFP displacement. Force can be monitored at the same time as fluorescence. RAD-51 displacement should be evident as the loss of blue fluorescence signal. In the absence of RAD-51, minimal loss of RPA-eGFP signal should be present for at least 20 min (Belan et al., 2021) as RPA binds very tightly to ssDNA (Ma et al., 2017). If rapid fluorescence loss is observed, this is likely due to extensive photobleaching - fresh batch of PCA/PCD should be prepared. Troubleshooting 3 and 4
Optional: To better resolve individual growing RAD-51 filaments, 10 nM free RPA-eGFP can be included with RAD-51 in channel 4. This will supress RAD-51 nucleation on ssDNA, resulting in sparser filaments that are better resolved. However, fewer filaments per single kymograph will be collected. This will prolong the time needed for sample collection.
Figure 5.

Monitoring bidirectional RAD-51 filament growth as an example of gapped DNA usage
(A) Kymograph showing the displacement of RPA-eGFP bound to λ2/5 gDNA by 500 nM RAD-51 in the presence of ATP.
(B) Examples of individual growing RAD-51 filaments (dark). Growth rate was measured as a slope of the border of RPA-eGFP displaced signal (illustrated by dashed white line in the right panel).
(C) Histogram of individual growth rates for indicated directions (n = 32 total filaments analyzed). 5’ to 3’ rates are shown as negative numbers, while 3’ to 5’ are indicated as positive. Black lines represent lognormal fit. Geometric mean for 5’ to 3’ direction: 9.7 ± 1.7 nm/min. Geometric mean for 3’ to 5’ direction: 7.6 ± 1.7 nm/min.
Image and force data analysis
This section briefly mentions a few options for imaging and force data analysis. Generally, raw kymographs are accessed through Pylake package and then multiple parameters can be exported and further processed in different software. Although the specific data analysis method heavily depends on the nature of the experiment, we provide several analysis pipelines for common readouts.
-
14.
The .h5 files containing kymographs, force data or 2D scans are accessed through Lumicks Pylake package. Imaging data are exported as TIFFs.
-
15.
The exported kymographs can be analyzed using Fiji by plotting integrated fluorescence intensity in individual channels. This can be done for specific genomic positions of DNA along x axis of the kymograph to provide time-resolved information.
-
16.
The force data can be exported through Lumicks Pylake package. If longer observation windows are employed (more than 5 min), we suggest to significantly down-sample the force data. For example, to 3 Hz.
-
17.
To analyze genomic positions of DNA-bound fluorescent molecules, previously described approaches can be used (Newton et al., 2019). Similar previously-described approaches can be used to track moving particles and analyze diffusion rates along gapped DNA molecules (Graneli et al., 2006).
-
18.
To determine the number of fluorophores within a single cluster of fluorescent particles bound to gDNA, a single-step photobleaching analysis can be employed as described previously (Autour et al., 2018; Belan et al., 2021). To find individual steps, a step-finding algorithm in Matlab is employed.
Expected outcomes
This protocol should yield λ gDNA precursors in good quantity (~50 μL of 1.35 nM precursor) and good quality (low frequency of additional nicks observed – roughly 1 in 10 molecules melted). The gaps within λ DNA molecules should be that of the expected size and length. The asymmetric position of the gap should be apparent as it is for λ2/5 gDNA. In case of λ2/5 gDNA, 3’ ss-dsDNA junction is found on the shorter arm, while 5’ ss-dsDNA junction is found on the longer arm. Upon movement to RAD-51 containing channel 4 overall decrease of RPA-eGFP fluorescence should be observed (Figure 5A). The RAD-51 filament formation should be evident from the emergence of RPA-eGFP-free or 'dark' zones over time in the kymograph (Figure 5B). The slopes of RPA-eGFP-containing ‘blue’ and RPA-eGFP-free ‘dark’ zone borders in the kymograph (Figure 5B) can be manually measured for individual well-resolved filaments and converted to growth rates. The distribution of measured growth rates towards either 5’ or 3’ direction can be plotted as histogram to assess growth symmetry of RAD-51 filaments on ssDNA (Figure 5C). Typically, 7–14 RAD-51 growing filaments can be analyzed for a single λ2/5 gDNA molecule. The gDNA substrates described in this protocol have a wide variety of applications for different enzymatic reactions including helicase unwinding, 3’ end extension by polymerases, nucleolytic processing, etc.
Limitations
Among the limitations of the protocol is the length of the gap within λ DNA construct. Although we were able to generate 18 knt-long gaps, it should be noted that increasing gap size increases the difficulty of generating such a gap using force-induced melting as the DNA molecules are more likely to snap when held over extended periods of time at higher force regimes. On the other hand, generation of short gaps (less than 200 nt) might face problems stemming from errors of gap length calculation. In such cases, it might be difficult to accurately assess whether these gaps are that of the expected length. Thus, short gaps should be engineered into shorter DNA constructs.
The other general limitation is the resolution of the system. Given the diffraction limit for confocal microscopy, growth rates of RAD-51 proteins moving on short ssDNA gaps (less than 5 knt which equates to ≈ 160 nm) may not be determined accurately. This limitation could be overcome by the use of a C-trap with STED imaging functionality. Furthermore, long ssDNA gaps (such as 18 knt in case of λ2/5 gDNA) have to be held at higher forces (10–15 pN) to keep the molecule in focus for confocal imaging due to the high portion of flexible ssDNA. The fluorescence imaging at low force regimes (1–5 pN) may be difficult using these constructs.
Another limitation is the use of SYTOX to visualize dsDNA as this may interfere with biological processes, similarly as RPA-eGFP can compete with protein of interest binding to ssDNA or potentially inhibit its enzymatic activity. If this is of concern, RPA-eGFP can be omitted and fluorescent labeled protein of interest can be visualized binding to bare (dark) ssDNA. It should be mentioned that any fluorescent dyes can be omitted altogether when processes such as exonucleolytic processing of DNA, DNA unwinding by a helicase or 3’ DNA end extension by DNA polymerase are investigated. During these enzymatic reactions, content of ssDNA in the gap changes and distance between the beads at a constant force can be measured and subsequently used to calculate rate of ssDNA content change within the construct.
Finally, we note that fairly high sample volumes have to be flown through the protein channel during bead capture and DNA tethering. The protein consumption can be slowed down by keeping protein-containing channels closed during bead capture, DNA tethering and DNA melting. However, this may result in buffers from protein-free channels being flown/diffusing into the protein channels, if bead capture, DNA tethering and melting steps take long, or high flow rates are employed, thus lowering effective protein concentration in the protein channel. In our experience with 5 knt gap, 250 μL of protein sample added to the syringe will be sufficient to visualize 3–4 DNA molecules. Thus, if the protein quantity is extremely low, a different approach might be needed, although this is rarely the case in our experience.
Troubleshooting
Problem 1
It is possible to encounter difficulties to melt gDNA precursors containing long gaps. It especially becomes more evident when the gap is partially present at GC-rich half of λ DNA. The complete re-annealing after incomplete melting may occur frequently forcing the researcher to restart the melting process from fully re-annealed dsDNA (step 7f).
Potential solution
To help melting these λ gDNA precursors, we suggest adding low concentrations of RPA (5 nM or less) to the melting channel. To further help the melting, salt concentration can be further decreased. Many studies use negligible amount of salt during DNA melting and some even use no salt at all. Previous work (Candelli et al., 2013) has explored this in great detail suggesting that dropping the salt from 50 mM NaCl to 5 mM leads to roughly double melting efficiency of λ DNA. Application of buffer flow can also aid the melting process (Wasserman et al., 2019). If not required, divalent cations can be omitted to further help the force-induced melting of DNA. As noted previously, gaps at GC-rich regions of λ DNA are generally more difficult to melt, however salt exclusion (10 mM and less) in the melting buffer with applied flow could significantly improve the melting of these regions. The higher force regimes (up to 150 pN) can also be utilized although probability of DNA snapping at these forces is also higher. More information on force-induced melting of DNA gaps from nicks together with formation of DNA hairpins near ds-ssDNA junction can also be found in (Okoniewski et al., 2017).
Problem 2
Due to high levels of oxygen radicals, gDNA precursors may break frequently if the amount of ssDNA content is high. This is particularly an issue for monitoring long-lived intermediates or slow reactions (step 7f).
Potential solution
To increase the lifetime of gDNA held between the traps, several adjustments can be made. Larger beads produce less radicals on their surface and hence their usage reduces the likelihood of DNA breakage. Similarly, the inclusion of an oxygen-scavenging system in the DNA channel can be beneficial. Using the lowest possible trapping laser power is also advisable. Finally, if the design allows, ssDNA gaps can be placed more centrally, further from the biotinylated end-caps that are in the contact with streptavidin-coated beads.
Problem 3
The failure of RAD-51 filaments formation (step 13).
Potential solution
If the reaction also contains free RPA-eGFP, remove it or add lower amounts to rule out prep-to-prep variations. Similarly, the optimal RAD-51 concentration where efficient assembly occurs should be determined empirically as variations between protein preparations may occur. If no assembly is observed even when high (1000 nM and more) RAD-51 concentrations are employed, PCA/PCD oxygen scavenger system should be checked. We suggest performing a test reaction where no oxygen scavenger system is used. Care should be taken not to use degraded PCA which is indicated by yellow/brown color of PCA solution. Fresh PCA should be prepared if this turns out to be an issue. If very inefficient filament assembly is observed on first captured DNA molecules, but gets slightly better over time, we suggest passivating the channel with 1 mL of 0.5% BSA in PBS, then repeating assembly experiments. Finally, the activity of RAD-51 preps should be always checked by bulk assays such as EMSA or D-loop formation, as RAD-51 tends to precipitate during final steps of purification, which reduces the concentration of active species.
Problem 4
RAD-51 filaments are too sparse or too dense to efficiently analyze growth rates (13).
Potential solution
We suggest adding RAD-51 in increasing concentrations to channel 4 (e.g., from 200 to 2000 nM protein) and performing a few assembly experiments for each concentration. The optimal concentration of RAD-51 should yield frequent nucleation events, but sparse enough to grow most filaments to at least 0.5–1.0 μm in length. In conditions where nucleation rate is much higher than growth rate, RPA-eGFP can can be added to channel 4 in addition to RAD-51 where efficient assembly is observed. We suggest starting from 1 nM RPA-eGFP and adding up to 20 nM RPA-eGFP for 500 nM RAD-51 to find optimal nucleation suppression point, where formation of well-resolved long filaments can be observed. Alternatively, higher salt can be employed as in the case for RecA growing on dsDNA (Galletto et al., 2006).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David S. Rueda (david.rueda@imperial.ac.uk).
Materials availability
Plasmids, recombinant proteins, DNA substrates and newly generated nematode strains are available without restriction upon requests, which should be directed to the lead contact, David S. Rueda (david.rueda@imperial.ac.uk) or Simon J Boulton (simon.boulton@crick.ac.uk).
Data and code availability
Custom-written data analysis scripts are available upon request from the lead contact, David S. Rueda (david.rueda@imperial.ac.uk).
Acknowledgments
We thank members of Boulton and Rueda labs for discussion and critical reading of the manuscript. We thank Dr. Mauro Modesti for RPA-eGFP expression plasmid. The work in the Boulton Lab is supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC0010048), the UK Medical Research Council (FC0010048), and the Wellcome Trust (FC0010048). S.J.B. also holds European Research Council (ERC) Advanced Investigator (TelMetab) and Wellcome Trust Senior Investigator grants. S.J.B. and D.S.R. are also funded by a Wellcome Trust Collaborative Grant (P67153). The Single-Molecule Imaging Group is also funded by a core grant of the MRC-London Institute of Medical Sciences (UKRI MC-A658-5TY10) and a BBSRC CASE studentship (to M.D.N.).
Author contributions
S.J.B., O.B., and D.S.R. conceived the study. O.B. and R.A. expressed and purified the proteins; O.B., G.M., and M.D.N. performed DNA construct preparation; O.B. and G.M. performed single-molecule imaging and force detection experiments; O.B. and G.M. analyzed the data; O.B., A.K., D.S.R., and S.J.B. wrote the manuscript.
Declaration of interests
S.J.B. is also scientific co-founder and VP Science Strategy at Artios Pharma Ltd., Babraham Research Campus, Cambridge, UK.
Contributor Information
Ondrej Belan, Email: ondrej.belan@crick.ac.uk.
Simon J. Boulton, Email: simon.boulton@crick.ac.uk.
David S. Rueda, Email: david.rueda@imperial.ac.uk.
References
- Aitken C.E., Marshall R.A., Puglisi J.D. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autour A., C Y Jeng S., D Cawte A., Abdolahzadeh A., Galli A., Panchapakesan S.S.S., Rueda D., Ryckelynck M., Unrau P.J. Fluorogenic RNA Mango aptamers for imaging small non-coding RNAs in mammalian cells. Nat. Commun. 2018;9:656. doi: 10.1038/s41467-018-02993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belan O., Barroso C., Kaczmarczyk A., Anand R., Federico S., O'Reilly N., Newton M.D., Maeots E., Enchev R.I., Martinez-Perez E. Single-molecule analysis reveals cooperative stimulation of Rad51 filament nucleation and growth by mediator proteins. Mol. Cell. 2021;81:1058–1073.e7. doi: 10.1016/j.molcel.2020.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell J.C., Plank J.L., Dombrowski C.C., Kowalczykowski S.C. Direct imaging of RecA nucleation and growth on single molecules of SSB-coated ssDNA. Nature. 2012;491:274–278. doi: 10.1038/nature11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelli A., Hoekstra T.P., Farge G., Gross P., Peterman E.J., Wuite G.J. A toolbox for generating single-stranded DNA in optical tweezers experiments. Biopolymers. 2013;99:611–620. doi: 10.1002/bip.22225. [DOI] [PubMed] [Google Scholar]
- Galletto R., Amitani I., Baskin R.J., Kowalczykowski S.C. Direct observation of individual RecA filaments assembling on single DNA molecules. Nature. 2006;443:875–878. doi: 10.1038/nature05197. [DOI] [PubMed] [Google Scholar]
- Graneli A., Yeykal C.C., Robertson R.B., Greene E.C. Long-distance lateral diffusion of human Rad51 on double-stranded DNA. Proc. Natl. Acad. Sci. U S A. 2006;103:1221–1226. doi: 10.1073/pnas.0508366103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King G.A., Burla F., Peterman E.J.G., Wuite G.J.L. Supercoiling DNA optically. Proc. Natl. Acad. Sci. U S A. 2019;116:26534–26539. doi: 10.1073/pnas.1908826116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.J., Gibb B., Kwon Y., Sung P., Greene E.C. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017;45:749–761. doi: 10.1093/nar/gkw1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modesti M. Fluorescent Labeling of Proteins. Methods Mol. Biol. 2018;1665:115–134. doi: 10.1007/978-1-4939-7271-5_6. [DOI] [PubMed] [Google Scholar]
- Newton M.D., Taylor B.J., Driessen R.P.C., Roos L., Cvetesic N., Allyjaun S., Lenhard B., Cuomo M.E., Rueda D.S. DNA stretching induces Cas9 off-target activity. Nat. Struct. Mol. Biol. 2019;26:185–192. doi: 10.1038/s41594-019-0188-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoniewski S.R., Uyetake L., Perkins T.T. Force-activated DNA substrates for probing individual proteins interacting with single-stranded DNA. Nucleic Acids Res. 2017;45:10775–10782. doi: 10.1093/nar/gkx761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik D.H., Perkins T.T. Overstretching DNA at 65 pN does not require peeling from free ends or nicks. J. Am. Chem. Soc. 2011;133:3219–3221. doi: 10.1021/ja108952v. [DOI] [PubMed] [Google Scholar]
- Taylor M.R.G., Spirek M., Chaurasiya K.R., Ward J.D., Carzaniga R., Yu X., Egelman E.H., Collinson L.M., Rueda D., Krejci L. Rad51 paralogs remodel pre-synaptic Rad51 filaments to stimulate homologous recombination. Cell. 2015;162:271–286. doi: 10.1016/j.cell.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman M.R., Schauer G.D., O'Donnell M.E., Liu S. Replication fork activation is enabled by a single-stranded DNA gate in CMG helicase. Cell. 2019;178:600–611.e16. doi: 10.1016/j.cell.2019.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Custom-written data analysis scripts are available upon request from the lead contact, David S. Rueda (david.rueda@imperial.ac.uk).