

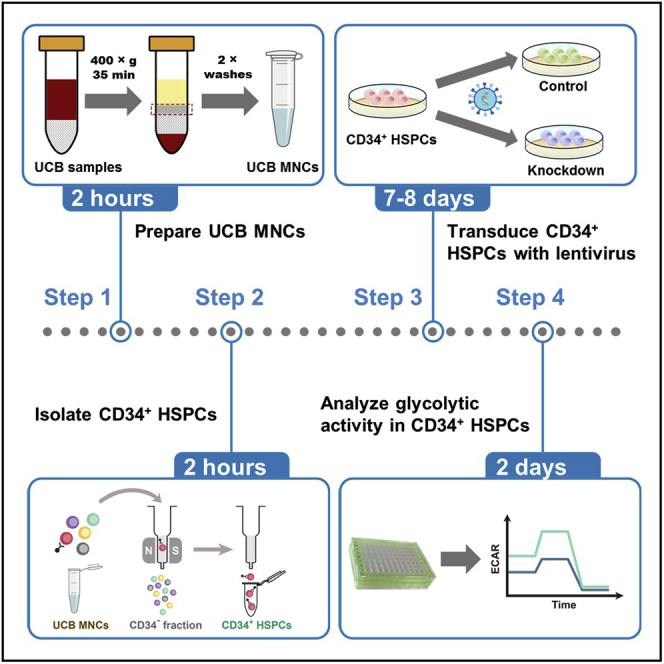
Summary
This protocol describes how to evaluate cellular metabolic state, including the glycolytic activity, in live CD34+ hematopoietic stem/progenitor cells (HSPCs) upon depletion of a specific target gene. We detail the procedure of enriching mononuclear cells from human umbilical cord blood samples, isolation of CD34+ HSPCs using positive immunomagnetic separation, and the analysis of glycolytic activity in lentivirally transduced CD34+ HSPCs using Seahorse Assay. This protocol can be applied to evaluate potential aerobic glycolysis gene targets for cancer therapy.
For complete details on the use and execution of this protocol, please refer to Qing et al. (2021).
Subject areas: Cell isolation, Cell-based Assays, Cancer, Metabolism, Molecular Biology, Gene Expression, Stem Cells
Graphical abstract
Highlights
-
•
Preparation of umbilical cord blood mononuclear cells (UCB MNCs) from human samples
-
•
Isolation of CD34+ HSPCs from UCB MNCs by positive immunomagnetic separation
-
•
Detailed protocol for lentiviral transduction of CD34+ HSPCs
-
•
Optimized procedure to assess glycolytic rates in CD34+ HSPCs via Seahorse Assay
This protocol describes how to evaluate cellular metabolic state, including the glycolytic activity, in live CD34+ hematopoietic stem/progenitor cells (HSPCs) upon depletion of a specific target gene. We detail the procedure of enriching mononuclear cells from human umbilical cord blood samples, isolation of CD34+ HSPCs using positive immunomagnetic separation, and the analysis of glycolytic activity in lentivirally transduced CD34+ HSPCs using Seahorse Assay. This protocol can be applied to evaluate potential aerobic glycolysis gene targets for cancer therapy.
Before you begin
The ultimate goal of cancer treatment is to eradicate tumor cells, while sparing normal cells. Therefore, in the development of new targeted cancer therapeutics, it is critical to evaluate the effects of targeted gene depletion on normal cells. Altered metabolism is a core hallmark of cancer, as tumor cells consume a markedly larger amount of glucose and secrete most glucose carbon as lactate even in normoxic conditions, a phenomenon called ‘aerobic glycolysis’ or ‘Warburg effect’ (Ward and Thompson, 2012). Recent remarkable advances have unveiled exciting therapeutic windows to effectively target cancer aerobic glycolysis, but the safety of anti-cancer metabolism therapies need to be carefully assessed before they can proceed to clinical applications. In this protocol, we use normal hematopoietic cells isolated from umbilical cord blood (UCB) as an example of normal cells, and assess the effects of silencing a specific gene (i.e., FTO alpha-ketoglutarate dependent dioxygenase [FTO], which is used as an example in the protocol) on glycolytic rates in normal hematopoietic stem/progenitor cells (HSPCs). For comparison, we have shown that genetic deletion of FTO significantly suppresses glycolytic rates in leukemia cells (Qing et al., 2021). This protocol will also be useful to assess the effects of depleting other candidate therapeutic target genes on glycolytic activity in both normal HSPCs and primary leukemia blast cells.
Prepare lentiviral particles
Timing: 4 days
Note: Prepare lentiviral particles in advance for effective target gene knockdown in CD34+ HSPCs via lentiviral transduction. Begin the packaging of lentiviral particles approximately one week prior to receiving fresh human UCB samples, so that the lentiviral particles are ready to be used for transduction when CD34+ HSPCs are ready.
-
1.
The HEK293T cell line (a highly transfectable derivative of human embryonic kidney 293 cells that expresses the SV40 T-antigen) is used for lentivirus packaging. On the day prior to transfection (day 0), seed 0.6 × 106 HEK 293T cells in 2 mL Dulbecco's Modified Eagle Medium (DMEM) complete medium per well in a tissue culture 6-well plate (seed 1.5× 106 HEK 293T cells in 4 mL DMEM complete medium for a 6 cm dish). Incubate the cells at 37 ˚C with 5% CO2 overnight (∼18 h) to allow cell attachment. The cells should be ∼70% confluent at time of transfection.
Note: To ensure efficient lentivirus packaging, the HEK 293T cells should be in good physiological state and be actively dividing.
-
2.On the day of transfection (day 1), bring the X-tremeGENE HP DNA Transfection Reagent, plasmid DNA, and the diluent (we used Opti-MEM I Reduced Serum Medium) to room temperature (20°C).
-
a.Dilute lentiviral packaging vectors (pMD2.G, pMDLg/PRRE, and pRSV-Rev) and pLKO.1 shRNA vectors (2 shRNAs targeting FTO and the control shNS [nonspecific shRNA coding plasmid] vectors in this protocol) with the diluent according to the following table.
Transfection complex
Reagent Amount for 6-well plate (1 well) Amount for 6 cm dish pMD2.G 0.5 μg 1.25 μg pMDLg/PRRE 0.3 μg 0.75 μg pRSV-Rev 0.7 μg 2.1 μg pLKO.1 shRNA for target gene 0.6 μg 1.8 μg Diluent (Opti-MEM I Reduced Serum Medium) 0.2 mL 0.5 mL -
b.Pipette 6 μL (for 6-well plate, 18 μL for 6 cm dish) of X-tremeGENE HP Reagent directly into the diluted mixture and avoid contacting the walls of the plastic tubes. Briefly vortex to mix and incubate at room temperature (20°C) for 15 min.
-
c.Add the DNA:Reagent complex to the cells and gently agitate plate to achieve a homogeneous distribution of compounds.
-
a.
Note: When adding the transfection complex, gently dispense it against the well wall and take care not to disrupt cells.
Note: Dilute DNA with at least 100 μL diluent, as transfection efficiency could be significantly impaired by lower volumes.
Alternatives: Other appropriate diluents, such as serum-free medium, may be used to dilute DNA.
Alternatives: Other transfection reagents, such as Lipofectamine 3000 and Effectene transfection reagent, may be used for transfection. Refer to the manufacturer’s instructions for efficient transfection and lentiviral packaging.
-
3.
At 6 h post transfection, carefully aspirate medium containing transfection complex from each well and slowly add 2 mL (for 6-well plate; 4 mL for 6 cm dish) pre-warmed DMEM complete medium.
CRITICAL: The removed medium should be handled as waste according to biosafety level 2 because it contains a small amount of lentivirus. Typically, we treat the removed medium with 10% bleach for at least 20 min prior to disposal. Discard the bleach-treated medium appropriately per institutional guidelines.
-
4.
Harvest the first batch of lentiviral supernatant at 30 h post transfection and replace with 2 mL (for 6-well plate; 4 mL for 6 cm dish) DMEM complete medium. Harvest the second batch lentiviral supernatant at 54 h post transfection. Combine the two batches and filter the lentiviral supernatant through a 0.45 μm filter to remove any cellular debris.
Note: The filtered lentiviral supernatant is ready to be used for transduction of CD34+ HSPCs. The lentiviral supernatant can be stored at 2°C–8°C for 1 week or be aliquoted and stored at −80°C. However, the virus titer could be significantly decreased during the freeze-thaw process. We highly recommend planning the experiment ahead of time so that fresh virus is used for infection of CD34+ HSPCs.
Prepare buffer solutions and cell culture medium
-
5.
Prepare the isolation buffer and MACS buffer for isolation of CD34+ HSPCs from human UCB via the magnetic labeling-separation method. Refer to materials and equipment for more details and complete recipe of the isolation buffer and MACS buffer.
Note: The MACS buffer needs to be degassed by vacuum, preferentially at room temperature (20°C), before use to get rid of air bubbles as the MACS column could be blocked by air bubbles.
-
6.
Prepare the culture medium for CD34+ HSPCs by supplementing SFEM medium (09650, STEMCELL Technologies) with human cytokines (SCF, IL-3, and IL-6). For detailed recipe of SFEM complete medium, refer to materials and equipment.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD34 FITC (4H11) | eBioscience | Cat# 11-0349-42; RRID: AB_1518732 |
| Biological samples | ||
| Fresh human umbilical cord blood | StemCyte | https://www.stemcyte.com/ |
| Chemicals, peptides, and recombinant proteins | ||
| Puromycin Dihydrochloride | Sigma-Aldrich | Cat# P8833; CAS: 58-58-2 |
| Bovine Serum Albumin solution | Sigma-Aldrich | Cat# A7034; CAS: 9048-46-8 |
| Recombinant Human IL-6 | PeproTech | Cat# 200-06 |
| Recombinant Human IL-3 | PeproTech | Cat# 200-03 |
| Recombinant Human SCF | PeproTech | Cat# 300-07 |
| L-Glutamine (200 mM) | Thermo Fisher Scientific | Cat# 25030-081 |
| Sodium Pyruvate (100 mM) | Thermo Fisher Scientific | Cat# 11360-070 |
| Penicillin Streptomycin | Thermo Fisher Scientific | Cat# 15-140-122 |
| Plasmocin prophylactic | InvivoGen | Cat# ant-mpp |
| M-PER Mammalian Protein Extraction Reagent | Thermo Fisher Scientific | Cat# 78501 |
| Ficoll Paque Plus | GE Healthcare | Cat# 17-1440-02 |
| DMEM, high glucose, with pyruvate | Life Technologies | Cat# 11995-073 |
| X-tremeGENE HP DNA Transfection Reagent | Sigma-Aldrich | Cat# 6366546001 |
| RetroNectin® Recombinant Human Fibronectin Fragment | Takara Bio Inc. | Cat# T100B |
| Trypsin-EDTA 0.05% | Fisher Scientific | Cat# 25-300-120 |
| Fetal bovine serum (FBS) | Gemini Bio-Products | Cat# 100-106 |
| Dimethyl Sulfoxide (DMSO) | Fisher Bioreagents | Cat# BP231-100; CAS: 67-68-5 |
| HEPES (1M) | Thermo Fisher Scientific | Cat# 15630080 |
| Polybrene | Sigma-Aldrich | Cat# H9268; CAS: 28728-55-4 |
| StemSpan SFEM medium | STEMCELL Technologies | Cat# 09650 |
| UltraPure 0.5M EDTA, pH 8.0 | Invitrogen | Cat# 15575020 |
| Cell-Tak Cell and Tissue Adhesive | Corning | Cat# 354240 |
| Seahorse XF RPMI Medium, pH 7.4 | Agilent Technologies | Cat# 103576-100 |
| Glucose Solution | Gibco | Cat# A2494001 |
| Sodium bicarbonate | Sigma-Aldrich | Cat# S5761-500G; CAS: 144-55-8 |
| Critical commercial assays | ||
| CD34 MicroBead Kit, human | Miltenyi Biotec | Cat# 130-046-702 |
| Seahorse XF Glycolytic Rate Assay Kit | Agilent Technologies | Cat# 103344-100 |
| Bio-Rad Protein (Bradford) Assay Dye Reagent | Bio-Rad | Cat# 5000006 |
| Seahorse XFe96 FluxPak mini | Agilent Technologies | Cat# 102601-100 |
| Experimental models: Cell lines | ||
| 293T | ATCC | (CRL-3216); RRID:CVCL_0063 |
| Recombinant DNA | ||
| pMD2.G | A gift from Dr. Didier Trono | Addgene plasmid # 12259; RRID: Addgene_12259 |
| pMDLg/pRRE | (Dull et al., 1998) | Addgene plasmid # 12251; RRID: Addgene_12251 |
| pRSV-Rev | (Dull et al., 1998) | Addgene plasmid # 12253; RRID: Addgene_12253 |
| pLKO.1-shNS | (Stewart et al., 2003) | Addgene plasmid # 8453 |
| pLKO.1-shFTO-1 | Sigma-Aldrich | TRCN0000246247 |
| pLKO.1-shFTO-2 | Sigma-Aldrich | TRCN0000246249 |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Seahorse Wave | Agilent Technologies | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-wave-desktop-software-740897 |
| Seahorse XF Glycolytic Rate Assay Report Generators | Agilent Technologies | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-xf-glycolytic-rate-assay-report-generators-740900 |
| FlowJo v10 | BD Life Sciences | https://www.flowjo.com/ |
| Other | ||
| Falcon polystyrene microplates, non-treated | Corning | Cat# 351146 |
| Seahorse XFe96 Analyzer | Agilent Technologies | n/a |
| Eppendorf Research plus 8-channel adjustable volume pipette, 30–300 μL | Eppendorf | Cat# 3125000052 |
| Eppendorf Research plus 8-channel adjustable volume pipette, 10–100 μL | Eppendorf | Cat# 3125000036 |
| Matrix Reagent Reservoirs | Thermo Scientific | Cat# 8094 |
Materials and equipment
Isolation buffer for preparation of UCB mononuclear cells (MNCs)
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| PBS, pH 7.4 | 137 mM NaCl 2.7 mM KCl 10 mM Na2HPO4 1.8 mM KH2PO4 |
137 mM NaCl 2.7 mM KCl 10 mM Na2HPO4 1.8 mM KH2PO4 |
498 mL |
| EDTA | 0.5 M | 2 mM | 2 mL |
| Total | n/a | n/a | 500 mL |
This buffer can be stored at 2°C–8°C for up to 24 months. Warm the buffer to 20°C before use.
Alternatives: Other anticoagulants can be used to replace EDTA, such as citrate phosphate dextrose (CPD) or citrate dextrose formula-A (ACD-A).
MACS buffer
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| PBS, pH 7.4 | 137 mM NaCl 2.7 mM KCl 10 mM Na2HPO4 1.8 mM KH2PO4 |
137 mM NaCl 2.7 mM KCl 10 mM Na2HPO4 1.8 mM KH2PO4 |
500.0 mL |
| EDTA | 0.5 M | 2 mM | 2.1 mL |
| BSA | 22% | 0.5% | 11.7 mL |
| Total | n/a | n/a | 513.8 mL |
The MACS buffer can be stored for up to 6 months at 2°C–8°C.
Alternatives: Other anticoagulants can be used to replace EDTA, such as citrate phosphate dextrose (CPD) or citrate dextrose formula-A (ACD-A).
Alternatives: Other proteins can be used to replace BSA, such as human serum albumin or fetal bovine serum (FBS).
Note: Ca2+- or Mg2+-containing buffers or media are not recommended for use.
SFEM complete medium
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| SFEM medium | n/a | n/a | 49.4 mL |
| SCF | 1000 μg/mL (100 μg/mL in the cytokine mixture) | 100 ng/mL | 50 μL of cytokine mixture |
| IL-3 | 100 μg/mL (10 μg/mL in the cytokine mixture) | 10 ng/mL | |
| IL-6 | 100 μg/mL (10 μg/mL in the cytokine mixture) | 10 ng/mL | |
| Penicillin-streptomycin | 10,000 U/mL | 100 U/mL | 500 μL |
| Plasmocin prophylactic | 2.5 mg/mL | 2.5 μg/mL | 50 μL |
| Total | n/a | n/a | 50 mL |
Prepare freshly, do not store.
Note: Human cytokines (SCF, IL-3, and IL-6) are purchased in powder form and reconstituted to stock solutions. Store cytokine stock solutions in aliquots of 10 μL at −20°C for up to 12 months and avoid freeze-thaw cycles. When preparing the SFEM complete medium, make a cytokine mixture that contains 100 μg/mL of SCF and 10 μg/mL of IL-3 and IL-6 by adding 10 μL of each cytokine stock solution to 70 μL PBS. Supplement 50 mL SFEM medium with 50 μL cytokine mixture to make the complete medium. To ensure optimal culture condition for CD34+ HSPCs, prepare a small amount of SFEM medium freshly every time. Store the remaining cytokine mixture at −20°C and use within 3 months.
DMEM complete medium
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| DMEM, high glucose, with pyruvate | n/a | n/a | 500.0 mL |
| FBS | n/a | 10% (vol/vol) | 56.0 mL |
| Penicillin-streptomycin | 10,000 U/mL | 100 U/mL | 5.6 mL |
| Plasmocin prophylactic | 2.5 mg/mL | 2.5 μg/mL | 562 μL |
| Total | n/a | n/a | 562.2 mL |
The DMEM complete medium can be stored for up to 3 months at 2°C–8°C.
Freezing medium
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| FBS | n/a | 90% (vol/vol) | 45 mL |
| Dimethyl sulfoxide (DMSO) | n/a | 10% (vol/vol) | 5 mL |
| Total | n/a | n/a | 50 mL |
The freezing medium can be stored for up to 6 months at 2°C–8°C.
0.1 M NaHCO3 solution, pH 8.0
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| NaHCO3, powder | n/a | 0.1 M | 420.05 mg |
| double-distilled H2O | n/a | n/a | adjust to a final volume of 50 mL |
| Total | n/a | n/a | 50 mL |
Adjust pH to 8.0 with HCl or NaOH and filter the solution through a 0.22 μm filter. The prepared solution can be stored at 2°C–8°C for at least one year.
Glycolytic rate assay medium
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| XF RPMI medium (with 1 mM HEPES) | n/a | n/a | 96.7 mL |
| HEPES | 1 M | 5 mM | 0.4 mL |
| L-Glutamine | 200 mM | 2 mM | 1.0 mL |
| Glucose solution | 200 g/L (1.11 M) | 10 mM | 0.9 mL |
| Sodium pyruvate | 100 mM | 1 mM | 1.0 mL |
| Total | n/a | n/a | 100.0 mL |
Prepare freshly, do not store.
Step-by-step method details
Enrichment of MNCs from UCB
UCB MNCs are isolated with Ficoll Paque Plus by density gradient centrifugation. Usually, about 2 × 108 UCB MNCs can be isolated from 100 mL fresh UCB samples.
Alternatives: In this protocol, fresh human UCB is used as the source of MNCs and CD34+ HSPCs. Other sample types that also contain CD34+ HSPCS, such as bone marrow (BM) or peripheral blood (PB), may be used to replace cord blood. The percentage of hematopoietic progenitor cells is about 0.1%–1% in UCB, 0.5%–5% in BM, and 0.05%–0.2% in PB.
-
1.
Disinfect the UCB bag with 70% ethanol and cut the end of tubing to allow UCB flow into a 50 mL tube.
-
2.
Dilute 1 volume of UCB with 3 volumes of isolation buffer. For example, dilute 10 mL UCB with 30 mL isolation buffer.
Note: The yield and purity of UCB MNCs depend largely on the removal efficiency of erythrocytes. When erythrocytes aggregate and form clumps in cord blood, some UCB MNCs can be trapped in the clumps, leading to their co-sedimentation with the erythrocytes and consequently a low yield of UCB MNCs. This tendency to trap UCB MNCs can be reduced by dilution of the cord blood sample, which decreases the size of erythrocyte clumps and gives a better yield of UCB MNCs.
-
3.
Add 15 mL Ficoll Paque Plus to a new 50 mL tube, and slowly layer the diluted cord blood on top of Ficoll Paque Plus while holding the tube in a tilt position. Take care not to disturb the interface between Ficoll Paque Plus media and the diluted cord blood; do not mix Ficoll Paque Plus with the sample (Figure 1A). The technique for layering sample on top of Ficoll Paque Plus is demonstrated in Methods video S1.
Figure 1.

Preparation of UCB MNCs from human cord blood samples
(A) Fresh UCB sample diluted with isolation buffer was layered onto Ficoll Paque Plus without disturbing the interface.
(B) Separation of the four fractions after density gradient centrifugation. From top to bottom: plasma + platelets, UCB MNCs, Ficoll Paque Plus, and granulocytes + erythrocytes.
(C) Assembly of the MACS MS column with the MACS Separator and the MACS MultiStand.
See also Methods video S1.
-
4.
Conduct density gradient centrifugation in a centrifuge with swinging bucket rotor without brake at 400 × g for 35 min at 20°C.
-
5.
Remove the upper layer (this fraction contains plasma and platelets and may be saved for later use if needed; Figure 1B) and leave the mononuclear cell layer undisturbed at the interface.
-
6.
Use a sterile pipette to transfer the layer of UCB MNCs to a new 50 mL centrifuge tube.
-
7.
Fill the centrifuge tube with isolation buffer to wash the cells. The volume of isolation buffer should be at least 3 times of that of the transferred mononuclear cells. For example, add at least 3 mL of isolation buffer to 1 mL of transferred mononuclear cells.
-
8.
Suspend the cells in isolation buffer and centrifuge with break on at 300 × g for 10 min at 20°C.
-
9.
Remove the supernatant and resuspend the cell pellet in 50 mL isolation buffer. Count the cells with a hemocytometer or an automated cell counter. Centrifuge with break on at 300 × g for 10 min at 20°C to wash cells.
Note: Although centrifugation at a higher speed can increase the mononuclear cell recovery, a lower centrifugation speed (60–100 × g) is recommended when it is critical to get rid of platelets.
-
10.
Carefully aspirate the supernatant completely and resuspend up to 108 cells in 300 μL cold (4°C) MACS buffer. Proceed to isolation of CD34+ HSPCs.
Note: If more than 108 cells are used for isolation of CD34+ HSPCs, scale up the buffer volume accordingly. For example, use 600 μL MACS buffer to resuspend up to 2 × 108 cells.
Optional: If the total number of UCB MNCs is greater than that needed for isolation of CD34+ HSPCs, the extra cells may be cryopreserved at a density of 10–50 × 106 cells per mL of freezing media at −80°C (for up to 1 month) or in liquid nitrogen (for long-term storage).
Isolation of CD34+ HSPCs via positive immunomagnetic separation
Isolation of CD34+ HSPCs from UCB MNCs is based on a magnetic labeling-separation method. CD34 antibody-conjugated MicroBeads are used to magnetically label CD34-expressing cells, including hematopoietic progenitor cells, endothelial progenitor cells, vascular endothelial cells, etc. (Dever et al., 2016).
Note: The reagent and total volumes given in the following steps are for isolation of up to 108 UCB MNCs. Scale up all volumes if more than 108 total cells are used for isolation of CD34+ HSPCs.
-
11.
For up to 108 cells resuspended in 300 μL MACS buffer, add 100 μL FcR blocking reagent and 100 μL CD34 MicroBeads. Mix well and incubate at 2°C–8°C (in the refrigerator) for 30 min. During incubation, flick the bottom of the tube every 10 min to mix contents.
Note: 2°C–8°C is the recommended incubation temperature and 30 min is the recommended incubation time. Non-specific cell labeling may be increased with higher temperatures and/or longer incubation times. If the incubation is conducted on ice, longer incubation times may be required.
-
12.
Wash the cells by adding 5–10 mL MACS buffer and centrifuge with break on at 300 × g for 10 min at 4°C.
Note: Pre-cool the centrifuge to 4°C to keep cells cold.
-
13.
Completely aspirate the supernatant and resuspend the cell pellet in 500 μL MACS buffer.
-
14.
Place a MS column in the magnetic field of a MACS Separator (Figure 1C) and rinse the column with 500 μL MACS buffer.
Note: The MS column can be used to isolate up to 2 × 108 total cells and up to 107 labeled CD34+ cells. For larger cell numbers, choose another appropriate MACS Column and MACS Separator for isolation of CD34+ cells. For example, the LS column can be used to isolate up to 2 × 109 total cells and up to 108 CD34+ cells, and the XS column can be used to isolate up to 2 × 1010 total cells and up to 109 CD34+ cells. Refer to the manufacturer’s instructions for appropriate volumes to be used if other columns are chosen.
-
15.
Add cell suspension onto the column and collect the flow-through. This cell fraction contains unlabeled cells that are CD34 negative and can be saved for later use if needed.
-
16.
Wash the column with 500 μL MACS buffer. Collect the flow-through and combine with the unlabeled cells collected in step 15 if needed. Repeat the wash twice for a total of three washes.
-
17.
Label a 1.5 mL Eppendorf tube with “CD34+ cells”, remove the column from the MACS separator and place it on the tube.
-
18.
Add 1 mL MACS buffer onto the column and immediately push the plunger firmly into the column to flush out the labeled CD34+ cells.
Optional: The eluted fraction can be enriched over a second MS Column to improve the purity of isolated CD34+ cells. Repeat the magnetic separation procedure from steps 14–18 once by using a new column.
Optional: The purity of the isolated hematopoietic progenitor cells can be evaluated by flow cytometry (Figure 2). The CD34 antibody used for direct immunofluorescent staining of the cells should recognize another epitope different from that recognized by QBEND/10, which is the CD34 monoclonal antibody conjugated to the magnetic beads. We use Anti-Human CD34 FITC (4H11) antibody from eBioscience in this protocol.
Figure 2.

Isolation of CD34+ HSPCs from UCB MNCs
The percentage of CD34+ cells was 0.36% in UCB MNCs, as tested in the sample used in this protocol. After immunomagnetic isolation of CD34+ HSPCs from UCB MNCs, the percentage of CD34+ cells was 90.1% in the isolated CD34+ HSPCs.
Pause point: If the lentiviral particles are not ready or the number of CD34+ HSPCs isolated is not enough, the cells could be cultured in SFEM complete medium to expand for a few days (no longer than one week as the CD34+ HSPCs have a limited lifespan in culture) before the lentiviral transduction. The isolated CD34+ HSPCs could also be cryopreserved at −80°C for up to one month or in liquid nitrogen for up to one year, if not immediately used for lentiviral transduction and functional analysis. However, we recommend using freshly isolated CD34+ HSPCs for the downstream analysis to achieve optimal and reliable results.
Lentiviral transduction of CD34+ HSPCs
-
19.
Dilute the RetroNectin stock solution (1 mg/mL) with sterilized PBS to achieve a concentration of 50 μg/mL.
-
20.
Dispense 1 mL of diluted RetroNectin solution (50 μg/mL) into each well of a non-treated 6-well plate with aseptic technique. Incubate overnight (∼18 h) at 4°C when coating the plate on the day prior to lentiviral transduction (or for at least 0.5 h at room temperature [20°C] when coating the plate on the same day of lentiviral transduction).
Note: The coating efficiency of RetroNectin and especially the transduction efficiency could be reduced when using a tissue culture-treated plate. Hence, it is necessary to use non-treated culture vessels for RetroNectin coating and lentiviral transduction.
-
21.
Aspirate the RetroNectin solution and block the plate with 2% bovine serum albumin (BSA) solution for 30 min at room temperature (20°C) to reduce non-specific protein-protein interactions.
-
22.
Remove the BSA solution and add 2 mL of PBS to each well to wash the plate. Remove PBS but do not allow the plate to dry. The RetroNectin-coated plate is now ready to be used for infection.
Note: If not used immediately, the RetroNectin-coated plate can be stored at 4°C for up to one week.
-
23.
Add 1 mL of lentiviral supernatants prepared in steps 1–4 of the “Before You Begin” section to each well of the RetroNectin-coated plate. In this protocol, we add lentiviral supernatants of shNS, shFTO-1, and shFTO-2 into three wells of the 6-well plate.
-
24.
Centrifuge the plate at 2000 × g for 2.5 h at 32°C. Remove the lentiviral supernatant from the plate. Do not allow the plate to dry.
-
25.
Pipette 1 × 106 CD34+ HSPCs in 2 mL SFEM complete medium into each well of the plate. Add polybrene to each well to a final concentration of 4 μg/mL. Centrifuge the plate with break on at 600 × g for 10 min at 32°C, and incubate for 48 h in a 37°C, 5% CO2 incubator.
-
26.
At 48 h post lentiviral transduction, change medium for the transduced cells and add puromycin to the cells to a final concentration of 2 μg/mL. Culture the cells in presence of puromycin for 3–4 days to select successfully-transduced CD34+ HSPCs.
-
27.
Use ∼1 × 106 transduced cells from each group to conduct protein extraction and Western Blot (or RNA extraction and RT-qPCR) to validate that the target gene expression has been efficiently knocked down. Troubleshooting 2
Note: At the time of Glycolytic Rate Assay, the cells have been cultured ex vivo for about one week and the percentage of CD34 expression will not be as high as freshly isolated CD34+ HSPCs. This protocol aims to evaluate the effects of shRNA-mediated FTO knockdown on glycolytic activity in normal healthy hematopoietic cells, as compared to its effects in leukemic cells. Thus, the expansion and potential differentiation of CD34+ HSPCs do not have significant impact on the results. However, if it is critical to keep high purity of CD34+ cells for the analysis, the cell culture condition should be further optimized to maintain the stemness and CD34+ expression of the cells (Jiang et al., 2018).
Analysis of glycolytic activity in lentivirally transduced CD34+ HSPCs
The basal and compensatory glycolytic activity in CD34+ HSPCs transduced with shNS, shFTO-1, and shFTO-2 are measured by the Seahorse XF Glycolytic Rate Assay. The CD34+ HSPCs are more delicate than cell lines and therefore it is important to limit the operation time to a minimum (within 2 h from steps 36–45b) to maintain the viability of CD34+ HSPCs. It is recommended to familiarize yourself with this assay using leukemia cell lines before conducting it with CD34+ HSPCs.
-
28.
On the day prior to the Glycolytic Rate Assay, unpack a Seahorse XFe96 sensor cartridge set in the cell culture hood, disassemble the sensor cartridge from the utility plate, and place the sensor cartridge upside down as shown in Figure 3A.
-
29.
Fill each well of the utility plate with 200 μL Seahorse XF calibrant solution. Put the sensor cartridge onto the utility plate and make sure sensors of the cartridge are completely submerged in the calibrant solution.
-
30.
Hydrate the assembled sensor cartridge at 37°C in a non-CO2 incubator overnight (∼18 h). To prevent evaporation of the calibrant, verify that the incubator is properly humidified. Troubleshooting 3
-
31.
Turn on the Seahorse XFe96 Analyzer, open the “Wave” software, and select “Heater on” (with the temperature set at 37°C) to allow the temperature to stabilize.
-
32.
On the day of the assay, dilute Cell-Tak stock solution with 0.1 M NaHCO3 (pH 8.0) to a final concentration of 22.4 μg/mL.
Note: The stock concentrations of Cell-Tak vary lot-to-lot; refer to the package for the exact concentration. For one Seahorse XF96 cell culture microplate, prepare 3 mL of diluted Cell-Tak solution.
Note: The optimal final pH of the diluted Cell-Tak solution is between 6.5 and 8.0. Once diluted into the neutral buffer (0.1 M NaHCO3, pH 8.0), apply to coat each well of the cell culture microplate within 10 min as adsorption begins immediately upon pH changing.
-
33.
Apply 25 μL of the diluted Cell-Tak solution to each well of a Seahorse XF96 cell culture microplate and let stand for at least 20 min at room temperature (20°C) to allow adsorption.
-
34.
Aspirate the solution and wash each well twice with 200 μL sterile water.
Alternatives: The Seahorse XF96 cell culture microplate can be coated on the day of the Glycolytic Rate Assay and used immediately after coating. Alternatively, follow steps 32–34 to coat the plate in advance and store the coated plate at 4°C for up to one week before using it in the assay. Warm the coated plate to room temperature (20°C) before cell seeding.
-
35.
Prepare the assay medium by supplementing the Seahorse XF RPMI medium (pH 7.4, with 1 mM HEPES) with 2 mM glutamine, 10 mM glucose, 1 mM pyruvate and 4 mM HEPES (to obtain a final concentration of 5 mM HEPES). Warm the assay medium to 37°C in a water bath.
Alternatives: The Seahorse XF RPMI medium can be replaced with other Seahorse XF medium without phenol red, such as the Seahorse XF DMEM medium (pH 7.4, Cat#103575-100). Choose the most appropriate medium for the cells used in the assay (please check other options here: https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-assay-kits-reagents-cell-assay-media/seahorse-xf-media-supplements-calibrant-740890).
-
36.
Count each group of the lentivirally transduced CD34+ HSPCs three times with an automated cell counter. Use the average cell number to calculate the volume needed to transfer 1.0 × 106 cells of each group to a 15 mL centrifuge tube.
-
37.
Pellet cells at 300 × g with break on for 10 min at room temperature (20°C) and resuspend the cell pellet of each group in 1.8 mL warm assay medium.
-
38.
Using an 8-channel multi-pipette and a sterile reagent reservoir, pipette 180 μL cell suspension to each sample well and 180 μL assay medium without cells to each background correction well of the Cell-Tak-coated XF96 cell culture microplate.
Note: Keep an angle (∼45°) while pipetting so that the cells can evenly distribute and adhere to the well.
Note: In this protocol, 8 wells in a row are assigned to each group as technical replicates to reduce measurement errors (Figure 3B). Similarly, 8 wells in a row are assigned as the background correction wells. It is recommended to keep at least 4 wells as technical replicates for each group to keep the measurement errors within the acceptable range.
-
39.
Centrifuge the plate with break on at 300 × g for 5 min at room temperature (20°C). Ensure that the centrifuge is properly balanced.
Note: Examine under a microscope to visually confirm adherence of the cells to the well bottom.
-
40.
Place the cell culture plate in a regular incubator (37°C, with 5% CO2) until the sensor cartridge is loaded with the assay compounds and ready for calibration. It usually takes 10–30 min to load the sensor cartridge with the assay compounds (steps 41–43). Only place the cell culture plate in a non-CO2 incubator when the sensor cartridge is loaded and the calibration process of Seahorse Glycolytic Rate Assay is ready to be started (step 44). The cells need to be stabilized in the non-CO2 incubator for 30 min before being measured in the Glycolytic Rate Assay, which can be performed during the calibration process.
-
41.
Open one foil pack from the Seahorse XF Glycolytic Rate Assay Kit and remove the Rotenone (Rot)/Antimycin A (AA) vial and the 2-deoxy-D-glucose (2-DG) vial. Tap down the vials to make sure the powder is on the tube bottom before opening the vials. Prepare the compound stock solutions with the assay medium according to Table 1. Vortex for ∼1 min to ensure that compounds are completely dissolved.
Note: Use compounds the same day they are reconstituted. It is not recommended to save the remaining compounds (at −20°C) because old compounds will not function the same as the freshly prepared ones and will cause a considerable increase in measurement errors. Troubleshooting 5
-
42.
According to Table 2, dilute the stock solutions to working solutions that are ready for loading the sensor cartridge.
-
43.Retrieve the hydrated sensor cartridge from the non-CO2 incubator and load the sensor cartridge by completing the following steps:
-
a.Align the A/D port loading guide (provided with each Seahorse XFe96 sensor cartridge set) with the sensor cartridge so that the letter “A” on the guide is in the top left-hand corner (Figure 4A).
- b.
-
c.Align the B/C port loading guide with the cartridge to place the letter “B” of the guide in the top left-hand corner (Figure 4B).
-
d.Repeat the loading procedure described in step 43b to load port B with 22 μL 2-DG working solution.
-
a.
Alternatives: The sensor cartridge can be loaded without using a port loading guide. If doing so, do not insert the entire pipet tips into the port to avoid unintended compound leaking caused by accidental forcing injection. Position the pipette tips as shown in Figure 4C. Troubleshooting 5
-
44.
Place the cell culture plate in a non-CO2 incubator for 30 min before being measured in the Glycolytic Rate Assay.
-
45.Run the Glycolytic Rate Assay by completing the following steps:
-
a.In the “Wave” software, select the Seahorse XF Glycolytic Rate Assay template and click “Design”. Tailor the template (groups/conditions, plate map, and instrument protocol) to specific assay design (see Figure 5 for an example).
-
b.When prompted, place the loaded sensor cartridge with the utility plate into the Seahorse XFe Analyzer and start the calibration process.
-
c.After calibration, replace the utility plate with the XF96 cell culture microplate seeded with CD34+ HSPCs (after 30 min of incubation in a non-CO2 incubator) when prompted, and begin the assay.
-
a.
Note: Remove the cartridge lid and verify correct plate orientation when putting the sensor cartridge into the XFe Analyzer; calibration will take approximately 20–30 min. Troubleshooting 3
Optional: If it is difficult to count the cell number accurately, an additional normalization step can be performed after the assay by measuring the total protein content in each well of the XF96 cell culture microplate. Use M-PER Mammalian Protein Extraction Reagent to lyse the cells and conduct Bradford Assay or BCA assay to determine the protein concentration.
-
46.
The assay result can be viewed in the “Wave” software. For further analysis and calculation of basal/compensatory glycolytic rates, export the data as “GraphPad Prism” file or “Seahorse XF Glycolytic Rate Assay Report Generator” file (download the “Seahorse XF Glycolytic Rate Assay Report Generator” using this link: https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-xf-glycolytic-rate-assay-report-generators-740900). The calculated basal/compensatory glycolytic rates are provided in the “Seahorse XF Glycolytic Rate Assay Report Generator” result file.
Figure 3.

Hydration of sensor cartridge and a typical plate map of Glycolytic Rate Assay
(A) (Top) Image of the Seahorse XFe96 sensor cartridge set with its lid, sensor cartridge, and utility plate labeled. (Bottom) When filling the utility plate with calibrant to hydrate the sensor cartridge, put the sensor cartridge with the lid upside down to avoid sensors touching work surface.
(B) A typical plate map with 8 wells in a row assigned to each experimental group in the Glycolytic Rate Assay. Similarly, a row of 8 wells are assigned to background control group and only assay medium without cells will be added to these wells.
Table 1.
Stock solutions of Rot/AA and 2-DG
| Compound | Stock concentration | Volume of assay medium |
|---|---|---|
| Rot/AA | 50 μM | 540 μL |
| 2-DG | 500 mM | 3000 μL |
Table 2.
Working solutions of Rot/AA and 2-DG
| Compound | Concentration in well | Working solution concentration | Stock solution volume | Assay medium volume |
|---|---|---|---|---|
| Rot/AA | 0.5 μM | 5 μM | 300 μL | 2700 μL |
| 2-DG | 50 mM | 500 mM | 3000 μL | 0 μL |
Figure 4.

Loading of Glycolytic Rate Assay compounds into the sensor cartridge
(A and B) With the help of loading guides, the Glycolytic Rate Assay compounds Rot/AA should be loaded into injection port A (A), and 2-DG should be loaded into injection port B (B).
(C) Different techniques for compound loading with (top) or without (bottom) using the loading guide.
Figure 5.

A typical running protocol for the Glycolytic Rate Assay
This running protocol includes an initialization measurement stage (calibration + equilibration), a basal glycolysis measurement step (3 measurement cycles of 6 min each), a compensatory glycolysis measurement step (after addition of Rot/AA; 3 measurement cycles of 6 min each), and a residual background acidification measurement stage (after addition of 2-DG; 3 measurement cycles of 6 min each).
Expected outcomes
Using the Ficoll Paque Plus-based density gradient centrifugation method described in this protocol, ∼2 × 108 UCB MNCs can be successfully isolated from ∼100 mL fresh cord blood samples, from which ∼0.5–2 × 106 CD34+ HSPCs can be enriched. The viability of the enriched CD34+ HSPCs should be around or above 90%. The purity of the enriched CD34+ HSPCs will be around or above 90%, as evaluated by flow cytometry with a CD34 FITC antibody recognizing an epitope other than that recognized by QBEND/10 (Figure 2).
The lentiviral transduction of CD34+ HSPCs facilitated by RetroNectin provides a feasible method to effectively manipulate target gene expression for further downstream analysis. After selection with puromycin, almost all the viable CD34+ HSPCs should have been successfully transduced and the target gene should have been efficiently knocked down given that the shRNAs used are effective to induce target gene silencing. In this protocol, the knockdown efficiency of FTO was confirmed by Western blot (Qing et al., 2021).
The Seahorse XF Glycolytic Rate Assay is expected to generate similar results to that shown in Figure 6A, with a basal glycolytic activity determined at the first stage, a compensatory glycolytic rate measured after the injection of Rot/AA (mitochondrial inhibitors), and a residual background acidification rate detected after the injection of 2-DG and inhibition of glycolysis. The accurate glycolytic rates are calculated by the Seahorse XF Glycolytic Rate Assay Report Generator using ECAR (extracellular acidification rate, mainly corresponding to glycolytic activity) and OCR (oxygen consumption rate, mainly corresponding to mitochondrial respiration activity) values measured during the assay. The example result shown in Figure 6A is obtained from a human leukemia cell line U937. Because glycolysis is generally running at a much higher rate in leukemia cells than in normal CD34+ HSPCs (the “Warburg effect”), the glycolytic rates in CD34+ HSPCs are expected to be much lower than that in leukemia cells, whereas the OCR values will demonstrate a much smaller difference between leukemia cells and normal CD34+ HSPCs (Figure 6A)(Qing et al., 2021). The calculated glycolytic rates will also be directly affected by the number of cells seeded per well (Figure 6B).
Figure 6.

Good and bad results from the Glycolytic Rate Assay
(A) A typical good result from Glycolytic Rate Assay demonstrating a 3-stage ECAR (extracellular acidification rate; corresponding to glycolytic activity) curve: the basal stage, the compensatory stage, and the post 2-DG stage. The glycolytic rates were calculated by the Seahorse XF Glycolytic Rate Assay Report Generator. The assay was performed with U937 cells and used as an example to show the expected pattern of Glycolytic Rate Assay.
(B) The number of cells seeded per well will directly affect the ECAR and OCR values measured during the assay. The assay was performed with U937 cells and used as an example to show the impact of different cell densities on the result.
(C) A bad result from Glycolytic Rate Assay due to incorrect or uneven loading of assay compounds into the injection ports. The assay was performed with U937 cells and used as an example to show the impact of bad loading technique on the results.
(D) A bad result from Glycolytic Rate Assay due to the use of assay compounds that are ineffective to inhibit mitochondrial respiration or glycolysis. The assay was performed with U937 cells and used as an example to show the impact of using old compounds on the result.
Data are represented as mean ± SD.
Limitations
The main purpose of this protocol is to evaluate the effects of a specific gene (we used the gene FTO as an example) on glycolytic activity in normal hematopoietic cells, as compared to the effects in leukemia cells (Qing et al., 2021). The UCB-derived CD34+ HSPCs are lentivirally transduced to achieve effective knockdown of the target gene. However, this will require additional culture time of CD34+ HSPCs to allow sufficient gene knockdown via selection with puromycin. By the time of Glycolytic Rate Assay, the CD34+ HSPCs will have been cultured ex vivo for about a week and will not be as primitive as freshly isolated ones. Herein, the purpose of our study is to determine the effect of a specific gene on metabolic state in normal hematopoietic cells, so the decrease in CD34 expression does not affect the results. However, if it is critical to measure the glycolytic activity in the pure stem/progenitor cell population, the cell culture condition need to be further modified to decrease differentiation/expansion and maintain stemness of the infected cells (Jiang et al., 2018).
While the Seahorse XF Glycolytic Rate Assay provides a convenient approach for measuring glycolytic activity in lentivirally transduced CD34+ HSPCs, the reliability of its results is highly dependent on the accurate counting of cell number of each group. It might be impossible to count the viable cells correctly for every group if the cell viability is significantly affected by the lentiviral transduction, leading to the unreliable results of Glycolytic Rate Assay. Although an additional protein content-based optimization step could be conducted to partially resolve this problem, cells with low viability are not appropriate to be measured by Glycolytic Rate Assay as they may go through apoptosis during the assay due to the harsh treatments of assay compounds. This would lead to results with unexpected patterns and big error bars, and thus are highly unreliable. Therefore, when working with genes that may significantly affect the viability of CD34+ HSPCs, the lentiviral transduction efficiency/gene knockdown efficiency need to be optimized and an alternative method to accurately measure the glycolytic activity (e.g., the radioactive glycolysis assay [Ashcroft et al., 1972; Tandon et al., 2011]) might be required.
Troubleshooting
Problem 1
Low UCB MNC or CD34+ HSPC yield/viability (step 4).
Potential solution
The freshness of UCB samples and appropriate temperature are essential to ensure efficient removal of erythrocytes and the yield/viability of isolated UCB MNCs and CD34+ HSPCs. Use fresh UCB samples that are not older than 4 hours and are kept at 4°C upon collection. The temperature should be maintained at 18°C–20°C during preparation of UCB MNCs and at 2°C–8°C during isolation of CD34+ HSPCs. Choose an appropriate MACS column for isolation of CD34+ HSPCs to ensure the efficiency of magnetic separation (e.g., MACS MS column for up to 2 × 108 total cells and MACS LS column for up to 2 × 109 total cells).
Problem 2
Knockdown efficiency is low (step 27).
Potential solution
Primary CD34+ HSPCs are more difficult to transduce compared to human leukemia cell lines. However, if the lentiviral particle preparation and CD34+ HSPC transduction are conducted strictly following the protocol, the target gene should be efficiently knocked down after selection with puromycin (Qing et al., 2021). If the knockdown efficiency is low for some shRNAs (but high for others), this indicates that those shRNAs are not effective to suppress the target gene expression, and more shRNAs targeting the same gene need to be screened to select those that can efficiently induce target gene silencing.
Problem 3
Calibration failure in the Glycolytic Rate Assay (steps 30 and 45).
Potential solution
The sensors embedded on the cartridge must be thoroughly hydrated for at least 6–8 h before the assay for the Seahorse sensor cartridge to function correctly. Shorter period of hydration or hydration longer than 48 h might impair the function of sensors and cause failure of the sensor cartridge to pass the calibration step. Hydrate the sensor cartridge overnight (∼18 h) to ensure the sensors function properly.
Problem 4
Low signal or unexpected pattern of the Glycolytic Rate Assay (step 40).
Potential solution
As shown in Figure 6B, different cell density would significantly affect the signal intensity measured during the assay. Since CD34+ HSPCs have much lower glycolytic activity than most leukemia cell lines, seeding too few CD34+ HSPCs per well might cause a low ECAR level not within the reliable detection range. In contrast, seeding too many cells will lead to the formation of multilayers, which may be disturbed and forced to be suspended during the injection and mixing steps of the assay, causing the number of cells being measured in the transient microchamber to change throughout the assay. Choose a cell density that would allow the cells to be ∼80%–90% confluent after being attached to the Cell-Tak coated plate and avoid seeding multilayers of cells. It is of equal importance to keep high viability of CD34+ HSPCs. Cells with low viability may not survive the harsh treatments during the assay and could not generate reliable results, and thus should not be used for the assay. Incubate CD34+ HSPCs for exactly 30 min in the non-CO2 incubator to prevent decrease in viability caused by longer incubation without CO2.
Problem 5
High variability among replicates in the Glycolytic Rate Assay (steps 41 and 43).
Potential solution
Variations in the Seahorse Glycolytic Rate Assay are affected by several factors: compound loading technique, effectiveness of the assay compounds, cell counting and seeding technique, and whether a monolayer of cells is seeded per well.
If assay compounds are not correctly loaded into the cartridge injection ports, the compound solution may not be injected by pressurized air into all wells during the assay, or a leakage of compound solution into the wells may occur before the assay starts, both leading to high variation and big error bars in the assay results (Figure 6C). Use the technique depicted in Figure 4C to correctly load assay compounds into the injection ports. Old or expired compounds that cannot effectively inhibit mitochondrial respiration and glycolysis in all wells will also lead to big error bars in the results (Figure 6D). Make sure the assay compounds are freshly prepared every time for the assay and correctly diluted to the desired concentrations.
Another important reason for high variability is uneven cell numbers among replicates in the same group. Make sure that cells are counted accurately, and that same number of cells is seeded evenly into each replicate well. As mentioned above, it is also critical that only a monolayer of cells is seeded per well to avoid disturbance of the cells during the assay to decrease the variability among replicates. Therefore, it is important to select an optimal cell density for the assay in a pilot experiment before conducting the actual one (Figure 6B).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jianjun Chen (jianchen@coh.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate/analyze new unique datasets/code.
Acknowledgments
This work was supported in part by the National Institutes of Health (NIH) grants R01 CA243386 (J.C.), R01 CA214965 (J.C.), R01 CA236399 (J.C.), R01 CA211614 (J.C.), and R01 DK124116 (J.C.), the Margaret Early Medical Research Trust (R.S.), and the Held Foundation Fellowship (Y.Q.). J.C. is a Leukemia & Lymphoma Society (LLS) Scholar.
Author contributions
Conceptualization, Y.Q., R.S., and J.C.; supervision, R.S. and J.C.; investigation, Y.Q., L.G., and L.H.; resources, R.S. and J.C.; funding acquisition, Y.Q., R.S., and J.C.; writing – original draft, Y.Q.; writing – review & editing, R.S. and J.C.
Declaration of interests
J.C. is a scientific founder of Genovel Biotech Corp. and holds equities with the company and is also a scientific advisor for Race Oncology.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100603.
Contributor Information
Ying Qing, Email: yqing@coh.org.
Rui Su, Email: rsu@coh.org.
Jianjun Chen, Email: jianchen@coh.org.
References
- Ashcroft S.J., Weerasinghe L.C., Bassett J.M., Randle P.J. The pentose cycle and insulin release in mouse pancreatic islets. Biochem. J. 1972;126:525–532. doi: 10.1042/bj1260525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M., Chen H., Lai S., Wang R., Qiu Y., Ye F., Fei L., Sun H., Xu Y., Jiang X. Maintenance of human haematopoietic stem and progenitor cells in vitro using a chemical cocktail. Cell Discov. 2018;4:59. doi: 10.1038/s41421-018-0059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing Y., Dong L., Gao L., Li C., Li Y., Han L., Prince E., Tan B., Deng X., Wetzel C. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m6A/PFKP/LDHB axis. Mol. Cell. 2021;81:922–939.e9. doi: 10.1016/j.molcel.2020.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart S.A., Dykxhoorn D.M., Palliser D., Mizuno H., Yu E.Y., An D.S., Sabatini D.M., Chen I.S., Hahn W.C., Sharp P.A. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon P., Gallo C.A., Khatri S., Barger J.F., Yepiskoposyan H., Plas D.R. Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency. Proc. Natl. Acad. Sci. U S A. 2011;108:2361–2365. doi: 10.1073/pnas.1013629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P.S., Thompson C.B. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze new unique datasets/code.