

Abstract
Eukaryotic protein phosphorylation modulates nearly every major biological process. Phosphorylation regulates protein activity, mediates cellular signal transduction, and manipulates cellular structure. Consequently, the dysregulation of kinase and phosphatase pathways has been linked to a multitude of diseases. Mass spectrometry-based proteomic techniques are increasingly used for the global interrogation of perturbations in phosphorylation-based cellular signaling. Strategies for studying phosphoproteomes require high-specificity enrichment, sensitive detection, and accurate localization of phosphorylation sites with advanced LC-MS/MS techniques and downstream informatics. Sample multiplexing with isobaric tags has also been integral to recent advancements in throughput and sensitivity for phosphoproteomic studies. Each of these facets of phosphoproteomics analysis present distinct challenges and thus opportunities for improvement and innovation. Here, we review current methodologies, explore persistent challenges, and discuss the outlook for isobaric tag-based quantitative phosphoproteomic analysis.
Keywords: automation, high-throughput, tandem-mass tag
1 |. INTRODUCTION
Protein phosphorylation is a post-translational modification (PTM) of central importance in cellular signaling as it is essential to numerous cellular functions. Protein phosphorylation serves as a primary mechanism of cellular signal transduction and modulates pathways, such as cell division, apoptosis, response to extracellular signals and growth factor stimulation, as a reversible and rapid means to transduce signals and modulate protein activity. Dysregulation of phosphorylation-associated signaling cascades has been implicated in many diseases including numerous cancers [1], diabetes [2, 3], and Alzheimer’s disease [4]. In fact, kinases and phosphatases, constitute approximately 2–4% of eukaryotic proteomes [5]. Moreover, estimates suggest that 33–75% of eukaryotic proteins are phosphorylated though it has been suggested that many of these phosphorylation events are biochemical noise [6]. It follows that determining the precise sites of phosphorylation, accurate abundance, and general roles of this PTM is vital to understanding cellular signaling and the dysregulation thereof.
Regulation of the phosphoproteome represents an important area for the development of drugs to treat human disease [7]. Over 40 drugs targeting protein kinases have been clinically approved over the past decade [8]. The first such drug, imatinib (Gleevec) [9], is a highly efficacious and relatively selective drug that inhibits the tyrosine kinase ABL and is used for treating chronic myeloid leukemia (CML) [10]. Other approved drugs include trametinib that targets MEK1/2 [11], ibrutinib targeting Bruton’s tyrosine kinase (BTK) [12], palbociclib targeting CDK4/6 [13], and fostamatinib that targets SYK [14]. Given the effectiveness of these drugs and the multitude of potential phosphorylation sites, future drug discovery efforts will likely be directed at specific phosphorylation sites and/or associated kinase.
Traditional biochemical techniques for studying phosphorylation are limited to assaying specific sites in a single protein of interest. However, ~97% of reported phosphorylation sites have no known regulatory kinase [15]. The identification and quantification of unknown phosphorylation sites in a manner that is unbiased, comprehensive, and high throughput continues to require novel strategies and innovative approaches.
The comprehensive analysis of protein phosphorylation, by bottom-up mass spectrometry-based proteomics is challenging with respect to 1) sample preparation, 2) data acquisition and 3) data analysis. While there have been many recent advances to address these standing issues—for example, data-independent acquisition (DIA), label-free, and sample multiplexed analyses [16–19], here, we discuss the wide array of current methodologies, persistent challenges, and future outlook specifically for isobaric tag-based quantitative phosphoproteomic analysis.
2 |. SAMPLE PREPARATION
Sample preparation may be considered the single most vital aspect of any proteomics experiment. Regardless of the advanced instrumentation and data analysis techniques, poor sample quality greatly affects all downstream processes. This concept is particularly true for phosphopeptides which are generally of low abundance and often of low stoichiometry. The low stoichiometry of protein phosphorylation has necessitated the development of specific enrichment techniques, which are typically applied on the peptide level (post-tryptic digest) to enrich phosphopeptides.
Strategies for phosphopeptide analysis typically include LC–MS/MS analysis of phosphopeptides that have been enriched from enzymatically digested proteins of lysed cells. Numerous schemas have been developed for phosphopeptide enrichment. Some of these strategies label peptides before enrichment (Figure 1A and B). Such strategies mix the differentially labeled peptides early and reduce the likelihood for variability and error by limiting individual sample preparation steps. limiting manipulation steps. However, this advantage is at the expense of the higher financial cost due to the vast majority of isobaric taglabeled peptides not being phosphorylated, and thus will be discarded following enrichment. Nonetheless, some of the cost may be recouped by using the enrichment flow-through for whole proteome analysis. Conversely, the isobaric labeling can be performed post-enrichment (Figure 1C and D). Although the amount of labeling reagent used is reduced (up to 100 times), variability may be introduced by multiple individual enrichments and associated desalting, that are avoided when the samples are mixed first and then enriched.
FIGURE 1.

General analytical schemes for phosphoproteomics sample processing. Several workflows are commonly used for phosphoproteomics analysis. Some workflows decrease the number of individual sample processing steps by labeling earlier in the workflow, but this is at the expense of additional use of labeling reagents (as the non-phosphorylated majority of peptides are also labeled). In these workflows, peptides are labeled first then either (A) enriched and fractionated or (B) fractionated and enriched prior to LC-MS3. We note that in each workflow, multiple enrichment and fractionation steps can be performed. Other workflows aim to conserve label at the expense of increased susceptibility to variability. Such workflows incorporate individual sample enrichments (C and D) prior to labeling and may include (B) fractionation post-labeling
Previously we described a simple, yet universally applicable isobaric-tag-based sample preparation strategy, which incorporated a phosphopeptide enrichment step, thereby allowing for deep proteome and medium-scale phosphoproteome analysis in a single preparation. We termed this method Streamlined-tandem mass tagging, or SL-TMT [20]. Specifically, incorporation of this phosphopeptide enrichment step (“mini-phos”) into a standard TMT workflow required only one additional sample preparation step, that being a 30-min long enrichment step. Importantly, this option allows for single-shot LC-MS/MS-based identification and quantification of phosphopeptides, and the ability to relate phosphorylation changes to protein changes in the same sample.
In brief, the process (outlined in Figure 2) is as follows: A) Cells are lysed in an 8 M urea-containing buffer, in which cysteine bonds are reduced and alkylated. Methanol-chloroform precipitation is performed to extract proteins which are digested using Lys-C followed by trypsin. B) The resulting peptides are labeled with TMT (or other isobaric tag reagent). The samples are mixed at a 1:1 protein ratio and desalted. C) The sample is then subjected to Ni-NTA-based phosphopeptide enrichment. D) The enriched phosphopeptides are analyzed by SPS-MS3, while the flow-through from this enrichment is desalted and maybe fractionated by basic pH reversed-phase (BPRP) HPLC. Each of these whole proteome fractions are desalted by StageTip [21] and analyzed by SPS-MS3. Moreover, having both total proteome and phosphoproteome data allows for meaningful interpretation of phosphorylation state changes in the context of alterations in protein abundance. For instance, a two-fold increase in the phosphorylation state of a peptide might have a different meaning if the protein abundance also increases two-fold or if the protein level remains constant. In multiplexed workflows, the use of SL-TMT with a mini-phos phosphopeptide enrichment step enables low-cost, efficient, accurate and precise measurements of phosphoproteome alterations.
FIGURE 2.

Streamlined TMT workflow for SPS-MS3-based phosphoproteomics analysis. (A) Samples are lysed in 8 M urea-containing buffer with protease and phosphatase inhibitors. Proteins are isolated via precipitation, and proteolytically digested with Lys-C and trypsin. (B) The samples are labeled with TMT or TMTpro reagents in the digestion buffer (200 mM EPPS, pH8.5) plus 30% acetonitrile. These TMT labels act as a chemical barcode to track the origin of each protein from each sample. (C) Phosphopeptides are enriched by one or several methods. The flow-through from the enrichment (which is isobaric-tag labeled) can be used to profile the whole proteome of the sample under investigation. (D) Samples may be analyzed using SPS/RTS-MS3. The MS2 stage can include either lower Energy HCD (CE = 30–32) or multistage activation (MSA)-CID for peptide fragmentation to be analyzed in the ion trap. The MS3 stage consists of HCD-based fragmentation for Orbitrap-based mass analysis
2.1 |. Phosphopeptide enrichment strategies
Although most sample preparation steps for phosphopeptide analysis are identical to those for whole proteome analysis, substantially more starting material is required for phosphorylation analysis. Whereas comprehensive proteomes can be performed with a few micrograms of protein, phosphoproteomic analyses typically require up to 100 times more material to enable the enrichment of low-abundance phosphopeptides (<2–3%) that are present in a vast background of unmodified peptides. The selection of a particular phosphopeptide enrichment strategy may introduce the most variation into a phosphoproteomic workflow. Workflows that minimize the number of desalting and enrichment steps can help eliminate variance that may be accrued during those steps if they were performed on individual (un-pooled) samples (Figure 2).
Studies comparing multiple enrichment protocols generally show modest overlap, which is potentially a consequence of the lack of phosphoproteome depth attained with these various methods and the multitude of phosphopeptides in each system. The most common enrichment strategies currently use immobilized metal ion affinity chromatography (IMAC) [22, 23] and metal oxide affinity chromatography (MOAC) [24], although other strategies, such as strong cation exchange chromatography (SCX) [25], electrostatic repulsion–hydrophilic interaction chromatography (ERLIC) [26], phosphotyrosine immunoprecipitation [27], pan-pS/pT antibody enrichment [28], and Ti4+ /Zr4+-IMAC [29] have been used successfully.
In early proteome-wide phosphorylation studies, SCX was the method of choice [30]. Here, peptides are retained on a column through the interaction of positively charged peptide side chains with negatively charged column resin. Peptides elute from the column using a salt concentration gradient in order of increasing isoelectric point (pI). The negatively charged phosphoryl group (at low pH) lends phosphopeptides a lower affinity for the negatively charged resin compared to corresponding non-phosphorylated peptides, as such phosphopeptides are enriched in the earlier-eluting fractions.
As an alternative to SCX, hydrophilic interaction chromatography (HILIC) has been used for pre-enrichment fractionation [31]. HILIC uses a polar stationary phase and an organic-to-aqueous gradient (i.e., the opposite of reversed-phase) as the mobile phase, with peptide retention being based on hydrophilicity. Several groups have used HILIC as an orthogonal dimension in phosphopeptidome studies, coupling it with IMAC enrichments and SCX separations [32]. HILIC and ERLIC use both hydrophilic and electrostatic interactions for separation of charged analytes by adjusting the pH, salt concentration, and organic solvent compositions in the mobile phase. The technique can separate singly from multiply phosphorylated peptides [26]. A recent study comparing ERLIC, weak and strong anion exchange (WAX and SAX) showed that ERLIC enriched and identified more than double the number of phosphopeptides as the ion exchange systems [33]. Moreover, ERLIC in combination with orthogonal separation or enrichment has been shown to increase phosphoproteomic depth [34].
MOAC and IMAC methods have become the most common means of enriching phosphorylated peptides. Titanium dioxide (TiO2) remains the most used MOAC affinity matrix. Here, phosphopeptides are loaded onto the metal oxide at acidic pH, with specific additives such as lactic acid, and eluted at pH > 10. TiO2 enrichment is most often performed offline in a column-based or in-batch format [23, 35]. Another enrichment strategy, uses positively charged 3+ metal ions, such as Fe(III) or Ga(III), that are chelated onto a solid phase nitrilotriacetic/iminodiacetic acid resin and as such, are presented for interaction with negatively charged phosphate groups. The sequential elution from IMAC (SIMAC) on the same sample [36] has also shown promise, as has SMOAC (Sequential enrichment of Metal Oxide Affinity Chromatography) which uses serial enrichment with TiO2 and Fe-NTA [37]. Modern IMAC (Fe-NTA) spin columns provide a balance between recovery, enrichment, throughput and efficiency [38]. These simple and efficient centrifugation-based phosphopeptide enrichment columns have been introduced commercially as well (e.g., Pierce Fe-NTA Phosphopeptide Enrichment Kit). The commercial options have also been shown to have a high recovery rate for phosphopeptides [39]. Furthermore, they enable processing of 50 μg to 5 mg of peptide and can yield up to 150 μg of phosphopeptides per column. Although many of these methods have been developed and widely used for label-free experiments, all may be applied to the enrichment of isobarically-labeled peptides without major procedural modifications.
2.2 |. Sample multiplexing with isobaric tags
Isobaric tag-based proteome profiling is a leading technology for multiplexed proteomic and phosphoproteomic analysis of complex peptide mixtures [40]. Sample multiplexing allows for improved statistical power by including replicates within a single experiment and limiting missing values which often occur across experiments. Yet, these advantages come at the cost of lower sensitivity for the detection of phosphopeptides [41].
Several chemistries have been developed to enable chemical-tag-based sample multiplexing. Early developments focused on iTRAQ and TMT reagents. These NHS-ester reactive molecules initially enabled multiplexing of four to six samples which has been further extended over the years. The latest commercial iteration of isobaric tags, TMTpro, allows for multiplexing of up to 16 samples for simultaneous identification and quantification of peptides and proteins [42]. This multiplexing limit can ostensibly be expanded upon as multiplexing of up to 21 samples has been reported for the DiLeu chemistries [43].
New tag designs are still actively being developed, to enable multiplexing. The recently described Easi-tag chemistry uses a sulfoxide group rather than an amide to enable tag release at lower collision energies [44]. Instead of measuring a released reporter ion at specific and consistent m/z values, the Easi-tag quantifying ions are larger and reduce the effects of ratio compression resulting from co-isolated precursors. This technology is similar to the TMT complement ion approach (TMTc) which uses the concomitantly released peptide bound to the balancer region of the TMT molecule to quantify relative difference among samples [45]. While the TMTc method reduces the number of samples that can be multiplexed it has been observed to improve quantitative accuracy [45]. These emerging chemical labeling strategies are evidence for the continuous improvements and innovations in isobaric labeling approaches.
2.3 |. Processing of multiplexed samples
In the case of the SL-TMT protocol, a mini-phos enrichment is seamlessly integrated into the workflow, which facilitates a single, medium-scale phosphopeptide enrichment as part of any TMT experiment [20]. Such analysis permits the survey of several thousand phosphorylation sites with nominal cost and with minimal effort [38]. Yet, the adoption of the “mini-phos” enrichment is still hampered by a lack of depth due to the relatively low amount of analyte compared to larger-scale studies that enrich from several milligrams of protein lysate [46]. As such, a need persists for additional developments to achieve analytical depth with high-efficiency phosphorylation workflows. In this review, we address advances in phosphoproteomics with any eye towards sample multiplexing; however, label-free approaches have also been well studied for phosphopeptide quantitation and are discussed proficiently elsewhere [47].
The plex level for isobaric tags has steadily increased over the past decade. Until recently, commercial isobaric tagging reagents were limited to a plex level of 11. However, the next-generation higher-throughput tandem mass tags, TMTpro, expanded the plex level to 16 with a redesigned mass normalization group and reporter group [42, 48, 49]. Using the SL-TMT protocol across three TMTpro16-plex mini-phos experiments, it was possible to quantify 9566 phosphosites across 48 cell line samples [42]. Quantified phosphosites were highly consistent among triplicates with a median coefficient of variation of approximately 6%. In addition, we showed that quantitative integrity was not compromised with the increase in plex level from 11 to 16. We stress that isobaric tagging is highly beneficial for phosphorylation analysis, in part, as it improves statistical power and decreases missing values across samples.
3 |. MASS SPECTROMETRY FOR PHOSPHOPROTEOMICS
3.1 |. Instrumentation
Sample multiplexing most often focuses on the use of high-resolution instrumentation to resolve nitrogen and carbon isotopic defects. Extensive work has been carried out with Orbitrap mass spectrometers owing to their high resolving power and ease-of-use. In general, to basepeak resolve the N/C tags of TMT requires resolution of ~45,000 in the range of the reporter m/z’s to accurately quantify all 16 channels in a TMTpro experiment. To achieve this resolution requires ~100 ms per high-resolution scan, equivalent to a 10 Hz scan speed on modern Orbitrap instruments (i.e., Exploris and Eclipse)[50]. By comparison, ion traps and time-of-flight traps can achieve scan speeds on the order to 50–100 Hz yet cannot achieve the resolving power necessary to differentiate all 16 reporter ions. Interestingly, the new TMTpro reagents can be analyzed at unit resolution—which can be resolved in ion traps and TOF instruments—with multiplexing of up to nine samples [42]. As instrumentation evolves, faster scan speeds and greater resolving power will massively improve phosphoproteomic depth and quantitative accuracy.
3.2 |. Spectral acquisition for sample multiplexing
Initially, methods for sample multiplexing focused on the use of time or scan based DDA methods with high-resolution MS2 (HRMS2) scans for identification and quantification [42]. While these methods can be quite fast, a well-known issue in multiplexed proteomics is the existence of reporter ‘interference’ caused by co-isolation of TMT-containing peaks the leads to compression of ‘real’ ratios to a median between all co-isolated precursors. The result is lower dynamic range for quantitation. This ratio compression led to the development of a sensitive and accurate ternary scan sequence that uses synchronous precursor selection (SPS) of TMT-containing peptide fragments at the MS2 stage and a second round of fragmentation to liberate reporter ions for peptide quantification at the MS3 stage [51]. This method was shown to dramatically improve quantitative accuracy for multiplexed workflows but requires longer duty cycles to acquire the high-resolution MS3 scan.
While HRMS2 and SPS-MS3 acquisition methods are used for the large majority of multiplexed studies to quantify released reporter ions, additional strategies have been reported. Namely, the TMTc and EASI-Tag methods mentioned above rely on quantitation of complement ions released during fragmentation prior to MS2 spectral acquisition [44, 45]. These methods require optimized fragmentation to efficiently release compliment ions and do not require effciencty retention of low m/z reporter ions. In general, TMTc and EASI-Tag experiments are analyzed using a modified HRMS2 scan sequence for quantitation. Further development in spectral acquisition methods, in conjunction with ion mobility and real time database searching, as discussed later, will greatly enhance the depth and quantitative accuracy of phosphopeptide analysis.
3.3 |. Phosphopeptide fragmentation
The quintessential sample preparation strategy would comprehensively extract phosphopeptides with high selectivity. However, even the best sample preparation requires comprehensive data acquisition as a subsequent step to successfully address a given scientific inquiry. Along with limited sample availability, the labile nature of the phosphoester bond can adversely affect phosphopeptide analysis. The addition of the phosphate active group can impair the fragmentation behavior when compared to non-modified peptides [52]. Studies have addressed the use of different fragmentation techniques in efforts to improve the analysis of phosphopeptides. Some fragmentation techniques that have been evaluated for phosphopeptide analysis include: electron transfer dissociation (ETD) [53], higher-energy collisional dissociation (ion-beam CID, HCD) [54], and ultraviolet photodissociation (UVPD) [55], as well as combinations thereof (such as electron-transfer/higher-energy collision dissociation, EThcD) [56], with HCD-type fragmentation providing the greatest performance in typical large-scale phosphoproteomics experiments [57]. Although many of these methodologies have been performed in label-free settings, all are readily applicable to isobaric tag-based experiments. The most common fragmentation strategies, particularly in the context of isobarically-tagged phosphopeptides, use CID, which is at times referred to as CAD, collision assisted dissociation. CID encompasses two subclasses, ion trap CID and beam-type CID (i.e., higher energy CID or HCD). In ion traps, peptides collide with an inert gas, typically helium, which transfers internal energy to the peptide. The transferred energy fragments a given peptide ion once, and does so at the most labile bond, which in the case of a phosphopeptide is the phosphoester bond. This break leads to neutral loss of phosphoric acid (98 Da) from the precursor. For many phosphorylated peptide ions, this fragmentation scheme is preferred as only low intensity b- and y-type ions are observed. We note that this neutral loss peak typically dominates phosphopeptide CID tandem MS spectra and only minor backbone fragmentation is observed. Methods incorporating MS3 fragmentation [58] and pseudoMS3 (multistage activation, MSA) [41, 59, 60] are frequently used to increase the number of peptide fragments available for peptide matching and thereby improve database searching (Figure 3A and B). Previously, a label-free phosphopeptide identification analysis that compared MS2-alone, MSA, and MS2/MS3 methodologies has shown that MSA performs optimally and does not result in significant loss of unique peptide identifications [61]. In addition, in beam-type CID (i.e., HCD), collisions are assisted by an electrical potential that accelerates peptide ions towards the inert gas, typically nitrogen, leading to higher energy collisions and the potential for multiple fragmentation events per precursor. As such, the intensity of the neutral loss precursor is considerably reduced resulting in a richer and more complex fragmentation patterns. We note that sequential acquisition of CID-MSA and HCD show complimentary phosphopeptide identification for medium scale mini-phos-based analyses [41].
FIGURE 3.

Acquisition and data analysis strategies specific to phosphoproteomics. (A) Low-resolution (ion-trap) and high-resolution (Orbitrap) spectra for the same phosphopeptide sequentially fragmented with CID, CID-MSA, or HCD activation. (B) Peptide spectrum match scores for the phosphopeptide spectra in A. (C) Generalized workflow for identifying and validating phosphorylation sites. (D) Illustration of the challenge of phosphopeptide localization. Often, a single ion can be responsible for determining site-localization and is thus dependent on the fragmentation scheme used for acquisition. (E) Estimation of site localization can be accomplished through the use of decoy amino acids (amino acids with no nucleophile capable of covalently binding the phosphate) during the search. This target-decoy strategy can be used to estimate and report the false localization rate. (F) Phosphosite stoichiometry can be an important metric for determining biological relevance of phosphorylation sites
In contrast, electron capture dissociation (ECD) [53] and electron transfer dissociation (ETD) [53], can preserve labile phosphate side chain and facilitate phosphopeptide identification. In ECD, a low-energy electron is captured by a multiply charged precursor ion. Whilst in ETD analysis, an electron is transferred from a radical anion with low electron affinity to the peptide precursor cation. The recently developed EThcD technique melds ETD and HCD to generate rich MS/MS spectra with both b/y and c/z ion pairs [56, 62]. Such spectra can yield higher peptide sequence coverage and more confident localization of phosphorylation sites [56]. However, both fragmentation schemes are slower scanning, slightly less sensitive, and only efficient for peptides with charge stages greater than 2. Although we can attest to the advantages of using MSA-CID and HCD for phosphopeptide analysis, we acknowledge that fledgling strategies, such as EThcD have potential benefits with advancements in instrument speed, as well as software enhancements.
4 |. DATA ANALYSIS
Data analysis strategies for proteomics studies are readily applicable to analogous phosphoproteomic analysis; however, several additional phosphorylation-specific steps required. These additional layers of analysis are necessary for accurate determination of phosphorylation sites and the compilation of reproducible datasets (Figure 3C). In the first layer, like a typical peptide analysis, the acquired spectra are still matched against a database of known peptides and false discovery rates are determined typically using the target-decoy strategy [63]. A major caveat to these searches is that variable modification searches on serines, threonines and tyrosines results in larger search databases and longer search times. In the second layer, specific sites are scored for the likelihood, or probability, of a phosphorylation event at that amino acid. This is ideally done with a concomitant assignment of the false-localization rate (FLR) for a modification at a given site, which is particularly important because multiple phosphorylatable sites can appear on a single peptide—these are often proximal amino acids, as well. In the third layer, often considered optional, the relative stoichiometry of phosphorylation is determined. Stoichiometry has a major effect on both identification and quantification, and, more generally, is of significant relevance biologically [64]. By addressing these three main analytical areas, it is possible to generate robust and interpretable datasets to garner greater biological insight.
4.1 |. Open database searching for identification and localization
Open database searching was pioneered in 2005 for the identification of diverse post-translational modifications [65]. The strategy consists of searching spectra with extremely large precursor tolerances. The mass error for scored peptides can then be assessed to determine if mass deviations are consistent with specific post-translational modifications. The development and deployment of modern frameworks—for example, MSFragger [66], pFind [67], TagGraph [68], and MetaMorpheous [69]—has extended the accessibility and efficiency of open database searching. These workflows enable the identification of diverse modifications within the same analysis, including the identification of phosphorylation. Recent work with MSFragger has highlighted its potential for localization-aware searching to further improve the identification and localization of post-translational modifications [70]. While these techniques have not been thoroughly integrated into common phosphoproteomic studies, they can potentially be used in concert with isobaric-tag based phosphoproteomics workflows to increase identification rates of phosphorylation events. This strategy is particularly appealing in the background of multiple diverse modifications on the same protein, for example, phosphoproteome analysis of histones (e.g., methylation and acetylation) or surface exposed proteins (e.g., glycosylation) [71].
4.2 |. Site localization and false localization rate
Determination of site localization is a key computational challenge associated with phosphoproteomics analysis (Figure 3C). Phosphopeptide searches are inherently more computationally demanding than those of unmodified peptides due to the increased database size incurred with dynamic modifications, as phosphorylation is possible at every serine, threonine, and tyrosine residue. An additional complication is site localization which typically requires the measurement of sequence-specific fragment ions comprising the intact phosphorylated moiety or those resulting from the cleavage of the phosphoester bond (Figure 3D). Precursor masses and fragments are not the only factors needed for accurate identification, as in general peptide identification. Instead, specific fragments are needed to pinpoint an exact site(s) of phosphorylation as well as the absence of any modification at potentially phosphorylatable residues. Most database search algorithms do not evaluate different peptide isoforms and so regardless of score or mass accuracy, no distinction can be made of a positional isomer with respect to the phosphorylated residue. To that end, several algorithms are available to aid in site localization, including Mascot delta score [72], AScore [73], PTM score [74], PhosphoScore [75], Phosphinator [76], PhosphoRS [77], and LuciPHOr [78], among others.
When providing a conditional compendium of phosphorylation sites, it is essential to include the relative confidence of the assignment of these reported sites. It is worth noting that many localization algorithms score the confidence of the localization, but use arbitrary score cut-offs instead of estimating the FLR [79] (Figure 3E). FLR estimation is important, however, as studies have reported that these rates can be as high as 20% [79]. One strategy for calculating FLR is to perform searches in which decoy phosphorylations are allowed on glutamic acid (E) and proline (P), the so-called SLIP approach [80]. Another is the LuciPHOr program which directly reports FLR estimations based on smoothed distributions of the localization delta scores [78]. While these methods and others are still being actively developed, they provide the essential first steps in addressing the issue of false localization. We emphasize that determining the precise localization of a phosphosite and associated FLR, particularly regarding positional isomers with multiple sequential phosphorylatable sites is essential for pinpointing residues to target or mutate for downstream applications.
4.3 |. Phosphopeptide stoichiometry
In a typical phosphoproteomics experiment, the abundance of phosphorylated peptides is orders of magnitude below that of the unphosphorylated peptides and as such, improvements in sample preparation, data collection, and analysis are needed to increase phosphopeptide coverage. Understanding the function of these sites with relation to the degree of phosphorylation, that is, stoichiometry, is important (Figure 3F).
Site stoichiometry refers to the percentage occupancy of a given phosphosite—this can be calculated as the phosphopeptide abundance compared to the total abundance for the phosphorylated and non-phosphorylated versions of the peptide. Isotopic labeling and enzymatic dephosphorylation may also be used to determine phosphorylation stoichiometry by measuring the abundance of the non-phosphorylated peptides with and without phosphatase treatment. This requires amending the general phosphoproteomics workflows to ensure that both the modified and unmodified versions of a given peptide can be quantitatively measured (Figure 1).
Interestingly, the stoichiometry of a site can represent a threshold that differentiates functional from non-functional phosphorylation events. Cellular conditions, such as cell cycle state, can alter phosphorylation site stoichiometry [81]. For this reason, several approaches have been developed to enable the global estimation of phosphorylation site occupancy [64, 82]. These studies revealed that in some cases, a substantial proportion of sites are phosphorylated to high stoichiometry. For instance, half of all mitotic phosphorylation sites had a stoichiometry occupancy of more than 75% during mitosis [81]. However, we stress that even considering sites with high site occupancy, only ~1–2% of the peptides in a typical peptide mixture are phosphorylated, imposing limits on both sensitivity and dynamic range. It follows that due to the general low abundance of phosphorylated peptides in a typical tryptic digest, the adaptation of novel methods and technologies to enhance phosphopeptide analysis is imperative to improve the sensitivity of the technique and expand our understanding of phosphorylation-related cellular signaling.
5 |. CURRENT STATE-OF-THE-FIELD DEPTH FOR MULTIPLEXED PHOSPHOPROTEOMICS
In general, we estimate 10,000–20,000 phosphorylation sites per sample can be quantified using sample multiplexing in conjunction with large-scale phosphorylation enrichment (i.e., 2–10 mg/channel of starting protein). Specifically, large scale, multiplexed phosphorylation analysis (12 basic pH fractions) has achieved the quantification of 10,562 phosphosites in mouse kidney tissue [3], 14,602 phosphosites in yeast [83], and 16,622 phosphosites in nicotine-treated pancreatic stellate cell line [84] using SPS-MS3 analysis on an Orbitrap Fusion or Lumos mass spectrometer. Methods using HRMS2 for quantitation have reported up to 37,000 phosphosites for large-scale phosphoproteome preparations from patient-derived xenografts, though no site level filtering was reported [17].
Using medium-scale sample preparation, we estimate that 4,000 to 10,000 phosphorylation sites can be quantified from at least 50–100 μg of starting material per channel. In practice for medium-scale phosphorylation enrichment, over 4,100 sites have been quantified in plasmodium [85], 6,622 phosphosites in a series of yeast deletion strains [86], and 7,140 phosphosites in human cell lines [42] using two shots of an unfractionated sample with SPS-MS3 analysis. By comparison quantitation of ~11,000 phosphosites has been observed with label-free quantitation of 500 μg of input protein mass or ~16,000 combining label-free quantitation with match-between runs [18]. We emphasize again that innovations in technology, both at the hardware and software levels, will greatly increase the depth of phosphoproteomic analyses.
6 |. RECENT ADVANCES AND FUTURE INNOVATIONS IN PHOSPHOPROTEOMICS ANALYSIS
We highlight four areas which hold promise for improving phosphoproteomics analysis (Figure 4). First, high-field asymmetric waveform ion mobility spectrometry (FAIMS), which interfaces between the liquid chromatograph and the mass spectrometer, can separate ions in the gas phase based on differences in mobility in varying electric fields. This adds an additional, orthogonal mode of separation that provides higher sensitivity and more accurate quantification. Second, real-time database searching-MS3 (RTS-MS3) further improves quantitative accuracy of phosphopeptide quantification by ensuring that synchronous precursor selection (SPS) ions originate from the precursor of interest and are not an artifact of a mixed spectrum. Third, high-throughput sample processing allows for hundreds of samples to be handled in parallel instead of processing samples in individual tubes. As such, time of sample processing is reduced, potentially leading to better reproducibility as samples are prepared in parallel. Fourth, automation represents the next step in high-throughput sample processing. The use of robotic liquid handling can greatly reduce human error, improve reproducibility, and allow methods to be replicated better across laboratories.
FIGURE 4.
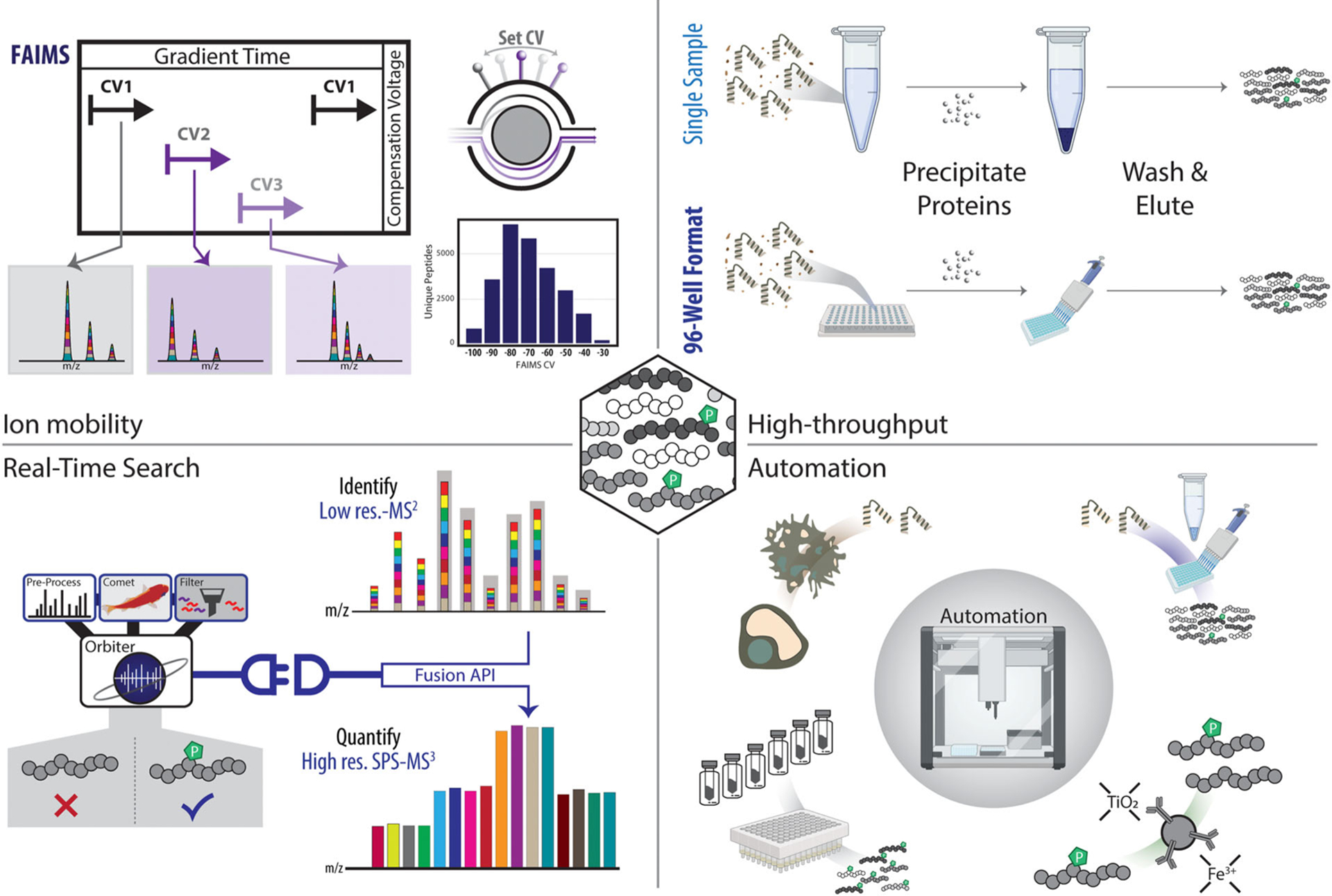
Recent advances and future innovations in phosphoproteomics analysis. (A) Ion mobility, specifically high-field asymmetric waveform ion mobility spectrometry (FAIMS), adds another dimension of fractionation. Here, ions are separated in the gas phase based on differences in mobility in high and low electric fields as they transit between an inner and outer electrode. (B) Real-time database searching (RTS) improves quantitative accuracy of phosphopeptide quantification when performing SPS-MS3 analysis. Here, the peptide fragment ions to be used for quantification are pre-selected (not simply the most intense in a given spectrum, as in traditional SPS-MS3), thereby reducing interference from precursors which are not those of interest. (C) High-throughput sample processing allows for hundreds of samples to be handled in parallel. Rather than processing samples in individual tubes, we anticipate the use of bead-based proteins extractions (e.g., SP3) and plate-based enrichment in which up to 96 samples can be processed in parallel. (D) Automation using laboratory robotics can greatly eliminate many of the repetitive manual steps associated with high-throughput sample processing. With the use of magnetic modules and/or vacuum filtration, all sample processing steps maybe accomplished in 96- or 384-well plates with limited human intervention. Automation can greatly reduce human error, improve reproducibility, and better allow methods to be replicated across laboratories
6.1 |. Ion mobility for phosphoproteomics
High-Field Asymmetric waveform Ion Mobility Spectrometry (FAIMS) can separate ions in the gas phase in a manner that is orthogonal to that of liquid chromatography-based fractionation (Figure 4 top left). FAIMS can discriminate among ions in the gas phase based on an ion’s mass, charge, size, and shape as it traverses an electrode and collides with the buffer gas molecules. FAIMS separation is based on differences in mobility in high and low electric fields as ions transit between an inner and outer electrode [87]. This asymmetric waveform applies a short high positive voltage- the peak which is termed ‘dispersion volage’ or ‘DV’- and a longer low negative voltage. The drift of an ion towards electrodes can be stopped by the application of a tunable small DC voltage (‘compensation voltage’ or ‘CV’) to either of the plates to compensate for ion drift. Alternating CVs can rapidly separate different types of ions as the ions traverse the plates at a specific characteristic CV [88].
We, and others, have recently applied FAIMS to isobaric tag-labeled, phosphorylated peptide-enriched samples using the FAIM-Spro interface [89, 90]. Notably it was shown that modern FAIMS devices were capable of improving the sensitivity of phosphopeptide quantification in stable isotope incorporated cellular samples [91]. Research with label-free phosphopeptide mixtures has similarly shown improved identification and quantification when incorporating FAIMS into the mass spectrometric acquisition process [92]. For isobarically tagged phosphopeptides, we furthermore defined the optimal CVs for phosphorylation-centric FAIMS analysis (i.e., −40, −60, −80 V in a single method). We also showed that consecutive analyses using MSA-CID (multistage activation collision-induced dissociation) and HCD (higher-energy collisional dissociation) fragmentation at the MS2 stage increased the depth of phosphorylation analysis [41]. The benefits in analytical depth and quantitative accuracy using FAIMS [89] led us to anticipate that future improvements to FAIMS interfaces will continue to enhance phosphopeptide analysis.
Another ion mobility technology with growing potential for proteomics and phosphoproteomics workflows is the trapped-IMS (TIMS), which was recently commercialized as part of the timsTOF pro mass spectrometer [93]. In TIMS, ions are pushed through an electric field by a carrier gas; ions become trapped when the ion drift velocity equals the carrier gas velocity. By serial reducing the electric field strength, ions are released to the mass spectrometer, which in the case of the timsTOF pro, is a quadrupole time-of-flight instrument. TIMS has been proposed as a means to reduce precursor co-isolation and improve quantitative accuracy, with the current limitation of unit resolution [93]. While the timsTOF system has not yet been widely applied to multiplex phosphoproteomics analysis, recent work has been proposed that high-sensitivity DIA-based phosphoproteomics is possible using TIMS [19].
We believe these IMS technological developments will be a boon for phosphoproteomics analyses going forward. Importantly, IMS developments have proven to be useful across different quantitative approaches (e.g., label-free, DIA, isobaric-tagging), suggesting that future improvements in sensitivity and resolution will bolster a diverse cadre of innovative and informative phosphoproteomic studies.
6.2 |. Real-time searching
In addition to FAIMS, real-time database searching-MS3 (RTS-MS3) can improve phosphopeptide analysis (Figure 4 bottom left). SPS-MS3 decouples peptide identification and quantification and has the advantage of higher quantitative accuracy compared to traditional MS2-only methods for isobaric tag-based analyses [48, 94]. However, SPS-MS3 suffers from long acquisition duty cycles, thereby affecting proteome depth, which is especially detrimental for low abundance samples that preclude fractionation, like small-scale (less than 1 mg of starting protein) phosphorylation analyses. The RTS strategy uses a client application on the external computer that receives MS2 spectra from the instrument and searches a complete protein database for peptide identification in real-time, that is, as the data are acquired. In the absence of a confident identification, no analysis time-intensive SPS-MS3 spectrum is collected. Using the RTS-MS3 method can typically comprehensively characterize a multiplexed proteome with 50% less data acquisition time [95, 96]. A single spectrum database search can be performed in less than 10 ms [95]. Quantitative accuracy is also improved as only SPS ions that originate from the precursor of interest are selected for the MS3 stage. One challenge anticipated for the use of RTS-MS3 with phosphopeptide database searching has been the possibility that the expanded database size necessary for phosphoproteome searching would hinder rapid spectral matching. Initially work with RTS-MS3 provided proof-of-principle analyses that even with the expanded database sizes, RTS was capable of rapidly matching phosphopeptides to spectra in real-time [95]. We expect that applying this strategy to multiplexed phosphoproteome analysis will show a strong improvement in data acquisition throughput and improved accuracy for multiplexed quantitation.
6.3 |. High throughput analysis
Reducing the time and associated costs of performing phosphoproteomics experiments can be accomplished using high-throughput sample processing techniques (Figure 4 top right). One challenge for high-throughput phosphoproteomic samples preparation is the reliance on chemical-based protein precipitation steps which are tedious, more susceptible to human error, and not amenable to automation. Common protein extraction methods, such as chloroform-methanol [20] or acetone precipitations [18], require careful collection of phase separated protein pellets and thereby are more prone to sample-to-sample variation. Several methods have been described recently with an eye towards high-throughput proteomic sample preparation, including Preomics iST, Thermo’s EasyPep system, the S-Trap by Protifi, and protein aggregation techniques (e.g., SP3), among others [97]. An important aspect of these methods is that they can be readily applied to not only sample multiplexed workflows, but also DIA and label-free methods.
Nanopartical-base protein aggregation is one such technology gaining traction in high-throughput workflows for everything from general proteomics to single-cell proteomics [98]. These methods enable plate-based sample preparation, including integrated lysis, digestion, labeling, and sample cleanup. The SP3 methods, in particular, were recently integrated with TMT workflows (SL-SP3-TMT [99]) to overcome the major bottlenecks that hinder the application of SL-TMT to automated sample processing, specifically sample cleanup and desalting. Likewise, throughput can be increased during the desalting and enrichment steps using a 96-well SPE and enrichment plates [18]. It follows that higher throughput will allow for more efficient sample multiplexing and the ready expansion of sample sets into the hundreds or thousands.
6.4 |. Automation
High-throughput sample processing typically lends itself to automation (Figure 4 bottom right). Automation and robotics, such as the use of liquid handlers can increase throughput and reproducibility, by eliminating human error in highly repetitive sample preparation steps [100]. Accessories for many automated liquid handlers, such as the opentrons OT-2 or the Agilent Bravo systems, include magnetic modules which can easily accommodate high-throughput processing, such as magnetic beads for bead-based precipitation (SP3) or phosphopeptide enrichment, many of which are available in a 96-well format. Additionally, isobaric tagging enables different pharmacological doses, time-series data, and positive and negative control samples to be analyzed together in a single experiment, but often require replicates for which manual processing becomes a daunting task. The ability to automate high-throughput tasks will have a positive effect on data reproducibility by ensuring samples are processed in parallel and by decreasing the potential for human error which may have implications in the quantitative integrity of the data.
Progress has already been made toward automation of streamlined phosphopeptide enrichment workflows, including the ‘rapid-robotic phosphoproteomics’ (R2-P2) and EasyPhos platforms18,106. The R2-P2 platform enabled identification of more than 11,000 unique phosphopeptides from 250 μg of yeast protein. EasyPhos reliably identified over 15,000 distinct phosphopeptides from 200 μg of human protein in less than 24 h in a single 96-well plate [18]. These concise and well-designed protocols are highly amenable to, and potentially enhanced through, coupling with isobaric tag-based multiplexing strategies.
7 |. REMAINING CHALLENGES AND OUTLOOK IN PHOSPHOPROTEOMICS
Modern phosphoproteomic approaches must move beyond the simple cataloguing of modified residues. In this work, we have indicated the many advances that enable identification and quantification of thousands of phosphosites from complex samples. Yet, challenges remain for the field of phosphoproteomics. New developments are needed across all frontiers of data analysis, hardware, and methods to address this issue. These improvements include further advances in bioinformatics and computational biology that integrate different omics fields that should provide useful information on kinase-regulated phosphorylation events. These developments are necessary to: (1) robustly profile phosphorylation stoichiometry, (2) extend applications to large scale studies (hundreds or thousands of samples), and (3) improve the sensitivity to ensure comprehensive and comparable study of sample cohorts.
The comprehensiveness of the reported phosphoproteome remains limited. The fact that enrichment methods, such as IMAC and MOAC, show moderate overlap rather than perfect redundancy reveals that further effort is necessary in the development of strategies to capture a larger portion of the phosphoproteome. The development of more sensitive and selective enrichment methods and the increase in the throughput of the phosphoproteomics workflows themselves will facilitate progressively more complex experimental designs for comprehensive sample analysis. Updated workflows will help to address known issues with missing values across multiplexed batches—when more than 16 samples need to be analyzed with TMTpro. As sample size increases, the intersection of consistently quantified phosphopeptides and phosphosites decreases. This inverse proportionality leads to smaller useful datasets with comprehensive coverage across all samples and limit potential network assembly and conclusion derivation. Improved instrument sensitivity will help by generating larger datasets per plex, and thereby larger intersections among batches [18]. Also, FAIMS-based workflows can help by exposing precursors for subsequent fragmentation more consistently across analytical runs, but the improvement remains imperfect [41]. Targeted workflows offer one potential mechanism to improve sample coverage using acquisition strategies such as Tomahto/TOMAHAQ, but these require synthetic peptides and can target only a small cohort of potential phosphopeptides [96, 101].
Moreover, other elusive phosphorylation sites may exist in the form of non-canonical phosphorylation. Although the phosphorylation of canonical residues (i.e., Ser, Thr, Tyr) has been widely investigated, phosphorylation of non-canonical amino acid residues is not commonly investigated and remains a largely unexplored landscape. Studies, including several using mass spectrometry, have shown the occurrence of histidine phosphorylation [102] in both eukaryotic and prokaryotic organisms. Another study using strong anion exchange noted the prevalence of histidine (His), arginine (Arg), lysine (Lys), aspartate (Asp), glutamate (Glu) and cysteine (Cys) phosphorylation, being one-third that of canonical protein phosphorylation [103]. The relative instability under acidic conditions and/or elevated temperature during sample preparation of non-canonical phosphorylated residues may result in standard methods of phosphoprotein characterization being unsuitable for the analysis of non-canonical phosphorylation.
Finally, a need exists to link mechanism-of-action to specific phosphorylation sites. Approximately 97% of reported phosphosites have no known regulatory kinase assignment [15]. Several bioinformatics approaches have been devised in efforts to assign kinases to a given phosphorylation site [104]. In addition, the KAYAK (Kinase ActivitY Assay for Kinome profiling) assay was designed to measure the phosphorylation rates of dozens of peptide substrates simultaneously, directly from cell lysates [105]. Expansion of this or similar assays with modern instrumentation, bioinformatic workflows, and higher-order sample multiplexing techniques offer potential for the exact assignment of a kinase to a measured phosphorylation event.
Future studies which can efficiently incorporate higher-order sample multiplexing and the latest advances in data acquisition and analysis will draw the field closer toward true comprehensive phosphoproteome analysis. Many of technologies and techniques highlighted in this review can be used to advance post-translational modifications beyond phosphorylation. Thus, advances that improve sensitivity, quantitative accuracy, and automation for proteomics will also enrich the larger field of PTM analysis.
ACKNOWLEDGMENTS
The authors would like to thank members of the Gygi and Schweppe labs for thoughtful discussion. The authors would also like to acknowledge the following funding: R01GM132129-02 to J.A.P.
Funding information
National Institute of General Medical Sciences, Grant/Award Number: R01 GM132129
Abbreviations:
- PTM
post-translational modification
- TMT
tandem mass tag
- IMAC
Immobilized metal affinity chromatography
- HILIC
hydrophilic interaction chromatography
- FAIMS
high-field asymmetric-waveform ion mobility spectrometry
- RTS
real-time search
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nar-done J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, Sullivan L, Mitchell J, Wetzel R, Macneill J, Ren JM, Yuan J, Bakalarski CE, Villen J, Kornhauser JM, Smith B, Li D, Zhou X, Gygi SP, Gu TL, Polakiewicz RD, Rush J, & Comb MJ (2007). Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell, 131, 1190–1203; [DOI] [PubMed] [Google Scholar]; Zanivan S, Meves A, Behrendt K, Schoof EM, Neilson LJ, Cox J, Tang HR, Kalna G, van Ree JH, van Deursen JM, Trempus CS, Machesky LM, Linding R, Wickstrom SA, Fassler R, & Mann M (2013). In vivo SILAC-based proteomics reveals phosphoproteome changes during mouse skin carcinogenesis. Cell Reports, 3, 552–66. [DOI] [PubMed] [Google Scholar]
- 2.Danielsson A, Ost A, Nystrom FH, & Stralfors P (2005). Cttenuation of insulin-stimulated insulin receptor substrate-1 serine 307 phosphorylation in insulin resistance of type 2 diabetes. Journal of Biological Chemistry, 280, 34389–34392. [DOI] [PubMed] [Google Scholar]
- 3.Paulo JA, McAllister FE, Everley RA, Beausoleil SA, Banks AS, & Gygi SP (2015). Effects of MEK inhibitors GSK1120212 and PD0325901 in vivo using 10-plex quantitative proteomics and phosphoproteomics. Proteomics, 15, 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eidenmuller J, Fath T, Maas T, Pool M, Sontag E, & Brandt R (2001). Phosphorylation-mimicking glutamate clusters in the proline-rich region are sufficient to simulate the functional deficiencies of hyperphosphorylated tau protein. Biochemical Journal, 357, 759–759; [DOI] [PMC free article] [PubMed] [Google Scholar]; Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, & Binder LI (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. PNAS, 83, 4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manning G, Plowman GD, Hunter T, & Sudarsanam S (2002). Evolution of protein kinase signaling from yeast to man. Trends in Biochemical Sciences, 27, 514–520. [DOI] [PubMed] [Google Scholar]
- 6.Cohen P (2000). The regulation of protein function by multisite phosphorylation–a 25 year update. Trends in Biochemical Sciences, 25, 596–601; [DOI] [PubMed] [Google Scholar]; Sharma K, D’Souza RC, Tyanova S, Schaab C, Wisniewski JR, Cox J, & Mann M (2014). Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell reports, 8, 1583–1594. [DOI] [PubMed] [Google Scholar]
- 7.Cohen P, & Alessi DR (2013). Kinase drug discovery–what’s next in the field? Acs Chemical Biology, 8, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson LJ, Linley A, Hammond DE, Hood FE, Coulson JM, MacEwan DJ, Ross SJ, Slupsky JR, Smith PD, Eyers PA, & Prior IA (2018). New Perspectives, Opportunities, and Challenges in Exploring the Human Protein Kinome. Cancer Research, 78, 15–29. [DOI] [PubMed] [Google Scholar]
- 9.Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, & Lydon NB (2000). Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. Journal of Pharmacology and Experimental Therapeutics, 295, 139–145. [PubMed] [Google Scholar]
- 10.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, & Lydon NB (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature Medicine, 2, 561–566. [DOI] [PubMed] [Google Scholar]
- 11.Shi-Lin D, Yuan X, Zhan S, Luo-Jia T, & Chao-Yang T (2015). Trametinib, a novel MEK kinase inhibitor, suppresses lipopolysaccharide-induced tumor necrosis factor (TNF)-α production and endotoxin shock. Biochemical and Biophysical Research Communications, 458, 667–673. [DOI] [PubMed] [Google Scholar]
- 12.Brown JR (2013). Ibrutinib (PCI-32765), the first BTK (Bruton’s tyrosine kinase) inhibitor in clinical trials. Curr Hematol Malig Rep, 8, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCain J (2015). First-in-Class CDK4/6 Inhibitor Palbociclib Could Usher in a New Wave of Combination Therapies for HR+, HER2-Breast Cancer. P T, 40, 511–520. [PMC free article] [PubMed] [Google Scholar]
- 14.Bajpai M (2009). Fostamatinib, a Syk inhibitor prodrug for the treatment of inflammatory diseases. Idrugs, 12, 174–185. [PubMed] [Google Scholar]
- 15.Needham EJ, Parker BL, Burykin T, James DE, & Humphrey SJ (2019). Illuminating the dark phosphoproteome. Science signaling, 12, eaau8645. [DOI] [PubMed] [Google Scholar]
- 16.Hogrebe A, von Stechow L, Bekker-Jensen DB, Weinert BT, Kelstrup CD, & Olsen JV (2018). Benchmarking common quantification strategies for large-scale phosphoproteomics. Nature communications, 9, 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mertins P, Tang LC, Krug K, Clark DJ, Gritsenko MA, Chen L, Clauser KR, Clauss TR, Shah P, Gillette MA, Petyuk VA, Thomas SN, Mani DR, Mundt F, Moore RJ, Hu Y, Zhao R, Schnaubelt M, Keshishian H, Monroe ME, Zhang Z, Udeshi ND, Mani D, Davies SR, Townsend RR, Chan DW, Smith RD, Zhang H, Liu T, & Carr SA (2018). Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nature Protocols, 13, 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humphrey SJ, Karayel O, James DE, & Mann M (2018). High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nature Protocols, 13, 1897–1916. [DOI] [PubMed] [Google Scholar]
- 19.Bekker-Jensen DB, Bernhardt OM, Hogrebe A, Martinez-Val A, Verbeke L, Gandhi T, Kelstrup CD, Reiter L, & Olsen JV (2020). Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nature communications, 11, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navarrete-Perea J, Yu Q, Gygi SP, & Paulo JA (2018). Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of Proteome Research, 17, 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rappsilber J, Ishihama Y, & Mann M (2003). Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Analytical Chemistry, 75, 663–670. [DOI] [PubMed] [Google Scholar]
- 22.Zhou H, Ye M, Dong J, Corradini E, Cristobal A, Heck AJ, Zou H, & Mohammed S (2013). Robust phosphoproteome enrichment using monodisperse microsphere-based immobilized titanium (IV) ion affinity chromatography. Nature Protocols, 8, 461–480. [DOI] [PubMed] [Google Scholar]
- 23.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, & White FM (2020). Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nature Biotechnology, 20, 301–305. [DOI] [PubMed] [Google Scholar]
- 24.Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, & Heck AJ (2004). Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Analytical Chemistry, 76, 3935–3943. [DOI] [PubMed] [Google Scholar]
- 25.Ballif BA, Villen J, Beausoleil SA, Schwartz D, & Gygi SP (2004). Phosphoproteomic analysis of the developing mouse brain. Molecular and Cellular Proteomics, 3, 1093–1101. [DOI] [PubMed] [Google Scholar]
- 26.Loroch S, Zahedi RP, & Sickmann A (2015). Highly sensitive phophoproteomics by tailoring solid-phase extraction to electrostatic repulsion-hydrophilic interaction chromatography. Analytical Chemistry, 87, 1596–1604. [DOI] [PubMed] [Google Scholar]
- 27.Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, & Comb MJ (2005). Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nature Biotechnology, 23, 94–101. [DOI] [PubMed] [Google Scholar]
- 28.Possemato AP, Paulo JA, Mulhern D, Guo A, Gygi SP, & Beausoleil SA (2017). Multiplexed Phosphoproteomic Profiling Using Titanium Dioxide and Immunoaffinity Enrichments Reveals Complementary Phosphorylation Events. Journal of Proteome Research, 16, 1506–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou H, Ye M, Dong J, Han G, Jiang X, Wu R, & Zou H (2008). Specific phosphopeptide enrichment with immobilized titanium ion affinity chromatography adsorbent for phosphoproteome analysis. Journal of Proteome Research, 7, 3957–3967. [DOI] [PubMed] [Google Scholar]
- 30.Zhai B, Villen J, Beausoleil SA, Mintseris J, & Gygi SP (2008). Phosphoproteome analysis of Drosophila melanogaster embryos. Journal of Proteome Research, 7, 1675–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McNulty DE, & Annan RS (2009). Hydrophilic interaction chromatography for fractionation and enrichment of the phosphoproteome. Methods in Molecular Biology, 527, 93–105. [DOI] [PubMed] [Google Scholar]
- 32.Villen J, & Gygi SP (2008). The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nature Protocols, 3, 1630–1638; [DOI] [PMC free article] [PubMed] [Google Scholar]; Trinidad JC, Specht CG, Thalhammer A, Schoepfer R, & Burlingame AL (2006). Comprehensive identification of phosphorylation sites in postsynaptic density preparations. Molecular and Cellular Proteomics, 5, 914–922; [DOI] [PubMed] [Google Scholar]; Meijer LA, Zhou H, Chan OY, Altelaar AF, Hennrich ML, Mohammed S, Bos JL, & Heck AJ (2013). Quantitative global phosphoproteomics of human umbilical vein endothelial cells after activation of the Rap signaling pathway. Molecular Biosystems, 9, 732–749; [DOI] [PubMed] [Google Scholar]; Double-day PF, & Ballif BA (2014). Developmentally-Dynamic Murine Brain Proteomes and Phosphoproteomes Revealed by Quantitative Proteomics. Proteomes, 2, 197–207; [DOI] [PMC free article] [PubMed] [Google Scholar]; Dephoure N, & Gygi SP (2011). A solid phase extraction-based platform for rapid phosphoproteomic analysis. Methods (San Diego, Calif.), 54, 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alpert AJ, Hudecz O, & Mechtler K (2015). Anion-exchange chromatography of phosphopeptides: weak anion exchange versus strong anion exchange and anion-exchange chromatography versus electrostatic repulsion-hydrophilic interaction chromatography. Analytical Chemistry, 87, 4704–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loroch S, Schommartz T, Brune W, Zahedi RP, & Sickmann A (2015). Multidimensional electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) for quantitative analysis of the proteome and phosphoproteome in clinical and biomedical research. Biochimica Et Biophysica Acta, 1854, 460–468. [DOI] [PubMed] [Google Scholar]
- 35.Ndassa YM, Orsi C, Marto JA, Chen S, & Ross MM (2006). Improved immobilized metal affinity chromatography for large-scale phosphoproteomics applications. Journal of Proteome Research, 5, 2789–2799. [DOI] [PubMed] [Google Scholar]
- 36.Thingholm TE, Jensen ON, Robinson PJ, & Larsen MR (2008). SIMAC (sequential elution from IMAC), a phosphoproteomics strategy for the rapid separation of monophosphorylated from multiply phosphorylated peptides. Molecular and Cellular Proteomics, 7, 661–671. [DOI] [PubMed] [Google Scholar]
- 37.Choi J, Snovida SI, Bomgarden RD, & Rogers JC, Sequential enrichment from Metal Oxide Affinity Chromatography (SMOAC), a phosphoproteomics strategy for the separation of multiply phosphorylated from monophosphorylated peptides. American Society for Mass Spectrometry Conference 2017, WP601. [Google Scholar]
- 38.Paulo JA, Navarrete-Perea J, Erickson AR, Knott J, & Gygi SP (2018). An Internal Standard for Assessing Phosphopeptide Recovery from Metal Ion/Oxide Enrichment Strategies. Journal of the American Society for Mass Spectrometry, 29, 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paulo JA, Navarrete-Perea J, Erickson AR, Knott J, & Gygi SP (2018). An Internal Standard for Assessing Phosphopeptide Recovery from Metal Ion/Oxide Enrichment Strategies. Journal of the American Society for Mass Spectrometry, 29, 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hogrebe A, von Stechow L, Bekker-Jensen DB, Weinert BT, Kelstrup CD, & Olsen JV (2018). Benchmarking common quantification strategies for large-scale phosphoproteomics. Nature communications, 9, 1045; [DOI] [PMC free article] [PubMed] [Google Scholar]; Rauniyar N, & Yates JR 3rd (2014). Isobaric labeling-based relative quantification in shotgun proteomics. Journal of Proteome Research, 13, 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schweppe DK, Rusin SF, Gygi SP, & Paulo JA (2020). Optimized Workflow for Multiplexed Phosphorylation Analysis of TMT-Labeled Peptides Using High-Field Asymmetric Waveform Ion Mobility Spectrometry. Journal of Proteome Research, 19, 554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Van Vranken JG, Pontano Vaites L, Schweppe DK, Huttlin EL, Etienne C, Nandhikonda P, Viner R, Robitaille AM, Thompson AH, Kuhn K, Pike I, Bomgarden RD, Rogers JC, Gygi SP, & Paulo JA (2020). TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nature Methods, 17, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frost DC, & Feng Y, Li L, 21-plex DiLeu Isobaric Tags for High-Throughput Quantitative Proteomics (2020). Analytical Chemistry, 92, 8228–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Virreira Winter S, Meier F, Wichmann C, Cox J, Mann M, & Meissner F (2018). EASI-tag enables accurate multiplexed and interference-free MS2-based proteome quantification. Nature Methods, 15, 527–530. [DOI] [PubMed] [Google Scholar]
- 45.Sonnett M, Yeung E, Wuhr M (2018). Accurate, Sensitive, and Precise Multiplexed Proteomics Using the Complement Reporter Ion Cluster. Analytical Chemistry, 90, 5032–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mertins P, Tang LC, Krug K, Clark DJ, Gritsenko MA, Chen L, Clauser KR, Clauss TR, Shah P, Gillette MA, Petyuk VA, Thomas SN, Mani DR, Mundt F, Moore RJ, Hu Y, Zhao R, Schnaubelt M, Keshishian H, Monroe ME, Zhang Z, Udeshi ND, Mani D, Davies SR, Townsend RR, Chan DW, Smith RD, Zhang H, Liu T, & Carr SA (2018). Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nature Protocols, 13, 1632–1661; [DOI] [PMC free article] [PubMed] [Google Scholar]; Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, & Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell, 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riley NM, & Coon JJ (2016). Phosphoproteomics in the Age of Rapid and Deep Proteome Profiling. Analytical Chemistry, 88, 74–94; [DOI] [PMC free article] [PubMed] [Google Scholar]; Nita-Lazar A, Saito-Benz H, & White FM (2008). Quantitative phosphoproteomics by mass spectrometry: past, present, and future. Proteomics, 8, 4433–4443; [DOI] [PMC free article] [PubMed] [Google Scholar]; Grimsrud PA, Swaney DL, Wenger CD, Beauchene NA, & Coon JJ (2010). Phosphoproteomics for the masses. Acs Chemical Biology, 5, 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gygi JP, Ramin R, Navarrete-Perea J, Younesi S, Gygi SP, & Paulo JA (2020). A Triple Knockout Isobaric-Labeling Quality Control Platform with an Integrated Online Database Search. Journal of the American Society for Mass Spectrometry, 31, 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson A, Wolmer N, Koncarevic S, Selzer S, Bohm G, Legner H, Schmid P, Kienle S, Penning P, Hohle C, Berfelde A, Martinez-Pinna R, Farztdinov V, Jung S, Kuhn K, & Pike I (2019). TMT-pro: Design, Synthesis, and Initial Evaluation of a Proline-Based Isobaric 16-Plex Tandem Mass Tag Reagent Set. Analytical Chemistry, 91, 15941–15950. [DOI] [PubMed] [Google Scholar]
- 50.Yu Q, Paulo JA, Naverrete-Perea J, McAlister GC, Canterbury JD, Bailey DJ, Robitaille AM, Huguet R, Zabrouskov V, Gygi SP, & Schweppe DK (2020). Benchmarking the Orbitrap Tribrid Eclipse for Next Generation Multiplexed Proteomics. Analytical Chemistry, 92, 6478–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, & Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Analytical Chemistry, 86, 7150–7158; [DOI] [PMC free article] [PubMed] [Google Scholar]; Ting L, Rad R, Gygi SP, Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nature Methods, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boersema PJ, Mohammed S, & Heck AJ (2009). Phosphopeptide fragmentation and analysis by mass spectrometry. Journal of Mass Spectrometry, 44, 861–878. [DOI] [PubMed] [Google Scholar]
- 53.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, & Hunt DF (2004). Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. PNAS, 101, 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olsen JV, Macek B, Lange O, Makarov A, Horning S, & Mann M (2007). Higher-energy C-trap dissociation for peptide modification analysis. Nature Methods, 4, 709–712. [DOI] [PubMed] [Google Scholar]
- 55.Robinson MR, Taliaferro JM, Dalby KN, & Brodbelt JS (2016). 193 nm Ultraviolet Photodissociation Mass Spectrometry for Phosphopeptide Characterization in the Positive and Negative Ion Modes. Journal of Proteome Research, 15, 2739–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frese CK, Zhou H, Taus T, Altelaar AF, Mechtler K, Heck AJ, & Mohammed S (2013). Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD). Journal of Proteome Research, 12, 1520–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frese CK, Altelaar AF, Hennrich ML, Nolting D, Zeller M, Griep-Raming J, Heck AJ, & Mohammed S (2011). Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. Journal of Proteome Research, 10, 2377–2388. [DOI] [PubMed] [Google Scholar]
- 58.Palumbo AM, & Reid GE (2008). Evaluation of gas-phase rearrangement and competing fragmentation reactions on protein phosphorylation site assignment using collision induced dissociation-MS/MS and MS3. Analytical Chemistry, 80, 9735–9747; [DOI] [PubMed] [Google Scholar]; Villen J, Beausoleil SA, & Gygi SP (2008). Evaluation of the utility of neutral-loss-dependent MS3 strategies in large-scale phosphorylation analysis. Proteomics, 8, 4444–4452; [DOI] [PMC free article] [PubMed] [Google Scholar]; Wan Y, Cripps D, Thomas S, Campbell P, Ambulos N, Chen T, & Yang A (2008). Phospho-Scan: a probability-based method for phosphorylation site prediction using MS2/MS3 pair information. Journal of Proteome Research, 7, 2803–2811; [DOI] [PMC free article] [PubMed] [Google Scholar]; Jiang X, Han G, Feng S, Jiang X, Ye M, Yao X, & Zou H (2008). Automatic validation of phosphopeptide identifications by the MS2/MS3 target-decoy search strategy. Journal of Proteome Research, 7, 1640–1649; [DOI] [PubMed] [Google Scholar]; Lee J, Xu Y, Chen Y, Sprung R, Kim SC, Xie S, & Zhao Y (2007). Mitochondrial phosphoproteome revealed by an improved IMAC method and MS/MS/MS. Molecular and Cellular Proteomics, 6, 669–6767 [DOI] [PMC free article] [PubMed] [Google Scholar]; Amoresano A, Monti G, Cirulli C, & Marino G (2006). Selective detection and identification of phosphopeptides by dansyl MS/MS/MS fragmentation. Rapid Communications in Mass Spectrometry: Rcm, 20, 1400–1404; [DOI] [PubMed] [Google Scholar]; Wolschin F, Lehmann U, Glinski M, & Weckwerth W (2005). An integrated strategy for identification and relative quantification of site-specific protein phosphorylation using liquid chromatography coupled to MS2/MS3. Rapid Communications in Mass Spectrometry: Rcm, 19, 3626–3632. [DOI] [PubMed] [Google Scholar]
- 59.Jiang X, Bomgarden R, Brown J, Drew DL, Robitaille AM, Viner R, & Huhmer AR (2017). Sensitive and Accurate Quantitation of Phosphopeptides Using TMT Isobaric Labeling Technique. Journal of Proteome Research, 16, 4244–4252; [DOI] [PubMed] [Google Scholar]; Fischer J, Dos Santos MDM, Marchini FK, Barbosa VC, Carvalho PC, & Zanchin NIT (2015). A scoring model for phosphopeptide site localization and its impact on the question of whether to use MSA. Journal of Proteomics, 129, 42–50; [DOI] [PubMed] [Google Scholar]; Wiese H, Kuhlmann K, Wiese S, Stoepel NS, Pawlas M, Meyer HE, Stephan C, Eisenacher M, Drepper F, & Warscheid B (2014). Comparison of alternative MS/MS and bioinformatics approaches for confident phosphorylation site localization. Journal of Proteome Research, 13, 1128–1137; [DOI] [PubMed] [Google Scholar]; Linke D, Hung CW, Cassidy L, & Tholey A (2013). Optimized fragmentation conditions for iTRAQ-labeled phosphopeptides. Journal of Proteome Research, 12, 2755–2763; [DOI] [PubMed] [Google Scholar]; Vandenbogaert M, Hourdel V, Jardin-Mathe O, Bigeard J, Bonhomme L, Legros V, Hirt H, Schwikowski B, & Pflieger D (2012). Automated phosphopeptide identification using multiple MS/MS fragmentation modes. Journal of Proteome Research, 11, 5695–6703; [DOI] [PubMed] [Google Scholar]; Palumbo AM, Smith SA, Kalcic CL, Dantus M, Stemmer PM, & Reid GE (2011). Tandem mass spectrometry strategies for phosphoproteome analysis. Mass Spectrometry Reviews, 30, 600–625. [DOI] [PubMed] [Google Scholar]
- 60.Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, & Coon JJ (2004). A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Analytical Chemistry, 76, 3590–3598. [DOI] [PubMed] [Google Scholar]
- 61.Ulintz PJ, Yocum AK, Bodenmiller B, Aebersold R, Andrews PC, & Nesvizhskii AI (2009). Comparison of MS(2)-only, MSA, and MS(2)/MS(3) methodologies for phosphopeptide identification. Journal of Proteome Research, 8, 887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu F, van Breukelen B, & Heck AJ (2014). Facilitating protein disulfide mapping by a combination of pepsin digestion, electron transfer higher energy dissociation (EThcD), and a dedicated search algorithm SlinkS. Molecular and Cellular Proteomics, 13, 2776–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elias JE, & Gygi SP (2010). Target-decoy search strategy for mass spectrometry-based proteomics. Methods in Molecular Biology, 604, 55–71; [DOI] [PMC free article] [PubMed] [Google Scholar]; Elias JE, & Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods, 4, 207–214. [DOI] [PubMed] [Google Scholar]
- 64.Lim MY, O’Brien J, Paulo JA, & Gygi SP (2017). Improved Method for Determining Absolute Phosphorylation Stoichiometry Using Bayesian Statistics and Isobaric Labeling. Journal of Proteome Research, 16, 4217–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsur D, Tanner S, Zandi E, Bafna V, & Pevzner PA (2005). Identification of post-translational modifications by blind search of mass spectra. Nature Biotechnology, 23, 1562–1567. [DOI] [PubMed] [Google Scholar]
- 66.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, & Nesvizhskii AI (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nature Methods, 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang LH, Li DQ, Fu Y, Wang HP, Zhang JF, Yuan ZF, Sun RX, Zeng R, He SM, & Gao W (2007). pFind 2.0: a software package for peptide and protein identification via tandem mass spectrometry. Rapid Communications in Mass Spectrometry: Rcm, 21, 2985–2891; [DOI] [PubMed] [Google Scholar]; Li D, Fu Y, Sun R, Ling CX, Wei Y, Zhou H, Zeng R, Yang Q, He S, & Gao W (2005). pFind: a novel database-searching software system for automated peptide and protein identification via tandem mass spectrometry. Bioinformatics, 21, 3049–3050. [DOI] [PubMed] [Google Scholar]
- 68.Devabhaktuni A, Lin S, Zhang L, Swaminathan K, Gonzalez CG, Olsson N, Pearlman SM, Rawson K, & Elias JE (2019). TagGraph reveals vast protein modification landscapes from large tandem mass spectrometry datasets. Nature Biotechnology, 37, 469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Solntsev SK, Shortreed MR, Frey BL, & Smith LM (2018). Enhanced Global Post-translational Modification Discovery with MetaMorpheus. Journal of Proteome Research, 17, 1844–1851. [DOI] [PubMed] [Google Scholar]
- 70.Tarasova IA, Chumakov PM, Moshkovskii SA, & Gorshkov MV (2019). Profiling modifications for glioblastoma proteome using ultra-tolerant database search: Are the peptide mass shifts biologically relevant or chemically induced? Journal of Proteomics, 191, 16–21. [DOI] [PubMed] [Google Scholar]
- 71.Yu FC, Teo GC, Kong AT, Haynes SE, Avtonomov DM, Geiszler DJ, & Nesvizhskii AI (2020). Identification of modified peptides using localization-aware open search.Nature communications, 11, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]; Lu L, Riley NM, Shortreed MR, Bertozzi CR, & Smith LM (2020). O-Pair Search with MetaMorpheus for O-glycopeptide characterization. Nature Methods, 17, 1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Savitski MM, Lemeer S, Boesche M, Lang M, Mathieson T, Bantscheff M, & Kuster B (2011). Confident phosphorylation site localization using the Mascot Delta Score. Molecular and Cellular Proteomics, 10, M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beausoleil SA, Villen J, Gerber SA, Rush J, & Gygi SP (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nature Biotechnology, 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- 74.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, & Mann M (2006). Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell, 127, 635–638. [DOI] [PubMed] [Google Scholar]
- 75.Ruttenberg BE, Pisitkun T, Knepper MA, & Hoffert JD (2008). PhosphoScore: an open-source phosphorylation site assignment tool for MSn data. Journal of Proteome Research, 7, 3054–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Swaney DL, Wenger CD, Thomson JA, & Coon JJ (2009). Human embryonic stem cell phosphoproteome revealed by electron transfer dissociation tandem mass spectrometry. PNAS, 106, 995–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, & Mechtler K (2011). Universal and confident phosphorylation site localization using phosphoRS. Journal of Proteome Research, 10, 5354–5362. [DOI] [PubMed] [Google Scholar]
- 78.Fermin D, Avtonomov D, Choi H, & Nesvizhskii AI (2015). LuciPHOr2: site localization of generic post-translational modifications from tandem mass spectrometry data. Bioinformatics, 31, 1141–1143; [DOI] [PMC free article] [PubMed] [Google Scholar]; Fermin D, Walmsley SJ, Gingras AC, Choi H, & Nesvizhskii AI (2013). LuciPHOr: algorithm for phosphorylation site localization with false localization rate estimation using modified target-decoy approach. Molecular and Cellular Proteomics, 12, 3409–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Potel CM, Lemeer S, & Heck AJR (2019). Phosphopeptide Fragmentation and Site Localization by Mass Spectrometry: An Update. Analytical Chemistry, 91, 126–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baker PR, Trinidad JC, & Chalkley RJ (2011). Modification site localization scoring integrated into a search engine. Molecular and Cellular Proteomics, 10, M111.008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, & Mann M (2010). Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Science signaling, 3, ra3. [DOI] [PubMed] [Google Scholar]
- 82.Wu R, Haas W, Dephoure N, Huttlin EL, Zhai B, Sowa ME, & Gygi SP (2011). A large-scale method to measure absolute protein phosphorylation stoichiometries. Nature Methods, 8, 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Paulo JA, & Gygi SP (2015). A comprehensive proteomic and phosphoproteomic analysis of yeast deletion mutants of 14-3-3 orthologs and associated effects of rapamycin. Proteomics, 15, 474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Paulo JA, Gaun A, & Gygi SP (2015). Global Analysis of Protein Expression and Phosphorylation Levels in Nicotine-Treated Pancreatic Stellate Cells. Journal of Proteome Research, 14, 4246–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ganter M, Goldberg JM, Dvorin JD, Paulo JA, King JG, Tripathi AK, Paul AS, Yang J, Coppens I, Jiang RH, Elsworth B, Baker DA, Dinglasan RR, Gygi SP, & Duraisingh MT (2017). Plasmodium falciparum CRK4 directs continuous rounds of DNA replication during schizogony. Nat. Microbiol, 2, 17017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li J, Paulo JA, Nusinow DP, Huttlin EL, & Gygi SP (2019). Investigation of Proteomic and Phosphoproteomic Responses to Signaling Network Perturbations Reveals Functional Pathway Organizations in Yeast. Cell reports, 29, 2092–2104.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hebert AS, Prasad S, Belford MW, Bailey DJ, McAlister GC, Abbatiello SE, Huguet R, Wouters ER, Dunyach JJ, Brademan DR, Westphall MS, & Coon JJ (2018). Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Analytical Chemistry, 90, 9529–9537; [DOI] [PMC free article] [PubMed] [Google Scholar]; Prasad S, Belford MW, Dunyach JJ, & Purves RW (2014). On an aerodynamic mechanism to enhance ion transmission and sensitivity of FAIMS for nanoelectrospray ionization-mass spectrometry. Journal of the American Society for Mass Spectrometry, 25, 2143–2153; [DOI] [PubMed] [Google Scholar]; Purves RW, Prasad S, Belford M, Vandenberg A, & Dunyach JJ (2017). Optimization of a New Aerodynamic Cylindrical FAIMS Device for Small Molecule Analysis. Journal of the American Society for Mass Spectrometry, 28, 525–538. [DOI] [PubMed] [Google Scholar]
- 88.Saba J, Bonneil E, Pomies C, Eng K, & Thibault P (2009). Enhanced sensitivity in proteomics experiments using FAIMS coupled with a hybrid linear ion trap/Orbitrap mass spectrometer. Journal of Proteome Research, 8, 3355–3566. [DOI] [PubMed] [Google Scholar]
- 89.Schweppe DK, Prasad S, Belford MW, Navarrete-Perea J, Bailey DJ, Huguet R, Jedrychowski MP, Rad R, McAlister G, Abbatiello SE, Woulters ER, Zabrouskov V, Dunyach JJ, Paulo JA, & Gygi SP (2019). Characterization and Optimization of Multiplexed Quantitative Analyses Using High-Field Asymmetric-Waveform Ion Mobility Mass Spectrometry. Analytical Chemistry, 91, 4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pfammatter S, Bonneil E, McManus FP, & Thibault P (2019). Accurate Quantitative Proteomic Analyses Using Metabolic Labeling and High Field Asymmetric Waveform Ion Mobility Spectrometry (FAIMS). Journal of Proteome Research, 18, 2129–2138; [DOI] [PubMed] [Google Scholar]; Pfammatter S, Bonneil E, McManus FP, Prasad S, Bailey DJ, Belford M, Dunyach JJ, & Thibault P (2018). A Novel Differential Ion Mobility Device Expands the Depth of Proteome Coverage and the Sensitivity of Multiplex Proteomic Measurements. Molecular and Cellular Proteomics, 17, 2051–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao H, Cunningham DL, Creese AJ, Heath JK, & Cooper HJ (2015). FAIMS and Phosphoproteomics of Fibroblast Growth Factor Signaling: Enhanced Identification of Multiply Phosphorylated Peptides. Journal of Proteome Research, 14, 5077–5087. [DOI] [PubMed] [Google Scholar]
- 92.Bridon G, Bonneil E, Muratore-Schroeder T, Caron-Lizotte O, & Thibault P (2012). Improvement of phosphoproteome analyses using FAIMS and decision tree fragmentation. application to the insulin signaling pathway in Drosophila melanogaster S2 cells. Journal of Proteome Research, 11, 927–940. [DOI] [PubMed] [Google Scholar]
- 93.Ogata K, & Ishihama Y (2020). Extending the Separation Space with Trapped Ion Mobility Spectrometry Improves the Accuracy of Isobaric Tag-Based Quantitation in Proteomic LC/MS/MS. Analytical Chemistry, 92, 8037–8040. [DOI] [PubMed] [Google Scholar]
- 94.Paulo JA, Navarrete-Perea J, Guha Thakurta S, & Gygi SP (2019). TKO6: A Peptide Standard To Assess Interference for Unit-Resolved Isobaric Labeling Platforms. Journal of Proteome Research, 18, 565–570; [DOI] [PMC free article] [PubMed] [Google Scholar]; Gygi JP, Yu Q, Navarrete-Perea J, Rad R, Gygi SP, & Paulo JA (2019). Web-Based Search Tool for Visualizing Instrument Performance Using the Triple Knockout (TKO) Proteome Standard. Journal of Proteome Research, 18, 687–693; [DOI] [PMC free article] [PubMed] [Google Scholar]; Paulo JA, O’Connell JD, & Gygi SP (2016). A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. Journal of the American Society for Mass Spectrometry, 27, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schweppe DK, Eng JK, Yu Q, Bailey D, Rad R, Navarrete-Perea J, Huttlin EL, Erickson BK, Paulo JA, & Gygi SP (2020). Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. Journal of Proteome Research, 19, 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Erickson BK, Mintseris J, Schweppe DK, Navarrete-Perea J, Erickson AR, Nusinow DP, Paulo JA, & Gygi SP (2019). Active Instrument Engagement Combined with a Real-Time Database Search for Improved Performance of Sample Multiplexing Workflows. Journal of Proteome Research, 18, 1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sacco F, Seelig A, Humphrey SJ, Krahmer N, Volta F, Reggio A, Marchetti P, Gerdes J, & Mann M (2019). Phosphoproteomics Reveals the GSK3-PDX1 Axis as a Key Pathogenic Signaling Node in Diabetic Islets. Cell metabolism, 29, 1422–1432.e3; [DOI] [PubMed] [Google Scholar]; HaileMariam M, Eguez RV, Singh H, Bekele S, Ameni G, Pieper R, & Yu YB (2018). S-Trap, an Ultrafast Sample-Preparation Approach for Shot-gun Proteomics. Journal of Proteome Research, 17, 2917–2924; [DOI] [PubMed] [Google Scholar]; Muller T, Kalxdorf M, Longuespee R, Kazdal DN, Stenzinger A, & Krijgsveld J (2020). Automated sample preparation with SP3 for low-input clinical proteomics. Molecular Systems Biology, 16, e9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hughes CS, Sorensen PH, & Morin GB (2019). A Standardized and Reproducible Proteomics Protocol for Bottom-Up Quantitative Analysis of Protein Samples Using SP3 and Mass Spectrometry. Methods in Molecular Biology, 1959, 65–87; [DOI] [PubMed] [Google Scholar]; Hughes CS, Moggridge S, Muller T, Sorensen PH, Morin GB, & Krijgsveld J (2019). Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature Protocols, 14, 68–85; [DOI] [PubMed] [Google Scholar]; Moggridge S, Sorensen PH, Morin GB, & Hughes CS (2018). Extending the Compatibility of the SP3 Paramagnetic Bead Processing Approach for Proteomics. Journal of Proteome Research, 17, 1730–1740; [DOI] [PubMed] [Google Scholar]; Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, & Krijgsveld J (2014). Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology, 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Paulo JA, Navarrete-Perea J, & Gygi SP (2020). Multiplexed proteome profiling of carbon source perturbations in two yeast species with SL-SP3-TMT. Journal of Proteomics, 210, 103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhao Y, Liu G, Zambito FC, Zhang YJ, DeSilva BS, Kozhich AT, & Shen JX (2017). A multiplexed immunocapture liquid chromatography tandem mass spectrometry assay for the simultaneous measurement of myostatin and GDF-11 in rat serum using an automated sample preparation platform. Analytica Chimica Acta, 979, 36–44; [DOI] [PubMed] [Google Scholar]; Ruelcke JE, Loo D, & Hill MM (2016). Reducing the cost of semi-automated in-gel tryptic digestion and GeLC sample preparation for high-throughput proteomics. Journal of Proteomics, 149, 3–6; [DOI] [PubMed] [Google Scholar]; Ippoliti PJ, Kuhn E, Mani DR, Fagbami L, Keshishian H, Burgess MW, Jaffe JD, & Carr SA (2016). Automated Microchromatography Enables Multiplexing of Immunoaffinity Enrichment of Peptides to Greater than 150 for Targeted MS-Based Assays. Analytical Chemistry, 88, 7548–7555. [DOI] [PubMed] [Google Scholar]
- 101.Yu Q, Xiao H, Jedrychowski MP, Schweppe DK, Navarrete-Perea J, Knott J, Rogers J, Chouchani ET, & Gygi SP (2020). Sample multiplexing for targeted pathway proteomics in aging mice. PNAS, 117, 9723–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kalagiri R, Adam K, & Hunter T (2020). Empirical Evidence of Cellular Histidine Phosphorylation by Immunoblotting Using pHis mAbs. Methods in Molecular Biology, 2077, 181–191; [DOI] [PMC free article] [PubMed] [Google Scholar]; Hardman G, & Eyers CE (2020). High-Throughput Characterization of Histidine Phosphorylation Sites Using UPAX and Tandem Mass Spectrometry. Methods in Molecular Biology, 2077, 225–235; [DOI] [PubMed] [Google Scholar]; Attwood PV (2020). A Quantitative Method for the Measurement of Protein Histidine Phosphorylation. Methods in Molecular Biology, 2077, 51–61; [DOI] [PubMed] [Google Scholar]; Potel CM, Lin MH, Heck AJR, & Lemeer S (2018). Widespread bacterial protein histidine phosphorylation revealed by mass spectrometry-based proteomics. Nature Methods, 15, 187–190; [DOI] [PubMed] [Google Scholar]; Fuhs SR, & Hunter T (2017). pHisphorylation: the emergence of histidine phosphorylation as a reversible regulatory modification. Current Opinion in Cell Biology, 45, 8; [DOI] [PMC free article] [PubMed] [Google Scholar]; Sun Q, & Julian RR (2011). Probing sites of histidine phosphorylation with iodination and tandem mass spectrometry. Rapid Communications in Mass Spectrometry: Rcm, 25, 2240–2246; [DOI] [PubMed] [Google Scholar]; Zu XL, Besant PG, Imhof A, & Attwood PV (2007). Mass spectrometric analysis of protein histidine phosphorylation. Amino Acids, 32, 347–357; [DOI] [PubMed] [Google Scholar]; Sickmann A, & Meyer HE (2007). Histidine Phosphorylation Site Identification by ESI-MS. CSH protocols, 2007, pdb prot4628. [DOI] [PubMed] [Google Scholar]
- 103.Hardman G, Perkins S, Brownridge PJ, Clarke CJ, Byrne DP, Campbell AE, Kalyuzhnyy A, Myall A, Eyers PA, Jones AR, & Eyers CE (2019). Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. Embo Journal, 38, e100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Linding R, Jensen LJ, Pasculescu A, Olhovsky M, Colwill K, Bork P, Yaffe MB, & Pawson T (2008). NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic Acids Research, 36, D695–D699; [DOI] [PMC free article] [PubMed] [Google Scholar]; Munk S, Refsgaard JC, Olsen JV, & Jensen LJ (2016). From Phosphosites to Kinases. Methods in Molecular Biology, 1355, 307–321; [DOI] [PubMed] [Google Scholar]; Yaffe MB, Leparc GG, Lai J, Obata T, Volinia S, & Cantley LC (2001). A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nature Biotechnology, 19, 348–353. [DOI] [PubMed] [Google Scholar]
- 105.Kunz RC, McAllister FE, Rush J, & Gygi SP (2012). A high-throughput, multiplexed kinase assay using a benchtop orbitrap mass spectrometer to investigate the effect of kinase inhibitors on kinase signaling pathways. Analytical Chemistry, 84, 6233–6239; [DOI] [PubMed] [Google Scholar]; Kubota K, Anjum R, Yu Y, Kunz RC, Andersen JN, Kraus M, Keilhack H, Nagashima K, Krauss S, Paweletz C, Hendrickson RC, Feldman AS, Wu CL, Rush J, Villen J, & Gygi SP (2009). Sensitive multiplexed analysis of kinase activities and activity-based kinase identification. Nature Biotechnology, 27, 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leutert M, Rodríguez-Mias RA, Fukuda NK, & Villén J (2019). R2-P2 rapid-robotic phosphoproteomics enables multidimensional cell signaling studies. Molecular Systems Biology, 15, 12, 10.15252/msb.20199021. [DOI] [PMC free article] [PubMed] [Google Scholar]