

Abstract
Macrophages play a central role in the pathogenesis of atherosclerosis. The inflammatory properties of these cells are dictated by their metabolism, of which the mechanistic target of rapamycin (mTOR) signaling pathway is a key regulator. Using myeloid cell-specific nanobiologics in apolipoprotein E-deficient (Apoe−/−) mice, we found that targeting the mTOR and ribosomal protein S6 kinase-1 (S6K1) signaling pathways rapidly diminished plaque macrophages’ inflammatory activity. By investigating transcriptome modifications, we identified Psap, a gene encoding for the lysosomal protein prosaposin, as closely related with mTOR signaling. Subsequent in vitro experiments revealed that Psap inhibition suppressed both glycolysis and oxidative phosphorylation. Transplantation of Psap−/− bone marrow to low-density lipoprotein receptor knock-out (Ldlr−/−) mice led to a reduction of atherosclerosis development and plaque inflammation. Finally, we confirmed the relationship between PSAP expression and inflammation in human carotid atherosclerotic plaques. Our findings provide mechanistic insights into the development of atherosclerosis, and identify prosaposin as a potential therapeutic target.
Single sentence summary:
Prosaposin mediates inflammation in experimental and human atherosclerosis and is associated with cellular metabolism and mTOR signaling.
Introduction
Atherosclerosis is a lipid-induced chronic inflammatory condition and the underlying cause of myocardial infarction and stroke. It is caused by the focal accumulation of lipoproteins in the arterial subendothelial space. After oxidative modification, lipoproteins act as danger associated molecular patterns (DAMPs) triggering an inflammatory response with macrophages as the main protagonists (1).
The activation of macrophages by oxidized low-density lipoprotein (oxLDL) and cholesterol crystals is an energy-demanding process and requires adjustment of their metabolism (2–4). Recent studies revealed that metabolic reprogramming dictates the phenotype and inflammatory response of macrophage subsets in plaques, and that the metabolic signature of macrophages is associated with the vulnerability of plaques for rupture (3–5). Unraveling the regulation of plaque macrophage metabolism is therefore of fundamental importance and may uncover new targets for therapy.
In the present study we investigated the mechanistic target of rapamycin (mTOR) signaling pathway in plaque macrophages. mTOR orchestrates cell metabolism and inflammatory activity in macrophages (6). However, mTOR’s role in regulating immunometabolism in atherosclerosis is poorly understood (6). Here, we investigated the role of mTOR signaling in atherosclerosis-prone apolipoprotein E-deficient (Apoe−/−) mice, through inhibition of mTOR or its downstream target ribosomal protein S6 kinase-1 (S6K1). To achieve specific inhibition, we intravenously administered two different myeloid cell-specific nanobiologics that respectively target mTOR or S6K1. We observed a consistent reduction of plaque inflammation across multiple modalities and readouts. Subsequently, we unraveled the molecular mechanisms underlying this anti-inflammatory effect by transcriptome analyses of myeloid cells isolated from plaques. Psap surfaced as a key-regulating gene. This gene encodes prosaposin, a highly conserved lysosomal protein involved in glycosphingolipid metabolism. As its role in atherosclerosis is unknown, we set out to study how prosaposin mediates inflammation in monocytes and macrophages. We performed in vitro metabolic analyses and studies on plaque development using Psap knockout mice. In addition, we investigated the role of PSAP in human atherosclerosis by functional assays, histology and single-cell transcriptome analysis of plaques.
Results
mTOR inhibitor and S6K1 inhibitor nanobiologic therapies reduce plaque inflammation
In addition to monocytes and macrophages, other cell types including T cells, endothelial cells and smooth muscle cells play pivotal roles in the pathogenesis of atherosclerosis (1). mTOR signaling is essential to cell metabolism (7) and systemic mTOR inhibition will affect all cell types involved in atherogenesis. We aimed to investigate the effect of inhibiting the mTOR pathway specifically in monocytes and macrophages. To achieve this, we used apolipoprotein A1-based nanobiologics that facilitate drug delivery to myeloid cells with high targeting efficiency (8, 9). We employed a nanobiologic containing the mTOR inhibitor rapamycin (mTORi-NB) and a newly developed nanobiologic containing the S6K1 inhibitor PF-4708671 (S6K1i-NB) (10) (fig. S1A). Ex vivo near-infrared fluorescence (NIRF) imaging performed 24 hours after intravenous administration in Apoe−/− mice showed that DiIC18(7) (DiR)-labeled nanobiologics primarily accumulated in the liver, spleen and kidneys (fig. S1, B and C). High DiR uptake was observed in the aortic sinus area (fig. S1D), which is the preferential site of plaque development in this mouse model. Immune cell specificity was evaluated by flow cytometry using mTORi-NB and S6K1i-NB labeled with the fluorophore DiOC18(3) (DiO). Similar to previous studies (8, 9), both nanobiologics were predominantly taken up by aortic macrophages, Ly6Chi monocytes, neutrophils, and dendritic cells (fig. S1, E and F). Non-myeloid cells (Lin+) took up a negligible amount of the nanobiologics. In blood, spleen and bone marrow, we identified a similar myeloid cell-biased uptake pattern (fig. S1G).
We studied the effect of mTORi- and S6K1i-NB treatment on plaque inflammation in 20-week old Apoe−/− mice that had been fed a Western diet (WD) for 12 weeks to develop atherosclerotic lesions. While they remained on a WD, all mice were treated with four intravenous injections of phosphate-buffered saline (PBS), unloaded nanobiologics, mTORi-NB (containing 5 mg/kg rapamycin), or S6K1i-NB (containing 5 mg/kg PF-4708671) over the course of one week (Fig. 1A). We verified that mTORi-NB treatment did not affect serum cholesterol (fig. S1H). Mice treated with mTORi-NB had a 14% (P<0.0001) and 9% (P=0.006) smaller plaque size as compared to animals treated with PBS and unloaded NB, respectively (Fig. 1, B to D). Plaque collagen content was not affected by mTORi-NB, whereas macrophage content was reduced by 33% (P = 0.013) and 34% (P = 0.004) as compared to both control groups (Fig. 1, E and F, fig. S2A). S6K1i-NB treatment showed a similar effect on plaque inflammation with a 20% (P = 0.046) reduction in plaque macrophage content as compared to PBS-treated mice (Fig. 1, E and F), whereas no effect on plaque size and collagen content was observed (Fig. 1, B to D and fig. S2A). These data indicate that mTORi-NB and S6K1i-NB treatment ameliorated plaque vulnerability by reducing macrophage-rich areas without affecting collagen content (fig. S2B).
Fig. 1. Myeloid-specific mTOR inhibition reduces atherosclerotic plaque inflammation.
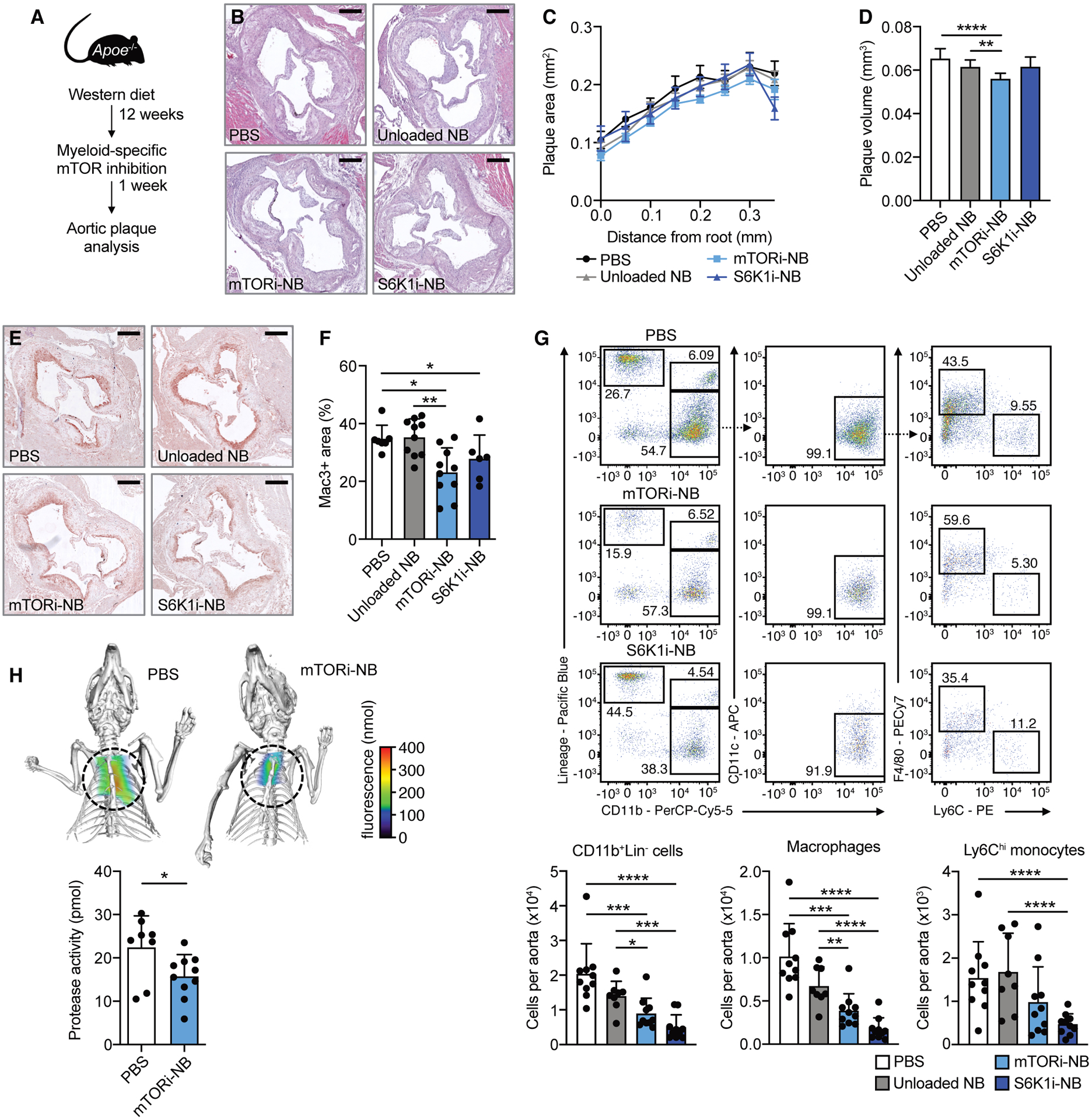
Apoe−/− mice were fed a Western diet for 12 weeks, followed by 1 week of treatment, while continuing the diet. Treatment consisted of 4 intravenous injections of PBS, mTORi-NB (rapamycin at 5 mg/kg), S6K1i-NB (PF-4708671 at 5 mg/kg) or unloaded nanobiologics (NB, at a comparable dose). See schematic in (A). (B) Representative images of H&E-stained aortic roots, scale bar = 250 μm. (C) Histologic quantification of plaque area at set distances from the aortic root, presented as mean ± SEM (n = 6–10 mice/group). (D) Lesion volume was calculated as area under the curve in C. (E) Representative Mac3-stained aortic roots (scale bar = 250 μm) and (F) quantification of Mac3+ area of treated mice (n = 6–10 mice/group). (G) Representative flow cytometry plots and quantification of CD11b+Lin− cells, macrophages (CD11b+Lin− CD11c−F4/80+Ly6Clo) and Ly6Chi monocytes (CD11b+Lin−CD11c−F4/80−Ly6Chi) in the aorta (n = 8–10 mice/group). (H) FMT/CT imaging of protease activity in the aortic root of PBS or mTORi-NB-treated mice (n = 8–10 mice/group). Experiments were performed once. Data are presented as mean ± SD unless otherwise stated. ANOVA with Dunnett’s correction was used in D, non-parametric Mann-Whitney U tests were applied in F, G and H. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The histology results were corroborated by flow cytometry of whole aortas. After one week of treatment, mTORi-NB reduced the number of aortic CD11b+Lin− cells (monocytes and macrophages) by 56% (P = 0.0005) and 36% (P = 0.027), as compared to PBS and unloaded nanobiologics, respectively (Fig. 1G and fig. S2C). This effect was mainly driven by a reduction in plaque macrophages. Aortic CD11b+Lin− cells were also markedly decreased in the S6K1i-NB treated mice, by 76% (P < 0.0001) and 65% (P = 0.0005) in comparison to PBS and unloaded nanobiologic-treated groups (Fig. 1G and fig. S2C). In this treatment group both macrophage and Ly6Chi monocyte numbers were reduced. Analysis of myeloid cell populations in the bone marrow, spleen and peripheral blood indicated that the inhibition of plaque inflammation could not be explained by suppressed myelopoiesis, as neutrophils, Ly6Clo and Ly6Chi monocytes were equal or increased in the nanobiologic-treated mice (fig. S3A to C).
To test the plaque’s inflammatory activity, we performed in vivo fluorescence molecular tomography with computed tomography (FMT-CT) imaging to quantify protease activity in the aortic sinus area. We used the same mouse model and treatment regimen as described above (Fig. 1A). Control- and mTORi-NB-treated Apoe−/− mice received a single injection of an activatable pan-cathepsin protease sensor 24 hours before imaging. The protease sensor is taken up by activated macrophages and cleaved in the endolysosome (11), yielding fluorescence as a function of enzyme activity. mTORi-NB reduced protease activity by 30% compared to PBS control (P = 0.03, Fig. 1H).
Taken together, these data show that myeloid-specific inhibition of the mTOR signaling pathway rapidly reduces inflammatory activity in atherosclerotic lesions. This incentivized us to unravel the underlying molecular mechanisms.
mTOR and S6K1 inhibition downregulate Psap in plaque macrophages
To gain insight into the mechanism by which mTOR-S6K1 signaling affects monocytes and macrophages in atherosclerosis, we used laser capture microdissection (LCM) to isolate CD68+ cells from aortic sinus plaques of Apoe−/− mice that were treated for one week with either PBS, mTORi-NB or S6K1i-NB. Total RNA of these cells was isolated for sequencing and whole transcriptome analysis.
First, we assessed whether the reduced plaque monocyte and macrophage burden could be explained by diminished monocyte recruitment or was potentially mediated by autophagy, since the latter can be induced by mTOR inhibition (12). Both mechanisms did not provide a satisfactory explanation. We found no inhibiting effect of our nanobiologic treatments on chemokine-related gene expression (tables S1 and S2). mTORi-NB treatment did not affect autophagy related gene expression (table S3). In the S6K1i-NB treated group, five autophagy-related genes were differentially expressed, of which two were up-regulated and three were down-regulated (table S4). The current data do not allow for definitive conclusions regarding the effects of our nanobiologics on autophagy. Autophagy plays a crucial role in atherosclerosis, and potential effects of our nanobiologics on this process require further investigation in future studies.
Subsequently, we adopted a systems biology approach of weighted gene co-expression network analysis (WGCNA) in which a co-expression network is constructed based on expression correlation between genes. We used topological overlap matrix (TOM) plots to show correlations among all genes, in which increased color intensity indicates strong correlation coefficients between genes. To identify modules with groups of strongly co-expressed genes, we used linkage hierarchical clustering to group genes based on their topological overlap with other genes. We then ranked the modules by the significance of enrichment with the differentially expressed genes (DEGs) between treatments and controls. For both the mTORi-NB- and S6K1i-NB-treated mice, the turquoise modules were of highest interest, as DEGs were most significantly enriched in these modules. The mTORi-NB turquoise module contained 1052 genes, which significantly enriched with 46% of the DEGs (fold enrichment = 5.90, adjusted P = 9.75×10−20, Fig. 2, A and B). The S6K1i-NB turquoise module consisted of 1825 genes, which significantly enriched with 51% of the DEGs (fold enrichment = 3.76, adjusted P = 1.80×10−154, Fig. 2, C and D). Gene ontology analysis of both turquoise modules showed the most pronounced enrichment of genes in cellular processes (GO:0009987) and metabolic processes (GO:0008152).
Fig. 2. Effect of mTOR inhibition on plaque macrophage transcriptome.
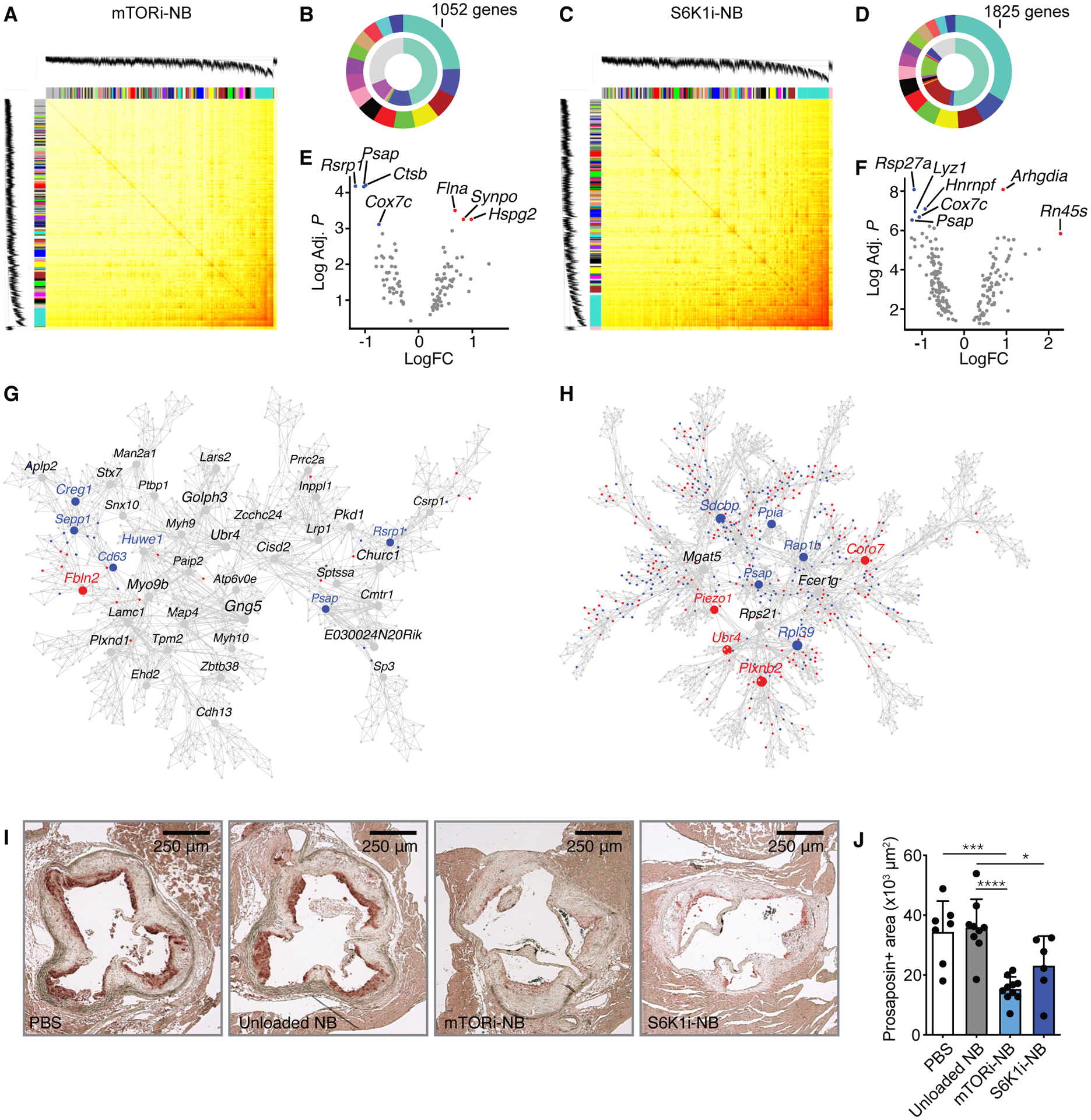
Transcriptome analysis was performed on CD68+ cells isolated from aortic roots of Apoe−/− mice after mTORi-NB (A,B,E,G) or S6K1i-NB (C,D,F,H) treatment, as compared to PBS. (n = 8–10 mice/group). (A,C) Topological overlap matrix. Each row and column of the heatmap represent a single gene, with the color intensity indicating the network connection strength. The dendrograms on the upper and left sides show the hierarchical clustering tree of genes. (B,D) The fifteen modules with the highest connectivity are ordered by size (outer ring). The inner ring shows differentially expressed genes within a module, as a percentage of total number of differentially expressed genes. (E,F) Volcano plot of genes within the turquoise module with the highest connectivity. Hub gene are identified based on p-value and fold change. The up- and downregulated hub genes are shown in red and blue, respectively. (G,H) Multiscale embedded gene co-expression network analysis (MEGENA) of the turquoise module. Up- and downregulated genes are shown in red and blue, respectively. (I,J) Apoe−/− mice were fed a Western diet for 12 weeks, followed by 1 week of treatment, while kept on a Western diet. Treatment consisted of 4 intravenous injections of PBS, mTORi-NB (rapamycin at 5 mg/kg), S6K1i-NB (PF-4708671 at 5 mg/kg) or unloaded nanobiologics (NB, at a comparable dose). Aortic roots were harvested for histological analysis. (I) Representative images of prosaposin staining of the aortic root and (J) quantification of prosaposin-positive areas within the plaque (n = 6–10 mice/group). Experiments were performed once. Data are presented as mean ± SD, non-parametric Mann-Whitney U test was used in J. *P < 0.05, ***P < 0.001, ****P < 0.0001.
Next, we identified intramodular hub genes in both the mTORi-NB and S6K1i-NB turquoise modules. For this purpose, we selected the top 10% genes of the turquoise module with the highest connectivity index. Of these highly connected genes, the ones with highest significance and fold change in expression (Fig. 2, E and F) were considered likely to be key regulators in the modules and may provide important biological insights (13). We identified four down-regulated hub genes (Psap, Cox7c, Rsrp1 and Ctsb) and three up-regulated hub genes (Flna, Synpo and Hspg2) (Fig. 2E, table S5) in the mTORi-NB turquoise module. In the S6K1i-NB turquoise module we identified five down-regulated hub genes (Psap, Cox7c, Hnmpf, Rps27a, Lyz1) and two up-regulated hub genes (Arhgdia, Rn45s) (Fig. 2F, table S6). Psap and Cox7c were consistently down-regulated by both mTOR and S6K1 inhibition. Psap encodes prosaposin, which is a proprotein for the saposins A to D, and is essential in lysosomal glycosphingolipid degradation (14). Cox7c encodes the cytochrome c oxidase subunit 7C which is a component of the mitochondrial respiratory chain. Next, we performed a multiscale embedded gene co-expression network analysis (MEGENA) of the turquoise modules, as an additional method to identify biologically meaningful hub genes. Although Cox7c was not identified by this analysis, Psap was confirmed as an important hub gene that was downregulated in response to both mTOR and S6K1 inhibition (Fig. 2, G and H), making Psap the prime candidate for further analysis. So far, its role in atherosclerosis is unknown.
The decrease in Psap transcription that we observed in the transcriptome analysis also translated into diminished protein expression of prosaposin. We performed histologic staining of prosaposin on cross-sections of the aortic sinus area of Apoe−/− mice with advanced lesions. Indeed, we observed widespread prosaposin expression in plaques (Fig. 2I), with a high degree of macrophage co-localization (fig. S4). Prosaposin expression in plaques of mice that were treated with either mTORi-NB or S6K1i-NB was reduced by 57% and 35%, respectively, as compared to unloaded nanobiologics (Fig. 2J).
Silencing Psap expression affects immunometabolism
Stimulation of monocytes with oxLDL upregulates aerobic glycolysis as well as oxidative phosphorylation (15). Since we found that the effects of mTOR inhibition were associated with Psap, we investigated whether Psap expression affects cellular metabolism. For this purpose, we formulated lipid nanoparticles containing small interfering RNA that targets Psap (Psap siRNA-LNPs), as previously described (16). The effect of Psap siRNA-LNPs on metabolic reprogramming was assessed in vitro in bone marrow-derived macrophages by extracellular flux analysis. Changes in extracellular acidification rate (ECAR) in response to glucose and oligomycin (OM) injection were used to calculate glycolysis parameters. Changes in oxygen consumption rate (OCR) in response to OM, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), and rotenone (ROT) + antimycin A (AA) injection were used to calculate oxidative phosphorylation parameters. We found that silencing of Psap expression suppressed glycolysis (Fig. 3A) as well as oxidative phosphorylation parameters (Fig. 3B), when compared to control (media only) and control (Ctrl) siRNA-LNPs. These results mirror the effects of mTOR inhibition (rapamycin) and S6K1 inhibition (PF-4708671) on glycolysis (Fig. 3C) and oxidative phosphorylation (Fig. 3D). Together these data indicate that Psap expression affects cell metabolism, which may explain the mechanism by which Psap regulates inflammatory activity of monocytes and macrophages.
Fig. 3. Psap affects immunometabolism.
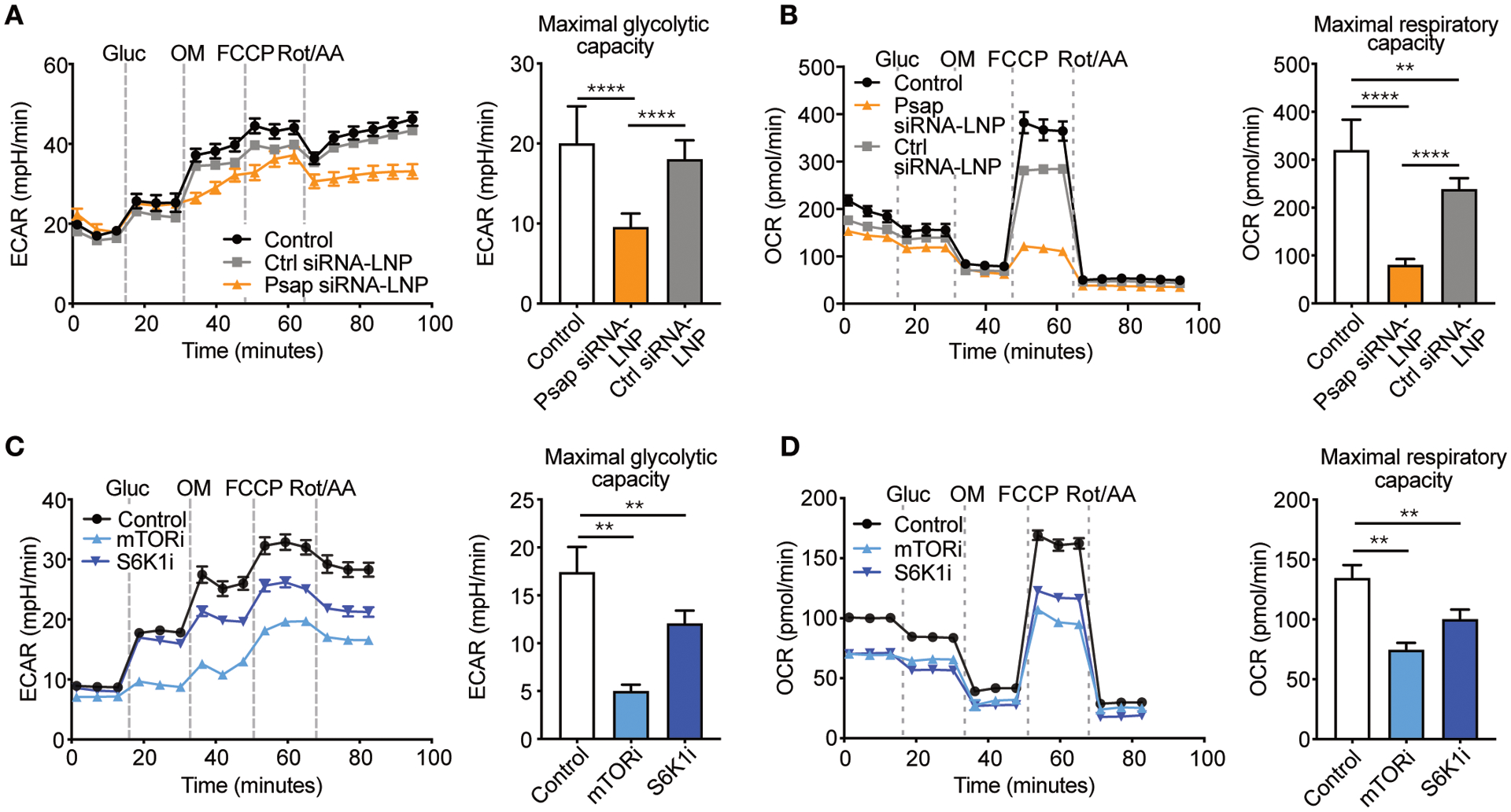
(A–B) Murine bone marrow-derived macrophages were incubated with Psap small interfering RNA lipid nanoparticles (Psap siRNA-LNPs) or control (Ctrl) siRNA-LNPs and subjected to a metabolic assay (n = 10 wells/condition). Maximal glycolytic capacity (A) and maximal respiratory capacity (B) of murine bone marrow-derived macrophages. (C–D) Murine bone marrow-derived macrophages were incubated with mTOR or S6K1 inhibitors (both 20 μM) and subjected to a metabolic assay (n = 6 wells/condition). Maximal glycolytic capacity (C) and maximal respiratory capacity (D). Experiments were performed once. Line graphs are presented as mean ± SEM, bar graphs are presented as mean ± SD, non-parametric Mann-Whitney U tests were used. **P < 0.01, ****P < 0.0001. Gluc, glucose; OM, oligomycin; FCCP, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; Rot/AA, roterone/antimycinA; ECAR, extracellular acidification rate; OCR, oxygen consumption rate.
Psap expression mediates inflammation in experimental atherosclerosis
To assess the role of Psap in atherosclerosis development and plaque inflammation, we transplanted bone marrow from Psap−/− mice into lethally irradiated Ldlr−/− mice. Apoe−/− mice are unsuitable for this purpose because transplantation of bone marrow producing apolipoprotein E would affect serum lipids content and prohibit the development of atherosclerosis (17). Mice that received Psap+/+ bone marrow cells served as controls (Figure 4A). Mice were fed a Western diet for 11 weeks to develop atherosclerotic lesions. Serum cholesterol was equal in both groups (fig. S5A). We performed quantitative histologic analysis of plaques in the aortic sinus area by serial cross sectioning at set distances from the aortic root. Cross-sections were stained with Hematoxylin and Eosin (H&E, Fig. 4B). Lesion volume was calculated from the area under the curve (Fig. 4C). Mice receiving Psap−/− bone marrow showed a 22.8% (P<0.0001) reduction in plaque volume as compared to mice transplanted with Psap+/+ bone marrow (Fig. 4D). There was no difference in the collagen content of the plaques as assessed by Sirius red staining (Fig. 4, E and F).
Fig. 4. Psap mediates atherosclerotic plaque inflammation in Ldlr−/− mice.
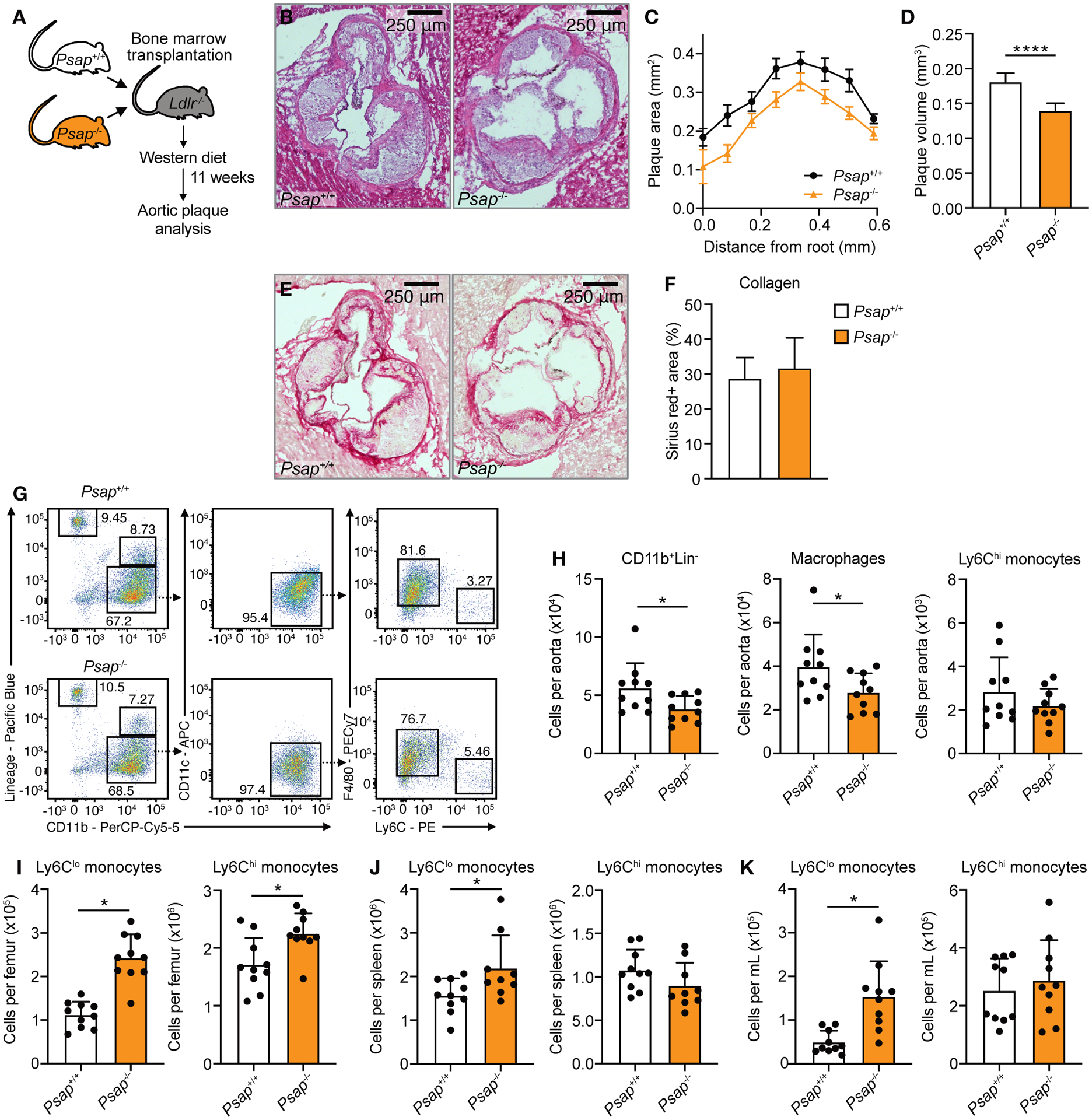
Ldlr−/− mice were lethally irradiated and transplanted with Psap+/+ or Psap−/− bone marrow cells. Mice were left to reconstitute for 6 weeks after which they were put on a Western diet for 11 weeks (n = 10 mice/group for all panels). (A) Schematic of experimental setup. (B) Representative images of H&E-stained aortic roots. (C) Histologic quantification of plaque area at set distances from the aortic root, presented as mean ± SEM. (D) Lesion volume was calculated as area under the curve in C. (E) Representative images of Sirius red-stained aortic roots. (F) Histologic quantification of plaque collagen content. (G) Representative flow cytometry plots and (H) quantification of CD11b+Lin− cells, macrophages (CD11b+Lin−CD11c−F4/80+Ly6Clo) and Ly6Chi monocytes (CD11b+Lin− CD11c−F4/80−Ly6Chi) in the aorta. Quantification of Ly6Clo and Ly6Chi monocytes in the bone marrow (I), spleen (J) and blood (K). Experiments were performed once. Data are presented as mean ± SD unless otherwise stated, non-parametric Mann-Whitney U tests were used. *P < 0.05, ****P < 0.0001.
Subsequently we focused on quantifying immune cells in atherosclerotic lesions by flow cytometry of whole aortas. Again, lethally irradiated Ldlr−/− mice received either Psap−/− or Psap+/+ bone marrow and were fed a Western diet for 11 weeks. Aortic plaques of mice receiving Psap−/− bone marrow contained 32.4% (P = 0.04) fewer CD11b+Lin− cells, primarily caused by a reduction in plaque macrophages, as well as 32.9% (P = 0.02) fewer neutrophils when compared to Psap+/+ transplanted animals (Fig. 4, G and H and fig. S5B). The number of non-myeloid cells (Lin+ cells) was unaffected (fig. S5B). These data indicate a reduction of plaque inflammation in mice that received Psap−/− bone marrow.
We investigated if these changes in plaque size and myeloid cell content were the result of systemic immune effects. There was no difference in numbers of Lin−Sca1+c-kit− (LSK) cells or proliferation rates of multipotent progenitors (MPP) (fig. S5, C to E). Ly6Chi and Ly6Clo monocyte counts were increased in the bone marrow (Fig. 4I), while Ly6Clo monocytes were increased and Ly6Chi monocytes unchanged in the blood and spleen (Fig. 4, J and K). Together, these results show that myelopoiesis in mice receiving Psap−/− bone marrow was not suppressed. The increase in Ly6Clo monocyte counts in the bone marrow, blood and spleen of Psap−/− bone marrow-transplanted mice (Fig. 4, I to K) may contribute to a beneficial effect on plaque inflammation since these cells play a critical role in tissue homeostasis and repair (18). Concerning the neutrophils, we did not observe changes in the circulation, the spleen and bone marrow which could explain the lower neutrophil number in the plaques (fig. S5F).
Together, our data show that plaque inflammation and atherosclerosis development is reduced in Ldlr−/− mice receiving Psap−/− bone marrow. This underscores the data from our transcriptome analysis, supporting that Psap in myeloid cells plays a key role in atherosclerosis.
Prosaposin and inflammation in human atherosclerosis
Prosaposin is highly conserved during evolution and is found in all bony vertebrates (19). Since the function of prosaposin is similar in mice and man, we were interested in the involvement of prosaposin in human atherosclerosis.
First, we investigated the role of prosaposin in oxLDL priming of human myeloid cells. We stimulated primary human monocytes in vitro with oxLDL or RPMI media (control) for 24 hours. Cells rested for 5 days after which they were re-stimulated with lipopolysaccharide (LPS) or Pam3Cys (fig. S6A). OxLDL-primed cells displayed a higher cytokine response as compared to control cells (Fig. 5A and fig. S6B). When oxLDL stimulation was combined with mTORi-NB or S6K1i-NB treatment, priming was prevented (Fig. 5B and fig. S6B), indicating that mTOR-S6K1 signaling is required for oxLDL priming.
Fig. 5. PSAP mediates atherosclerotic plaque inflammation in humans.
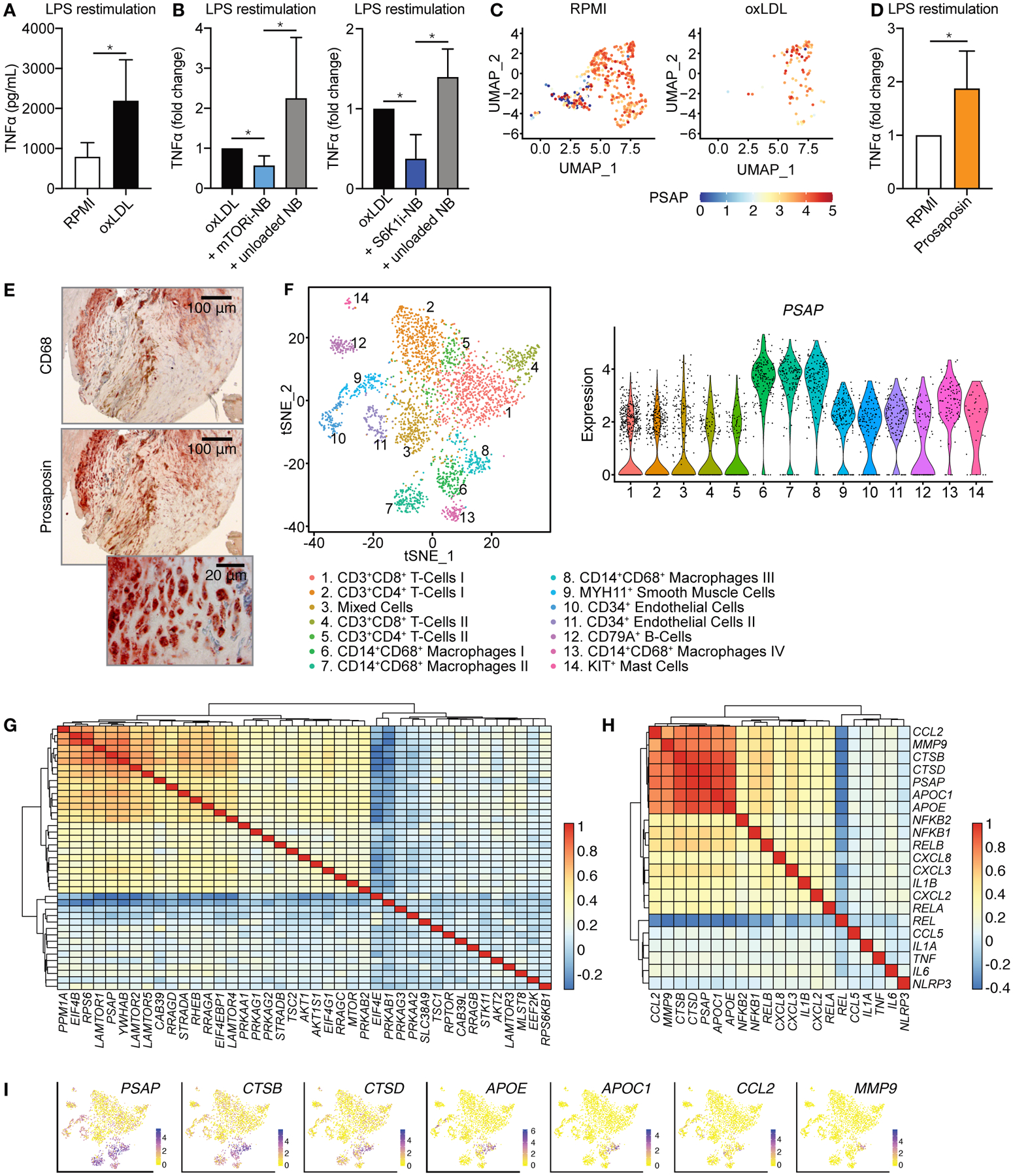
(A-D) Human primary monocytes were incubated with oxidized LDL (oxLDL) or prosaposin for 24 hours. Media only (RPMI) was used as control. After a 5-day rest, cells were restimulated with LPS. (A) TNFα production upon LPS stimulation, as measured by ELISA (n = 6). (B) TNFα production of human monocytes primed with oxLDL in combination with mTORi-NB or S6K1i-NB as compared to unloaded NB or oxLDL only (n = 6). (C) Single cell transcriptome analysis of adherent human monocytes after oxLDL priming and LPS restimulation. Uniform Manifold Approximation and Projection (UMAP) plot shows the different monocyte clusters and PSAP expression is shown for each cell (n = 3). (D) Human monocytes were primed with prosaposin or RPMI (negative control) for 24 hours. After a 5-day rest, cells were restimulated with LPS and TNFα production was measured by ELISA, (n = 5). (E) Representative images of CD68 (top) and prosaposin (middle and bottom) staining on a human carotid endarterectomy sample (n = 4, see also fig. S7). (F) Single-cell RNA sequencing of human atherosclerotic plaques identifies 14 leukocyte subsets. (n = 18). (G,H) Transcriptomic analyses were performed on human atherosclerotic plaques (n = 620). Heatmap depicting co-expression of PSAP and genes involved in (G) the mTOR signaling pathway or (H) atherosclerotic plaque macrophages, clustered based on co-expression values. (I) Expression of 6 inflammatory genes, as compared to PSAP expression, based on single-cell RNA sequencing, also presented in (F). Experiments were performed once. Bar graphs are presented as mean ± SD, Wilcoxon signed-rank test were used in A, B and D. *P < 0.05.
Subsequently, we performed single-cell RNA sequencing of non-primed (RPMI) and oxLDL-primed adherent monocytes that were subsequently stimulated with LPS. We identified cells with low PSAP expression in the non-primed monocytes, whereas PSAP expression was high in nearly all monocytes primed with oxLDL (Fig. 5C). We also observed that the prosaposin protein itself was capable of priming human primary monocytes, as evidenced by the enhanced cytokine production upon re-stimulation with LPS (Fig. 5D).
To investigate prosaposin’s involvement in human atherosclerosis, we obtained carotid plaque specimens from patients undergoing elective endarterectomy and stained them for prosaposin. We confirmed prosaposin’s presence in human plaques and its colocalization with plaque macrophages (Fig. 5E and fig. S7A). We further explored this by transcriptome analysis, utilizing single-cell RNA sequencing of 18 human plaques(20). Here, 14 distinct leukocyte populations were identified, among which we observed four different CD14+CD68+ macrophage subtypes (Fig. 5F). The highest PSAP expression was found in the macrophage populations, with relatively lower expression across the other leukocytes (Fig. 5F).
Given the strong relationship between mTOR signaling and Psap expression in mouse plaques, we set out to study this in human atherosclerosis. To this aim, we analyzed transcriptome data from 620 carotid plaques(20). In these tissues, PSAP was highly expressed, as compared to the average expression of a random sample of other genes (fig. S7B). Among the 40 genes assigned to the mTOR signaling pathway, we found PSAP to correlate with elements of the Ragulator complex (LAMTOR1, LAMTOR2 and LAMTOR5), a component of active mTORC1 (Fig. 5G, fig. S7C, fig. S8 and table S7). Furthermore, we found co-expression of PSAP and RPS6, which provides a link to S6K1 signaling, as S6K1 catalyzes the phosphorylation of ribosomal protein S6, encoded by RPS6.
Recently published single-cell transcriptional data from atherosclerotic plaques of mice and humans identified gene signatures of distinct macrophage populations (21, 22). We investigated the relationship of PSAP with gene expression related to these previously identified signatures. APOE, APOC1, CCL2, CTSB, CTSD and MMP9 displayed the highest co-expression with PSAP (Fig. 5H, fig. S9 and table S8). These genes were identified by Fernandez et al. (22) as markers of the same macrophage cluster in human atherosclerosis. APOE and APOC1 are related to cholesterol uptake and believed to be markers for foam cells. CCL2, CTSB, CTSD and MMP9 are important inflammatory markers and involved in matrix degradation. CTSB, encoding for cathepsin B, was also recognized as one of the downregulated hub genes in our murine plaque transcriptome analysis (Fig. 2C), and is the molecular target of FMT-CT imaging, which was reduced by mTORi-NB treatment. When we evaluated the expression of these six genes in our own single-cell transcriptional data, CTSB and CTSD mostly resembled the cell-specific expression pattern of PSAP, further affirming their connectivity (Fig. 5I). Collectively, these data demonstrate that the expression of PSAP is related to mTOR signaling and inflammation in human atherosclerotic lesions.
Discussion
Atherosclerosis is a cholesterol-induced inflammatory disease in which monocytes and macrophages are the main protagonists. The mTOR signaling network is fundamental for balancing anabolic and catabolic pathways in response to the nutritional status in all eukaryotic cells and plays a dominant role in regulating inflammatory activity in immune cells. In this study we showed that myeloid cell-specific mTOR and S6K1 inhibition rapidly suppressed plaque inflammation in atherosclerotic mice. We identified prosaposin as a mediator of these anti-inflammatory effects and revealed prosaposin’s regulatory role in immunometabolism. In humans we confirmed high PSAP expression in plaque macrophages and found it to be related to mTOR signaling and inflammation.
Prosaposin is the precursor of four similar proteins named saposin A, B, C and D (23, 24) and this protein is highly conserved in evolution (19). Saposins are essential for lysosomal degradation of glycosphingolipids by facilitating the access of the degrading enzymes to their substrates (25). Deleterious genetic mutations in any of the saposin domains lead to lysosomal storage disease (26). Besides the intracellular function of prosaposin in lysosomes, the protein is also excreted and can be detected in various body fluids including serum (27). Concerning hematopoietic cells, prosaposin is predominantly expressed in monocytes and macrophages, and much lower expression is found in lymphocytes (28). Prosaposin and the individual saposins are known to have specific immunological functions in innate immune cells (14). Saposins are indispensable for lipid antigen presentation to CD1-restricted T cells, as they mobilize lipids from lysosomal membranes to facilitate their association with CD1d (14). CD1 lipid antigen presentation is of relevance in infectious diseases such as Mycobacterium tuberculosis (29). CD1 is also important in atherosclerosis, exemplified by the fact that CD1d−/−Apoe−/− mice, which are incapable of lipid antigen presentation, showed markedly reduced atherosclerosis development (30, 31). Furthermore, prosaposin is related to progranulin, with which it interacts to facilitate its lysosomal targeting (32). Progranulin is of importance in the innate immune response and was previously found to be highly expressed in atherosclerotic plaque macrophages (33–35).
In our current study we revealed prosaposin to be an important mediator of the anti-inflammatory effect of mTOR and S6K1 inhibition in plaque macrophages. This may be mediated in part through above mentioned effects on CD1 lipid antigen presentation and/or progranulin. Another mechanism may be prosaposin’s central role in sphingolipid metabolism (14). Sphingolipids are universal building blocks of cell membranes and include ceramide, sphingomyelin and many different forms of glycosphingolipids (36). Sphingolipid metabolites, particularly ceramide and sphingosine-1-phosphate (S1P), modulate a wide variety of cellular processes involved in inflammation, cell cycle and metabolism (36–38). Ceramides affect cellular metabolism by inhibiting uptake of amino acids (39, 40) and glucose (41), leading to utilization of fatty acids for energy production (38). Besides this, ceramides influence mitochondrial activity by changing the mitochondrial membrane potential (42), which is required for efficient ATP production through oxidative phosphorylation. Furthermore, the respiratory chain activity can be modified by ceramides (43, 44).
The precise role of PSAP in human atherosclerosis has not been studied previously. The rapid progress of single-cell technologies, such as single-cell RNA sequencing and CyTOF (cytometry by time of flight), aids in unraveling cellular subsets, phenotypes, and also the underlying cellular processes of a complex disease such as atherosclerosis. Recently, Fernandez et al. (22) defined human plaque macrophage clusters based on their gene expression signatures. PSAP appeared in one of the clusters in their data, which corroborates our data from both mice and human atherosclerosis. Similar to the observation made by Fernandez et al. (22), we showed that PSAP is coexpressed with other genes known to play an important role in plaque inflammation, namely APOE, APOC1, CCL2, CTSB, CTSD and MMP9.
In this study we revealed the role of prosaposin in atherosclerosis in both mice and humans. This suggests it may be possibile to target prosaposin or the individual saposin domains for the treatment of atherosclerosis. RNA interference with siRNA could be a way to achieve this, as we showed in our current study. siRNA therapy is highly specific to its target. Yet, the off-target effects on PSAP suppression in other tissues could be a limitation. This may be overcome by targeting the therapy specifically to myeloid cells (45), using nanotherapeutic siRNA delivery (46). Another form of PSAP-targeted treatment could be the application of small molecules binding to prosaposin or the saposin domains. To our knowledge no specific small molecule inhibitor has been developed for this purpose. However, from literature we found that the anti-malarial drug hydroxychloroquine binds to saposin B (47). Hydroxychloroquine is an immunomodulating drug used to treat rheumatoid arthritis and systemic lupus erythematosus (SLE), and recent studies showed anti-atherosclerotic effects in patients with SLE (48, 49). Another potential strategy may be to interfere with sphingolipid signaling. S1P receptor 1 modulators and agonists are currently approved for the treatment of multiple sclerosis, an auto-immune disease of the central nervous system in which macrophages play a central role (50). S1P receptor 1 modulators and agonists were also observed to reduce atherosclerosis in experimental studies by modulating macrophage function (51, 52).
Collectively, our findings advance several concepts. First, we show that mTOR and S6K1 inhibition in myeloid cells rapidly reduces plaque inflammation. Second, we found prosaposin to be an important mediator of these anti-inflammatory effects, which likely relates to prosaposin’s effect on cell metabolism. Third, we show that prosaposin is associated with plaque inflammation in human atherosclerosis. Our data identify prosaposin and the individual saposin domains as potential therapeutic targets for the treatment of atherosclerosis.
Materials and methods
Study design
We designed and formulated two myeloid cell-specific nanobiologics to selectively inhibit mTOR signaling. We treated Apoe−/− mice with these nanobiologics for one week and used flow cytometry and histology to study the systemic immune status and plaque inflammation (n = 20 mice/group). By investigating transcriptome modifications, we identified Psap, a gene encoding for prosaposin, to be closely related with mTOR signaling (n = 10 mice/group). Subsequently, Psap siRNA lipid nanoparticles were designed and formulated to study the influence of Psap on immune cell metabolism. Here we used murine bone marrow-derived macrophages and a metabolic flux assay (n = 10 wells/condition). To study the direct effects of Psap on atherosclerotic inflammation, we transplanted Psap−/− or Psap+/+ bone marrow in lethally irradiated Ldlr−/− mice and again studied the systemic immune status and plaque inflammation (n = 20 mice/group). Finally, we corroborated our findings in human monocytes through in vitro assays (n = 6 donors), single cell RNA sequencing (n = 3 donors) and in human atherosclerotic plaque specimens through histology (n = 4 patients), single cell RNA sequencing (n = 18 patients) and bulk RNA sequencing (n = 620 patients).
Mice
Female Apoe−/− mice (B6.129P2-Apoetm1Unc/J), female Ldlr−/− mice (B6.129S7-Ldlrtm1Her/J) and male and female Psap+/− mice (B6.129P2-Psaptm1Suz/J) were purchased from The Jackson Laboratory. Eight-week-old Apoe−/− mice were fed a Western diet (0.2% weight cholesterol; 15.2% kcal protein, 42.7% kcal carbohydrate, 42.0% kcal fat; Harlan TD. 88137) for 12 weeks. Male and female Psap+/− mice were bred to obtain Psap−/− and Psap+/+ mice. After bone marrow transplantation and reconstitution, Ldlr−/− mice were fed a Western diet for 11 weeks. Animal care and procedures were based on an approved institutional protocol from Icahn School of Medicine at Mount Sinai.
In vitro experiments were performed on murine bone marrow-derived macrophages (BMDMs). BMDMs were cultured in cell culture dishes, in Roswell Park Memorial Institute medium (RPMI) supplemented with 10% fetal bovine serum, 2mM L-Glutamine, 100 U/mL Penicillin-Streptomycin and 15% L929-cell-conditioned medium. All cells were incubated at 37 °C in a 5% CO2 atmosphere.
Human subjects
For in vitro studies on human monocytes, buffy coats from healthy donors were obtained after written informed consent (Sanquin blood bank, Nijmegen, The Netherlands). For histologic analysis, human atherosclerotic plaque samples were obtained from four patients. All four patients had an indication for carotid endarterectomy. Gender of the included subjects for both studies is known, although gender association cannot be analyzed due to small group sizes. Subject allocation to groups is not applicable.
For bulk RNA-seq and single-cell RNA-seq analysis of human carotid atherosclerotic plaques from the Athero-Express Biobank Study, research was executed according to the principles of the Declaration of Helsinki and its later amendments (53) all patients provided informed consent and the study was approved by the medical ethics committee of the UMC Utrecht.
Synthesis of nanobiologics
Nanobiologic formulations were synthesized according to previously published methods (54, 55). For mTORi-NB, the mTORC1-complex inhibitor rapamycin (3 mg, 3.3 μmol), was combined with 1-myristoyl-2-hydroxy-sn-glycero-phosphocholine (MHPC) (6 mg, 12.8 μmol) and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) (18 mg, 26.6 μmol) (Avanti Polar Lipids). For S6K1i-NB, the S6K1 inhibitor PF-4708671 (1.5 mg, 4.6 μmol) was combined with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (18 mg, 23.7 μmol) and 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (PHPC) (6 mg, 12.1 μmol). The inhibitors and lipids were dissolved in methanol and chloroform, mixed, and then dried in a vacuum, yielding a thin lipid film. A PBS solution containing human apolipoprotein A1 (APOA1) (4.8 mg in 5 mL PBS) was added to the lipid film. The mixture was incubated in an ice-cold sonication bath for 15–30 minutes. Subsequently, the solution was sonicated using a tip sonicator at 0 °C for 20 minutes to form APOA1 based nanobiologics. The obtained solution was concentrated by centrifugal filtration using a 100k MWCO Vivaspin tube at 3000 rpm to obtain a volume of ~1 mL, and then washed twice with fresh PBS (5 mL). The concentrated solution (~1 mL) was filtered through a 0.22 μm PES syringe filter to obtain the final nanobiologic solution. For targeting and biodistribution experiments, analogs of mTORi-NB and S6K1i-NB were prepared by incorporating the fluorescent dyes DiIC18(7) (1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindotricarbocyanine Iodide) or DiOC18(3) (3,3′-Dioctadecyloxacarbocyanine perchlorate) (Invitrogen).
Nanobiologic treatment
Twenty-week-old Apoe−/− received either PBS, unloaded nanobiologics, mTORi-NB (mTORi at 5 mg/kg), or S6K1i-NB (S6K1i at 5 mg/kg) through lateral tail vein injections. Mice were treated every other day during one week (total of 4 injections), while kept on a Western diet. For the targeting and biodistribution experiments, mice received a single intravenous injection. All animals were euthanized 24 hours after the last injection.
Histology and immunohistochemistry
For Mac3, CD68 and prosaposin staining, tissues were fixed in formalin, embedded in paraffin and sectioned into 4 μm slices. To perform immunohistochemical staining, mouse aortic roots and human carotid endarterectomy (CEA) sections were deparaffinized, blocked using 4% fetal calf serum (FCS, Gibco) in PBS for 30 minutes, and incubated in antigen-retrieval solution (DAKO) at 95 °C for 10 minutes. Mouse aortic root sections were immunolabeled with rat anti-mouse Mac3 monoclonal antibody (1:30, BD Biosciences, 553322). CEA samples were stained for macrophages using a mouse anti-human CD68 primary antibody (1:300, Abcam, Ab201340) in combination with a biotinylated donkey anti-mouse secondary antibody (1:300, Jackson ImmunoResearch, 715-065-150). Both mouse aortic roots and CEA samples were stained for prosaposin using a rabbit anti-human prosaposin primary antibody (1:500, Abcam, Ab180751) in combination with a biotinylated goat anti-rabbit secondary antibody (1:300, DAKO, E0432). Antibody staining was visualized by either Immpact AMEC red (Vectorlabs) or diaminobenzidine (DAB). Sections were analyzed using a Leica DM6000 microscope (Leica Microsystems) or the VENTANA iScan HT slide scanner (Ventana).
Aortic root samples from Ldlr−/− mice were harvested, embedded in Tissue-Tek O.C.T., and sectioned into 7 μm slices. To acquire lesion volume sections were collected starting at the beginning of the artic root until the aortic valves were no longer visible. After staining with H&E the lesion area was measured in intervals of 84 μm using Adobe Photoshop. The generated lesion area was plotted against the distance from the artic root after which the lesion volume was obtained by calculating the area under the curve. Sirius red staining was used for the analysis of collagen content.
RNA sequencing of murine plaque macrophages
The CD68+ cells collected by laser capture microdissection were used for RNA isolation (PicoPure RNA Isolation Kit, Arcturus) and subsequent RNA amplification and cDNA preparation according to the manufacturer’s protocols (Ovation Pico WTA System, NuGEN). The quality and concentration of the collected samples were measured using an Agilent 2100 Bioanalyzer. For RNA sequencing, pair-end libraries were prepared and validated. The purity, fragment size, yield, and concentration were determined. During cluster generation, the library molecules were hybridized onto an Illumina flow cell. Subsequently, the hybridized molecules were amplified using bridge amplification, resulting in a heterogeneous population of clusters. The data set was obtained using an Illumina HiSeq 2500 sequencer.
Bone marrow transplantation
Nine-week-old Ldlr−/− mice were lethally irradiated (2× 600 cGy). Subsequently, bone marrow cells were harvested from 3-week old Psap−/− and Psap+/+ mice and transplanted in Ldlr−/− recipients (5 × 106 cells/recipient). Mice were kept on polymyxin B sulfate and neomycin, administered through drinking water at 600 U/mL and 0.1 mg/mL, respectively, for 6 weeks.
Athero-Express human sample collection
The procedure of obtaining biomaterial of patients elected for endarterectomy within the Athero-Express Biobank Study has been described before (56). In short, arterial plaque material is obtained during endarterectomy. Each plaque is dissected into segments of 0.5-cm. From these, the culprit lesion is reserved for histological assessment. Surrounding segments are either frozen in liquid nitrogen without delay and stored at −80 °C for later use (bulk RNA-seq) or used immediately (single-cell RNA-seq).
Single-cell RNA-sequencing analysis Athero-Express samples
Prior to processing, reads were filtered for mitochondrial and ribosomal genes, MALAT1, KCNQ1OT1, UGDH-AS1, and EEF1A. Then, remaining single-cell sequencing data was processed as described previously (57) in an R 3.5 environment using Seurat (version 2.3.4) (58). Cells expressing between 500 and 10.000 genes and genes expressed in at least 3 cells were used for further analysis. Data was log-normalized and scaled with the exclusion of unique molecular identifiers (UMIs). Canonical correlation analysis (CCA) reduction was performed, resolution set to 1.2 for 15 dimensions, in order to identify clusters and to perform t-distributed stochastic neighbor embedding (t-SNE). Cell clusters were annotated by evaluating differential gene expression of individual cell clusters (Wilcoxon rank sum test) and analyzing against BLUEPRINT (59) reference data using SingleR (60).
Statistical analysis
Data are shown as mean ± SD, unless otherwise stated. For plaque volume analysis, either an unpaired t-test or a one-way ANOVA with Dunnett’s correction were applied depending on the number of groups. For in vitro human monocyte experiments, normality checks were performed using gg-plots and a normality assay. Non-parametric parameters were analyzed pairwise using a Wilcoxon signed-rank test. Correlations between genes in human bulk RNA sequencing were calculated by Spearman coefficients. Significance of differences in all other experiments was calculated using non-parametric Mann-Whitney U tests. Two-sided testing was used and a P value below 0.05 was considered statistically significant. For mouse transcriptome analyses, false discovery rate control was applied and adjusted p-values were reported. All data were analyzed using Graphpad Prism version 8.4.3.
Supplementary Material
Fig. S1. Characteristics of mTORi-NB and S6K1i-NB.
Fig. S2. Myeloid cell-specific mTOR inhibition reduces atherosclerotic plaque inflammation.
Fig. S3. Systemic effects of mTORi and S6K1i-NB treatment.
Fig. S4. Prosaposin colocalizes with macrophages in murine plaques.
Fig. S5. Psap mediates atherosclerotic plaque inflammation in Ldlr−/− mice.
Fig. S6. Nanobiologics inhibit oxLDL priming in human monocytes.
Fig. S7. PSAP mediates atherosclerotic plaque inflammation in humans.
Fig. S8. Correlation between PSAP expression and genes involved in mTOR signaling.
Fig. S9. Correlation between PSAP expression and genes involved in macrophage inflammation.
Table S1. Expression of genes coding for chemokines after mTORi-NB treatment
Table S2. Expression of genes coding for chemokines after S6K1i-NB treatment
Table S3. Expression of autophagy-related genes after mTORi-NB treatment.
Table S4. Expression of autophagy-related genes after S6K1i-NB treatment.
Table S5. Hub genes of the mTORi-NB-related turquoise module.
Table S6. Hub genes of the S6K1i-NB-related turquoise module.
Table S7. Correlation between PSAP expression and genes involved in mTOR signaling.
Table S8. Correlation between PSAP expression and genes involved in macrophage inflammation.
Data File S1. Data file.
Acknowledgments
The authors thank the Icahn School of Medicine and the following Mount Sinai’s core facilities: flow cytometry core, quantitative PCR core and the preclinical imaging core of the BioMedical Engineering and Imaging Institute.
Funding
This work was supported by National Institutes of Health grants R01 HL118440, R01 HL125703, and P01 HL131478 (all to W.J.M.M.), R01 HL084312 (to E.A.F.), R01 EB009638 (to Z.A.F.), and R01 HL144072 (to W.J.M.M. and Z.A.F.); National Heart, Lung, and Blood Institute R35 HL139598 (to M.N.); NIH Program of Excellence in Nanotechnology (PEN) Award (HHSN368201000045C to Z.A.F.), as well as the Harold S. Geneen Charitable Trust Award (Z.A.F.), the Netherlands Organisation for Scientific Research (NWO) grant ZonMW Veni 016156059 (R.D.), ZonMW Vidi 91713324 (W.J.M.M.), ZonMW Vici 91818622 (W.J.M.M.). E.L received funding from the European Research Council (ERC consolidator grant 681493) and the Deutsche Forschungsgemeinschaft (CRC 1123-A5) M.G.N. was supported by a Spinoza grant of the Netherlands Organization for Scientific Research. N.P.R., L.A.B.J. and M.G.N. received funding from the European Union’s Horizon 2020 research and innovation program (No 667837), and a grant from the Dutch Heart Foundation (CVON2012-03 and CVON2018-27). L.A.B.J. was supported by a Competitiveness Operational Programme grant of the Romanian Ministry of European Funds (HINT, P_37_762, MySMIS 103587) A.B. received funding from the European Union’s Horizon 2020 research and innovation program (No 755320). N.P.R. is recipient of a grant of the ERA-CVD Joint Transnational Call 2018, which is supported by the Dutch Heart Foundation (JTC2018, project MEMORY; 2018T093). S.B. is supported by the Dutch Heart Foundation (2018T028). M.M.T.v.L. received funding from the American Heart Association (19PRE34380423).
Competing interest
Z.A.F., W.J.M.M., M.G.N., L.A.B.J. are founders of Trained Therapeutix Discovery. M.N. has received funds or material research support from Novartis, GlaxoSmithKline, Pfizer, GlycoMimetics, Medtronic, Biotronik, Alnylam, and CSL Behring and consulting fees from Verseau Therapeutics, Sigilon, Molecular Imaging Inc, IFM Therapeutics, and Biogen.
Footnotes
Data and materials availability
All data associated with this study are in the paper or Supplementary Materials. Mouse RNA sequencing data are available at the Gene Expression Omnibus (GEO) database (GEO series number GSE104777). Human atherosclerotic plaque R scripts are available through GitHub [https://github.com/AtheroExpress/MicroanatomyHumanPlaque_scRNAseq].
References
- 1.Tabas I, Lichtman AH, Monocyte-Macrophages and T Cells in Atherosclerosis, Immunity 47, 621–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Neill LAJ, Kishton RJ, Rathmell J, A guide to immunometabolism for immunologists, Nat. Rev. Immunol 16, 553–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ketelhuth DFJ, Lutgens E, Bäck M, Binder CJ, Van Den Bossche J, Daniel C, Dumitriu IE, Hoefer I, Libby P, O’Neill L, Weber C, Evans PC, Immunometabolism and atherosclerosis: Perspectives and clinical significance: A position paper from the Working Group on Atherosclerosis and Vascular Biology of the European Society of Cardiology, Cardiovasc. Res 115, 1385–1392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koelwyn GJ, Corr EM, Erbay E, Moore KJ, Regulation of macrophage immunometabolism in atherosclerosis, Nat. Immunol 19, 526–537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomas L, Edsfeldt A, Mollet IG, Matic LP, Prehn C, Adamski J, Paulsson-Berne G, Hedin U, Nilsson J, Bengtsson E, Gonçalves I, Björkbacka H, Altered metabolism distinguishes high-risk from stable carotid atherosclerotic plaques, Eur. Heart J 39, 2301–2310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weichhart T, Hengstschläger M, Linke M, Regulation of innate immune cell function by mTOR, Nat. Rev. Immunol 15, 599–614 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saxton RA, Sabatini DM, mTOR Signaling in Growth, Metabolism, and Disease, Cell 168, 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duivenvoorden R, Tang J, Cormode DP, Mieszawska AJ, Izquierdo-Garcia D, Ozcan C, Otten MJ, Zaidi N, Lobatto ME, van Rijs SM, Priem B, Kuan EL, Martel C, Hewing B, Sager H, Nahrendorf M, Randolph GJ, Stroes ESG, Fuster V, Fisher EA, Fayad ZA, Mulder WJM, A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation., Nat. Commun 5, 3065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lameijer M, Binderup T, van Leent MMTT, Senders ML, Fay F, Malkus J, Sanchez-Gaytan BL, Teunissen AJPP, Karakatsanis N, Robson P, Zhou X, Ye Y, Wojtkiewicz G, Tang J, Seijkens TTPP, Kroon J, Stroes ESGG, Kjaer A, Ochando J, Reiner T, Pérez-Medina C, Calcagno C, Fisher EA, Zhang B, Temel RE, Swirski FK, Nahrendorf M, Fayad ZA, Lutgens E, Mulder WJMM, Duivenvoorden R, Fischer EA, Zhang B, Temel RE, Swirski FK, Nahrendorf M, Fayad ZA, Lutgens E, Mulder WJMM, Duivenvoorden R, Efficacy and safety assessment of a TRAF6-targeted nanoimmunotherapy in atherosclerotic mice and non-human primates, Nat. Biomed. Eng 2, 279–292 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, Hwang C, Alessi DR, Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1), Biochem. J 431, 245–255 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Nahrendorf M, Waterman P, Thurber G, Groves K, Rajopadhye M, Panizzi P, Marinelli B, Aikawa E, Pittet MJ, Swirski FK, Weissleder R, Hybrid in vivo FMT-CT imaging of protease activity in atherosclerosis with customized nanosensors, Arterioscler. Thromb. Vasc. Biol 29, 1444–1451 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF, Autophagy Links Inflammasomes to Atherosclerotic Progression, Cell Metab. 15, 534–544 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langfelder P, Mischel PS, Horvath S, Ravasi T, Ed. When Is Hub Gene Selection Better than Standard Meta-Analysis?, PLoS One 8, e61505 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Darmoise A, Maschmeyer P, Winau F, The immunological functions of saposins (Elsevier Inc., ed. 1, 2010; 10.1016/S0065-2776(10)05002-9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keating ST, Groh L, Thiem K, Bekkering S, Li Y, Matzaraki V, Van Der Heijden CDCC, Van Puffelen JH, Lachmandas E, Jansen T, Oosting M, & Charlotte L, De Bree J, Koeken VACM, Moorlag SJCFM, Mourits VP, Van Diepen J, Strienstra R, Novakovic B, Stunnenberg HG, Van Crevel R, Joosten LAB, Netea MG, Riksen NP, Rewiring of glucose metabolism defines trained immunity induced by oxidized low-density lipoprotein, J. Mol. Med (2020), doi: 10.1007/s00109-020-01915-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulkarni JA, Witzigmann D, Chen S, Cullis PR, van der Meel R, Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics, Acc. Chem. Res 52, 2435–2444 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Van Eck M, Herijgers N, Yates J, Pearce NJ, Hoogerbrugge PM, Groot PHE, Van Berkel TJC, Bone Marrow Transplantation in Apolipoprotein E–Deficient Mice, Arterioscler. Thromb. Vasc. Biol 17, 3117–3126 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Thomas G, Tacke R, Hedrick CC, Hanna RN, Nonclassical Patrolling Monocyte Function in the Vasculature, Arterioscler. Thromb. Vasc. Biol 35, 1306–1316 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hazkani-Covo E, Altman N, Horowitz M, Graur D, The Evolutionary History of Prosaposin: Two Successive Tandem-Duplication Events Gave Rise to the Four Saposin Domains in Vertebrates, J. Mol. Evol 54, 30–34 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Depuydt MAC, Prange KHM, Slenders L, Örd T, Elbersen D, Boltjes A, de Jager SCA, Asselbergs FW, de Borst GJ, Aavik E, Lönnberg T, Lutgens E, Glass CK, den Ruijter HM, Kaikkonen MU, Bot I, Slütter B, van der Laan SW, Yla-Herttuala S, Mokry M, Kuiper J, de Winther MPJ, Pasterkamp G, Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics, Circ. Res 127, 1437–1455 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willemsen L, de Winther MP, Macrophage subsets in atherosclerosis as defined by single-cell technologies., J. Pathol 250, 705–714 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, Khan NS, Wong CK, Shamailova R, Hill CA, Wang Z, Remark R, Li JR, Pina C, Faries C, Awad AJ, Moss N, Bjorkegren JLM, Kim-Schulze S, Gnjatic S, Ma’ayan A, Mocco J, Faries P, Merad M, Giannarelli C, Single-cell immune landscape of human atherosclerotic plaques, Nat. Med 25, 1576–1588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fürst W, Machleidt W, Sandhoff K, The precursor of sulfatide activator protein is processed to three different proteins., Biol. Chem. Hoppe. Seyler 369, 317–28 (1988). [DOI] [PubMed] [Google Scholar]
- 24.O’Brien JS, Kretz KA, Dewji N, Wenger DA, Esch F, Fluharty AL, Coding of two sphingolipid activator proteins (SAP-1 and SAP-2) by same genetic locus., Science (80-.) 241, 1098–101 (1988). [DOI] [PubMed] [Google Scholar]
- 25.Sandhoff K, Kolter T, Biochemistry of glycosphingolipid degradation., Clin. Chim. Acta 266, 51–61 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Ferreira CR, Gahl WA, Lysosomal storage diseases, Transl. Sci. Rare Dis 2, 1–71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hineno T, Sano A, Kondoh K, Ueno S, Kakimoto Y, Yoshida K, Secretion of sphingolipid hydrolase activator precursor, prosaposin, Biochem. Biophys. Res. Commun 176, 668–674 (1991). [DOI] [PubMed] [Google Scholar]
- 28.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA-K, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist P-H, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F, Tissue-based map of the human proteome, Science (80-.) 347, 1260419–1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB, Recognition of a lipid antigen by CD1-restricted αβ+ T cells, Nature 372, 691–694 (1994). [DOI] [PubMed] [Google Scholar]
- 30.Getz GS, Reardon CA, Natural killer T cells in atherosclerosis, Nat. Rev. Cardiol 14, 304–314 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Tupin E, Nicoletti A, Elhage R, Rudling M, Ljunggren H-G, Hansson GK, Berne GP, CD1d-dependent Activation of NKT Cells Aggravates Atherosclerosis, J. Exp. Med 199, 417–422 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou X, Sun L, de Oliveira FB, Qi X, Brown WJ, Smolka MB, Sun Y, Hu F, Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin, J. Cell Biol 210, 991–1002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He Z, Bateman A, Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis, J. Mol. Med 81, 600–612 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Kawase R, Ohama T, Matsuyama A, Matsuwaki T, Okada T, Yamashita T, Yuasa-Kawase M, Nakaoka H, Nakatani K, Inagaki M, Tsubakio-Yamamoto K, Masuda D, Nakagawa-Toyama Y, Nishida M, Ohmoto Y, Nishihara M, Komuro I, Yamashita S, Deletion of progranulin exacerbates atherosclerosis in ApoE knockout mice, Cardiovasc. Res 100, 125–133 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A, Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice, J. Exp. Med 207, 117–128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacEyka M, Spiegel S, Sphingolipid metabolites in inflammatory disease, Nature 510, 58–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hannun YA, Obeid LM, Sphingolipids and their metabolism in physiology and disease, Nat. Rev. Mol. Cell Biol 19, 175–191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Summers SA, Chaurasia B, Holland WL, Metabolic Messengers: ceramides, Nat. Metab 1, 1051–1058 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL, Ceramide starves cells to death by downregulating nutrient transporter proteins, Proc. Natl. Acad. Sci. U. S. A 105, 17402–17407 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hyde R, Hajduch E, Powell DJ, Taylor PM, Hundal HS, Ceramide downregulates System A amino acid transport and protein synthesis in rat skeletal muscle cells, FASEB J 19, 1–24 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Summers SA, Garza LA, Zhou H, Birnbaum MJ, Regulation of Insulin-Stimulated Glucose Transporter GLUT4 Translocation and Akt Kinase Activity by Ceramide, Mol. Cell. Biol 18, 5457–5464 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arora AS, Jones BJ, Patel TC, Bronk SF, Gores GJ, Ceramide induces hepatocyte cell death through disruption of mitochondrial function in the rat, Hepatology 25, 958–963 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Kogot-Levin A, Saada A, Ceramide and the mitochondrial respiratory chain, Biochimie 100, 88–94 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Zigdon H, Kogot-Levin A, Park J-W, Goldschmidt R, Kelly S, Merrill AH, Scherz A, Pewzner-Jung Y, Saada A, Futerman AH, Ablation of Ceramide Synthase 2 Causes Chronic Oxidative Stress Due to Disruption of the Mitochondrial Respiratory Chain, J. Biol. Chem 288, 4947–4956 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Novobrantseva TI, Borodovsky A, Wong J, Klebanov B, Zafari M, Yucius K, Querbes W, Ge P, Ruda VM, Milstein S, Speciner L, Duncan R, Barros S, Basha G, Cullis P, Akinc A, Donahoe JS, Narayanannair Jayaprakash K, Jayaraman M, Bogorad RL, Love K, Whitehead K, Levins C, Manoharan M, Swirski FK, Weissleder R, Langer R, Anderson DG, de Fougerolles A, Nahrendorf M, Koteliansky V, Systemic RNAi-mediated Gene Silencing in Nonhuman Primate and Rodent Myeloid Cells, Mol. Ther. - Nucleic Acids 1, e4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duivenvoorden R, Senders ML, van Leent MMT, Pérez-Medina C, Nahrendorf M, Fayad ZA, Mulder WJM, Nanoimmunotherapy to treat ischaemic heart disease, Nat. Rev. Cardiol 16, 21–32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tinklepaugh J, Smith BM, Hanlon E, Zubieta C, Bou-Abdallah F, Doyle RP, Exploring the multiligand binding specificity of saposin B reveals two binding sites, ACS Omega 2, 7141–7145 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schrezenmeier E, Dörner T, Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology, Nat. Rev. Rheumatol 16, 155–166 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Floris A, Piga M, Mangoni AA, Bortoluzzi A, Erre GL, Cauli A, Protective effects of hydroxychloroquine against accelerated atherosclerosis in systemic lupus erythematosus, Mediators Inflamm. 2018 (2018), doi: 10.1155/2018/3424136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Derfuss T, Mehling M, Papadopoulou A, Bar-Or A, Cohen JA, Kappos L, Advances in oral immunomodulating therapies in relapsing multiple sclerosis., Lancet. Neurol 19, 336–347 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Nofer J-R, Bot M, Brodde M, Taylor PJ, Salm P, Brinkmann V, van Berkel T, Assmann G, Biessen EAL, FTY720, a Synthetic Sphingosine 1 Phosphate Analogue, Inhibits Development of Atherosclerosis in Low-Density Lipoprotein Receptor–Deficient Mice, Circulation 115, 501–508 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Potì F, Gualtieri F, Sacchi S, Weißen-Plenz G, Varga G, Brodde M, Weber C, Simoni M, Nofer J-R, KRP-203, Sphingosine 1-Phosphate Receptor Type 1 Agonist, Ameliorates Atherosclerosis in LDL-R −/− Mice, Arterioscler. Thromb. Vasc. Biol 33, 1505–1512 (2013). [DOI] [PubMed] [Google Scholar]
- 53.World Medical Association, World Medical Association Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects, JAMA 310, 2191–2194 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Jonas A, Reconstitution of high-density lipoproteins., Methods Enzymol. 128, 553–82 (1986). [DOI] [PubMed] [Google Scholar]
- 55.Braza MS, van Leent MMT, Lameijer M, Sanchez-Gaytan BL, Arts RJW, Pérez-Medina C, Conde P, Garcia MR, Gonzalez-Perez M, Brahmachary M, Fay F, Kluza E, Kossatz S, Dress RJ, Salem F, Rialdi A, Reiner T, Boros P, Strijkers GJ, Calcagno CC, Ginhoux F, Marazzi I, Lutgens E, Nicolaes GAF, Weber C, Swirski FK, Nahrendorf M, Fisher EA, Duivenvoorden R, Fayad ZA, Netea MG, Mulder WJM, Ochando J, Inhibiting Inflammation with Myeloid Cell-Specific Nanobiologics Promotes Organ Transplant Acceptance, Immunity 49, 819–828.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van der Laan SW, Foroughi Asl H, van den Borne P, van Setten J, van der Perk MEM, van de Weg SM, Schoneveld AH, de Kleijn DPV, Michoel T, Björkegren JLM, den Ruijter HM, Asselbergs FW, de Bakker PIW, Pasterkamp G, Variants in ALOX5, ALOX5AP and LTA4H are not associated with atherosclerotic plaque phenotypes: The Athero-Express Genomics Study, Atherosclerosis 239, 528–538 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Muraro MJ, Dharmadhikari G, Grün D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJPP, van Oudenaarden A, A Single-Cell Transcriptome Atlas of the Human Pancreas, Cell Syst. 3, 385–394.e3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species, Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martens JHA, Stunnenberg HG, BLUEPRINT: mapping human blood cell epigenomes, Haematologica 98, 1487–1489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M, Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage, Nat. Immunol 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2, Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anders S, Pyl PT, Huber W, HTSeq-A Python framework to work with high-throughput sequencing data, Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mudge JM, Harrow J, Creating reference gene annotation for the mouse C57BL6/J genome assembly, Mamm. Genome 26, 366–378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies., Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang B, Horvath S, A General Framework for Weighted Gene Co-Expression Network Analysis, Stat. Appl. Genet. Mol. Biol 4 (2005), doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 66.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, Cytoscape: a software environment for integrated models of biomolecular interaction networks., Genome Res. 13, 2498–504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Song W-M, Zhang B, Wang E, Ed. Multiscale Embedded Gene Co-expression Network Analysis, PLOS Comput. Biol 11, e1004574 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bekkering S, Blok BA, Joosten LAB, Riksen NP, Van Crevel R, Netea MG, In Vitro Experimental Model of Trained Innate Immunity in Human, Clin. vaccine Immunol 23, 926–934 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Tits LJH, Stienstra R, Van Lent PL, Netea MG, Joosten LAB, Stalenhoef AFH, Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages : A crucial role for Krüppel-like factor 2, Atherosclerosis 214, 345–349 (2011). [DOI] [PubMed] [Google Scholar]
- 70.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data., Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hashimshony T, Wagner F, Sher N, Yanai I, CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification, Cell Rep. 2, 666–673 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li S, Livak KJ, Rozenblatt-Rosen O, Dor Y, Regev A, Yanai I, CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq, Genome Biol. 17, 77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ferraz MAMM, Rho HS, Hemerich D, Henning HHW, van Tol HTA, Hölker M, Besenfelder U, Mokry M, Vos PLAM, Stout TAE, Le Gac S, Gadella BM, An oviduct-on-a-chip provides an enhanced in vitro environment for zygote genome reprogramming, Nat. Commun 9, 4934 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform, Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Characteristics of mTORi-NB and S6K1i-NB.
Fig. S2. Myeloid cell-specific mTOR inhibition reduces atherosclerotic plaque inflammation.
Fig. S3. Systemic effects of mTORi and S6K1i-NB treatment.
Fig. S4. Prosaposin colocalizes with macrophages in murine plaques.
Fig. S5. Psap mediates atherosclerotic plaque inflammation in Ldlr−/− mice.
Fig. S6. Nanobiologics inhibit oxLDL priming in human monocytes.
Fig. S7. PSAP mediates atherosclerotic plaque inflammation in humans.
Fig. S8. Correlation between PSAP expression and genes involved in mTOR signaling.
Fig. S9. Correlation between PSAP expression and genes involved in macrophage inflammation.
Table S1. Expression of genes coding for chemokines after mTORi-NB treatment
Table S2. Expression of genes coding for chemokines after S6K1i-NB treatment
Table S3. Expression of autophagy-related genes after mTORi-NB treatment.
Table S4. Expression of autophagy-related genes after S6K1i-NB treatment.
Table S5. Hub genes of the mTORi-NB-related turquoise module.
Table S6. Hub genes of the S6K1i-NB-related turquoise module.
Table S7. Correlation between PSAP expression and genes involved in mTOR signaling.
Table S8. Correlation between PSAP expression and genes involved in macrophage inflammation.
Data File S1. Data file.