

Abstract

Data-independent acquisition (DIA) is an increasingly used approach for quantitative proteomics. However, most current isotope labeling strategies are not suitable for DIA as they lead to more complex MS2 spectra or severe ratio distortion. As a result, DIA suffers from a lower throughput than data-dependent acquisition (DDA) due to a lower level of multiplexing. Herein, we synthesized an isotopically labeled acetyl-isoleucine-proline (Ac-IP) tag for multiplexed quantification in DIA. Differentially labeled peptides have distinct precursor ions carrying the quantitative information but identical MS2 spectra since the isotopically labeled Ac-Ile part leaves as a neutral loss upon collision-induced dissociation, while fragmentation of the peptide backbone generates regular fragment ions for identification. The Ac-IP-labeled samples can be analyzed using general DIA liquid chromatography–mass spectrometry settings, and the data obtained can be processed with established approaches. Relative quantification requires deconvolution of the isotope envelope of the respective precursor ions. Suitability of the Ac-IP tag is demonstrated with a triplex-labeled yeast proteome spiked with bovine serum albumin that was mixed at 10:5:1 ratios, resulting in measured ratios of 9.7:5.3:1.1.
Introduction
Multiplexing with isotopic labels is an attractive alternative to label-free quantification in mass spectrometry-based proteomics since it increases throughput and reduces run-to-run variability.1 Remarkable advancements have been made in terms of multiplexing capacity over the last decades.2,3 Current multiplexing approaches can be classified into MS1-based methods4,5 that rely on isotopic labeling and quantify peptides based on distinct precursor ion masses and MS2-based methods that rely on isobaric labeling strategies and quantify peptides based on reporter ions,6,7 fragments of the peptide backbone,8,9 or peptide-coupled reporter ions.10−12 In the widely used data-dependent acquisition (DDA) mode, where a certain number of precursor ions are selected for fragmentation during a liquid chromatography–mass spectrometry (LC–MS) run in the order of decreasing intensity,13 current MS1-based quantification methods analyze up to three samples in a single experiment since higher multiplexing is constrained by the increasing complexity of MS1 spectra and the need for a mass difference of at least 4 Da between labeling channels to avoid overlap of isotope envelopes.14 In addition, the stochastic nature of selecting precursor ions for tandem MS in DDA leads to an imperfect overlap in peptide identifications, which means that there is a considerable likelihood that some peptides might be missed in some LC–MS runs depending on their relative abundance in given samples.15 Since only the peptides that are measured in all runs can be reliably quantified and thus compared across many runs in studies comprising hundreds of samples, it would be beneficial to use a strategy that is not data-dependent and thus avoids the stochastic precursor ion selection. This has led to the development of data-independent acquisition (DIA).
Since the sequential windowed acquisition of all theoretical fragment ion mass spectra (SWATH-MS)16 were proposed, DIA has become more widely used due to the advancement of mass spectrometry technology and sophisticated data processing algorithms.16−19 Instead of sampling a certain number of precursor ions, DIA selects all precursors within a predefined isolation window for fragmentation. This results in better reproducibility and avoids the stochastic nature of precursor ion selection in DDA. However, most DIA-based approaches can only analyze one sample per LC–MS run and most current isotope labeling-based quantification strategies, which are commonly used in DDA, complicate the MS2 spectra or severely distort the quantitative ratio in DIA. For example, reporter ion-based approaches, such as TMT7 or iTRAQ,6 are not suitable for DIA since most peptides are cofragmented with others. For the approaches generating a set of distinct fragment ions for different labeling channels, such as isotopic labeling using stable isotope labeling by amino acids in cell culture (SILAC)20 and fragment ion-based methods based on isobaric peptide termini labeling (IPTL),8 the complexity of MS2 spectra in DIA is multiplied by the number of labeled samples. This makes it extremely challenging to identify peptides since the MS2 spectra of DIA are already highly convoluted. As a result, DIA has a lower throughput than DDA due to a lower level of multiplexing. To increase the multiplexing capacity of DIA, a number of approaches have been reported, such as NeuCoDIA,21 MdFDIA,22 and mdDiLeu.23 These approaches rely on neutron encoding,24 which is based on the mDa mass difference due to incorporating either 13C or 15N, and require an ultrahigh resolution (>120 k) at the MS2 level, resulting in a reduced data acquisition rate on Orbitrap instruments. Recently, we reported a novel isobaric Ac-AG tag that reveals quantification information based on peptide-coupled reporter ions at a modest resolution of 17.5 k at the MS2 level in DIA mode.25 Since in DDA mode, only a limited number of precursor ions can be isolated for tandem MS within the time window of a chromatographic peak, isobaric labeling is advantageous since it enables multiplex labeling without increasing the complexity of MS1 spectra. This constraint does not apply to DIA mode where precursor selection for MS2 is independent of the complexity of the corresponding MS1 spectra, which means that isobaric labeling provides no particular advantage in this case, while the need for complementary isotope distributions between balancer and reporter ions complicates synthesis and limits the ultimately achievable multiplexing capacity.
Since isobaric labeling is not required in DIA, we developed a collision-induced dissociation (CID) cleavable, isotopically labeled acetyl-isoleucine-proline (Ac-IP) tag and demonstrated it with a triplex-labeled yeast-BSA (bovine serum albumin) mixed sample. While quantification is achieved at the precursor ion (MS1) level, differentially Ac-IP-labeled peptides generate identical MS2 spectra independent of the number of labeled samples and thus do not increase the complexity of MS2 spectra in DIA.
Experimental Section
Details of chemicals and materials, the synthesis of triplex acetyl-isoleucine-proline-N-hydroxysuccinimide (Ac-IP-NHS tags, see Figure 1A), reduction/alkylation and LysC digestion, selective N-terminal dimethylation of peptides, and LC–MS/MS analysis can be found in the Supporting Information.
Figure 1.

Design and concept of quantification based on the Ac-IP tag in DIA mode. (A) Molecular structure of the triplex Ac-IP tag. (B) Sample preparation workflow: protein digestion with LysC followed by selective N-terminal dimethylation and labeling with the Ac-IP tag. (C) Schematic representation of sample mixing and results at the MS1 and the MS2 level prior to and after deconvolution for a mixture of triplex-labeled samples. The differentially labeled samples are pooled prior to LC–MS/MS in DIA mode.
Triplex Labeling of N-Terminally Dimethylated Peptides
N-terminally dimethylated GTDWLANK (5 μg), LysC peptides of BSA, or LysC peptides of yeast proteins were dissolved in 50 μL of 200 mM triethylammonium bicarbonate (TEAB) buffer of pH 8.5. Then, 4 μL of 50 mM Ac-IP-NHS, 13C1-Ac-IP-NHS, and 13C2-Ac-IP-NHS in DMF was added to the three peptide solutions. The reaction mixtures were shaken for 2 h at room temperature. To ensure complete labeling, 2 μL of the respective Ac-IP-NHS reagent was added again and incubated for an additional 1 h. Esterification of the hydroxyl groups of Ser, Thr or Tyr, and excess N-hydroxysuccinimide (NHS) ester was hydrolyzed in the presence of 5% hydroxylamine hydrate at 55 °C for 5 min, and samples were then desalted by SPE using the STAGE (STop And Go Extraction) TIPS desalting procedure26 prior to LC–MS analysis. Acetonitrile (600 μL, 2%) in water with 0.1% trifluoroacetic acid (TFA) was added to remove excess Ac-IP-COOH before eluting peptides from the STAGE tips with 60% acetonitrile in water with 0.1% TFA (Figure S1, Supporting Information).
Database Searching and Quantification
To create the spectral library, LC–MS/MS raw files measured in DDA mode were analyzed with PEAKS X+ (Bioinformatics Solutions Inc., Waterloo, Ontario, Canada) and searched against the UniProt reference database of yeast (UP000002311, 6049 entries, downloaded on Jan. 20, 2020) to which the BSA entry (P02769) was added manually. LysC was selected as the enzyme, the digestion mode was set to specific, and the maximal number of missed cleavage sites was set to 1. A tolerance of 20 ppm for the precursor ion and 0.02 Da for the MS/MS fragment ions was applied. Carbamidomethylation (+57.02) on cysteine and dimethylation (+28.03) on the N-terminus were set as fixed modifications and oxidation (+15.99) on methionine as variable modification. The neutral losses of triplex Ac-IP tags were manually added in the “PTMNeutralLossScorer.properties” file as follows: “Ac-IP on Lys@K_155.094629_TRUE, Ac-IP (13C+1) on Lys@K_156.097983_TRUE, and Ac-IP (13C+2) on Lys@K_157.101338_TRUE.” For triplex labeling experiments, variable modifications on Lys were set as Ac-IP (+252.1474), 13C1-Ac-IP (+253.1508), and 13C2-Ac-IP (+254.1542). The identification results were filtered with a false discovery rate (FDR) of 0.5% for peptides. The filtered results were exported as the spectral library for DIA analysis using the default parameters of PEAKS X+.
For identification of peptides in DIA, the raw files measured in DIA mode were searched against the prepared spectral library and the UniProt reference database of yeast (UP000002311) complemented with BSA was selected as the PEAKS reference database. A tolerance of 20 ppm for the precursor ion and 0.02 Da for the MS/MS fragment ions was applied. The DIA results were also filtered with an FDR of 0.5% for peptides.
The PEAKS output of the peptides matched to proteins (protein-peptides.csv), and the peptide-spectrum matches (DB search psm.csv) were exported from the DIA identifications. All the following steps were performed using in-house built Python scripts (available at https://github.com/tianxiaobo002/Ac-IP_tag_scripts_and_quantification_outputs). Only the unique peptides extracted from the protein-peptides.csv files were used for quantification. For every peptide-spectrum match (PSM) that was derived from unique peptides in the DB search psm.csv files, the theoretical precursor masses were calculated and grouped by the scan number. The MS1 spectra of DIA raw data were converted to the mgf format with RawConverter27 for processing in Python. All MS1 spectra were extracted from the resulting mgf file and also grouped by the scan number. For a given PSM (based on the scan number of that PSM), the corresponding nearest preceding MS1 spectrum was selected from which the measured peak intensities of precursor masses were used to determine ratios for that PSM. Examples of deconvolution and ratio calculation at the PSM level can be found in Figure S2. When more than one PSM matched to the same peptide, the ratio derived from the PSM with the highest “–10lgP” value was used to represent the ratio for that peptide. The top three peptides with the highest −10lgP were used to calculate the ratio for the corresponding protein. Examples of calculating ratios for peptides and proteins can be found in Figure S3.
The quantification scripts and quantification outputs of triplex Ac-IP-labeled BSA, yeast, and BSA-yeast samples at the PSM, peptide, and protein level are available at https://github.com/tianxiaobo002/Ac-IP_tag_scripts_and_quantification_outputs. The raw mass spectrometry data of the yeast and the BSA-yeast samples have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD022606.
Results and Discussion
Design of the Ac-IP Tag and Labeling Strategy
The Ac-IP tag consists of three parts: (i) Ac-Ile, which contains distinct isotopes and will leave as a neutral loss fragment upon CID, (ii) proline, which facilitates dissociation of the Ile-Pro bond, and (iii) amine-reactive NHS ester reacting with the epsilon-amino group of the C-terminal Lys residues in peptides generated by LysC digestion (see Figure 1A for the structure design, Scheme 1 for the synthesis route, Figure S4 for LC–MS/MS results, and Figures S5–S7 for NMR analyses of the tags in the Supporting Information). Since the Ile-Pro bond fragments easily,9,28 the isotopically labeled Ac-Ile part of the tag leaves as a neutral loss fragment upon CID in addition to the fragmentation of the peptide backbone. Precursors of peptides derived from different samples thus have distinct masses carrying the quantitative information while generating identical MS2 spectra that can be analyzed by established DIA identification approaches.29 It is worth pointing out that the signals from the same peptide backbone fragment ions of every labeling channel add up, resulting in a better signal-to-noise (S/N) ratio in the MS2 spectra and thus facilitating peptide identification.
Scheme 1. Synthesis Approach for the Triplex Ac-IP Tag.

(i) 0.8 M DIPEA in DMF and Fmoc-Pro-COOH; (ii) 20% piperidine in DMF; (iii) 0.4 M HATU, 0.8 M DIPEA in DMF, and Fmoc-Ile-COOH; (iv) 20% piperidine in DMF; (v) acetic anhydride or acetic anhydride-1,1′-13C2 or acetic anhydride-13C4; (vi) 20% trifluoroethanol in DCM; (vii) EDC·HCl and N-hydroxysuccinimide. More detailed steps can be found in the Supporting Information.
Since labeling with the Ac-IP tag removes a high proton affinity site (epsilon-amino group of Lys), which might reduce the ionization efficiency of modified peptides (see Figure S8), we first dimethylated the N-terminal amine group30 by converting it into a tertiary amine to facilitate ionization31 and to prevent it from reacting with the Ac-IP tag (Figure 1B). Another advantage of N-terminal dimethylation is that it leads to a more complete b-ion series upon CID,31 which aids in the interpretation of the MS2 spectra. Since the mass difference between the Ac-IP tags is 1 Da, differentially labeled peptides will form overlapping isotope envelopes with a changing intensity profile according to the different quantitative ratios of a given peptide in the different samples (Figure 1C). Once a peptide has been identified, its composition is used to calculate the theoretical intensity distribution of the isotope envelope, which serves to deconvolute the overlapping precursor isotope envelopes from the MS1 spectra to retrieve the underlying quantitative information (see Figure S2 for details).
Derivatization and Quantification at the Peptide Level
GTDWLANK was selected as the model peptide to assess the labeling efficiency of the Ac-IP approach and investigate the influence of modifications on ionization efficiency and the charge state. As expected, ionization efficiency increased after N-terminal dimethylation31 (N-dime) while the intensities of precursor ions with or without the Ac-IP modification were comparable (Figure S9). Both reactions had yields exceeding 98%. The charge state distribution of N-dime-GTDWLANK-PI-Ac remained almost unchanged with the doubly charged ion being the most abundant (97.4%) (Table S1). Fragmentation of N-dime-GTDWLANK-PI-Ac was evaluated at different normalized collision energies (NCEs) ranging from 18 to 32. As shown in Figure S10, in the range from 18 to 22, fragmentation occurred only at the fragile Ile-Pro bond of the Ac-IP tag, giving only the “precursor minus Ac-Ile” fragment ion in MS2 spectra. With increasing the NCE from 24 to 32, fragment ions of the peptide backbone became stronger accompanied by a gradual decrease in intensity of the ion precursor minus Ac-Ile. Neutral loss of Ac-Ile is complete, and peptide backbone fragmentation is efficient at a NCE of 30, which was therefore selected for further experiments.
N-dime-GTDWLANK-PI-Ac (+0, +1, and +2) was analyzed individually or mixed at a ratio of 1:1:1. Differential labeling resulted in distinct peptide masses but identical MS2 spectra (Figure S11). The triplex-labeled N-dime-GTDWLANK-PI-Ac peptides were then mixed at ratios of 1:1:1, 1:2:5, 1:10:20, 5:2:1, and 20:10:1 followed by LC–MS/MS analysis. As shown in Figure 2A–F, the shape of the isotope envelope of the precursor ions changed with different mixing ratios while all mixtures produced identical MS2 spectra (Figure 2G). Quantification information was retrieved from the isotope envelopes of the precursor ions after deconvolution (see the example in Figure S2), and the measured ratios were in agreement with the actual mixing ratios.
Figure 2.
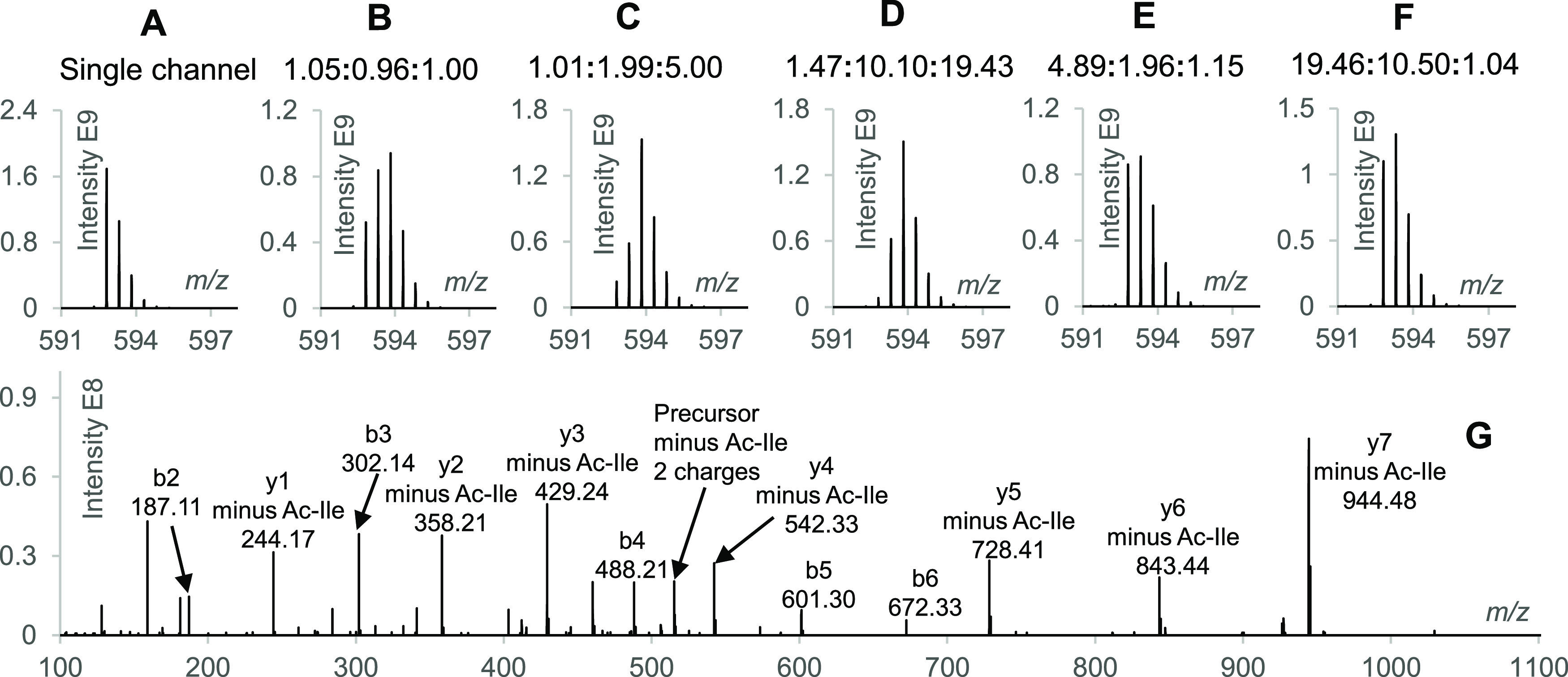
Investigations with triplex-labeled N-dime-GTDWLANK-PI-Ac. (A) Precursor isotope envelope of the single channel N-dime-GTDWLANK-PI-Ac-(+0). (B–F) Precursor isotope envelopes of triplex-labeled N-dime-GTDWLANK-PI-Ac (+0, +1, and +2) mixed at ratios of 1:1:1, 1:2:5, 1:10:20, 5:2:1, and 20:10:1, respectively (measured ratios are shown on the top of each panel). (G) MS2 spectrum of the triplex-labeled N-dime-GTDWLANK-PI-Ac mixed at 1:1:1 with a NCE of 30.
Preparation of a Spectral Library and DIA Quantification of the Triplex-Labeled BSA Sample
Triplex-labeled LysC BSA peptides (+0, +1, and +2) were analyzed with DDA, and the corresponding identifications, reported by PEAKS sofware,32 were exported as a spectral library. Subsequently, triplex-labeled BSA peptides were mixed at ratios of 1:1:1, 1:5:10, and 10:5:1 and analyzed with DIA followed by searching data against the spectral library. The DIA scanning scheme includes a survey scan acquired at a resolution of 70 k and 20 sequential precursor ion selection windows covering an m/z range of 450–950 acquired within a cycle time of about 2 s at a resolution of 17.5 k at the MS2 level, making the method accessible to most high-resolution mass spectrometers. The log 2-normalized ratios of the triplex-labeled BSA sample mixed at a ratio of 1:1:1 converge to zero for peptide-spectrum matches (PSMs) with a higher significance (−10lgP values) (Figure 3A and Figure S12) and for more intense peptides (Figure 3B). Consequently, the three peptides with the highest significance values, provided by the PEAKS identification algorithm, were selected and the total intensities of their respective labeling channels were used to calculate the normalized protein ratios (see Figure S3 for details). Applying these rules, the BSA samples mixed at 1:1:1, 10:5:1, and 1:5:10 were measured to be 1.02:1.00:0.98, 9.75:5.25:1.00, and 1.02:5.04:9.94, respectively (Figure 3C,D).
Figure 3.

Evaluations of the quantification performance based on triplex-labeled LysC peptides of BSA. (A) Log 2-normalized ratio distribution of peptide-spectrum matches (PSMs) of BSA peptides prepared by LysC digestion and mixed at a ratio of 1:1:1 in relation to PSM significance (−10lgP values). Only the PSM with the highest significance value for each peptide, as provided by the PEAKS identification algorithm, was used. (B) Log 2-normalized ratio distribution of BSA peptides prepared by LysC digestion and mixed at a ratio of 1:1:1 in relation to precursor intensity. (C, D) Ratio distributions at the peptide level of BSA peptides prepared from LysC digestions and mixed at ratios of 10:5:1 and 1:5:10, respectively. The dotted lines represent the expected values of log 2-normalized ratios.
Quantification of a Specific Protein within a Complex Proteomics Background
To assess the labeling efficiency and the quantification accuracy in complex samples, triplex-labeled LysC peptides of yeast proteins were prepared. For N-terminal dimethylation,9 more than 98% of LysC yeast peptides had a labeling yield exceeding 95%, as calculated from DDA data with variable modification searches. The Ac-IP labeling efficiency was high with 2292 out of 2313 peptides (99%) having a labeling yield of more than 98%. Only 0.03% of the identified peptides were also identified when the modification at the C-terminal Lys residue was set to proline instead of Ac-IP, which indicates that the in-source fragmentation of the Ile-Pro bond was negligible. The labeled peptide precursor ions were mostly doubly charged (Figure S13), and the numbers of identified peptides were comparable before and after labeling. Notably, ∼90% (24,531 out of 27,938) of matched y-ions had a neutral loss of Ac-Ile exceeding 98%. The spectra from the DDA runs of 908 identified proteins in the triplex Ac-IP-labeled LysC yeast sample were exported as a spectral library, and 688 proteins were identified by PEAKS when searching the DIA data of the 1:1:1 triplex-labeled sample. Out of these, 608 proteins were quantified based on the rules outlined above. Eighty identified proteins could not be quantified due to peptide precursor ions with incomplete isotope envelopes. The medians of protein ratios of the 1:1:1 mixed yeast sample were measured to be 1.01:0.96:1.02 (Figure 4A). In the mixed BSA (10:5:1)-yeast (5:2:1) sample, the medians of protein ratios of the LysC yeast sample were measured to be 4.96:1.95:1.09 (Figure 4B) and the ratios of triplex-labeled BSA were measured to be 9.67:5.26:1.07 (Figure 4C).
Figure 4.

Analysis of Ac-IP-tagged LysC yeast samples and BSA-yeast mixed samples. (A) Box plots showing the distribution of measured protein ratios in the log 2 transformation of triplex-labeled LysC yeast samples mixed at 1:1:1; (B, C) quantification results of a mixed BSA (10:5:1)-yeast (5:2:1) proteomics sample, in which triplex-labeled BSA peptides prepared from LysC digestion were mixed at 10:5:1 and yeast peptides prepared from LysC digestion were mixed at 5:2:1. (B, C) Box plots showing the distribution of measured protein ratios in the log 2 transformation of triplex-labeled LysC yeast samples and triplex-labeled BSA, respectively. The dotted lines represent the expected values of log 2-normalized ratios.
Based on the aforementioned results, the Ac-IP tag has a number of features that make it particularly suitable for multiplex DIA. It allows us to quantify peptides based on distinct precursor ions, which results in identical fragment ion spectra and thus avoids further complicating MS2 spectra in DIA. Compared to previously reported approaches, the Ac-IP tag has the potential to be extended to a higher multiplexing capacity, again without rendering MS2 spectra being more complex. While the proof-of-principle study was performed with triplex-labeled samples, multiplexing can be extended to 10-plex based on commercially available 13C- or 15N-labeled acetic anhydride and isoleucine following the same synthetic procedure (see Figure S14). Compared to other tags that have been proposed for multiplex DIA, such as the MdFDIA, mdDiLeu, and Ac-AG tag, the Ac-IP tag opens the possibility to obtain more accurate quantification by increasing the resolution at the MS1 level without extending the cycle time to a point that is no longer commensurate with DIA,33 as would be the case for tags that quantify peptides at the MS2 level (Table S2). The mass shift between the Ac-IP tags is 1 Da, which allows us to work at a customary resolution of 70 k for MS1 and 17.5 k for MS2 spectra in an Orbitrap. Resorting to ultrahigh resolution for MS1 spectra and making use of neutron encoding24 would further boost the multiplexing capacity. In addition, while the Ac-IP tag exhibits good quantification capability across a 10-fold dynamic range in DIA mode, it should be pointed out that the deconvolution method used in this manuscript is based on the assumption of expected intensities for individual peaks in the isotope envelope of the precursor ions, which means that inaccurate ratios are produced when more than one unresolved precursor isotope envelope overlaps or when there is interference from background ions. In these cases, it is necessary to resolve the isotope envelopes and assign them to different peptide precursor ions and a higher resolution at the MS1 level may be needed for accurate quantification. Furthermore, in this study, we chose the widely used DIA identification method based on a spectral library search, which inherently limits the number of DIA identifications to the number of proteins that are in the library. Using a DIA identification algorithm that does not require spectral libraries and that recognizes fragment ions with specific neutral losses would further enhance the potential of the Ac-IP tag approach.
Conclusions
In this paper, we describe a novel CID cleavable isotope tag for multiplexed quantification in DIA mode based on MS1 spectra. The peptides labeled with Ac-IP tags have distinct precursor masses containing the relative quantification information while having identical MS2 spectra. The Ac-IP approach neither complicates the interpretation of MS2 spectra nor relies on an ultrahigh-resolution power and indeed enhances the fragment ion intensity. The feasibility of quantifying peptides/proteins in DIA mode with the Ac-IP tag was demonstrated by the quantification of a triplex-labeled BSA sample with 10-fold dynamic changes that was spiked in a complex background of a triplex-labeled LysC yeast sample. The distinctive feature of producing the same MS2 spectra from the different precursor ions, the potential for a higher multiplexing capacity in DIA, and straightforward synthesis are features of the Ac-IP tag that are expected to attract more attention to multiplexed DIA.
Acknowledgments
We gratefully acknowledge the China Scholarship Council (CSC) for a PhD fellowship to X.T. X.T. thanks Jos Hermans for helping with the LC–MS analyses. X.T. thanks B. Zhang and C. Ye for help with the NMR analyses. This work is partially funded by the Open Technology Programme of Toegepaste en Technische Wetenschappen (TTW) with project number 15230, which is financed by the Netherlands Organisation for Scientific Research (NWO).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.1c00453.
Chemicals and materials, the synthesis of triplex Ac-IP tags, reduction/alkylation and LysC digestion, selective N-terminal dimethylation of peptides, and LC–MS/MS analysis; charge state distribution of different forms of modified GTDWLANK (Table S1); the influence of increasing resolution at the MS1 and MS2 level on the cycle time for the Q Exactive Plus (Table S2); removal of excess Ac-IP-COOH (Figure S1); example of deconvoluting the precursor ion isotope envelope and calculating the normalized ratio (Figure S2); example of calculating peptide and protein ratios (Figure S3); LC–MS/MS of the triplex Ac-IP-NHS tag (Figure S4); 1H NMR of Ac-IP-NHS (Figure S5); 13C NMR of Ac-IP-NHS (Figure S6); COSY, HSQC, and HMBC NMR of Ac-IP-NHS (Figure S7); the influence of double Ac-IP labeling on ionization efficiency (Figure S8); derivatization of GTDWLANK (Figure S9); MS2 spectra of N-dime-GTDWLANK-PI-Ac at various NCEs (Figure S10); comparison of (+0)-Ac-IP, (+1)-Ac-IP, (+2)-Ac-IP-labeled, and 1:1:1 mixed triplex-labeled N-dime-GTDWLANK-PI-Ac (Figure S11); comparison of all PSMs and of those PSMs with the highest −10lgP value (Figure S12); charge state distribution of identified unlabeled and Ac-IP-labeled LysC yeast peptides (Figure S13); and isotope distribution of a hypothetical 10-plex Ac-IP-NHS tag (Figure S14) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Dayon L.; Affolter M. Progress and pitfalls of using isobaric mass tags for proteome profiling. Expert Rev. Proteomics 2020, 17, 149–161. 10.1080/14789450.2020.1731309. [DOI] [PubMed] [Google Scholar]
- Li J. M.; Van Vranken J. G.; Vaites L. P.; Schweppe D. K.; Huttlin E. L.; Etienne C.; Nandhikonda P.; Viner R.; Robitaille A. M.; Thompson A. H.; Kuhn K.; Pike I.; Bomgarden R. D.; Rogers J. C.; Gygi S. P.; Paulo J. A. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 2020, 17, 399–404. 10.1038/s41592-020-0781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Shan Y.; Zhou Y.; Liang Z.; Zhang L.; Zhang Y. Advances and applications of stable isotope labeling-based methods for proteome relative quantitation. TrAC Trends Anal. Chem. 2020, 124, 115815. 10.1016/j.trac.2020.115815. [DOI] [Google Scholar]
- Gygi S. P.; Rist B.; Gerber S. A.; Turecek F.; Gelb M. H.; Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- Jiang H.; English A. M. Quantitative analysis of the yeast proteome by incorporation of isotopically labeled leucine. J. Proteome Res. 2002, 1, 345–350. 10.1021/pr025523f. [DOI] [PubMed] [Google Scholar]
- Ross P. L.; Huang Y. N.; Marchese J. N.; Williamson B.; Parker K.; Hattan S.; Khainovski N.; Pillai S.; Dey S.; Daniels S.; Purkayastha S.; Juhasz P.; Martin S.; Bartlet-Jones M.; He F.; Jacobson A.; Pappin D. J. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Dayon L.; Hainard A.; Licker V.; Turck N.; Kuhn K.; Hochstrasser D. F.; Burkhard P. R.; Sanchez J. C. Relative Quantification of Proteins in Human Cerebrospinal Fluids by MS/MS Using 6-Plex Isobaric Tags. Anal. Chem. 2008, 80, 2921–2931. 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Strozynski M.; Kozielski F.; Treumann A.; Thiede B. Isobaric Peptide Termini Labeling for MS/MS-Based Quantitative Proteomics. J. Proteome Res. 2009, 8, 4333–4341. 10.1021/pr900425n. [DOI] [PubMed] [Google Scholar]
- Tian X.; de Vries M. P.; Permentier H. P.; Bischoff R. A Collision-Induced Dissociation Cleavable Isobaric Tag for Peptide Fragment Ion-Based Quantification in Proteomics. J. Proteome Res. 2020, 19, 3817–3824. 10.1021/acs.jproteome.0c00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wühr M.; Haas W.; McAlister G. C.; Peshkin L.; Rad R.; Kirschner M. W.; Gygi S. P. Accurate Multiplexed Proteomics at the MS2 Level Using the Complement Reporter Ion Cluster. Anal. Chem. 2012, 84, 9214–9221. 10.1021/ac301962s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnett M.; Yeung E.; Wühr M. Accurate, Sensitive, and Precise Multiplexed Proteomics Using the Complement Reporter Ion Cluster. Anal. Chem. 2018, 90, 5032–5039. 10.1021/acs.analchem.7b04713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S. V.; Meier F.; Wichmann C.; Cox J.; Mann M.; Meissner F. EASI-tag enables accurate multiplexed and interference-free MS2-based proteome quantification. Nat. Methods 2018, 15, 527–530. 10.1038/s41592-018-0037-8. [DOI] [PubMed] [Google Scholar]
- Pappireddi N.; Martin L.; Wühr M. A Review on Quantitative Multiplexed Proteomics. ChemBioChem 2019, 20, 1210–1224. 10.1002/cbic.201800650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo J. A.; Gygi S. P. mTMT: An Alternative, Nonisobaric, Tandem Mass Tag Allowing for Precursor-Based Quantification. Anal. Chem. 2019, 91, 12167–12172. 10.1021/acs.analchem.9b03162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenes A.; Hukelmann E.; Bensaddek D.; Lamond A. I. Multibatch TMT Reveals False Positives, Batch Effects and Missing Values. Mol. Cell. Proteomics 2019, 18, 1967–1980. 10.1074/mcp.RA119.001472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet L. C.; Navarro P.; Tate S.; Röst H.; Selevsek N.; Reiter L.; Bonner R.; Aebersold R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics 2012, 11, O111.016717. 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern M.; Finney G.; Hoopmann M. R.; Merrihew G.; Toth M. J.; MacCoss M. J. Deconvolution of Mixture Spectra from Ion-Trap Data-Independent-Acquisition Tandem Mass Spectrometry. Anal. Chem. 2010, 82, 833–841. 10.1021/ac901801b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins B. C.; Gillet L. C.; Rosenberger G.; Röst H. L.; Vichalkovski A.; Gstaiger M.; Aebersold R. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system. Nat. Methods 2013, 10, 1246–1253. 10.1038/nmeth.2703. [DOI] [PubMed] [Google Scholar]
- Rardin M. J.; Schilling B.; Cheng L. Y.; MacLean B. X.; Sorensen D. J.; Sahu A. K.; MacCoss M. J.; Vitek O.; Gibson B. W. MS1 Peptide Ion Intensity Chromatograms in MS2 (SWATH) Data Independent Acquisitions. Improving Post Acquisition Analysis of Proteomic Experiments. Mol. Cell. Proteomics 2015, 14, 2405–2419. 10.1074/mcp.O115.048181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- Minogue C. E.; Hebert A. S.; Rensvold J. W.; Westphall M. S.; Pagliarini D. J.; Coon J. J. Multiplexed Quantification for Data-Independent Acquisition. Anal. Chem. 2015, 87, 2570–2575. 10.1021/ac503593d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Y.; Zhang Y.; Zhang L.; Tao T.; Lu H. MdFDIA: A Mass Defect Based Four-Plex Data-Independent Acquisition Strategy for Proteome Quantification. Anal. Chem. 2017, 89, 10248–10255. 10.1021/acs.analchem.7b01635. [DOI] [PubMed] [Google Scholar]
- Zhong X.; Frost D. C.; Yu Q.; Li M.; Gu T.-J.; Li L. Mass Defect-Based DiLeu Tagging for Multiplexed Data-Independent Acquisition. Anal. Chem. 2020, 92, 11119–11126. 10.1021/acs.analchem.0c01136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert A. S.; Merrill A. E.; Bailey D. J.; Still A. J.; Westphall M. S.; Strieter E. R.; Pagliarini D. J.; Coon J. J. Neutron-encoded mass signatures for multiplexed proteome quantification. Nat. Methods 2013, 10, 332–334. 10.1038/nmeth.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X.; de Vries M. P.; Permentier H. P.; Bischoff R. A Versatile Isobaric Tag Enables Proteome Quantification in Data-Dependent and Data-Independent Acquisition Modes. Anal. Chem. 2020, 92, 16149–16157. 10.1021/acs.analchem.0c03858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J.; Ishihama Y.; Mann M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem. 2003, 75, 663–670. 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- He L.; Diedrich J.; Chu Y.-Y.; Yates J. R. III Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. Anal. Chem. 2015, 87, 11361–11367. 10.1021/acs.analchem.5b02721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Triscari J. M.; Pasa-Tolic L.; Anderson G. A.; Lipton M. S.; Smith R. D.; Wysocki V. H. Dissociation Behavior of Doubly-Charged Tryptic Peptides: Correlation of Gas-Phase Cleavage Abundance with Ramachandran Plots. J. Am. Chem. Soc. 2004, 126, 3034–3035. 10.1021/ja038041t. [DOI] [PubMed] [Google Scholar]
- Muntel J.; Gandhi T.; Verbeke L.; Bernhardt O. M.; Treiber T.; Bruderer R.; Reiter L. Surpassing 10 000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol. Omics 2019, 15, 348–360. 10.1039/C9MO00082H. [DOI] [PubMed] [Google Scholar]
- Qin H.; Wang F.; Zhang Y.; Hu Z.; Song C.; Wu R.; Ye M.; Zou H. Isobaric cross-sequence labeling of peptides by using site-selective N-terminus dimethylation. Chem. Commun. 2012, 48, 6265–6267. 10.1039/c2cc31705b. [DOI] [PubMed] [Google Scholar]
- Fu Q.; Li L. De Novo Sequencing of Neuropeptides Using Reductive Isotopic Methylation and Investigation of ESI QTOF MS/MS Fragmentation Pattern of Neuropeptides with N-Terminal Dimethylation. Anal. Chem. 2005, 77, 7783–7795. 10.1021/ac051324e. [DOI] [PubMed] [Google Scholar]
- Tran N. H.; Qiao R.; Xin L.; Chen X.; Liu C.; Zhang X.; Shan B.; Ghodsi A.; Li M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat. Methods 2019, 16, 63–66. 10.1038/s41592-018-0260-3. [DOI] [PubMed] [Google Scholar]
- Scheltema R. A.; Hauschild J.-P.; Lange O.; Hornburg D.; Denisov E.; Damoc E.; Kuehn A.; Makarov A.; Mann M. The Q Exactive HF, a Benchtop Mass Spectrometer with a Pre-filter, High Performance Quadrupole and an Ultra-High Field Orbitrap Analyzer. Mol. Cell. Proteomics 2014, 3698. 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.