

Abstract
Mutations in the secreted metalloproteinase ADAMTS10 cause recessive Weill-Marchesani syndrome (WMS), comprising ectopia lentis, short stature, brachydactyly, thick skin and cardiac valve anomalies. Dominant WMS caused by FBN1 mutations is clinically similar and affects fibrillin-1 microfibrils, which are a major component of the ocular zonule. ADAMTS10 was previously shown to enhance fibrillin-1 assembly in vitro. Here, Adamts10 null mice were analyzed to determine the impact of ADAMTS10 deficiency on fibrillin microfibrils in vivo. An intragenic lacZ reporter identified widespread Adamts10 expression in the eye, musculoskeletal tissues, vasculature, skin and lung. Adamts10−/− mice had reduced viability on the C57BL/6 background, and although surviving mice were slightly smaller and had stiff skin, they lacked brachydactyly and cardiovascular defects. Ectopia lentis was not observed in Adamts10−/− mice, similar to Fbn1−/− mice, most likely because the mouse zonule contains fibrillin-2 in addition to fibrillin-1. Unexpectedly, in contrast to wild-type eyes, Adamts10−/− zonule fibers were thicker and immunostained strongly with fibrillin-2 antibodies into adulthood, whereas fibrillin-1 staining was reduced. Furthermore, fibrillin-2 staining of hyaloid vasculature remnants persisted post-natally in Adamts10−/− eyes. ADAMTS10 was found to cleave fibrillin-2, providing an explanation for persistence of fibrillin-2 at these sites. Thus, analysis of Adamts10−/− mice led to identification of fibrillin-2 as a novel ADAMTS10 substrate and defined a proteolytic mechanism for clearance of ocular fibrillin-2 at the end of the juvenile period.
Keywords: Weill-Marchesani syndrome, Acromelic dysplasia, Metalloprotease, Extracellular matrix, Ectopia lentis, Zonule
Introduction
Tissue microfibrils are supramolecular complexes in the extracellular matrix (ECM) that are evident ultrastructurally as filamentous beaded or non-beaded structures with a diameter of 10–12 nm [1,2]. They typically form bundles associated with elastic fibers [3], but occur independently of them in the ocular zonule. The zonule is an acellular structure comprising microfibrils that extends from the pars plana and ciliary body to the equatorial region of the lens and transmits ciliary muscle contraction and relaxation during visual accommodation [4]. The principal components of microfibrils in the zonule and elsewhere are fibrillins, which are large secreted glycoproteins [5–7]. A recent proteomic analysis of the zonule and other studies of microfibrils have indicated inclusion of several components besides fibrillins, including microfibril-associated glycoproteins, latent TGFβ-binding proteins and ECM-modifying enzymes [8,9]. The products of three fibrillin genes in humans and two in mice [5] appear to readily form both homotypic and heterotypic microfibrils [7,10]. Whereas fibrillin-1 is the major microfibril protein expressed post-natally, fibrillin-2 and fibrillin-3 seem to be the major embryonic isoforms, with a transition to fibrillin-1 predominance occurring from the late gestational period into the juvenile period [11–13]. In addition to their structural and elastogenic roles, microfibrils bind to growth factors of the TGFβ superfamily directly as well as indirectly via latent TGFβ-binding proteins to regulate their activity [14,15]. These diverse roles were elucidated by analysis of human genetic disorders affecting fibrillins, especially Marfan syndrome [16], and mouse fibrillin mutants [17].
Marfan syndrome (MFS, MIM 154700) is an inherited connective tissue disorder caused by dominant FBN1 mutations and affects 1:5000–1:10,000 individuals. Its characteristic manifestations are skeletal overgrowth with spidery hands and feet (arachnodactyly), lens dislocation (ectopia lentis), stretchy skin, and aneurysms affecting the aortic root and ascending aorta, which commonly undergo dissection [17]. Infrequently, FBN1 mutations cause Weill-Marchesani syndrome (WMS2, MIM 608328) and two related rare conditions, acromicric dysplasia (AD, MIM 102370) and geleophysic dysplasia 2 (GD2, MIM 614185) [18–20]. These disorders are categorized as acromelic dysplasias and share short stature, disproportionately short hands and feet (brachydactyly), cardiac valve anomalies and thick skin with reduced joint flexibility, characteristics which are the opposite of MFS [21,22]. In contrast to these opposing manifestations, ectopia lentis is present in both MFS and WMS. Unlike MFS, WMS does not typically predispose to aortic aneurysm. Whereas AD and GD mutations affect the TGFβ-binding domain 5 of fibrillin-1, the FBN1 mutations known in WMS affect other regions [18,19].
Autosomal recessive WMS is similar to the dominant form [23] and is caused by mutations affecting the secreted metalloprotease, ADAMTS10 (WMS1, MIM 277600) [24], a member of a large protein family with diverse roles in embryonic development and adult disorders [25,26]. In dogs, ADAMTS10 mutations lead to primary open angle glaucoma, without extra-ocular features of WMS [27,28]. A WMS-like syndrome (WMSL, MIM 613195) is caused by mutations of a related protease, ADAMTS17 [29]. Collectively, these disorders suggested a functional link between ADAMTS10, ADAMTS17 and fibrillin-1 [21,30]. Subsequent work showed that ADAMTS10 associated with microfibrils in the zonule [31], and that both ADAMTS10 and ADAMTS17 bound fibrillin-1 in vitro [31,32]. ADAMTS10 undergoes inefficient proteolytic excision of its propeptide owing to a suboptimal furin recognition site [31]. However, the fraction that is furin-processed can cleave fibrillin-1, albeit inefficiently, which was shown using an ADAMTS10 mutant of the furin recognition site that rendered it efficiently furin-cleavable [31]. ADAMTS10 was shown to enhance assembly of fibrillin-1 microfibrils in vitro, demonstrating that the proteolytic activity against fibrillin-1 was less significant than an ability to promote its assembly [31,33]. In the present work, we analyzed mice lacking Adamts10 to obtain additional cellular and molecular insights on its relationship with microfibrils in an in vivo context. These studies suggested a novel and unexpected relationship of ADAMTS10 with fibrillin-2 turnover in the eye and subsequent biochemical analysis identified fibrillin-2 as a novel ADAMTS10 substrate.
Results
Disruption of the Adamts10 locus leads to a null allele
Adamts10 was disrupted using homologous recombination in mouse embryonic stem cells. The targeting vector was designed to generate a frameshift mutation by deleting 41 nt of exon 5 and insertion of IRES-lacZ-Neo at the deletion site (Fig. 1A). Correct targeting was confirmed by PCR of genomic DNA (Fig. 1B). In contrast to cDNA generated from wild-type mouse tissues, RT-PCR of Adamts10−/− tissues did not provide an amplicon bridging exon 3 with the deleted sequence in exon 5, demonstrating gene disruption by the IRES-LacZ-Neo cassette (Fig. 1C). A primer pair bridging exon 3 to the targeting cassette in Adamts10−/− tissues amplified the expected fusion transcript of Adamts10 mRNA and IRES-lacZ, i.e., sequencing of this amplicon confirmed the Adamts10 frameshift mutation and continuity of Adamts10 and lacZ transcripts (Fig. 1C). qRT-PCR with a primer pair upstream of the targeting site showed comparable mutant RNA levels in null and wild-type mice (Fig. 1C), indicating that the Adamts10-IRES-lacZ fusion transcript was stable. This validated the use of β-galactosidase (β-gal) staining as a useful surrogate for Adamts10 mRNA expression. β-gal staining was only seen in Adamts10+/− and Adamts10−/− embryos and adult tissues but not in the wild-type (Fig. 1D and Supplemental Fig. 1). Consistent with inclusion of transcription termination and poly-A signals in the inserted cassette, exons downstream of the targeting site were not included in the mutant fusion transcript (Fig. 1C, Exon 5–7). The fusion transcript arising from the targeted allele encodes only the ADAMTS10 propeptide and the N-terminal 13 residues of the catalytic domain. Thus, production of catalytically active ADAMTS10 from the targeted allele is eliminated, although this could not be confirmed at the protein level in the absence of a suitable antibody. Although the ADAMTS10 propeptide could be involved in some binding interactions or other functions, none of the ADAMTS propeptides are known to be independently functional, rendering this possibility unlikely.
Fig. 1.
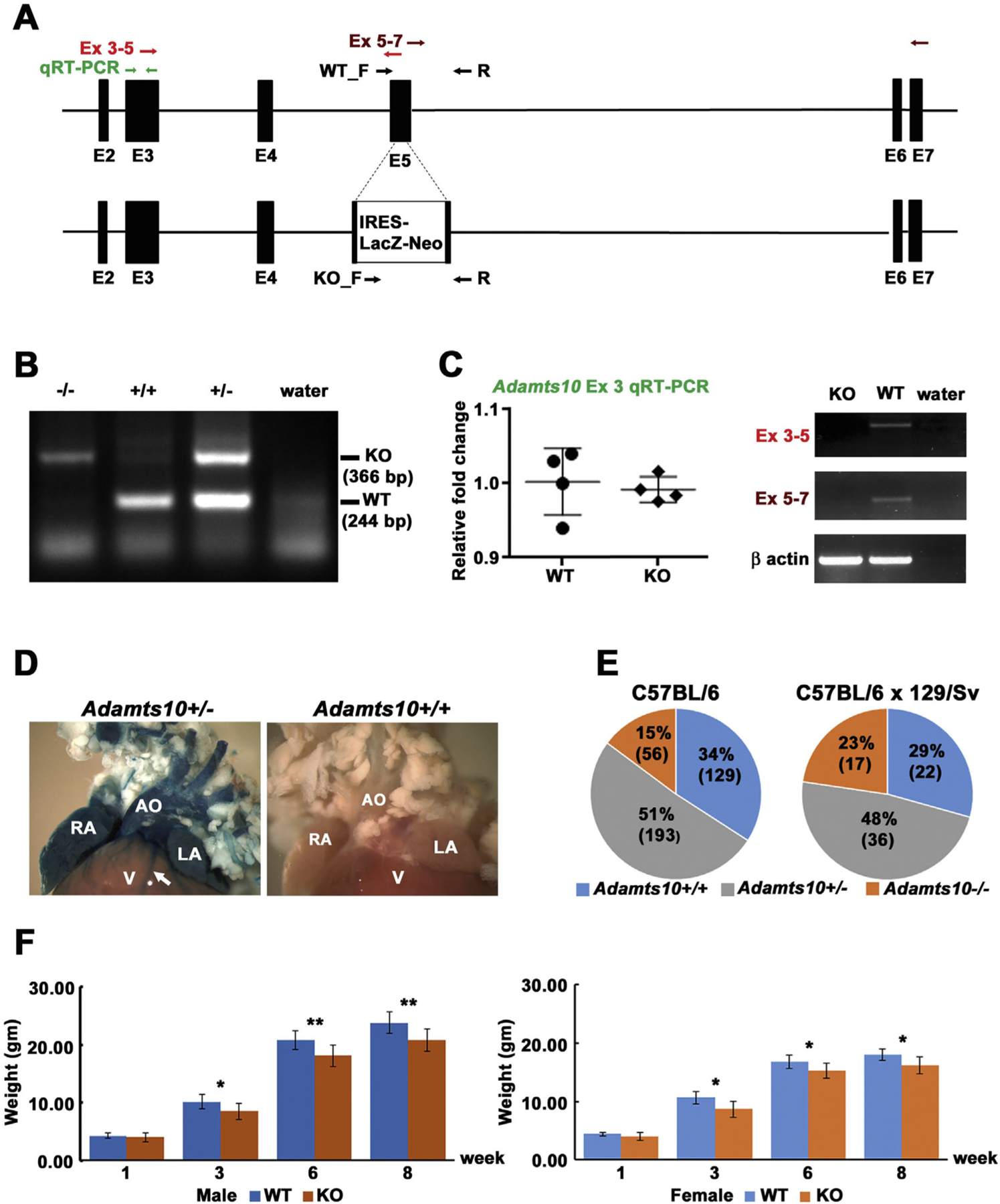
Generation and characterization of Adamts10−/− mice. A. Gene targeting strategy: the structure of the wild-type Adamts10 allele inclusive of exon 2 through exon 7 (top) and the targeted Adamts10 allele (bottom) are illustrated. The primers used for genotyping are shown by black arrows (WT_F/R and KO_F/R). The primers used for RT-PCR are shown as pairs of red, maroon or green arrows indicating their location. B. Agarose gel electrophoresis of genomic PCR products shows the distinction between wild type (WT) and knockout (KO) alleles. Water was used as the control target. C. Left: q-RT-PCR shows comparable upstream RNA levels in wild-type and Adamts10−/− mice (KO). Right: RT-PCR (exons 3–5) shows interruption of exon 5 by insertion of the IRES-lacZ-neo cassette and loss of RNA downstream of the targeting site (exons 5–7). β-actin PCR shows comparable source RNA. D. Consistent with generation of an Adamts10-IRES-lacZ transcript, aorta (AO), right atrium (RA), left atrium (LA) and coronary vessels (arrow), but not the ventricular myocardium (V) from an Adamts10+/− mouse, but not a wild-type mouse demonstrated robust β-gal staining (blue). E. Pie chart of the genotypes detected at 10 days of age from intercrosses of Adamts10+/− mice in the indicated strains. Note reduced viability of Adamts10−/− mice in the C57BL/6 strain. F. Adamts10−/− mice segregated by sex, as shown, weighed significantly less than wild-type littermates. *p < 0.01, **p < 0.001, n = 7 male wild-type mice, 6 female wild type mice; 13 male Adamts10−/− mice and 11 female Adamts10−/− mice, error bars indicate S.D.
Intercrosses of Adamts10+/− mice provided progeny in the expected Mendelian ratio in the hybrid 129/Sv X C57BL/6 background, but in the C57BL/6 strain, in which the mice were subsequently maintained, a Chi square calculation showed significant departure from the expected Mendelian ratio (p < 0.05) (Fig. 1E). Because neonatal lethality was not observed, this suggests partial embryonic lethality in the C57BL/6 strain. Both male and female Adamts10−/− mice were externally normal other than having lower body weight than wild-type littermates (Fig. 1F).
An intragenic lacZ reporter identifies widespread ocular and non-ocular Adamts10 expression
Analysis of Adamts10 mRNA expression during embryonic development using RNA in situ hybridization had previously shown broad expression in extra-ocular tissues [34], but ocular expression was not analyzed. Since disruption of the ocular zonule leading to ectopia lentis is a major primary pathology in WMS, we analyzed Adamts10 expression by β-gal staining at various times through embryonic ocular development and in the adult eye. No β-gal staining was seen in the developing eye until 12.5 days of embryo age (E12.5), but a consistent pattern was seen between E12.5 until birth. At E16.5, which exemplified the embryonic expression, there was nuclear staining of the ciliary margin zone, primary lens fibers, and ganglion cell layer of the retina that persisted into the adult eye (Fig. 2). Once development of the iris, ciliary body and drainage apparatus was initiated, intense Adamts10 expression was detected in the non-pigmented ciliary epithelium and the limbal region, including corneal and scleral stroma cells, cells around Schlemm’s canal and in the lens epithelium (Fig. 2). Corneal and scleral fibroblasts and extraocular muscle myofibers consistently expressed Adamts10 (data not shown) and corneal endothelium of adult eyes was also positive (Fig. 2). In extra-ocular tissues, strong expression was seen throughout the limb in cartilage, bone, skeletal muscle and tendons and the dermis of skin (Supplemental Fig. 1A). Widespread β-gal staining was present in lung, but in the heart and liver, it was primarily present in the large blood vessels, such as aorta, coronary, pulmonic and hepatic vessels, and in atria and cardiac valves, with ventricular myocardium lacking expression (Fig. 1D and Supplemental Fig. 1).
Fig. 2.
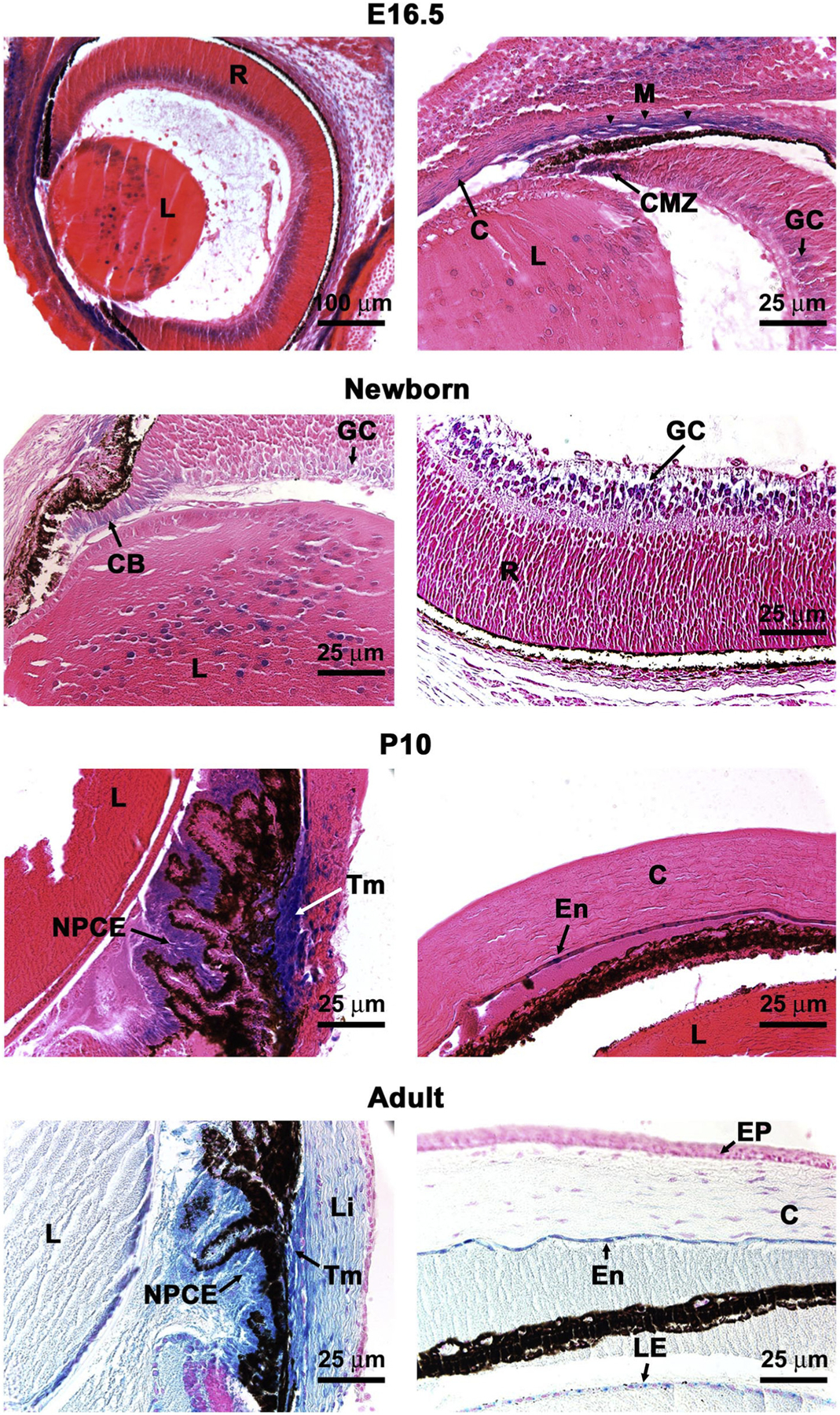
An Adamts10 reporter is expressed at specific sites in the mouse eye. β-gal staining (blue) of Adamts10+/− eyes indicates sites of Adamts10 mRNA expression in the 16.5-day old embryo (E16.5), newborn, and post-natal day 10 (P10) or adult (8 week) eyes as labeled. C, cornea; CB, ciliary body; CMZ, ciliary margin zone; En, corneal endothelium; EP, corneal epithelium; GC, ganglion cell layer of retina; L, lens; LE, lens epithelium; Li, limbus; M, primordial trabecular meshwork; NPCE, non-pigmented ciliary epithelium; Tm, trabecular meshwork; R, retina. Scale bars are 100 μm at top left and 25 μm in other panels.
ADAMTS10 deficiency in mice does not lead to the extra-ocular manifestations of Weill-Marchesani syndrome
Analysis of the lungs, based on the observed strong Adamts10 expression consistently identified a distinct anomaly in Adamts10−/− lungs with increased alveolarization of the alveolar ducts that are normally seen at the distal end of the terminal bronchiole (Supplemental Fig. 1B). However this anomaly was subtle and did not result in a significant change in mean linear intercept analysis of lung parenchyma (data not shown). Adamts10−/− mice had stiffer skin as determined by its reduced stretchiness when the mice were scruffed. To determine the mechanism of this further, we isolated dermal fibroblasts from Adamts10−/− mice and found that they continued to express Adamts10 mRNA after isolation (Supplemental Fig. 1C). Accordingly, dermal fibroblasts were also isolated from wild-type littermates and compared with Adamts10−/− cells for their contractile ability in a collagen gel contraction assay. The Adamts10−/− dermal fibroblasts contracted the gel to a greater extent than wild-type (Supplemental Fig. 1C). Adamts10−/− mice had normal skeletal morphology, including normal distal skeletal elements and lacked shortening of long bones as well as brachydactyly, a hallmark of WMS (Supplemental Fig. 2). Their aortic and mitral valves and cardiac function were normal up to a year of age as determined by echocardiography and histology (data not shown).
An intact zonule and undisplaced lens in the Adamts10−/− eye are accompanied by enhanced immunochemical signal for FBN2
Adamts10−/− eyes appeared externally normal throughout the first two years of life, although optical coherence tomography (OCT) identified a statistically significant reduction in anterior chamber depth (Supplemental Fig. 3A). No anomalies of the cornea, iris or lens were noted upon slit lamp examination (data not shown). Histological comparison of the Adamts10−/− eyes and wild type eyes by hematoxylin and eosin staining did not demonstrate anomalies (Supplemental Fig. 3B). Of specific relevance to WMS, the ocular lens appeared to be normal. No retinal anomalies were observed. As visualized by oxytalan stain for microfibrils, the ocular zonule was intact, and spanned the pars plana region to the lens equator explaining lack of ectopia lentis in null mice. Moreover, the oxytalan stain showed stronger staining of the Adams10−/− zonule than the corresponding wildtype at all times analyzed (8 days, 6 weeks and 6 months) (Fig. 3A). Because the mouse zonule contains both FBN1 and FBN2, [4,35] we immunostained the eyes with the respective antibodies. We consistently observed stronger FBN2 staining in the Adamts10−/− zonule from the juvenile into the adult period (Fig. 3B). In contrast, FBN1 staining varied, being reduced in the null zonule at 6 weeks, but not 1 year of age (Fig. 3C). The hyaloid vasculature normally undergoes complete regression in the mouse by 16 days after birth [36]. Persistent FBN2 fibril staining was observed in the vitreous in the presumed remnants of the hyaloid vessel matrix at both 8 days and 24 days after birth in Adamts10−/− eyes (Fig. 4).
Fig. 3.
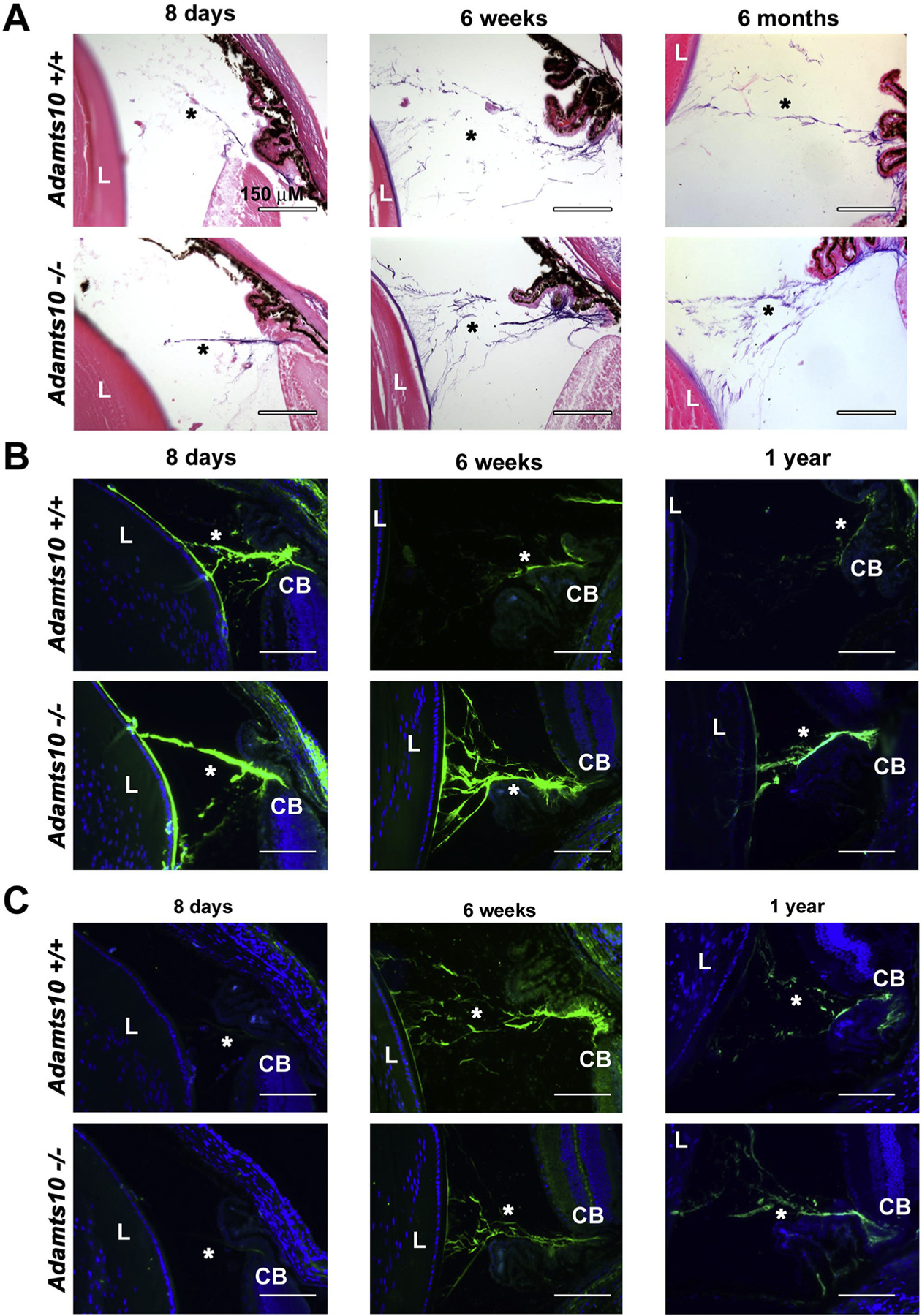
Increased microfibrils and stronger fibrillin-2 staining in the Adamts10−/− zonule. A. Oxytalan stain at various ages indicated showing consistently stronger staining (purple) of the Adamts10−/− zonule. B. Fibrillin-2 immunostaining is stronger post-natally in the Adamts10−/− zonule. C. Fibrillin-1 staining was reduced in the 6 week Adamts10−/− zonule, but was comparable thereafter to the wild-type. Asterisks indicate the zonule. L, lens, CB, ciliary body. Scale bars = 150 μm.
Fig. 4.
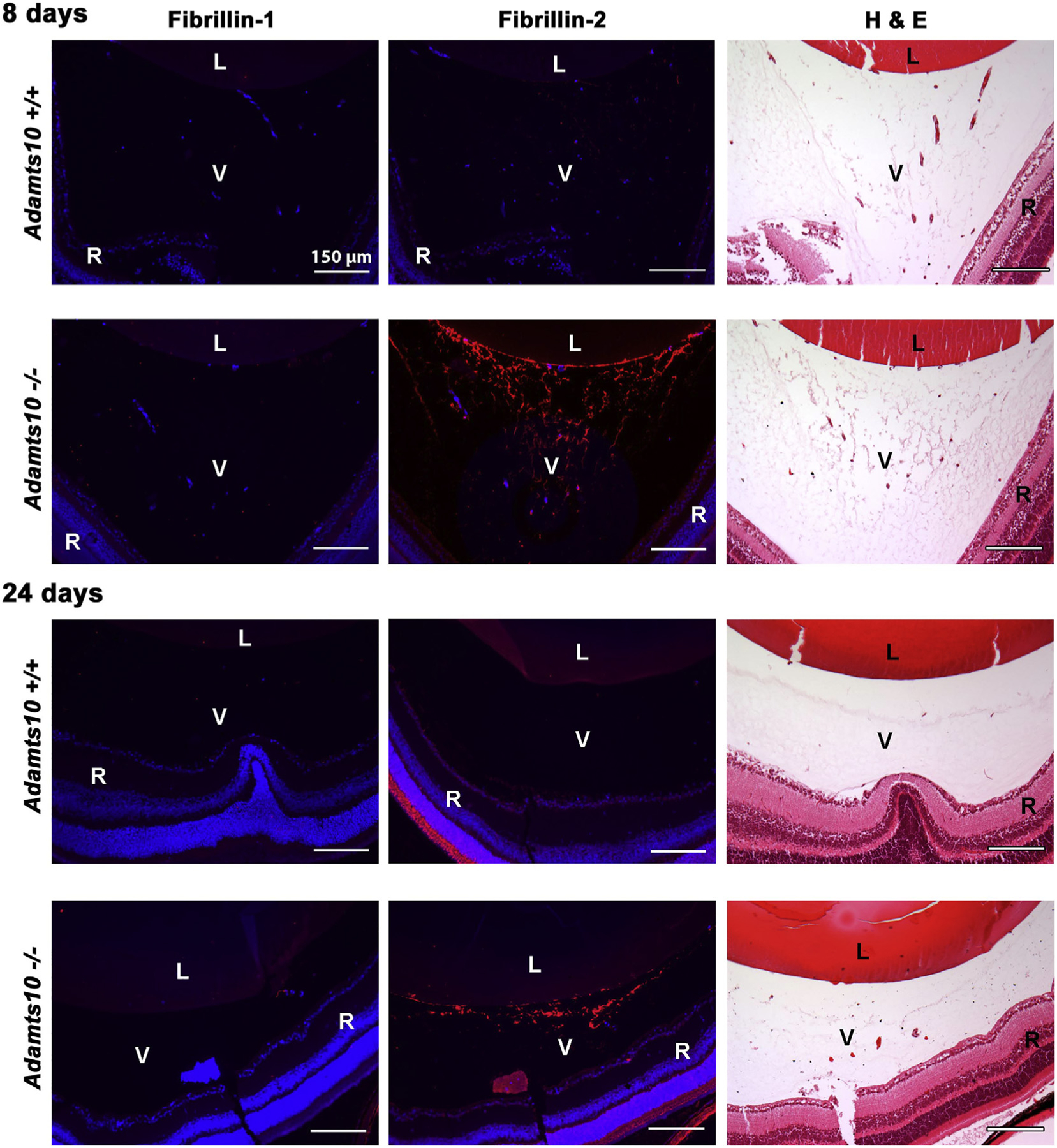
Persistent postnatal fibrillin-2 stained extracellular matrix strands in the Adamts10−/− vitreous. The figure shows fibrillin-1 and fibrillin-2 immunofluorescence staining (red, left-hand and center column) and H&E staining of an adjacent section (right-hand column) from juvenile wild-type and Adamts10−/− eyes. Note the relative abundance of H&E stained strands in the Adamts10−/− vitreous (V) at 8 days and their persistence at 24 days accompanied by increased fibrillin-2 staining but no change in fibrillin-1 staining. DAPI staining (blue) showed no intra-vitreal nuclei at 24 days, indicating that Adamts10−/− eyes had persistence of microfibrils but not hyaloid vasculature. Images are representative of staining observed in 5 wild-type and 5 Adamts10−/− eyes. L, lens, R, retina Scale bar = 150 μm.
ADAMTS10 cleaves fibrillin-2
The observed ocular accumulation of fibrillin-2 motivated investigation of its possible cleavage by ADAMTS10. Prior analysis had shown that only a limited proportion of ADAMTS10 was activated by removal of the propeptide, which was attributed to a suboptimal processing site (GLKR) for the action of proprotein convertases such as furin [31]. We had previously mutated the furin cleavage site to RRKR to render ADAMTS10 efficiently cleavable by furin, enabling analysis of the activity of this activated fraction [31]. This plasmid, or its catalytically inactive version, was co-transfected into HEK293F cells with a plasmid expressing His6-tagged N and C-terminal halves of fibrillin-2. Analysis of the conditioned medium from these experiments using anti-His6 antibody clearly identified a fibrillin-2 fragment arising from its C-terminal half in the presence of catalytically active ADAMTS10-RRKR, but not the corresponding inactive mutant ADAMTS10-RRKREA (Fig. 5A). We also observed a cleaved fibrillin-2 fragment using the anti-fibrillin-2 antibody Fbn2–1 in the presence of ADAMTS10-RRKR but not ADAMTS10-RRKREA (Fig. 5B). Antibody Fbn2–1 was generated against the entire C-terminal half of fibrillin-2 and its precise epitope is not known. Thus, it does not allow us to draw conclusions about the possible number and location of cleavage sites. Western blot using anti-His and anti-myc showed comparable levels of ADAMTS10-RRKR and ADAMTS10-RRKREA (Fig. 5A, C).
Fig. 5.
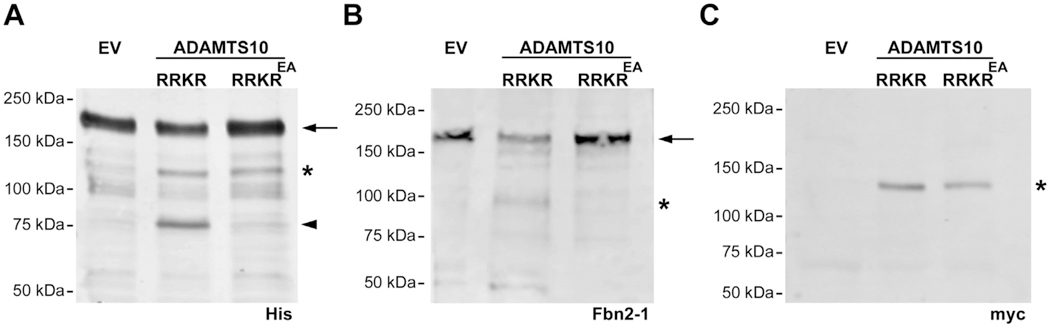
ADAMTS10 cleaves fibrillin-2. A. Representative Western blot using anti-His6 shows a major 185-kDa band in conditioned medium that corresponds to the intact fibrillin-2 C-terminal half (FBN2-1 (arrow)) and a 120-kDa band corresponding to ADAMTS10 (asterisk), which like FBN2-1, also has a C-terminal His6 tag. The arrowhead indicates a 75-kDa band corresponding to cleaved FBN2-1 in the presence of active ADAMTS10 (ADAMTS10 RRKR), but not a catalytically inactive form (ADAMTS10 RRKREA). B. Western blot using polyclonal antibody Fbn2-1 shows a major 185-kDa band in conditioned medium that corresponds to FBN2-1 (arrow) and a 100-kDa fragment (asterisk) that appears specifically in the presence of active ADAMTS10 but not ADAMTS10 RRKREA. C. Anti-myc Western blot confirming expression of active and inactive ADAMTS10 (120-kDa). EV, empty vector.
Discussion
For the present study we have generated and characterized Adamts10−/− mice. Their analysis demonstrated ocular fibrillin-2 accumulation, which is explained by its biochemical characterization here as a novel ADAMTS10 substrate. We had previously generated recombinant ADAMTS10 and characterized high-affinity binding of ADAMTS10 to fibrillin-1 [31]. Indeed, ADAMTS10 was localized to fibrillin microfibrils in human skin and ocular zonule [31]. Addition of recombinant ADAMTS10 to fibroblast cultures demonstrated increased fibrillin-1 microfibril staining, suggesting that it enhanced or accelerated fibrillin microfibril assembly or protected microfibrils from turnover [31,33]. The previous work had logically focused on fibrillin-1, given the strong genetic relationship with ADAMTS10 established by the similar clinical consequence of recessive ADAMTS10 and dominant FBN1 mutations [21,30].
A relationship with fibrillin-2 was not previously considered, but is relevant in developmental contexts as well as post-natally, because of the occurrence of heterotypic fibrils containing both fibrillin-1 and fibrillin-2 [10,35,37]. In mice, fibrillin-2 is strongly expressed during the embryonic period and is the major component of the zonule around birth [4,35]. Consistent with this, prior work has shown that during mouse eye development, Fbn2 mRNA is dominant, with Fbn1 mRNA expression commencing in the late embryonic period and dominating in adult tissues [13]. Fibrillin-2 protein is detected in both the human and mouse embryonic zonule [35] and in mice both fibrilllin-1 and fibrillin-2 comprise the adult zonule [35]. During human eye development, fibrillin-2 is present in the zonule region during early embryogenesis [38]. Fbn1−/− eyes do not have ectopia lentis, apparently reflecting the presence of an intact zonule comprising fibrillin-2 [35]. Fbn2−/− eyes also have an intact zonule but have a high incidence of morphogenetic defects especially iris coloboma [39]. Such morphogenetic defects do not appear in Adamts10−/− eyes, suggesting that excess fibrillin-2 is not as deleterious as its absence. Our analysis suggests that fibrillin-2 is disproportionately increased with respect to fibrillin-1, suggesting its selectively reduced turnover in the absence of ADAMTS10.
ADAMTS10 may thus contribute to maintaining an appropriate content of FBN1 and FBN2 in microfibrils in two ways, i.e., by enhancing FBN1 assembly, and cleaving FBN2. Intriguingly, lack of an ADAMTS-like protein, ADAMTSL2, in mice, also led to increased FBN2 staining in the peribronchial ECM of Adamtsl2−/− lungs [40]. ADAMTSL2 lacks proteolytic activity and is strongly expressed in the late embryonic period [40], coinciding with the switch from Fbn2 mRNA to Fbn1 mRNA dominance. Furthermore, ADAMTSL4 and ADAMTSL5 bind to and enhance the assembly of fibrillin-1 [41,42]. Thus, several ADAMTS proteins may participate in maintenance of the proper balance of fibrillin-1 and fibrillin-2 in tissues [30].
Adamts10−/− eyes also had postnatal accumulation of fibrillin-2 microfibrils in the vitreous, which may be remnants of the supporting ECM of hyaloid (vitreous) vessels. Fbn2 mRNA is expressed in the hyaloid tissue [4], although Adamts10 expression by β-gal staining was consistently seen in the ganglion cell layer of the retina, which abuts the vitreous. Thus, one role of the ganglion cell production of ADAMTS10 may be to degrade fibrillin-2 fibrils in the vitreous. The assembly and disassembly of the ECM supporting the hyaloid blood vessels is poorly understood. Persistent hyaloid vasculature is a recognized clinical problem in infants; however persistence of hyaloid ECM structures is not a known clinical disorder, and whether the remnant fibrillin-2 fibrils might impair vision is unknown.
Despite the noted anomalies in the eyes, lung and skin fibroblasts of Adamts10−/− mice, skeletal, skin and cardiac structure were unaffected, and this could be a result of compensation by other ADAMTS proteases. Recently, Mularczyk et al. reported analysis of mouse model of a human WMS-associated ADAMTS10 mutation which showed similarities with the present analysis, including fibrillin-2 accumulation in the zonule and skeletal muscle; however, they did not test whether fibrillin-2 was an ADAMTS10 substrate [43]. In contrast with our model, they observed reduced skeletal growth, and identified anomalies of muscle, which was not a focus of the present analysis [43]. Our novel finding that fibrillin-2 turnover requires ADAMTS10 explains the fibrillin-2 accumulation seen in both mouse strains.
In mice, the presence of two fibrillins instead of three in humans, and inclusion of fibrillin-2 in the mouse ocular zonule provides a possible explanation for differing phenotypes in humans and mice lacking ADAMTS10. Another explanation for the relatively mild phenotype of Adamts10 mutants could be functional overlap with and compensation by its closest homolog Adamts6, which has an apparently more important role in mouse development, demonstrated by embryonic lethality of Adamts6−/− embryos and severe cardiac malformations [44,45]. ADAMTS17 has the same domain structure as ADAMTS10 and ADAMTS6, but a lower sequence similarity to ADAMTS10 than ADAMTS6. Adamts17 is expressed in the ciliary margin zone as well as the lens fiber cells [32]. In contrast to ADAMTS10, the ADAMTS6 and ADAMTS17 furin processing sites (RQKR and RERR respectively) satisfy the required consensus for efficient activation. A recent analysis showed that ADAMTS17 is both furin-processed and proteolytically active, but did not cleave fibrillin-1 [32]. Thus, ADAMTS10, ADAMTS6 and ADAMTS17 may have overlapping as well as distinct roles vis-à-vis fibrillin fibrils as has been shown for multiple ADAMTS proteases that cleave versican [46–50]. Fibrillin-2 accumulation in ADAMTSL2-deficient lungs [40], and the reported suppression of fibrillin-2 mRNA by ADAMTS17 [32], further illustrate that the relationship between the ADAMTS proteins and microfibrils is complex and not confined solely to fibrillin-1. Consistent with a recent discussion in this journal [51], the data also illustrate how metalloproteases serve essential roles in biology and are not solely destructive entities in disease.
Methods
Transgenic mice
The Adamts10 locus was targeted by homologous recombination in embryonic stem cells by Deltagen Inc. The targeting construct was generated by deletion of 41 bp from exon 5 ( 903cgacaagatgat…gatgtgga944, Adamts10 cDNA sequence, GenBank accession number NM_172619.3), and insertion of an IRES-lacZ-neo cassette (specifically, IRES-lacZ-stop-SV40 poly A-Pgk1 promoter-Neomycin resistance (Neo)-stop-SV40 pA [52]) at the deletion site. ES clones hemizygous for the targeted mutation were injected into blastocysts for generation of chimeric mice. High percentage chimeras were used to obtain germline transmission of the targeted allele (Allele t1233, Deltagen Inc., San Carlos, CA). Heterozygous transgenic mice in the C57Bl/6J + 129/Sv mixed background were backcrossed for 10 generations in the C57Bl/6J strain for subsequent analysis. The genotype ratio obtained after heterozygote intercrosses was compared with the expected ratio using a Chi square calculator (GraphPad QuickCalcs, http://graphpad.com/quickcalcs/chisquared1.cfm).
Genotyping was performed using genomic DNA isolated from tail biopsies. Genotyping, RT-PCR primers and qRT-PCR primers used to analyze the impact of gene interruption are in Supplemental Table 1. qRT-PCR was done as previously described [48]. The eyes were analyzed by OCT essentially as previously described [53] to determine anterior segment morphology and measure the depth of the anterior chamber. Mice were weighed weekly using a laboratory scale. All mouse procedures were approved by the Cleveland Clinic Institutional Animal Care and Use Committee (protocol 2016–1458) and euthanasia was in accordance with the guidelines of the American Veterinary Association. Skeletal radiographs were obtained as previously described [49].
Histology and immunohistochemistry
Histologic analysis was performed using paraffin sections of eyes fixed in 4% paraformaldehyde (PFA) for 24–48 h. Hematoxylin & eosin and oxytalan fiber staining were performed using routine procedures. Fibrillin-1 and fibrillin-2 rabbit polyclonal antibodies were previously described [35] and used for immunofluorescence in an indirect staining method with Alexa Fluor 488-labeled (green) or Alexa fluor 568-labeled (red) secondary antibodies. For β-gal staining, tissues were fixed overnight in 4% PFA and stained using a previously described procedure [54] and subsequently paraffin-embedded for sectioning.
Western blot analysis
HEK293 cells stably expressing a fibrillin-2 C-terminal fragment 2-1 [55] were maintained in medium supplemented with 10% fetal bovine serum and 100 U/mL penicillin and 100 μg/mL streptomycin. Plasmid vectors for expression of active ADAMTS10 designated ADAMTS10-RRKR and inactive ADAMTS10, designated ADAMTS10-RRKREA were previously described [31]. The constructs were transfected using Lipofectamine 3000 Transfection Kit (catalog no. L3000; Invitrogen) following manufacturer’s instructions. After 72 h in serum-free medium, the medium was recovered and cell lysates were collected in lysis buffer (0.1% NP-40, 0.01% sodium dodecyl sulfate and 0.05% sodium deoxycholate in phosphate buffered saline), pH 7.4 (PBS). Medium and lysate were electrophoresed by reducing SDS-PAGE on 4–20% gradient Tris-glycine gels. Proteins were electro-blotted to Immobilon-FL membranes (IPFL00010, EMD Millipore), incubated with primary antibodies anti-myc (1:1000; Cleveland Clinic Hybridoma core facility), anti-His6 (catalogno. MAB050; 1:1000; R&D Systems) and anti-fibrillin 2-1 Dieter [55] (1:500), in 5% milk in PBS overnight at 4 °C, followed by IRDye secondary antibodies goat anti-mouse or anti-rabbit (926-68170, 827-08365; 1:10000; LI-COR) in PBS for 1 h at room temperature and visualized by Odyssey CLx (LI-COR).
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health award EY021151 (to S. Apte), the Marfan Foundation (award to S. Apte), the Knights Templar Eye Foundation (Pediatric Ophthalmology Career-Starter Research Grant to L. Beene) and by the Allen Distinguished Investigator Program, through support made by The Paul G. Allen Frontiers Group and the American Heart Association (to S. Apte). D.P. Reinhardt was supported by the Canadian Institutes of Health Research (MOP-106494). We thank Dr. Elias Traboulsi and Apte laboratory members for valuable discussion.
Abbreviations used:
- ADAMTS
a disintegrin-like and metalloprotease domain with thrombospondin type 1 motifs
- AD
acromicric dysplasia
- ECM
extracellular matrix
- GD
geleophysic dysplasia
- MFS
Marfan syndrome
- PVDF
polyvinyldifluoridene
- qRT-PCR
quantitative real-time PCR
- WMS
Weill-Marchesani syndrome
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.matbio.2018.09.004.
References
- [1].Davis EC, Roth RA, Heuser JE, Mecham RP, Ultrastructural properties of ciliary zonule microfibrils, J. Struct. Biol 139 (2) (2002) 65–75. [DOI] [PubMed] [Google Scholar]
- [2].Reinhardt DP, Keene DR, Corson GM, Poschl E, Bachinger HP, Gambee JE, Sakai LY, Fibrillin-1: organization in microfibrils and structural properties, J. Mol. Biol 258 (1) (1996) 104–116. [DOI] [PubMed] [Google Scholar]
- [3].Visconti RP, Barth JL, Keeley FW, Little CD, Codistribution analysis of elastin and related fibrillar proteins in early vertebrate development, Matrix Biol 22 (2) (2003) 109–121. [DOI] [PubMed] [Google Scholar]
- [4].Shi Y, Tu Y, De Maria A, Mecham RP, Bassnett S, Development, composition, and structural arrangements of the ciliary zonule of the mouse, Invest. Ophthalmol. Vis. Sci 54 (4) (2013) 2504–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Corson GM, Charbonneau NL, Keene DR, Sakai LY, Differential expression of fibrillin-3 adds to microfibril variety in human and avian, but not rodent, connective tissues, Genomics 83 (3) (2004) 461–472. [DOI] [PubMed] [Google Scholar]
- [6].Sakai LY, Keene DR, Engvall E, Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils, J. Cell Biol 103 (6 Pt 1) (1986) 2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang H, Apfelroth SD, Hu W, Davis EC, Sanguineti C, Bonadio J, Mecham RP, Ramirez F, Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices, J. Cell Biol 124 (5) (1994) 855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cain SA, Morgan A, Sherratt MJ, Ball SG, Shuttleworth CA, Kielty CM, Proteomic analysis of fibrillin-rich microfibrils, Proteomics 6 (1) (2006) 111–122. [DOI] [PubMed] [Google Scholar]
- [9].De Maria A, Wilmarth PA, David LL, Bassnett S, Proteomic analysis of the bovine and human ciliary zonule, Invest. Ophthalmol. Vis. Sci 58 (1) (2017) 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Charbonneau NL, Dzamba BJ, Ono RN, Keene DR, Corson GM, Reinhardt DP, Sakai LY, Fibrillins can co-assemble in fibrils, but fibrillin fibril composition displays cell-specific differences, J. Biol. Chem 278 (4) (2003) 2740–2749. [DOI] [PubMed] [Google Scholar]
- [11].Mariencheck MC, Davis EC, Zhang H, Ramirez F, Rosenbloom J, Gibson MA, Parks WC, Mecham RP, Fibrillin-1 and fibrillin-2 show temporal and tissue-specific regulation of expression in developing elastic tissues, Connect. Tissue Res 31 (2) (1995) 87–97. [DOI] [PubMed] [Google Scholar]
- [12].Sabatier L, Miosge N, Hubmacher D, Lin G, Davis EC, Reinhardt DP, Fibrillin-3 expression in human development, Matrix Biol 30 (1) (2011) 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang H, Hu W, Ramirez F, Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils, J. Cell Biol 129 (4) (1995) 1165–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ramirez F, Rifkin DB, Extracellular microfibrils: contextual platforms for TGFbeta and BMP signaling, Curr. Opin. Cell Biol 21 (5) (2009) 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB, Latent TGF-beta-binding proteins, Matrix Biol 47 (2015) 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ramirez F, Caescu C, Wondimu E, Galatioto J, Marfan syndrome; a connective tissue disease at the crossroads of mechanotransduction, TGFbeta signaling and cell stemness, Matrix Biol 71–72 (2017) 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ramirez F, Dietz HC, Marfan syndrome: from molecular pathogenesis to clinical treatment, Curr. Opin. Genet. Dev 17 (3) (2007) 252–258. [DOI] [PubMed] [Google Scholar]
- [18].Faivre L, Gorlin RJ, Wirtz MK, Godfrey M, Dagoneau N, Samples JR, Le Merrer M, Collod-Beroud G, Boileau C, Munnich A, et al. , In frame fibrillin-1 gene deletion in autosomal dominant Weill–Marchesani syndrome, J. Med. Genet 40 (1) (2003) 34–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sengle G, Tsutsui K, Keene DR, Tufa SF, Carlson EJ, Charbonneau NL, Ono RN, Sasaki T, Wirtz MK, Samples JR, et al. , Microenvironmental regulation by fibrillin-1, PLoS Genet 8 (1) (2012), e1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Le Goff C, Mahaut C, Wang LW, Allali S, Abhyankar A, Jensen S, Zylberberg L, Collod-Beroud G, Bonnet D, Alanay Y, et al. , Mutations in the TGFbeta binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias, Am. J. Hum. Genet 89 (1) (2011) 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hubmacher D, Apte SS, Genetic and functional linkage between ADAMTS superfamily proteins and fibrillin-1: a novel mechanism influencing microfibril assembly and function, Cell. Mol. Life Sci 68 (19) (2011) 3137–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Le Goff C, Cormier-Daire V, Genetic and molecular aspects of acromelic dysplasia, Pediatr. Endocrinol. Rev 6 (3) (2009) 418–423. [PubMed] [Google Scholar]
- [23].Faivre L, Dollfus H, Lyonnet S, Alembik Y, Megarbane A, Samples J, Gorlin RJ, Alswaid A, Feingold J, Le Merrer M, et al. , Clinical homogeneity and genetic heterogeneity in Weill–Marchesani syndrome, Am. J. Med. Genet 123A (2) (2003) 204–207. [DOI] [PubMed] [Google Scholar]
- [24].Dagoneau N, Benoist-Lasselin C, Huber C, Faivre L, Megarbane A, Alswaid A, Dollfus H, Alembik Y, Munnich A, Legeai-Mallet L, et al. , ADAMTS10 mutations in autosomal recessive Weill–Marchesani syndrome, Am. J. Hum. Genet 75 (5) (2004) 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dubail J, Apte SS, Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics, Matrix Biol 44–46 (2015) 24–37. [DOI] [PubMed] [Google Scholar]
- [26].Mead TJ, Apte SS, ADAMTS proteins in human disorders, Matrix Biol 71–72 (2018) 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ahonen SJ, Kaukonen M, Nussdorfer FD, Harman CD, Komaromy AM, Lohi H, A novel missense mutation in ADAMTS10 in Norwegian elkhound primary glaucoma, PLoS One 9 (11) (2014), e111941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kuchtey J, Olson LM, Rinkoski T, Mackay EO, Iverson TM, Gelatt KN, Haines JL, Kuchtey RW, Mapping of the disease locus and identification of ADAMTS10 as a candidate gene in a canine model of primary open angle glaucoma, PLoS Genet 7 (2) (2011), e1001306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Morales J, Al-Sharif L, Khalil DS, Shinwari JM, Bavi P, Al-Mahrouqi RA, Al-Rajhi A, Alkuraya FS, Meyer BF, Al Tassan N, Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature, Am. J. Hum. Genet 85 (5) (2009) 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hubmacher D, Apte SS, ADAMTS proteins as modulators of microfibril formation and function, Matrix Biol 47 (2015) 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kutz WE, Wang LW, Bader HL, Majors AK, Iwata K, Traboulsi EI, Sakai LY, Keene DR, Apte SS, ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts, J. Biol. Chem 286 (19) (2011) 17156–17167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hubmacher D, Schneider M, Berardinelli SJ, Takeuchi H, Willard B, Reinhardt DP, Haltiwanger RS, Apte SS, Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease, Sci. Rep 7 (2017), 41871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cain SA, Mularczyk EJ, Singh M, Massam-Wu T, Kielty CM, ADAMTS-10 and -6 differentially regulate cell-cell junctions and focal adhesions, Sci. Rep 6 (2016), 35956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Somerville RP, Jungers KA, Apte SS, ADAMTS10: discovery and characterization of a novel, widely expressed metalloprotease and its proteolytic activation, J. Biol. Chem 279 (2004) 51208–51217. [DOI] [PubMed] [Google Scholar]
- [35].Beene LC, Wang LW, Hubmacher D, Keene DR, Reinhardt DP, Annis DS, Mosher DF, Mecham RP, Traboulsi EI, Apte SS, Nonselective assembly of fibrillin 1 and fibrillin 2 in the rodent ocular zonule and in cultured cells: implications for marfan syndrome, Invest. Ophthalmol. Vis. Sci 54 (13) (2013) 8337–8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ito M, Yoshioka M, Regression of the hyaloid vessels and pupillary membrane of the mouse, Anat. Embryol. (Berl.) 200 (4) (1999) 403–411. [DOI] [PubMed] [Google Scholar]
- [37].Quondamatteo F, Reinhardt DP, Charbonneau NL, Pophal G, Sakai LY, Herken R, Fibrillin-1 and fibrillin-2 in human embryonic and early fetal development, Matrix Biol 21 (8) (2002) 637–646. [DOI] [PubMed] [Google Scholar]
- [38].Hubmacher D, Reinhardt DP, Plesec T, Schenke-Layland K, Apte SS, Human eye development is characterized by coordinated expression of fibrillin isoforms, Invest. Ophthalmol. Vis. Sci 55 (12) (2014) 7934–7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shi Y, Tu Y, Mecham RP, Bassnett S, Ocular phenotype of Fbn2-null mice, Invest. Ophthalmol. Vis. Sci 54 (12) (2013) 7163–7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hubmacher D, Wang LW, Mecham RP, Reinhardt DP, Apte SS, Adamtsl2 deletion results in bronchial fibrillin microfibril accumulation and bronchial epithelial dysplasia—a novel mouse model providing insights into geleophysic dysplasia, Dis. Model. Mech 8 (5) (2015) 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bader HL, Wang LW, Ho JC, Tran T, Holden P, Fitzgerald J, Atit RP, Reinhardt DP, Apte SS, A disintegrin-like and metalloprotease domain containing thrombospondin type 1 motif-like 5 (ADAMTSL5) is a novel fibrillin-1-, fibrillin-2-, and heparin-binding member of the ADAMTS superfamily containing a netrin-like module, Matrix Biol 31 (7–8) (2012) 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gabriel LA, Wang LW, Bader H, Ho JC, Majors AK, Hollyfield JG, Traboulsi EI, Apte SS, ADAMTSL4, a secreted glycoprotein widely distributed in the eye, binds fibrillin-1 microfibrils and accelerates microfibril biogenesis, Invest. Ophthalmol. Vis. Sci 53 (1) (2012) 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mularczyk EJ, Singh M, Godwin ARF, Galli F, Humphreys N, Adamson AD, Mironov A, Cain SA, Sengle G, Boot-Handford RP, et al. , ADAMTS10-mediated tissue disruption in Weill–Marchesani syndrome, Hum. Mol. Genet (2018) 10.1093/hmg/ddy276 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- [44].Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, et al. , Global genetic analysis in mice unveils central role for cilia in congenital heart disease, Nature 521 (7553) (2015) 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Prins BP, Mead TJ, Brody JA, Sveinbjornsson G, Ntalla I, Bihlmeyer NA, van den Berg M, Bork-Jensen J, Cappellani S, Van Duijvenboden S, et al. , Exome-chip meta-analysis identifies novel loci associated with cardiac conduction, including ADAMTS6, Genome Biol 19 (1) (2018) 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Enomoto H, Nelson C, Somerville RPT, Mielke K, Dixon L, Powell K, Apte SS, Cooperation of two ADAMTS metalloproteases in closure of the mouse palate identifies a requirement for versican proteolysis in regulating palatal mesenchyme proliferation, Development 137 (2010) 4029–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].McCulloch DR, Nelson CM, Dixon LJ, Silver DL, Wylie JD, Lindner V, Sasaki T, Cooley MA, Argraves WS, Apte SS, ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression, Dev. Cell 17 (5) (2009) 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mead TJ, Du Y, Nelson CM, Gueye NA, Drazba J, Dancevic CM, Vankemmelbeke M, Buttle DJ, Apte SS, ADAMTS9-regulated pericellular matrix dynamics governs focal adhesion-dependent smooth muscle differentiation, Cell Rep 23 (2) (2018) 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mead TJ, McCulloch DR, Ho JC, Du Y, Adams SM, Birk DE, Apte SS, The metalloproteinase-proteoglycans ADAMTS7 and ADAMTS12 provide an innate, tendon-specific protective mechanism against heterotopic ossification, JCI Insight 3 (7) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nandadasa S, Nelson CM, Apte SS, ADAMTS9-mediated extracellular matrix dynamics regulates umbilical cord vascular smooth muscle differentiation and rotation, Cell Rep 11 (10) (2015) 1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Apte SS, Parks WC, Metalloproteinases: a parade of functions in matrix biology and an outlook for the future, Matrix Biol 44–46 (2015) 1–6. [DOI] [PubMed] [Google Scholar]
- [52].Bjursell M, Admyre T, Goransson M, Marley AE, Smith DM, Oscarsson J, Bohlooly YM, Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet, Am. J. Physiol. Endocrinol. Metab 300 (1) (2011) E211–E220. [DOI] [PubMed] [Google Scholar]
- [53].Dubail J, Vasudevan D, Wang LW, Earp SE, Junkins MW, Haltiwanger RS, Apte SS, Impaired ADAMTS9 secretion: a potential mechanism for eye defects in Peters plus syndrome, Sci. Rep 6 (2016) (In Press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].McCulloch DR, Goff CL, Bhatt S, Dixon LJ, Sandy JD, Apte SS, Adamts5, the gene encoding a proteoglycan-degrading metalloprotease, is expressed by specific cell lineages during mouse embryonic development and in adult tissues, Gene Expr. Patterns 9 (5) (2009) 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lin G, Tiedemann K, Vollbrandt T, Peters H, Batge B, Brinckmann J, Reinhardt DP, Homo- and heterotypic fibrillin-1 and -2 interactions constitute the basis for the assembly of microfibrils, J. Biol. Chem 277 (52) (2002) 50795–50804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.