

Abstract
We describe the total synthesis of tutuilamide A, a potent porcine pancreatic elastase (PPE) inhibitor and a representative member of the 3-amino-6-hydroxy-2-piperidone (Ahp) cyclodepsipeptide family, isolated from marine cyanobacteria. The Ahp unit serves as a pharmacophore and the adjacent 2-amino-2-butenoic acid (Abu) is a main driver of the selectivity among serine proteases. We adapted our previous convergent strategy to generate the macrocycle, common with lyngbyastatin 7 and related elastase inhibitors, and then appended the tutuilamide A-specific side chain bearing a vinyl chloride. Tutuilamide A and lyngbyastatin 7 were evaluated side by side for the inhibition of the disease-relevant human neutrophil elastase (HNE). Tutuilamide A and lyngbyastatin 7 were approximately equipotent against HNE, while tutuilamide A was previously shown to be more active against PPE compared with lyngbyastatin 7, further demonstrating that the side chain provides opportunities to not only modulate potency but also selectivity among proteases of the same function from different organisms. Profiling of tutuilamide A against mainly human serine proteases revealed high selectivity for HNE (IC50 0.73 nM) and pleiotropic activity against kallikrein 7 (KLK7, IC50 5.0 nM), without affecting other kallikreins, similarly to lyngbyastatin 7 (IC50 0.85 nM for HNE and 3.1 nM for KLK7). A comprehensive molecular docking study for elastases and KLK7 afforded deeper insight into the intricate differences between inhibitor interactions with HNE and PPE, accounting for the differential activities for both compounds. The synthesis and molecular studies serve as a proof-of-concept that the macrocyclic scaffold can be diversified to fine-tune the activity of serine protease inhibitors
Keywords: Human neutrophil elastase, Pulmonary diseases, HNE inhibitors, Total synthesis, Tutuilamide A
Graphical Abstract
1. Introduction
Proteases play a critical role in the regulation of various biological processes such as cell proliferation, tissue remodeling, blood coagulation, digestion, immune function defensing pathogens, and other processes.1,2 Dysregulation of protease function would result in disruption of homeostasis and lead to various diseases, such as inflammation, cancer, pulmonary diseases, neurodegeneration, and angiogenesis disorder.1–3 Consequently, proteases are important therapeutic targets.1,4 Among the major five classes of proteases, serine proteases make up 30%, and elastases and kallikreins (KLKs) are two important sub-categories of serine proteases.5–7
Human neutrophil elastase (HNE) is a serine protease stored mainly in neutrophil azurophilic granules and released when neutrophils respond to inflammation.8 Normally, the proteolytic activity of HNE is tightly regulated by endogenous inhibitors such as α1-PI, serpin B1, SLPI, and elafin. However, imbalance of HNE and its endogenous inhibitors can lead to overactivity of HNE which then would degrade normal extracellular matrix (ECM), cleave inflammatory mediators, receptors, lung surfactant and induce cytokines, chemokines, and growth factors.9,10 The proteolytic cleavage of these molecular targets may promote a variety of inflammatory conditions, including chronic obstructive pulmonary disease (COPD), cystic fibrosis, acute lung injury, and acute respiratory distress syndrome (ARDS).1,9–12 HNE has also been linked to the progression of cancer, particularly cell migration and metastasis.9,13
Hence, the discovery and development of small molecule HNE inhibitors might be a promising therapeutic approach, particularly for pulmonary diseases. Compared with endogenous inhibitors, low-molecular weight inhibitors offer better physiological properties.9 However, currently only one small molecule HNE inhibitor is on the market, sivelestat (ONO-5046, Elaspol 100)1,12 (Figure 1A).
Figure 1.
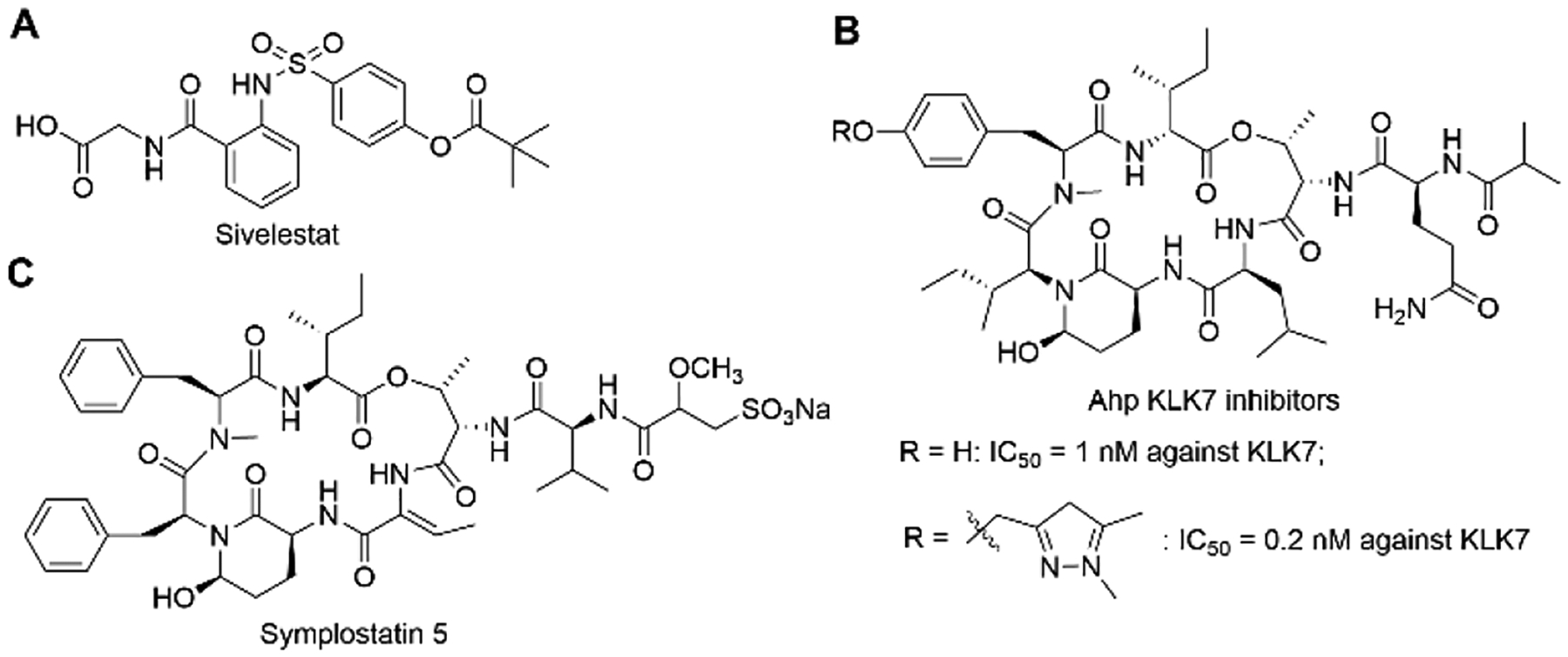
Chemical structures of sivelestat, reported Ahp-containing KLK7 inhibitor, and symplostatin 5.
Human tissue kallikreins (hKs) are secreted serine proteases, comprised of 15 family members and encoded by the largest contiguous protease gene cluster (80% of 178 serine proteases).7,14,15 KLK proteases play crucial roles in the regulation of skin desquamation, kidney function, seminal liquefaction, tooth enamel formation, synaptic neural plasticity and brain function.7,14,15 There are numerous diseases that result from dysfunctions of KLKs, for example, pathological inflammation, respiratory diseases, cancer, neurodegeneration, skin-barrier dysfunction, and others. Thus, KLKs have recently emerged as attractive therapeutic targets.7,14,15 Several drugs that target plasma kallikrein are already approved for the treatment of hereditary angioedema.1 KLK7, a stratum corneum chymotryptic enzyme, regulates epidermal desquamation by degrading corneodesmosome proteins. The overexpression of KLK7 would lead to severe skin inflammation and atopic dermatitis.7,16–18 Currently, two potent natural product-based KLK7 specific inhibitors with low-nanomolar IC50 were reported (Figure 1B).7,15,19
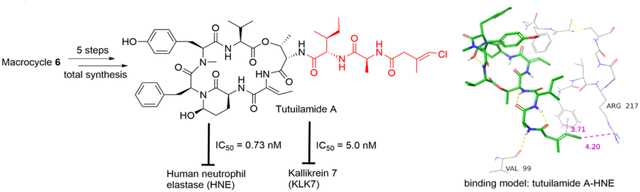
Marine cyanobacteria produce a class of potent serine protease inhibitors, featuring a 19-membered cyclic hexadepsipeptide and a varied pendant side chain. The 3-amino-6-hydroxy-2-piperidone (Ahp) is essential for the serine protease-inhibitory activity and the adjacent (Z)-2-amino-2-butenoic acid (Abu) confers selectivity for elastase. These two moieties are conserved in the cyclic core of lyngbyastatins 4–10,20–22 and these two moieties are also present in other marine natural products including symplostatins 2 and 5–10 (e.g., Figure 1C),12,23 somamides A and B,24 and molassamide.25 Compounds with the conserved macrocyclic scaffold have shown potent inhibition against HNE, especially when containing Abu over other hydrophobic amino acid.9,12–14, 20–27 There have also been reports of potent KLK7 inhibition by Ahp-cyclodepsipeptides without the Abu moiety.15,19 As macrocyclic molecules, they combine both advantages of peptide and small molecule inhibitors of proteases, i.e., increased binding surface area and good bioavailability.28 Lyngbyastatin 7 (Figure 2B) is one of the most potent HNE inhibitors of this structural class.12,29,30 Recently, a related series named tutuilamides A-C was reported, sharing the same macrocyclic core featuring Ahp-Abu units.31 Among them, tutuilamide A (Figure 2A) exhibited the greatest potency against porcine pancreatic elastase (PPE), with an IC50 of 1.2 nM and several-fold better activity than lyngbyastatin 7. Tutuilamide A is distinguished from lyngbyastatin 7 by its pendant side chain that is a vinyl chloride-containing tripeptide. We have previously established the total synthesis route for lyngbyastatin 7 and improved its synthetic strategy for process chemistry and side-chain diversification.29,30 Based on our previous investigation on Ahp- and Abu-containing compounds, we established that the side chain has significant impact on the antiprotease activity and that we can fine-tune the activities against HNE and also modulate the physicochemical properties. Here, we apply our synthetic route to the total synthesis of tutuilamide A through late-stage diversification, which was developed in the synthesis of lyngbyastatin 7 (Figure 2C).29,30 We then proceeded to test tutuilamide A (1) against the more relevant, human protease (HNE) and assessed global selectivity and comparative activity with lyngbyastatin 7, including the molecular basis as selective and tunable HNE and KLK7 inhibitors.
Figure 2.
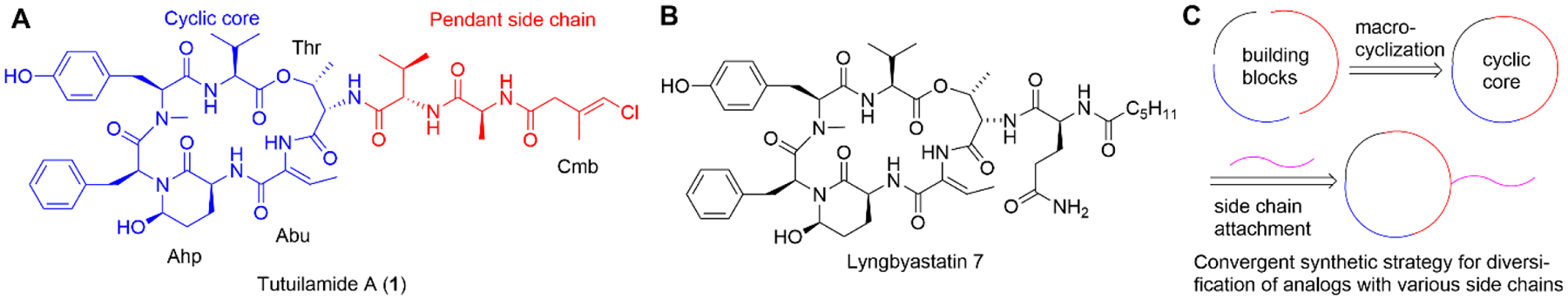
The chemical structures of tutuilamide A (1) and lyngbyastatin 7, and the convergent synthetic strategy for diversification of the macrocycle to access analogs.
2. Results and Discussion
2.1. Chemistry.
The synthetic route for the total synthesis of tutuilamide A is depicted in Scheme 1. Dipeptide 232 and (E)-4-chloro-3-methylbut-3-enoic acid 3 (Cmb)33 were synthesized using published protocols. The N-Boc group of 2 was removed with TFA and then the subsequent coupling of deprotected 2 with acid 3 gave tripeptide 4 under the mediation of EDCI/HOBt in presence of base DIEA. Free acid 5 was obtained by hydrolysis of 4 using trimethyltin hydroxide under refluxing in 1,2–dichloroethane.34
Scheme 1.
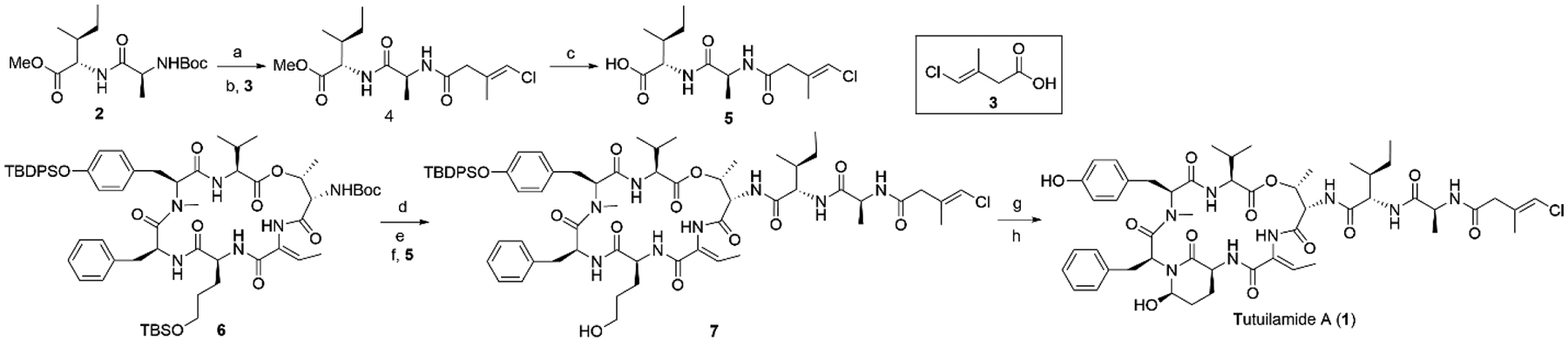
Total synthesis of tutuilamide A (1). Reagents and conditions: a) TFA, CH2Cl2, 0 °C, 1 h; b) 3, EDCI, HOBt, DIEA, DMF, rt, overnight, 60% 2 steps; c) Me3SnOH, 1,2-dichloroethane, reflux, overnight, 70%; d) TMSOTf, 2,6-lutidine, CH2Cl2, rt, 1.5 h; e) TFA, THF-H2O, rt, 2 h; f) 5, DEPBT, DIEA, DMF, rt, 17 h, 52% 3 steps; g) IBX, DMSO, rt, overnight; h) TBAF, THF, 0 °C, 1 h, 20% 2 steps.
The synthesis of macrocyclic core 6 was previously reported in our initial total synthesis of lyngbyastatin 7 and its analogs.29,30 N-Boc group of 6 were selectively removed with TMSOTf in the presence of 2,6-lutidine, and then TBS group was cleaved by the 15% TFA in the mixture of TFA-H2O. The resulting TFA salt from 6 was coupled with acid 5 using DEPBT as coupling reagent to give free alcohol 7 in 52% yield for 3 steps. However, we found TBS protected 7 was sensitive to acid when we tried to install tripeptide side chain 5 prior TBS cleavage (with 15% TFA). The primary alcohol of 7 was oxidized by IBX to provide aldehyde intermediate. The construction of Ahp moiety and TBDPS cleavage were achieved simultaneously when aldehyde intermediate was exposed to TBAF solution in THF. Finally, tutuilamide A (1) was obtained in 20% yield for final 2 steps. The NMR data of synthetic tutuilamide A (1) were identical to those of the natural product reported and optical rotation was similar31 ([α]20D −26.0 and lit31 −22.6 in MeOH, NMRs see Table S1, Figures S12 and S13).
2.2. Inhibition of serine proteases by synthetic tutuilamide A
Based on our previous research on related compounds that were tested against PPE and HNE20–27,29–31, tutuilamide A was expected to possess similar activity against HNE as PPE, and good selectivity for HNE among other human serine proteases. We tested its antiproteolytic activity against a panel of mostly human serine proteases (23 proteases, Figure 3 and Table 1; 7-amino-4-methylcoumarin (AMC) substrates were used in this assay, Table S2). It showed potent inhibition against elastase (HNE) and kallikrein 7 with low nanomolar IC50 (0.73 nM and 5 nM, respectively) and good potency against bacterial proteinase A and fungal proteinase K with nanomolar IC50. Tutuilamide A also exhibited strong inhibition of bovine chymotrypsin (97.4 nM), although 100-fold less that HNE and we expect that the activity against human chymotrypsin is much weaker, as reported for symplostatin 5 (Figure 1C) and lyngbyastatin 7.12 Based on the profiling results (Table 1, Figure 3), tutuilamide A displayed excellent selectivity towards HNE and human kallikrein 7. Particularly, it discriminated KLK7 from other KLK homologs with excellent selectivity. Compared with positive controls, the antiproteolytic activity of tutuilamide A was 5-, 5680-, 4000-, and 15-fold higher than those of positive controls sivelestat (HNE), gabexate mesylate (KLK7), leupeptin (proteinase A), and proteinase K inhibitor, respectively. For comparison, lyngbystatin 7 was also tested against HNE and KLK7 side by side and the inhibitory activity against HNE was virtually identical (IC50 0.85 nM), and activity against KLK7 slightly (1.6-fold) better (IC50 3.1 nM) than that of tutuilamide A. Figures 4A and 4B depict the dose- response curves for tutuilamide A, lyngbyastatin 7, and controls against HNE and KLK7, respectively. Importantly, the superior activity of tutuilamide A over lyngbyastatin 7 against PPE disappeared against HNE, suggesting that different mammalian enzyme sources might be differentially inhibited, dependent on the pendant side chain.
Figure 3.

Profiling of tutuilamide A against a panel of serine proteases. Heatmap represents the IC50 (nM) range of enzyme activity at 10 μM final concentration of tutuilamide A, but for elastase and kallikrein 7, tutuilamide A highest final concentration is 1 μM. The number in each scale represents the logarithm of highest IC50; for scale 4, it means highest IC50 equal or over 10 μM. Enzyme source: chymotrypsin and trypsin, bovine pancreas; proteinase A, Bacillus licheniformis; proteinase K, Tritirachium album limber; all others, human origin. Detailed reaction conditions see Table S2.
Table 1.
IC50 values (nM) of tutuilamide A and lyngbyastatin 7 against hits identified in screening and comparison with positive controls
| Protease | HNE | Human kallikrein 7 | Proteinase A | Proteinase K | chymotrypsin |
|---|---|---|---|---|---|
| Tutuilamide A | 0.73 | 5.0 | 31.3 | 12.3 | 97.4 |
| Lyngbyastatin 7 | 0.85 | 3.1 | |||
| Control | sivelestat | GM | leupeptin | PKi | chymostatin |
| 3.5 | 28400 | 129000 | 180 | 1.1 |
Note: GM, Gabexate mesylate; PKi, proteinase K inhibitor; enzyme source and detailed reaction conditions see Table S2. 95% confidence interval for IC50 values
Figure 4.
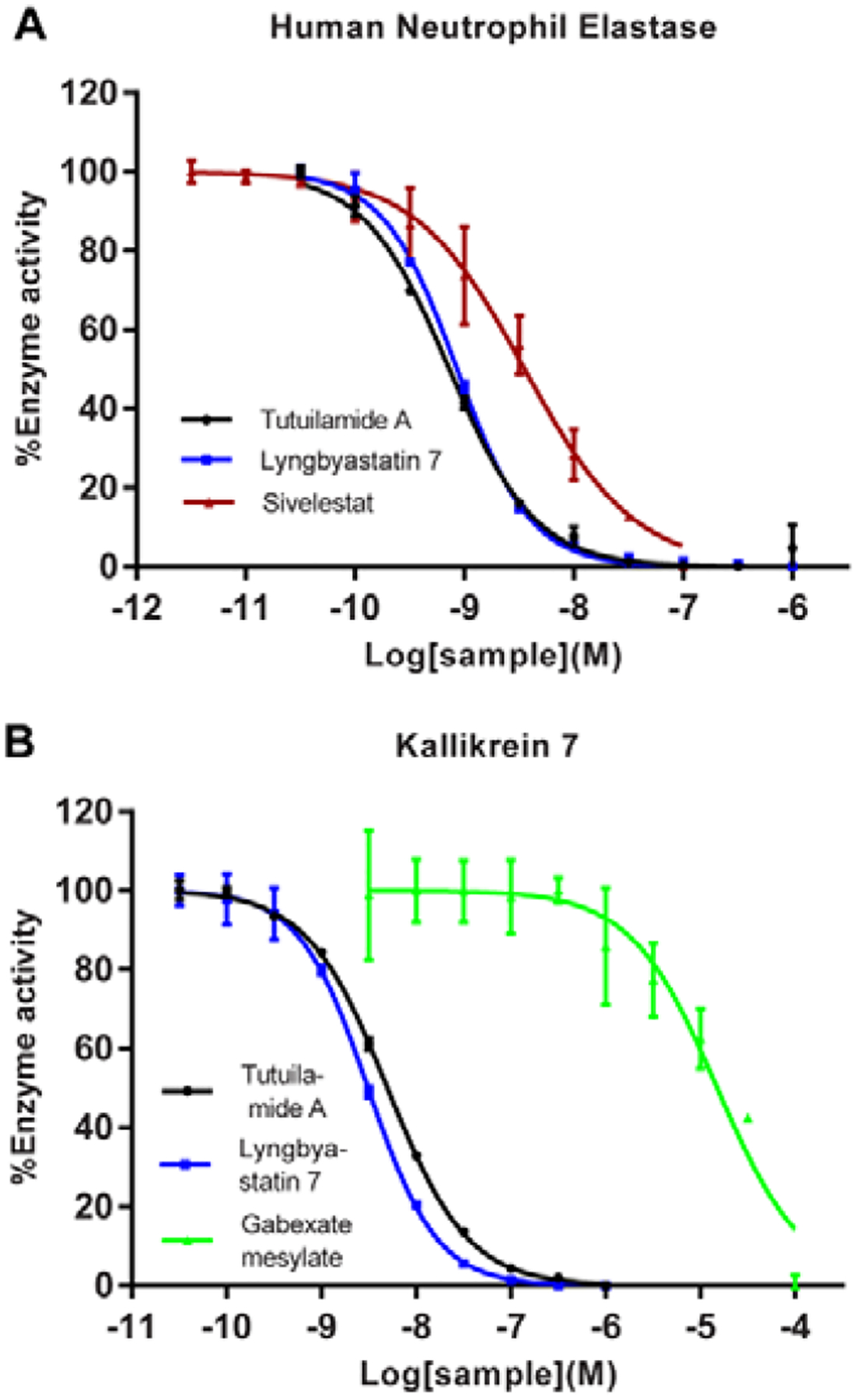
Protease inhibitory activity of tutuilamide A and lyngbyastatin 7 against HNE and KLK7. (A) Activity against HNE. Positive control: sivelestat; substrate: MeOSucAAPV-AMC; (B) Activity against KLK7. Positive control: gabexate mesylate; substrate: MCA-PKPVE-Nval-WRK (Dnp)-NH2. For reaction conditions, see Table S2. Data represent mean ± SD (n = 2), relative to 0.5% DMSO treatment (vehicle).
Assay results depend on specific different conditions and substrates (including AMC vs pNa (p-nitroanilide)) and side-by-side comparison is critical. In order to compare the potency of tutuilamide A against HNE with other analogs under previously published testing conditions, we also carried out dose-response assay with pNa substrates (Figure 5). Here, we found that tutuilamide A was 2.9-fold and 2.1-fold more potent than sivelestat and symplostatin 5, respectively, but 1.5-fold less active than lyngbyastatin 7 (IC50 values: 44 nM for tutuilamide A, and 28, 93, 128 nM for lyngbyastatin 7, symplostatin 5, sivelestat, respectively). This data validated the role of the pendant side chain in modulating the antiprotease activity, but also that the advantage of tutuilamide A observed for PPE is apparently lost with respect to HNE interactions. From dose-response assays, we conclude the potency of lyngbyastatin 7 is equal or better than tutuilamide A against HNE.
Figure 5.
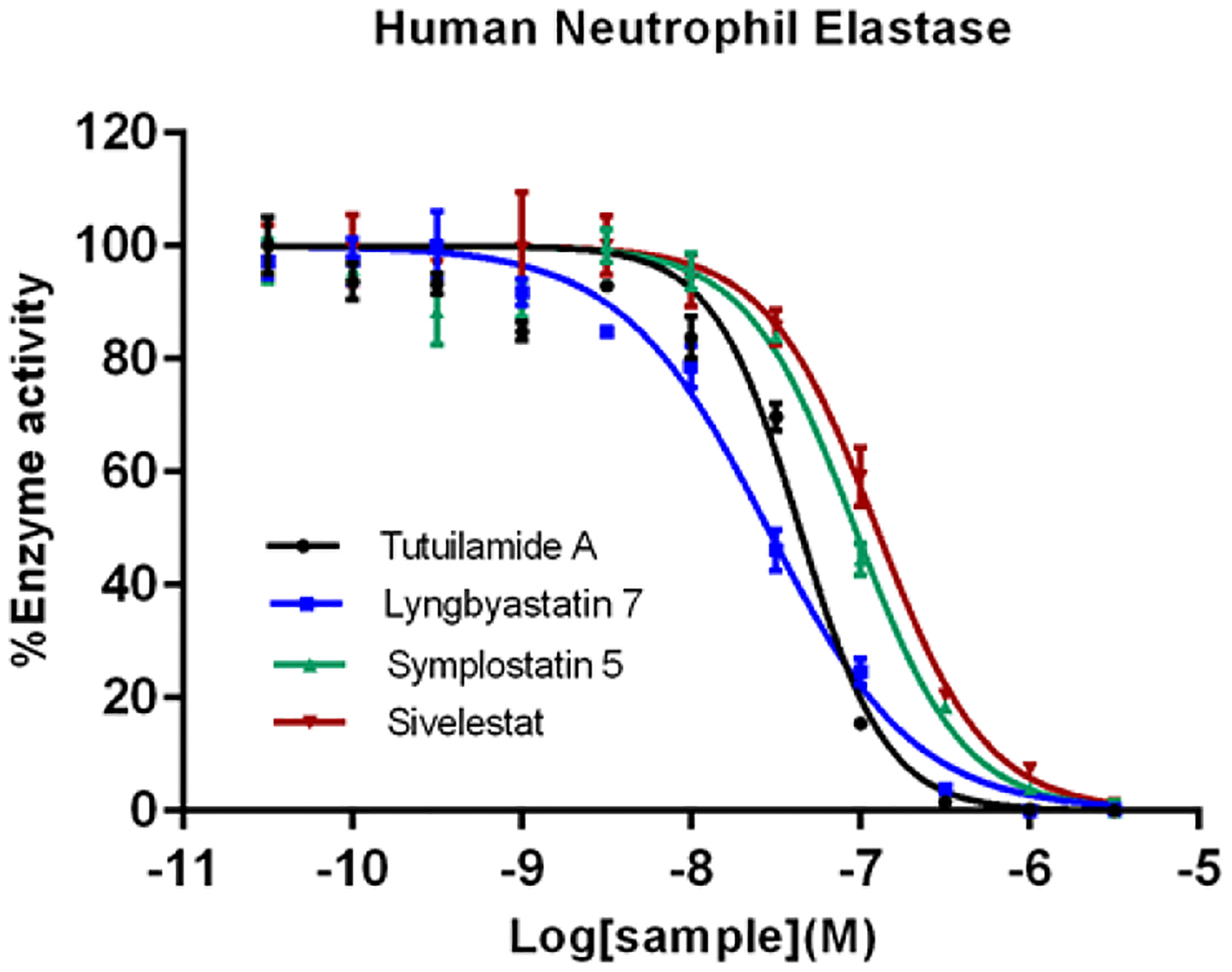
In vitro HNE enzyme assay of tutuilamide A, lyngbyastatin 7, symplostatin 5, and sivelestat. HNE was first incubated with compounds for 15 min in 0.1 M Tris−0.5 M NaCl (pH 7.5), and then N-(OMe-succinyl)-Ala-Ala-Pro-Val-p-nitroanilide was used as substrate to monitor the enzyme activity. The IC50 value for tutuilamide A, lyngbyastatin 7, symplostatin 5 and sivelestat is 44, 28, 93, and 128 nM, respectively. Data are presented as mean ± SD (n = 3), relative to 0.5% DMSO treatment (vehicle).
2.3. Molecular docking
To further understand the molecular basis leading to the strong antiproteolytic ability of tutuilamide A and lyngbyastatin 7 against HNE and KLK7, we carried out molecular docking. We aimed to compare the binding mode to the mode in the crystal structures obtained with PPE, which shares 37% identity (51% similarity) to HNE, and 26% identity (42% similarity) to KLK7 (Figure S1). The published crystal structures of proteases were used in this study.
Figures 6A and 6B show the crystal structures of tutuilamide A and lyngbyastatin 7 with PPE, respectively (PDBID: 6TH7 and 4GVU, respectively). Compared to the binding structure of lyngbyastatin 7 with PPE (Figure 6B, Figure S2), one extra H-bond was formed between the Cmb moiety of tutuilamide A with residue Arg217 of PPE at the binding pocket (Figure 6A, Figure S2), which probably resulted in the stronger potency of tutuilamide A against PPE than that of lyngbyastatin 7 to PPE, as previously hypothesized.31 An intramolecular H-bond for lyngbyastatin 7 between Gln of the side chain and Tyr in the cyclic core may prevent the Cmb H-bond by locking the ligand in an unfavorable conformation for binding to Val 215 and Arg217.
Figure 6.
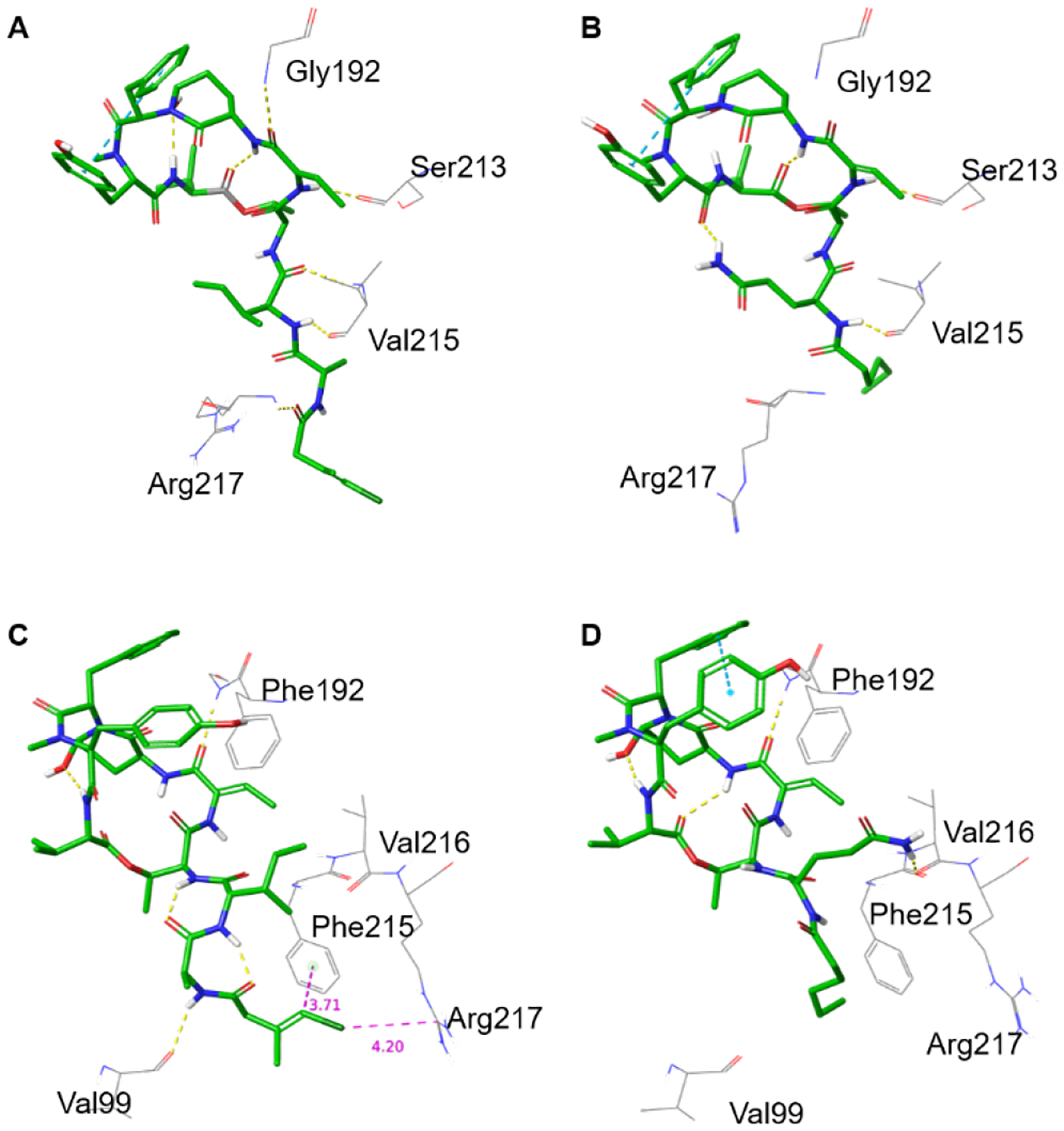
(A,B) Crystal structures of tutuilamide A (A) and lyngbyastatin 7 (B) co-crystallized with PPE (PDBIDs 6TH7 and 4GVU, respectively). (C,D) Docked structures of tutuilamide A (C) and lyngbyastatin 7 (D) into the crystal structure of HNE (PDBID: 3Q76). See the respective ligand interaction diagrams in the supporting information, figures S2–S4.
Figures 6C and 6D show the best binding modes of tutuilamide A and lyngbyastatin 7 obtained from docking to HNE (PDB ID: 3Q76) (also see Figure S4A,B). At the cyclic core binding pocket, both of them showed a similar binding mode as other “Ahp-Abu” compounds.12,13,29,30 At the pocket side chain locations, Gln of lyngbyastatin 7 forms strong H-bond with Val216. However, the loop containing Arg217 in HNE is one-residue shorter, lacking Ser216 (Figure S1), so that Arg217 points towards the outside of the binding pocket (Figure S5). As a result, for the binding of tutuilamide A with HNE, there is no H-bond formed between its side chain and Val216 and Arg217 (Figure 6C, Figure S4A). The absence of those bonds is energetically compensated by the formation of two intramolecular H-bonds in the side chain, one between the Ala carbonyl and Thr amide, and the second between the carbonyl on Cmb and the Ile amide (Figure 6C). Those last H-bonds also lock the tutuilamide A side chain in a position that favors the formation of a new H-bond between Ala and Val99, as well as CH-π, π-π and Cl-π interactions between Cmb and Phe215,12,35,36 and a charge interaction between Cl and Arg217.37 Those additional interactions make up for the absence of Gln, potentially rationalizing the equipotency of tutuilamide A and lyngbyastatin 7. Lyngbyastatin 7 does not seem to form intramolecular H-bonds in the linear side chain. The role of intramolecular H-bonds of this scaffold formed with the side chain to modulate binding will be tested in future SAR studies.
The strong antiproteolytic ability of tutuilamide A and lyngbyastatin 7 against KLK7 can also be rationalized by docking models (Figure 7, Figures S4C,D). Roughly, the H-bonds density of tutuilamide A with KLK7 is similar to that lyngbyastatin 7 with KLK7 (Figure 7A vs 7B, and Figure S4C vs S4D). Specifically, for tutuilamide A, there are three H-bonds between side chain and KLK7 residues, i.e., Cmb with Phe218 (1), Ile with Gly216 (2); additionally, five clear H-bonds and one π-π interaction are also observed between tutuilamide A cyclic core and other KLK7 residues, Asn192 (2), Gly193, Ser195, Ser214 and Phe218, respectively (Figure 7A and Figure S4B). However, in other KLK homologs, Asn192 and Phe218 are not conserved: Asn192 only exists in KLK7, and Phe218 only in KLK7 and KLK13 and it is replaced by a Tyr in KLK5 (Figure S1). Thus, this unique combination is only present in KLK7, which might contribute to the observed KLK7 selectivity. For lyngbyastatin 7, its H-bonds with KLK7 are almost the same as tutuilamide A with KLK7 except that Gln formed two intramolecular H-bonds with the Ahp moiety (Figure 7B, Figure S4D). To the best of our knowledge, tutuilamide A and lyngbyastatin 7 are the first examples of “Abu-Ahp” compounds as KLK7 inhibitors.
Figure 7.
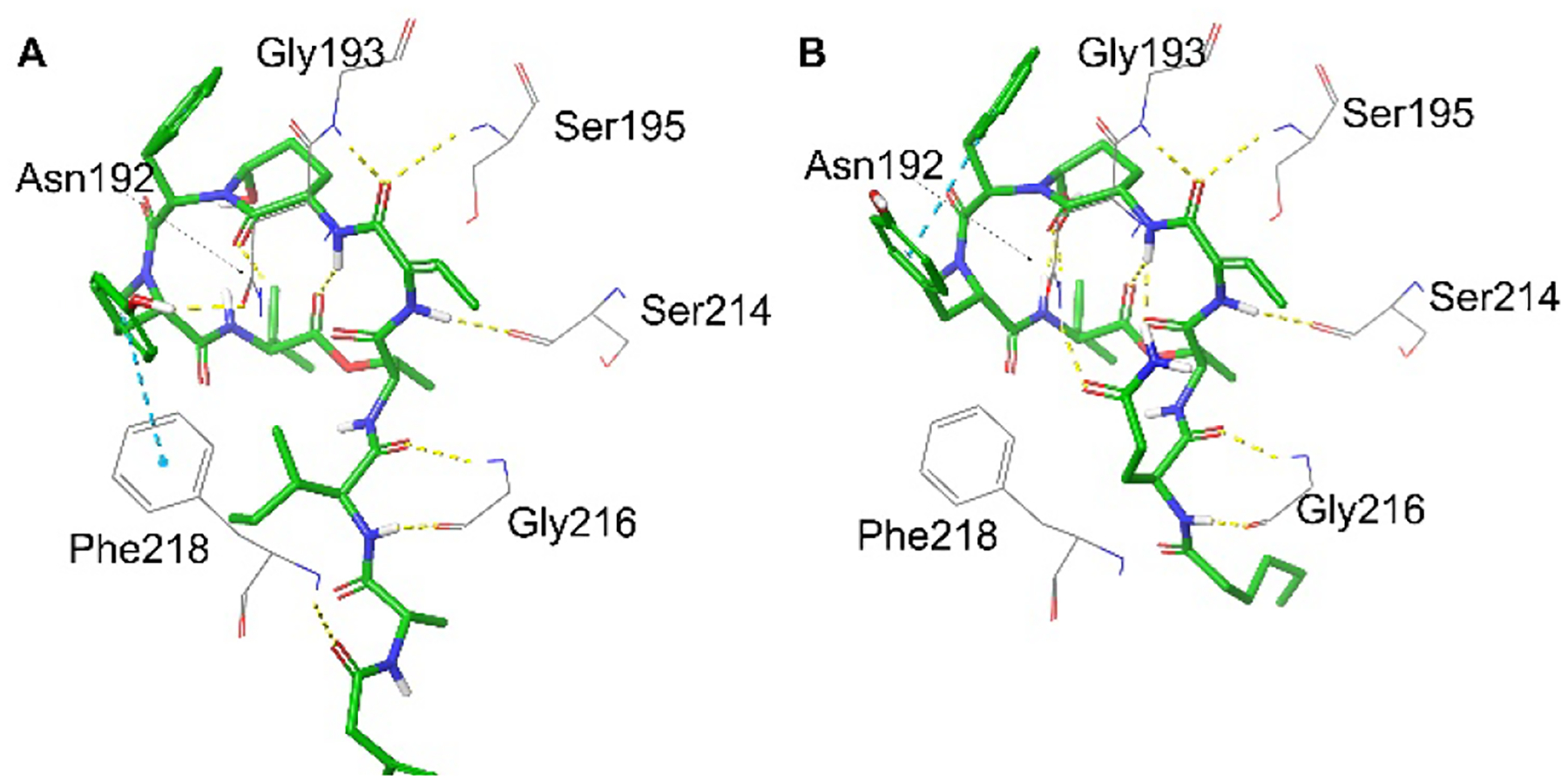
Docking model structures of tutuilamide A (A) and lyngbyastatin 7 (B) into KLK7 (PDBID:2QXI).
3. Conclusion
The successful completion of the total synthesis of tutuilamide A further expands the application of our convergent synthetic strategy, macrocyclic core plus pendant side chain, which not only provides easy access to focused libraries for SAR studies, but also offers efficient construction of newly discovered natural product for structure validation and bioactivity study. In general, convergent synthesis gave good yields for most steps, except for the final steps of Ahp formation (cyclization) and deprotection, which requires improvement. The protease assay revealed that tutuilamide A is a potent inhibitor of two human proteases, neutrophil elastase (HNE) and kallikrein 7. The docking studies of tutuilamide A and lyngbyastatin 7 with HNE, PPE and KLK7 provided insight into the molecular basis for the differential potency, even among the same proteases from different mammalian sources (HNE versus PPE) and contributed to our understanding of the SAR and the design of more potent and selective protease inhibitors with Ahp-cyclodepsipeptide structure, particularly against HNE.
4. Experimental Section
4.1. General experimental procedure
All commercial reagents were used without further purification unless otherwise noted. Solvents THF, CH2Cl2, DMF were purified by IT MD5 solvent purification system. DMSO was dried with 3Å molecular sieves all reactions were performed in heat-gun dried flasks (400°C under reduced pressure) under an inert atmosphere of anhydrous Ar unless otherwise noted. Thin layer chromatography was performed on EMD silica gel 60Å F254 glass plates and preparative thin layer chromatography was performed on Whatman silica gel 60Å F254 glass plates (layer thick 1000 μm). Flash column chromatography was performed with Fisher 170–400 mesh silica gel. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance II 600 MHz as indicated in the data list. Chemical shifts for proton nuclear magnetic resonance (1H NMR) spectra are reported in parts per million relative to the signal residual CDCl3 at 7.26 ppm, DMSO-d6 at 2.5 ppm; Chemicals shifts for carbon nuclear magnetic resonance (13C NMR) spectra are reported in parts per million relative to the center line of the CDCl3 triplet at 77.16 ppm, DMSO-d6 at 39.5 ppm; The abbreviations s, d, dd, ddd, t, q, br and m stand for the resonance multiplicity singlet, doublet, doublet of doublets, doublet of doublet of doublets, triplet, quartet, broad and multiplet, respectively. Optical rotation was measured on Rudolph Research Analytical Autopol III automatic polarimeter using a microcell of 1-dm path length. High resolution mass spectra (HRMS) data were obtained using a Q Exactive Focus with electrospray ionization (ESI) at UF’s Center for Natural Products, Drug Discovery and Development (CNPD3).
4.2. Synthesis
4.2.1. Tripeptide 4.
Trifluoroacetic acid (TFA) (2 mL) was added to the solution of dipeptide 2 (211.6 mg, 0.669 mmol) in anhydrous CH2Cl2 (4 mL) at 0 °C. After stirring for 30 min at 0 °C, the reaction mixture was diluted with toluene (6 mL) and evaporated, then it was azeotroped again with toluene (3×6 mL) and dried with oil pump for 2 h to give the free amino acid intermediate as TFA salt, which was used in the next step without further purification. The TFA salt from dipeptide 2 was dissolved in anhydrous DMF (5 mL). To the above solution were added acid 3 (89.7 mg, 0.669 mmol), EDCI·HCl (192.1 mg, 1 mmol), HOBt·H2O (163.8 mg, 1.072 mmol) and DIEA (117 μL, 1.336 mmol). The reaction mixture was stirred at room temperature overnight, then the solvent was evaporated in vacuo and the resulting residue was purified by flash chromatography column on silica gel (eluted by mixture of EtOAc/hexane 2:3, v/v) to give tripeptide 4 (133.5 mg, 61%). [α]20D −56.0 (c 0.2, MeOH). 1H NMR (600 MHz, CDCl3): δ 6.65 (d, J = 8.5 Hz, 1H), 6.32 (d, J = 7.5 Hz, 1H), 6.01 (dq, J = 2.4, 1.3 Hz, 1H), 4.57–4.52 (m, 2H), 3.74 (s, 3H), 2.99 (br dd, J = 1.4, 1.4 Hz, 2H), 1.92–1.86 (m, 1H), 1.83 (d, J = 1.4 Hz, 3H), 1.44–1.36 (m, 4H), 1.17 (ddq, J = 14.7, 9.2, 7.4 Hz, 1H), 0.92–0.88 (m, 6H) ppm. 13C NMR (150 MHz, CDCl3): δ 172.2, 172.0, 169.4, 132.8, 117.0, 56.8, 52.3, 49.0, 44.6, 37.8, 25.2, 18.4, 16.8, 15.6, 11.7 ppm. HRMS (ESI) m/z calcd for C15H25ClN2O4 (M+H)+ 333.1581, found 333.1561.
4.2.2. Acid 5.
Trimethyltin hydroxide (Me3SnOH) (743 mg, 4.082 mmol) was added to a solution of tripeptide 4 (131.5 mg, 0.408 mmol) in 1,2-dichloroethane (9 mL). The mixture was refluxed under heating for 15 h, then cooled down and concentrated in vacuo. The residue was dissolved with ethyl acetate (20 mL), washed with 2% KHSO4 (aq.) (3×5 mL) and brine (2×5 mL), and dried with anhydrous MgSO4. The dried ethyl acetate fraction was filtered and concentrated under reduced pressure to give white solid, which was re-suspended in small volume of CH2Cl2 (~2 mL). The suspension was filtered under reduced pressure and the collection of filtered solid to give pure acid 5 as white solid (90 mg, 70%). [α]20D −43.0 (c 0.13, MeOH). 1H NMR (600 MHz, DMSO-d6): δ 12.59 (br s, 1H), 8.13 (d, J = 7.5 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H), 6.11 (s, 1H), 4.37 (qd, J = 7.1, 7.1 Hz, 1H), 4.16 (dd, J = 8.4, 5.8 Hz, 1H), 2.95 (s, 2H), 1.80–1.73 (m, 1H), 1.71 (br s, 3H), 1.39 (dqd, J = 14.8, 7.5, 4.3 Hz, 1H), 1.20–1.13 (m, 4H), 0.85–0.83 (m, 6H) ppm. 13C NMR (150 MHz, DMSO-d6): δ 172.8, 172.3, 168.6, 134.2, 114.3, 56.2, 47.9, 42.6, 40.1, 36.4, 24.6, 18.1, 16.4, 15.5, 11.3 ppm. HRMS (ESI) m/z calcd for C14H23ClN2O4 (M–H)− 317.1268, found 317.1268.
4.2.3. Alcohol 7.
To the solution of macrocyclic core 6 (15 mg, 0.013 mmol) in anhydrous CH2Cl2 (1.0 ml) was added 2,6-lutidine (9.0 μL, 0.077 mmol) and trimethylsilyl trifluoro-methanesulfonate (TMSOTf) (11.6 μL, 0.064 mmol) dropwise at room temperature under argon. After being stirred at the same temperature for 1.5 h, the reaction mixture was quenched with MeOH (0.2 mL) and water (0.5 mL) at 0°C, and extracted with ethyl acetate (3×3 mL). The combined organic layer was washed with brine, dried over anhydrous MgSO4 and evaporated in vacuo and purified by preparative TLC plate on silica gel to give the crude free amine (12 mg, 88%, ethyl acetate /n-hexane 2:1, v/v, Rf = 0.1), which was used in the next step without further purification and characterization.
To the free amine crude of 6 (12 mg, 0.011 mmol) in the mixed solvent THF-H2O (1.14 mL −0.06 mL) was added TFA (180 μL). After stirred at room temperature for 2 h, the reaction mixture was evaporated in vacuo, co-evaporated with toluene (3×2 mL) and dried with oil pump for 2 h to give the free amino acid intermediate as TFA salt, which was used in the next step without further purification.
To the solution of the above TFA salt crude in anhydrous DMF (0.6 mL) was added DIEA (10 μL, 0.056 mmol), acid 5 (5.4 mg, 0.017 mmol), and DEPBT (6.7 mg, 0.022 mmol) at room temperature. After being stirred at room temperature overnight, the reaction mixture was evaporated in vacuo and purified preparative TLC plate on silica gel to give alcohol 7 as white solid (8.5 mg, 52% for 3 steps, MeOH /CH2Cl2 1:15, v/v, Rf = 0.25). [α]20D −20.0 (c 0.09, MeOH). 1H NMR (600 MHz, DMSO-d6): δ 9.39 (br s, 1H), 8.89 (br s, 1H), 8.21–8.14 (m, 3H), 7.81 (d, J = 8.8 Hz, 1H), 7.56 (d, J = 7.5 Hz, 2H), 7.50 (d, J = 7.4 Hz, 2H), 7.45 (dd, J = 7.4, 7.4 Hz, 2H), 7.38–7.33 (m, 4H), 7.22–7.16 (m, 3H), 7.11–7.09 (m, 3H), 6.97 (d, J = 8.1 Hz, 2H), 6.63 (q, J = 7.1 Hz, 1H), 6.55 (d, J = 8.1 Hz, 2H), 6.10 (s, 1H), 5.30 (br s, 1H), 5.10 (br m, 1H), 4.66 (d, J = 8.5 Hz, 1H), 4.60–4.56 (br m, 1H), 4.44 (br s, 1H), 4.37–4.32 (m, 3H), 4.23 (t, J = 7.2 Hz, 1H), 3.35–3.33 (m, 2H), 2.95 (s, 2H), 2.88 (dd, J = 13.7, 5.5 Hz, 1H), 2.68 (dd, J = 13.7, 9.9 Hz, 1H), 2.62 (s, 3H), 2.26 (br m, 1H), 2.18–2.15 (m,1H), 2.11–2.06 (m, 1H), 1.84–1.80 (br m, 1H), 1.77–1.71 (m, 5H), 1.52 (d, J = 7.0 Hz, 3H), 1.50–1.45 (m, 2H), 1.42–1.38 (m, 1H), 1.35–1.30 (m, 1H), 1.17–1.15 (m, 6H), 0.93 (s, 9H), 0.82–0.76 (m, 12H) ppm. 13C NMR (150 MHz, DMSO-d6): δ 172.4, 172.1, 171.7, 169.5.68.6, 168.3, 167.9, 162.8, 153.5, 136.9, 134.9, 134.1, 132.1, 132.0, 130.5, 130.3, 130.1, 129.7, 129.0, 128.0, 128.0, 127.9, 126.5, 119.1, 114.3, 70.5, 61.5, 60.4, 58.4, 56.5, 54.9, 54.6, 50.7, 49.6, 48.6, 48.1, 42.6, 40.1, 36.7, 35.7, 32.9, 31.3, 30.0, 29.7, 29.1, 28.7, 26.3, 26.2, 24.0, 22.1, 18.8, 18.6, 18.0, 16.4, 15.2, 14.0, 13.0, 11.0 ppm. HRMS (ESI) m/z calcd for C67H89ClN8O12Si (M+Na)+ 1283.5955, found 1283.5964.
4.2.3. Tutuilamide A (1).
To a solution of free primary alcohol 7 (8.4 mg, 0.0067 mmol) in anhydrous DMSO (0.3 mL) was added IBX (45%) (16.6 mg, 0.0267 mmol), and the resulting mixture was stirred for 15 h under argon at room temperature. Then the reaction mixture was diluted with ethyl acetate (10 mL), washed with water (2×3 mL). The organic layer was evaporated in vacuo, the resulting residue was dried by oil pump and the purification of the dried crude to give aldehyde intermediate (6.9 mg, 81%, MeOH /CH2Cl2 5:95, v/v, Rf = 0.3), which was used in the next step without further purification and characterization.
To the solution of above crude aldehyde (6.9 mg, 0.0055 mmol) in anhydrous THF (1.0 mL) was added TBAF (1.0 M in THF) (16.5 μL, 0.0165 mmol) at 0 °C. The resulting reaction mixture was stirred at 0 °C for 1 h, then quenched by water (3 mL). The quenched reaction mixture was partitioned with ethyl acetate (10 mL). The ethyl acetate layer was washed again by water (2×3 mL), evaporated in vacuo, the resulting residue was dried by oil pump and purified by analytical reversed-phase HPLC to give final product 1 as white solid (1.4 mg, 20% for 2 steps). The structure was confirmed by NMR spectra which are identical with those of natural tutuilamide. HPLC conditions: Phenomenex Luna 5u C18(2), 250×4.6 mm, 5 μm, 0.5 mL/min, UV detection at 200/220 nm; mobile phase MeOH-H2O; MeOH: 60–70% within 0–10 min, 70% within 10–30 min, 70–100% within 30–35 min; retention time for product 1, 26.8 min. [α]20D −26.0 (c 0.077, MeOH) (lit31 −22.6 (c 0.80, MeOH). HRMS (ESI) m/z calcd for C51H69ClN8O12 (M+H)+ 1021.4802, found 1021.4802. For 1H NMR and 13C NMR data and spectra and their comparison with the isolated natural product, please refer to Table S1 and Figures S12 and S13.
4.3. Protease assays
The enzyme assays for a panel of 23 proteases were performed by Reaction Biology Corp. The reaction conditions are listed in Table S2. In brief, tutuilamide A and the corresponding controls were tested in 10-dose IC50 with 3-fold serial dilution starting at 10 μM against 23 proteases in singlet. For HNE and KLK7, the starting concentration of tutuilamide A and lyngbyastatin 7 was 1 μM (duplicate measurements). AMC substrates were used in this screening (Table S2). The protease activities were monitored as a time-course measurement of the increase in fluorescence signal from fluorescently labeled peptide substrate, and the initial linear portion of the slope (signal/min) was analyzed.
In a complementary protease assay with pNa substrates, HNE was dissolved in 0.1 M Tris−0.5 M NaCl (pH 7.5) to give a concentration of 100 μg/mL. N-(OMe-succinyl)-Ala-Ala-Pro-Val-p-nitroanilide was also dissolved in 0.1 M Tris−0.5 M NaCl (pH 7.5) to give a 2 mM substrate solution. Varying doses (1 μL) of compound tutuilamide A, lyngbyastatin 7, symplostatin 5, sivelestat, or solvent control (DMSO), 5 μL of elastase solution, and 79 μL of 0.1 M Tris−0.5 M NaCl (pH 7.5) were preincubated at room temperature for 15 min in a 96-well plate. At the end of the incubation, 15 μL of substrate solution was added to each well using a multichannel pipet, and the reaction was monitored by recording the absorbance at 405 nm every 30 s for 30 min on a SpectraMax M5. Enzyme activity in each well was calculated on the basis of the initial slope of the reaction curve, expressed as a percentage of the initial slope of the uninhibited reaction. IC50 calculations were done by GraphPad Prism 6 based on triplicate experiments.
4.4. Docking study
All calculations were done within Schrödinger’s Maestro program.38 The initial structures of tutuilamide A and lyngbyastatin 7 were downloaded from the Protein Data Bank (www.rcsb.org), obtained at resolutions 2.20 Å and 1.55 Å with PDBID codes 6TH731 and 4GVU12, respectively. We also extracted the two PPE (6TH7 and 4GVU) structures for validation. The structure for the HNE target was extracted from the crystal structure for the Apo HNE obtained at 1.86 Å resolution (PDBID:3Q76).39 For the KLK7 target, we used the structure obtained at 1.0 Å resolution of KLK7 complexed with the Suc-Ala-Ala-Pro-Phe-chloromethylketone inhibitor (PDBID:2QXI).40
For all proteins, Schrödinger Maestro’s Protein Preparation Wizard was used to add hydrogens, using Epik to generate states at pH 7.0±2.0 and the H-bond networks optimized.38 Finally, the whole protein was submitted to two steps of restrained minimization: first, only the hydrogens are allowed to move, and then all atoms move restricted to a heavy-atom RMSD to the original structure of 0.30 Å, both using the OPLS3e force field.
All proteins were aligned to the PPE backbone from 6TH7, and the tutuilamide A from this last structure was used to define a cubic docking grid, centered on the tutuilamide A centroid (−18.14, 28.19, −4.75) and with sides 31.70 Å. The midpoint of the ligand was further restricted to a smaller box of 10 Å side around the center. The ligands were docked using Glide with SP precision, and the best pose (selected by lowest binding energy) was further optimized with MM-GBSA using Prime, where residues within 3.0 Å of the ligands are allowed to move. The docking strategy was validated by redocking and cross docking of tutuilamide A and lyngbyastatin 7 into the PPE crystal structures, as described in the Supporting Information (Figures S2 and S3), and was capable of adequately reproducing the crystal structures.
Supplementary Material
Acknowledgments
We acknowledge financial support from the National Institutes of Health (NCI Grant No. R01CA172310) and the Debbie and Sylvia DeSantis Chair professorship (H.L.).
Footnotes
Declaration of Competing Interest
None.
Supplementary Material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2020.115756.
References
- 1.Al-Awadhi FH, Luesch H. Targeting eukaryotic proteases for natural products-based drug development. Nat. Prod. Rep 2020;37:827–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nature Rev. Drug Discov 2010;9:690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi KY, Swierczewska M, Lee S, Chen X. Protease-activated drug development. Theranostics 2012;2:156–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turk B Targeting proteases: successes, failures and future prospects. Nature Rev. Drug Discov 2006;5:785–799. [DOI] [PubMed] [Google Scholar]
- 5.Tyndall JDA, Nall T, Fairlie DP. Proteases universally recognize beta strands in their active sites. Chem. Rev 2005;105:973–999. [DOI] [PubMed] [Google Scholar]
- 6.Veltri CA. Proteases: nature’s destroyers and the drugs that stop them. Pharm Pharmacol Int J 2015;2:222–230. [Google Scholar]
- 7.Prassas I, Eissa A, Poda G, Diamandis EP. Unleashing the therapeutic potential of human kallikrein-related serine proteases. Nature Rev. Drug Discov 2015;15:183–202. [DOI] [PubMed] [Google Scholar]
- 8.Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev 2010;62:726–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crocetti L, Quinn MT, Schepetkin IA, Giovannoni MP. A patenting perspective on human neutrophil elastase (HNE) inhibitors (2014–2018) and their therapeutic applications. Expert Opin. Ther. Pat 2019;29:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.William C Groutas WC, Doua D, Allistona KR. Neutrophil elastase inhibitors. Expert Opin Ther Pat 2011;21:339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Resch S, Kessenbrock T, Schrapp L, Ehrmann M, Kaiser M. From dolastatin 13 to cyanopeptolins, micropeptins, and lyngbyastatins: the chemical biology of Ahp-cyclodepsipeptides. Nat. Prod. Rep 2020;37:163–174. [DOI] [PubMed] [Google Scholar]
- 12.Salvador LA, Taori K, Biggs JS, Jakoncic J, Ostrov DA, Paul VJ, Luesch H. Potent elastase inhibitors from cyanobacteria: structural basis and mechanisms mediating cytoprotective and anti-inflammatory effects in bronchial epithelial cells. J. Med. Chem 2013;56:1276–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Awadhi FH, Paul VJ, Luesch H. Structural diversity and anticancer activity of marine-derived elastase inhibitors: key features and mechanisms mediating the antimetastatic effects in invasive breast cancer. ChemBioChem 2018;19:815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalinska M, Meyer-Hoffert U, Kantyka T, Potempa J. Kallikreins-the melting pot of activity and function. Biochimie 2106;122:270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sotiropoulou G, Pampalakis G. Targeting the kallikrein-related peptidases for drug development. Trends in Pharmacological Sciences 2012;33:623–634. [DOI] [PubMed] [Google Scholar]
- 16.Murafujia H, Sugawaraa H, Gotob M, Oyamac Y, Sakaib H, Imajo S, Tomood T, Muto T. Structure-based drug design to overcome species differences in kallikrein 7 inhibition of 1,3,6-trisubstituted 1,4-diazepan-7-ones. Bioorganic & Medicinal Chemistry 2018;26:3639–3653. [DOI] [PubMed] [Google Scholar]
- 17.Murafujia H, Mutoa T, Gotoa M, Imajoa S, Sugawaraa H, Oyamaa Y, Minamitsujib Y, Miyazaki S, Muraib K, Fujiok H. Discovery and structure-activity relationship of imidazolinylindole derivatives as kallikrein 7 inhibitors. Bioorganic & Medicinal Chemistry Letters 2019;29:334–338. [DOI] [PubMed] [Google Scholar]
- 18.Veer SJ, Furio L, Swedberg JE, Munro CA, Brattsand M, Clements JA, Hovnanian A, Harris JM. Selective substrates and inhibitors for kallikrein-related peptidase 7 (KLK7) shed light on KLK proteolytic activity in the stratum corneum. J. Invest. Dermatol 2017;137:430–439. [DOI] [PubMed] [Google Scholar]
- 19.Krastel P, Liechty BM, Meingassner JG, Schmitt E, Schreiner EP. Cyclic depsipeptides 2012; US 8,178,650 B2
- 20.Matthew S, Ross C, Rocca JR, Paul VJ, Luesch H. Lyngbyastatin 4, a dolastatin 13 analogue with elastase and chymotrypsin inhibitory activity from the marine cyanobacterium Lyngbya confervoides. J. Nat. Prod 2007;70:124–127. [DOI] [PubMed] [Google Scholar]
- 21.Taori K, Matthew S, Rocca JR, Paul VJ, Luesch H. Lyngbyastatins 5–7, potent elastase inhibitors from Floridian marine cyanobacteria, Lyngbya spp. J. Nat. Prod 2007;70:1593–1600. [DOI] [PubMed] [Google Scholar]
- 22.Kwan JC, Taori K, Paul VJ, Luesch H. Lyngbyastatins 8–10, elastase inhibitors with cyclic depsipeptide scaffolds isolated from the marine cyanobacterium Lyngbya semiplena. Mar. Drugs 2009;7:528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrigan GG, Luesch H, Yoshida WY, Moore RE, Nagle DG, Paul VJ. Symplostatin 2: a dolastatin 13 analogue from the marine cyanobacterium Symploca hydnoides. J. Nat. Prod 1999; 62:655–658. [DOI] [PubMed] [Google Scholar]
- 24.Nogle LM, Williamson RT, Gerwick WH. Somamides A and B, two new depsipeptide analogues of dolastatin 13 from a Fijian cyanobacterial assemblage of Lyngbya majuscula and Schizothrix Species. J. Nat. Prod 2001;64:716–719. [DOI] [PubMed] [Google Scholar]
- 25.Gunasekera SP, Miller MW, Kwan JC, Luesch H, Paul VJ. Molassamide, a depsipeptide serine protease inhibitor from the marine cyanobacterium Dichothrix utahensis. J. Nat. Prod 2010;73:459–462. [DOI] [PubMed] [Google Scholar]
- 26.Kang SH, Krunic A, Orjala J. Stigonemapeptin, an Ahp-containing depsipeptide with elastase inhibitory activity from the bloom-forming freshwater cyanobacterium Stigonema sp. J. Nat. Prod 2012;75:807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmad S, Saleem M, Riaz N, Lee YS, Diri R, Noor A, Almasri D, Bagalagel A, Elsebai MF. The natural polypeptides as significant elastase inhibitors. Front. Pharmacol 2020;11;688(1–19) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farady CJ, Craik CS. Mechanisms of macromolecular protease inhibitors. ChemBioChem 2010:11:2341–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo D, Luesch H. Ahp-cyclodepsipeptide inhibitors of elastase: lyngbyastatin 7 stability, scalable synthesis, and focused library analysis. ACS Med. Chem. Lett 2020;11: 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo D, Chen Q-Y, Luesch H. Total synthesis of the potent marine-derived elastase inhibitor lyngbyastatin 7 and in vitro biological evaluation in model systems for pulmonary diseases. J. Org. Chem 2016;81:532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keller L, Canuto KM, Liu C, Suzuki BM, Almaliti J, Sikandar A, Naman CB, Glukhov E, Luo D, Duggan BM, Luesch H, Koehnke J, O’Donoghue AJ, Gerwick WH. Tutuilamides A–C: vinyl-chloride-containing cyclodepsipeptides from marine cyanobacteria with potent elastase inhibitory properties. ACS Chem. Biol 2020;15:751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dahiya R, Gautam H. Toward the synthesis and biological screening of a cyclotetrapeptide from marine bacteria. Mar. Drugs 2011;9:71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu L,Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, kansson KH, Wipf P, Smith JL, Gerwick WH, Sherman DH. Metamorphic enzyme assembly in polyketide diversification. Nature 2009;459:731–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BS. A mild and selective method for the hydrolysis of esters with trimethyltin hydroxide. Angew. Chem. Int. Ed 2005;117:1402–1406. [DOI] [PubMed] [Google Scholar]
- 35.Imai Y, Inoue Y, Nakanishi I, Kitaura K. Cl–π interactions in protein–ligand complexes. Protein Science 2008;17:1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishio M, Umezawa Y, Fantini J, Weiss MS, Chakrabartie P. CH–π hydrogen bonds in biological macromolecules. Phys. Chem. Chem. Phys 2014;16:12648–12683. [DOI] [PubMed] [Google Scholar]
- 37.Lin F-Y, MacKerell AD Jr. Do halogen–hydrogen bond donor interactions dominate the favorable contribution of halogens to ligand–protein binding? J. Phys. Chem. B 2017;121:6813–6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrödinger LLC. Schrödinger Release 2020–1 2020.
- 39.Hansen G, Gielen-Haertwig H, Reinemer P, Schomburg D, Harrenga A, Niefind K. Unexpected active-site flexibility in the structure of human neutrophil elastase in complex with a new dihydropyrimidone inhibitor. J. Mol. Biol 2011;409(5):681–691. [DOI] [PubMed] [Google Scholar]
- 40.Debela M, Hess P, Magdolen V, Schechter N, Steiner T, Huber R, Bode W, Goettig P. Chymotryptic specificity determinants in the 1.0 Å structure of the zinc-inhibited human tissue kallikrein 7. Proc. Natl. Acad. Sci. USA 2007;104(41):16086–16091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.