

SUMMARY
The aging liver is affected by several disorders, including steatosis, that can lead to a decline of liver functions. Here, we present evidence that the cdk4-C/EBPα-p300 axis is a critical regulator of age-associated disorders, including steatosis. We found that patients with non-alcoholic fatty liver disease (NAFLD) have increased levels of cdk4 and that cdk4-resistant C/EBPα-S193A mice do not develop hepatic steatosis with advancing age. Underlying mechanisms include a block in C/EBPα activation and subsequent failure in activation of enzymes involved in the development of NAFLD. Inhibition of cdk4 in aged wild-type (WT) mice by a specific cdk4 inhibitor, PD-0332991, reduces C/EBPα-p300 complexes and eliminates hepatic steatosis. Moreover, the inhibition of cdk4 in aged mice reverses many age-related disorders. Mechanisms of correction include elimination of cellular senescence and alterations in the chromatin structure of hepatocytes. Thus, the inhibition of cdk4 might be considered as a therapeutic approach to correct age-associated liver disorders.
In Brief
Nguyen et al. show that nuclear elevation of cdk4 leads to age-associated disorders, such as hepatic steatosis, and to age-dependent decline of liver functions and morphology. Elevation of cdk4 changes multiple molecular aspects of liver biology. Inhibition of cdk4 in old mice eliminates hepatic steatosis and corrects age-associated liver disorders.
Graphical Abstract
INTRODUCTION
Aging is the most common cause for the progression of non-alcoholic fatty liver disease (NAFLD). The earliest stage of NAFLD, hepatic steatosis, is characterized by an accumulation of triglycerides (TGs) in the cytoplasm of hepatocytes and is seen in mice from 12 months of age. In patients with NAFLD, hepatic steatosis progresses to non-alcoholic steatohepatitis (NASH), which results in an increased risk for development of cirrhosis and hepatocellular carcinoma (Cohen et al., 2011; Hebbard and George, 2011; Schmucker, 2005; Timchenko, 2011; Engin, 2017). Several recent reports have described the essential role of the hedgehog pathway and gut microbiome in the development of NAFLD (Machado and Diehl, 2016; Verdelho Machado and Diehl, 2016; Boursier and Diehl, 2016; Boursier et al., 2016). Recent papers have reported that the microsomal triglyceride transfer protein is involved in development of NAFLD (Newberry et al., 2015, 2017). Although mechanisms of developing NAFLD under high-fat diet conditions are well investigated, little is known regarding the key events that determine the development of age-associated NAFLD. A number of papers uncovered that hepatocytes telomeres were shorter in NAFLD patients than in controls and that a marker of senescence, p21, was upregulated in old patients with NAFLD (Aravinthan et al., 2013, 2014; Valenti and Dongiovanni, 2014). The contribution of telomere dysfunction to the age-associated steatosis was further confirmed (Laish et al., 2016). A recent paper showed that cellular senescence drives age-associated hepatic steatosis (Ogrodnik et al., 2017).
C/EBPα is a strong inhibitor of liver proliferation and a tumor suppressor protein that is commonly eliminated in animal models of liver cancer and in patients with hepatocellular carcinoma (HCC) and hepatoblastoma (HBL) (Timchenko, 2009; Wang et al., 2006, 2010; Jiang et al., 2013; Valanejad et al., 2017). Ser193 is a key amino acid that regulates biological functions of C/EBPα (Jin et al., 2010, 2015; Wang et al., 2010). Ser193 is phosphorylated by cdk4, which is activated by type D cyclins (Wang et al., 2006, 2010). Phosphorylation of C/EBPα at Ser193 blocks the interactions of C/EBPα with chromatin remodeling proteins HDAC1 and p300. C/EBPα-p300 and C/EBPα-HDAC1 complexes are able to activate (complexes with p300) or repress (complexes with HDAC1) different C/EBPα-dependent genes (Jin et al., 2010,2015; Wang et al., 2010). Cdk4-dependent phosphorylation of C/EBPα at Ser193 is involved in the development of NAFLD under high-fat diet protocol (Jin et al., 2013, 2016). Phosphorylation of S193 also regulates stability of C/EBPα. Under conditions of liver cancer, the oncogene gankyrin (Gank) triggers degradation of S193-ph C/EBPα; however, Gank cannot degrade C/EBPα if it is dephosphorylated at Ser193 (Wang et al., 2010). This regulation might also exist in fatty livers because Gank is elevated in advanced stages of NAFLD (Jin et al., 2016). Cyclin-dependent kinase 4 (cdk4) was initially discovered as a strong driver of cell proliferation that phosphorylates retinoblastoma protein, Rb, at Ser780 (Baker and Reddy, 2012). However, further investigations revealed that cyclin D3 is elevated in several differentiation settings and activates cdk4 during aging (Timchenko, 2009). Because cdk4 is involved in the promotion of HCC, inhibition of cdk4 by small-molecule drugs is under investigation and development. It has been shown that a specific drug, PD-0332991, functions as a potent inhibitor of cdk4 (Flaherty et al., 2012; Endicott et al., 1999). The inhibition of cdk4 by PD-0332991 has been shown to block the development of hepatic steatosis under conditions of high fat diet (HFD) (Jin et al., 2016).
In this paper, we examined cdk4-dependent pathways of NAFLD in patients of different ages with fatty liver disease and in biologically relevant animal models of aging. We found that the elevation of cdk4 in patients is age dependent and is observed mainly in nuclei of older patients, which correlates with the severity of NAFLD. Moreover, we found that cdk4-resistant C/EBPα-S193A mice do not develop hepatic steatosis with age and that inhibition of cdk4 in old wild-type (WT) mice disrupts C/EBPα-p300 pathway and eliminates hepatic steatosis. We surprisingly found that, in addition to elimination of steatosis, the inhibition of cdk4 in aged mice also corrects pathways that cause many age-related disorders in the liver.
RESULTS
Elevation of cdk4 in Patients with NAFLD Correlates with the Severity of the Disease
Our previous studies found that cdk4 is activated by cyclin D3 in the liver under conditions of high-fat-diet-mediated NAFLD in mouse models (Jin et al., 2016). To further examine whether the elevation of cdk4 is associated with the severity of NAFLD and with age, we have obtained paraffin-embedded liver biopsy samples from control patients (healthy, 4 samples: two below and two above 60), from NAFLD patients with hepatic steatosis (12 patients of different age), and from NASH patients (17 patients of varying age). The biopsies were grouped into samples from patients under 60 years old and above 60 years old. These biopsies were immunostained for cdk4 expression (Figures 1A and 1B; images of cdk4 immunostaining for additional patients are shown in Figure S1), and the percent of cdk4-positive hepatocytes was correlated with age. In control, non-steatosis samples, we detected elevation of cdk4-positive hepatocytes in patients above 60 (Figure 1C). We also found that hepatic cdk4 levels are increased in patients under 60 with steatosis and with NASH (Figure 1D). Elevation of hepatic cdk4 in patients above 60 with steatosis and NASH is much higher than in patients under 60 (Figures 1B and 1D). We found that steatosis and NASH in patients above 60 are characterized by much stronger intensity of cdk4 staining in nucleus (Figure 1B, red arrows). The nuclear accumulation of cdk4 in patients with severe NAFLD is in agreement with previously found nuclear accumulation of cdk4 in animal models with severe NAFLD (Jin et al., 2016). In summary, we found that the elevation of cdk4 in patients with NAFLD correlates with age and with the severity of the disease and that NAFLD patients above 60 have much stronger nuclear accumulation of cdk4. Based on these results, we hypothesized that the elevation of nuclear cdk4 results in the development of age-associated NAFLD and that localization of elevated cdk4 and age of patients determine the severity of NAFLD (Figure 1E). To test this hypothesis, we performed experiments in cdk4-resistant C/EBPα-S193A mice.
Figure 1. Hepatic Levels of cdk4 Are Increased in Nuclei of Patients with NAFLD.
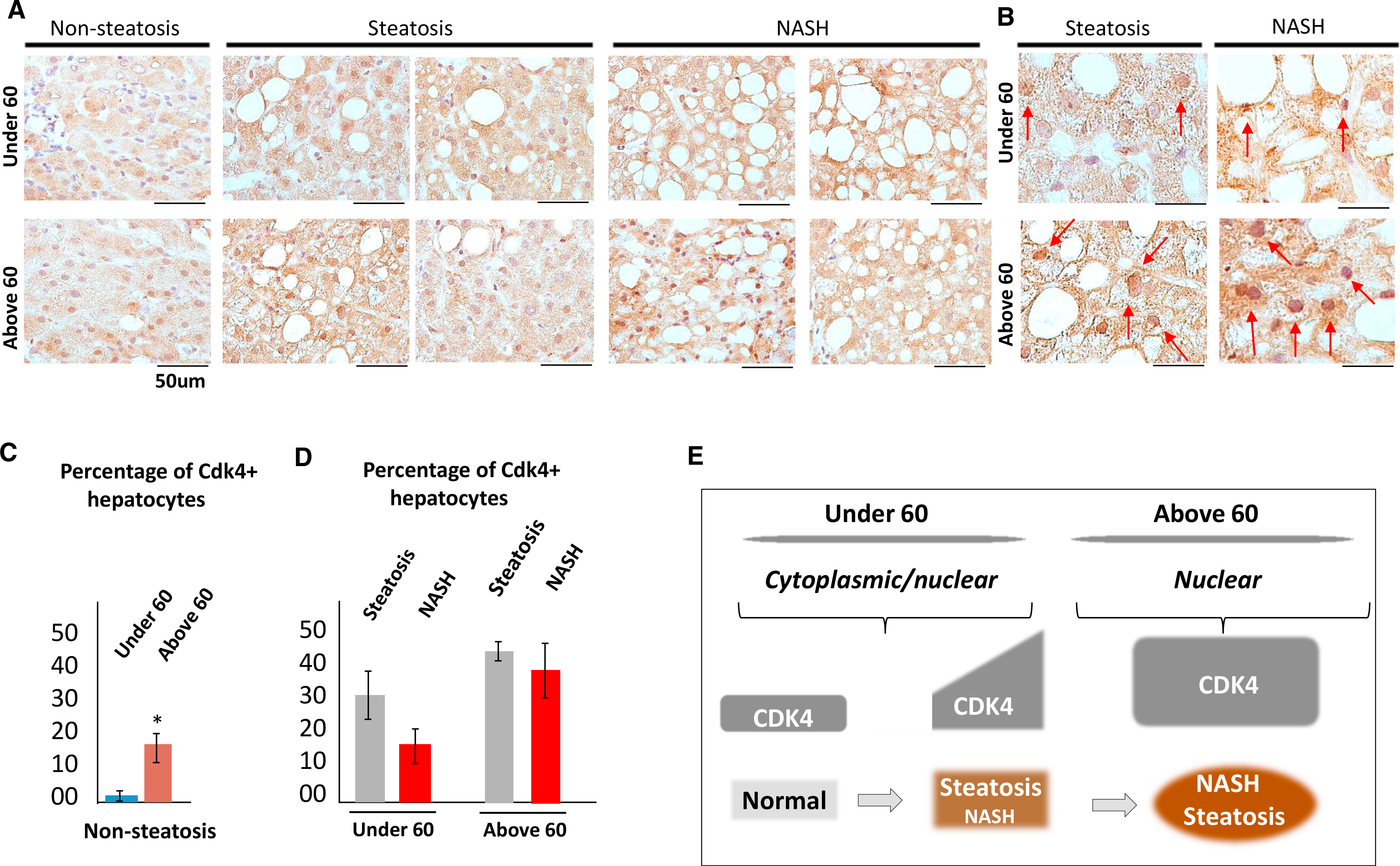
(A) Immunostaining of the human liver biopsies from patients with hepatic steatosis and NASH with antibodies to cdk4. Data for 2 controls, 4 patients with hepatic simple steatosis, and 4 patients with NASH are shown. Cdk4 staining of additional NAFLD patients is shown in Figure S1. The scale bars represent 50 μm.
(B) Images of cdk4 staining under high magnification. Hepatocytes with strong nuclear cdk4 staining are shown by red arrows. Scale bars represent 50 μm.
(C) Calculations of cdk4-positive hepatocytes in control patients under 60 and above 60 years old. *p < 0.05.
(D) Bar graphs show % of cdk4-positive hepatocytes in livers of patients with hepatic steatosis and NASH. Patients were grouped by age under 60 and above 60 years.
(E) A diagram showing the main hypothesis of this paper, which is based on data with human biopsies.
CDK4-Resistant C/EBPα-S193A Mice Do Not Develop Hepatic Steatosis with Age
The main target of cdk4 in the development of fatty liver diseases is the C/EBPα-p300 complex, which activates expression of genes of fat metabolism and glucose metabolism (Jin et al., 2013, 2016). This complex is elevated in livers of old mice due to increased phosphorylation of C/EBPα at Ser193, which is mediated by cdk4 (Jin et al., 2009; Guillory et al., 2018). To examine the consequences of an elevation of cdk4 in older models, we examined C/EBPα-S193A knockin mice (S193A), which are resistant to cdk4 activation (Jin et al., 2015). WT and S193A mice were aged to 22 months, and the development of hepatic steatosis was investigated. Oil red O staining revealed that all examined 22-month-old WT mice (6 mice) develop severe liver steatosis; and all examined young and old S193A mice (6 mice) do not develop hepatic steatosis (Figure 2A). Examination of levels of enzymes of fatty liver DGAT1, DGAT2, ACC, SCD1, and SREBP1 revealed that these proteins are elevated in old WT mice; however, they are not increased in livers of 22-month-old S193A (Figures 2B and 2C). Thus, these studies showed that the age-associated development of hepatic steatosis is inhibited in cdk4-resistant C/EBPα-S193A mice.
Figure 2. Cdk4-Resistant C/EBPα-S193A Mice Do Not Develop Hepatic Steatosis with Age.
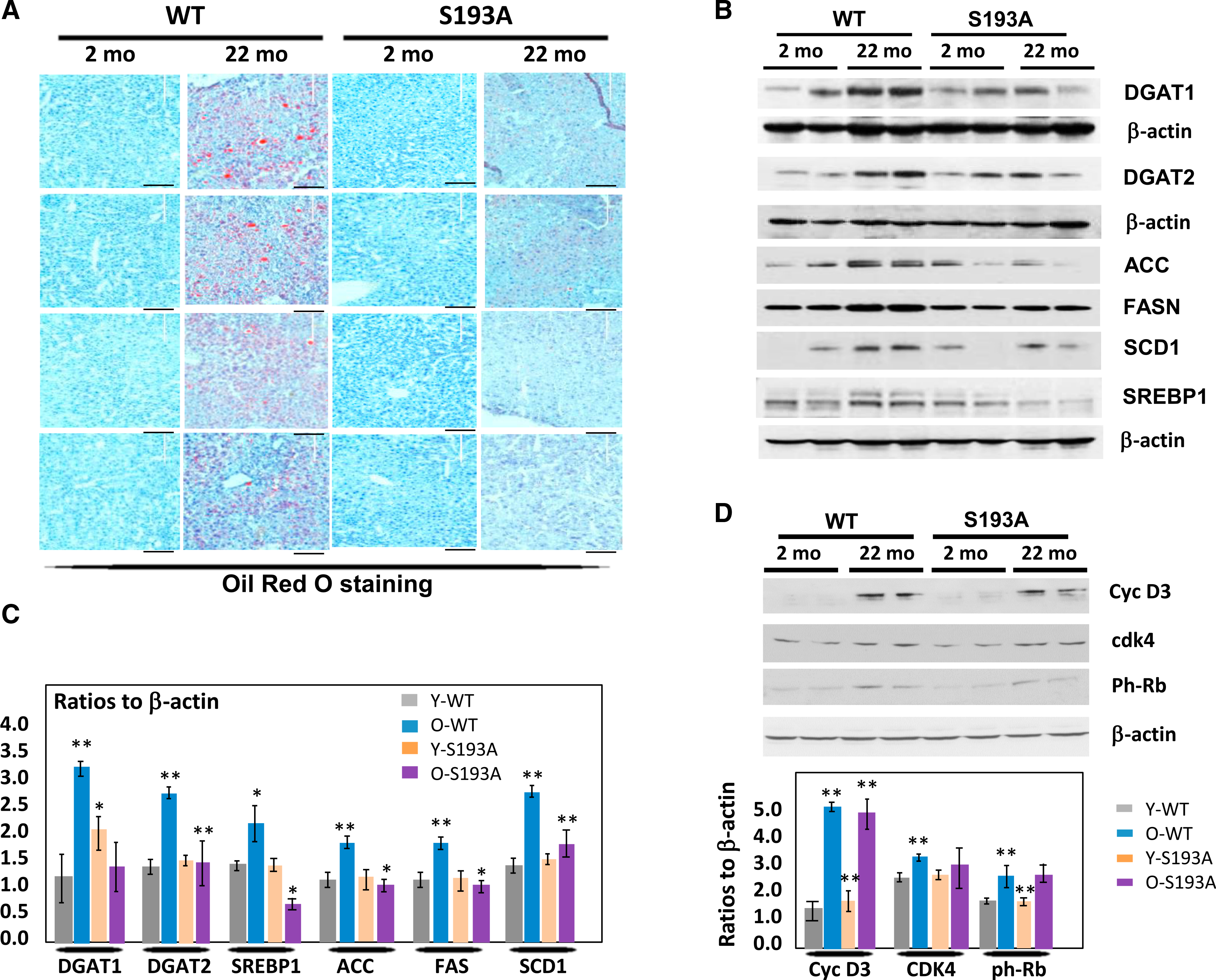
(A) Oil red O staining of livers of young and old WT and C/EBPα-S193A mice. The scale bars represent 200 μm.
(B) Levels of enzymes of fatty liver development DGAT1, DGAT2, ACC, SCD1, SREBP1, and FAS in livers of WT and S193A mice.
(C) Levels of proteins on (B) were calculated as ratios to β-actin.
(D) cycD3-cdk4 pathway is activated by age in both WT and S193A mice. Western blotting was performed with Abs shown on the right. Bar graphs show levels of proteins as ratios to β-actin.
For (C) and (D), *p < 0.05, **p < 0.01.
A Failure of C/EBPα-p300 Complex Activation by cdk4 Results in the Inhibition of Age-Dependent Hepatic Steatosis in C/EBPα-S193A Mice
Because the cdk4 controls C/EBPα-p300 complexes under HFD conditions (Jin et al., 2016), we next examined whether this pathway might be involved in the age-associated development of hepatic steatosis in WT mice and whether this pathway might be blocked in cdk4-resistant C/EBPα-S193A mice. Prior to examining the C/EBPα-p300 pathway, we asked whether cdk4 is activated with age to the same degree in WT and S193A mice. Because cdk4 is activated in livers of old mice by elevation of cyclin D3 (Wang et al., 2006; Jin et al., 2016), we examined expression of these proteins in livers of WT and S193A mice. Figure 2D shows that cyclin D3 is identically increased in both groups and that cdk4 is also elevated but to a lower degree. Phosphorylation of a known substrate of cdk4, Rb, is also increased to the same degree in old WT and S193A mice (Figure 2D). Thus, these studies revealed that cdk4 and cyclin D3 are identically elevated with age in WT and in S193A mice, suggesting that the subsequent inhibition of hepatic steatosis in S193A mice is mediated by pathways that are downstream and dependent on C/EBPα.
To test this suggestion, we examined the phosphorylation status of C/EBPα. Western blotting with antibodies that specifically recognize ph-S193 isoform of C/EBPα revealed that phosphorylation of C/EBPα is increased in WT mice with age; however, C/EBPα is not phosphorylated in S193A mice, regardless of age (Figure 3A). We next examined C/EBPα-p300 complexes by co-immunoprecipitation (coIP) and found that these complexes are elevated in WT 22-month-old mice compared to young 2-month-old mice. In S193A mice, these complexes are much weaker or are not detected at any age (Figures 3A and 3B). These data indicate that cdk4-resistant S193A mice failed to form C/EBPα-p300 complexes with age; and WT mice have increased C/EBPα-p300 complexes in 22-month-old mice compared to 2-month-old mice. It is interesting to note that total levels of p300 and C/EBPα are also elevated with age in WT mice, but not in S193A mice. We suggest that the lack of activation of C/EBPα with age in S193A mice is due to the fact that the C/EBPα promoter contains a C/EBP binding site and it is the subject of auto-regulation by C/EBPα-p300 (Jin et al., 2015). These complexes are reduced in S193A mice (Figure 2A), and this reduction is likely to cause low levels of C/EBPα in the liver of S193A mice. Because a recent report described that cellular senescence is involved in the development of age-associated steatosis (Ogrodnik et al., 2017), we next asked whether cellular senescence might also be inhibited in S193A mice. To test this, we examined levels of the marker of senescence, p21. Western blotting showed an elevation of p21 in WT aged mice. S193A mice also exhibited elevation of p21 but to a lesser degree (Figures 3A and 3B). To confirm this observation, we performed immunostaining and qRT-PCR experiments. Figure 3C illustrates that protein levels of p21 detected by immunostaining are elevated in both young and old WT and S193A mice, although the degree of elevation is slightly lower in old S193A mice. Surprisingly, we detected much higher levels of p21 mRNA in young S193Amice; however, old S193A and WT mice have almost similar levels of p21 mRNA. In the course of these studies, we noted that levels of p21 mRNA do not always correlate with levels of p21 protein, suggesting additional mechanisms of regulation of the p21 protein. Taken together, we conclude that the elevation of p21 protein is lower in S193A mice, suggesting a partial inhibition of cellular senescence.
Figure 3. The Failure of S193A Miceto Increase C/EBPα-p300 Complexes with Age Leads to a Failure to Increase Levels of EnzymesthatAre Involved in Fatty Liver Development.
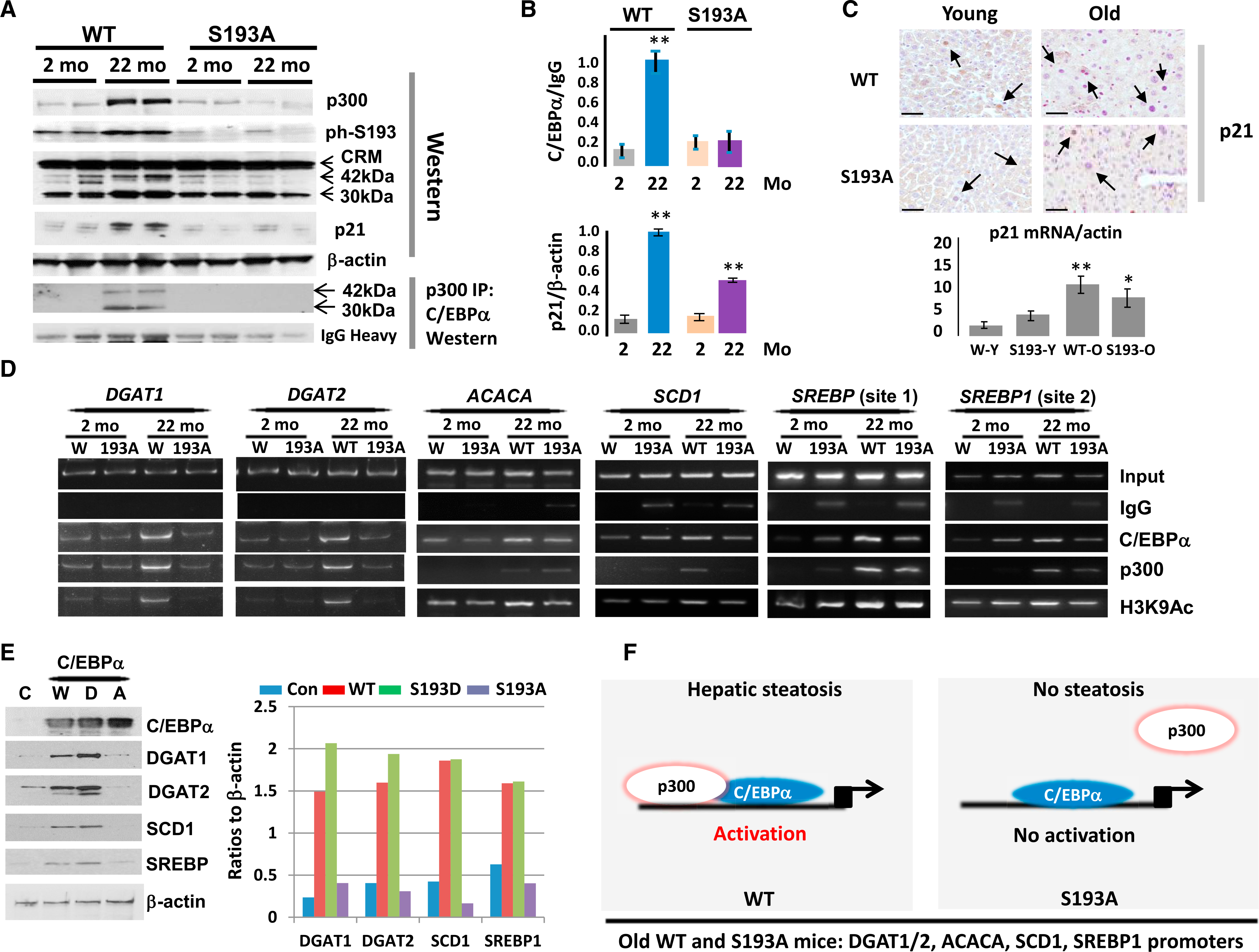
(A) (Upper) Western blotting of p300, phS193-C/EBPα, C/EBPα, and p21 in young (2-month-old) and old (22-month-old) WT and S193A mice. (Lower) Co-IP of p300 and western blot of C/EBPα in young and old WT and S193A mice is shown.
(B) (Upper) C/EBPα-p300 complexes were calculated as ratios of C/EBPα in p300 IPs to signals of immunoglobulin G (IgG). (Bottom) Levels of p21 were calculated as ratios to β-actin.
(C) Examination of p21 expression in livers of young and old S193A mice by immunostaining (upper images; scale bars 50 μm) and by qRT-PCR (bottom image).
(D) ChIP assay of the promoters of DGAT1, DGAT2, ACACA, SCD1, and SREBP1 genes. C/EBPα, p300, and H3K9Ac were immunoprecipitated from chromatin solutions of young and old WT and S193A mice. IgGs are negative controls.
(E) C/EBPα-S193Afailsto activate expression ofenzymes ofTG synthesis and lipogenic genes. Hepa-1c17 cells were transfected with WT (W), C/EBPα-S193D (D), and C/EBPα-S193A (A) mutants, and expression of DGAT1, DGAT2, SCD1, and SREBP1 proteins was examined by western blotting (left part). Bar graphs show levels of proteins as ratios to β-actin control.
(F) A schematic diagram showing activation of genes of fatty liver development in aged WT mice and the alterations of this process in aged S193A mice.
For (B) and (C), *p < 0.05, **p < 0.01.
We next examined the consequences of an age-associated accumulation of C/EBPα-p300 complexes in WT mice and the failure of S193A mice to increase these complexes. As shown in Figure 2, DGAT1, DGAT2, ACC, SCD1, and SREBP1 are increased in WT mice with age, but they are not elevated in old S193A mice. We have previously shown that the promoters of DGAT1 and DGAT2 are activated by C/EBPα-p300 under conditions of high-fat diet (Jin et al., 2013, 2016). Therefore, we searched the promoters of ACC, SCD1, and SREBP1 and found strong consensuses for C/EBPα in close proximity to the transcription start site (Figure S2). For SREBP1, we have found two binding sites for C/EBPα. Therefore, we next examined (1) whether C/EBPα-p300 complex binds to these promoters, (2) whether this binding is increased in old WT mice, and (3) whether the p300-C/EBPα complexes are detected on these promoters in S193A mice. For this goal, we have used a chromatin immunoprecipitation (ChIP) approach. To monitor the activity of the promoters, we examined acetylation of K9 at histone H3 (H3K9) because this modification increases activity of the promoters. The binding of C/EBPα-p300 complexes to all examined promoters is increased in livers of 22-month-old WT mice (Figure 3D). However, binding of C/EBPα-p300 complexes to these promoters is weak in S193A mice, and it is not increased with age. Examination of H3K9 acetylation revealed that these promoters are activated in old WT mice, but not in the S193A mice. Note that we observed slight differences in the occupation of different promoters by C/EBPα and p300. We hypothesize that this might be because p300 also interacts with another member of the C/EBP family, C/EBPβ (Jin et al., 2015), which might bind to the C/EBP sites within these promoters.
To further examine the role of cdk4-dependent phosphorylation of C/EBPα at Ser193 in the regulation of lipid-regulating genes containing C/EBPα site, we examined whether there is a difference in activation of these genes between WT C/EBPα and S193D and S193A mutants. WT, S193A, and S193D mutants were transfected in mouse Hepa-1c17 cells, and expression of the DGAT1/2, SCD1, and SREBP proteins was examined. Figure 3E demonstrates that WT C/EBPα activates expression of these genes and that phosphor-mimicking mutation S193D increases this activation. However, S193A mutation blocks the activity of C/EBPα to activate expression of these genes. Because S193A does not form complexes with p300 (Figure 3A), these results suggest that the lack of activation of lipogenic genes in S193A mice and in transient transfection studies is due to reduction of p300-C/EBPα complexes. Thus, our data demonstrate that the cdk4-dependent phosphorylation of C/EBPα in WT old mice and subsequent elevation of C/EBPα-p300 complexes lead to the binding to and activation of the DGAT1, DGAT2, ACC, SCD1, and SREBP1 promoters. However, in cdk4-resistant S193A mice, this pathway is inhibited and the promoters of DGAT1, DGAT2, ACC, SCD1, and SREBP1 genes are not activated. We suggest that this failure to activate the promoters re sults in the inhibition of age-associated hepatic steatosis in livers of S193A mice (Figure 3F).
Inhibition of cdk4 Activity by the Specific Inhibitor PD-0332991 Eliminates Hepatic Steatosis in Livers of Old Mice
To further determine the role of the cdk4-C/EBPα-p300 axis in age-associated hepatic steatosis, we have inhibited cdk4 by PD-0332991 in WT old mice using two doses of PD-0332991: 150 mg/kg and 450 mg/kg in chow. Consistent with previous reports, liver to body weight ratio is increased in aged WT mice (Figures 4A and 4B). Surprisingly, we found that the inhibition of cdk4 by a higher dose of PD-0332991 normalizes this ratio (Figure 4C), suggesting that the inhibition of cdk4 has an overall therapeutic effect on the mice. Examination of blood parameters revealed that inhibition of cdk4 also corrected cholesterol levels (Figure S3).
Figure 4. Inhibition of cdk4 by PD-0332991 Eliminates Hepatic Steatosis in WT Mice.
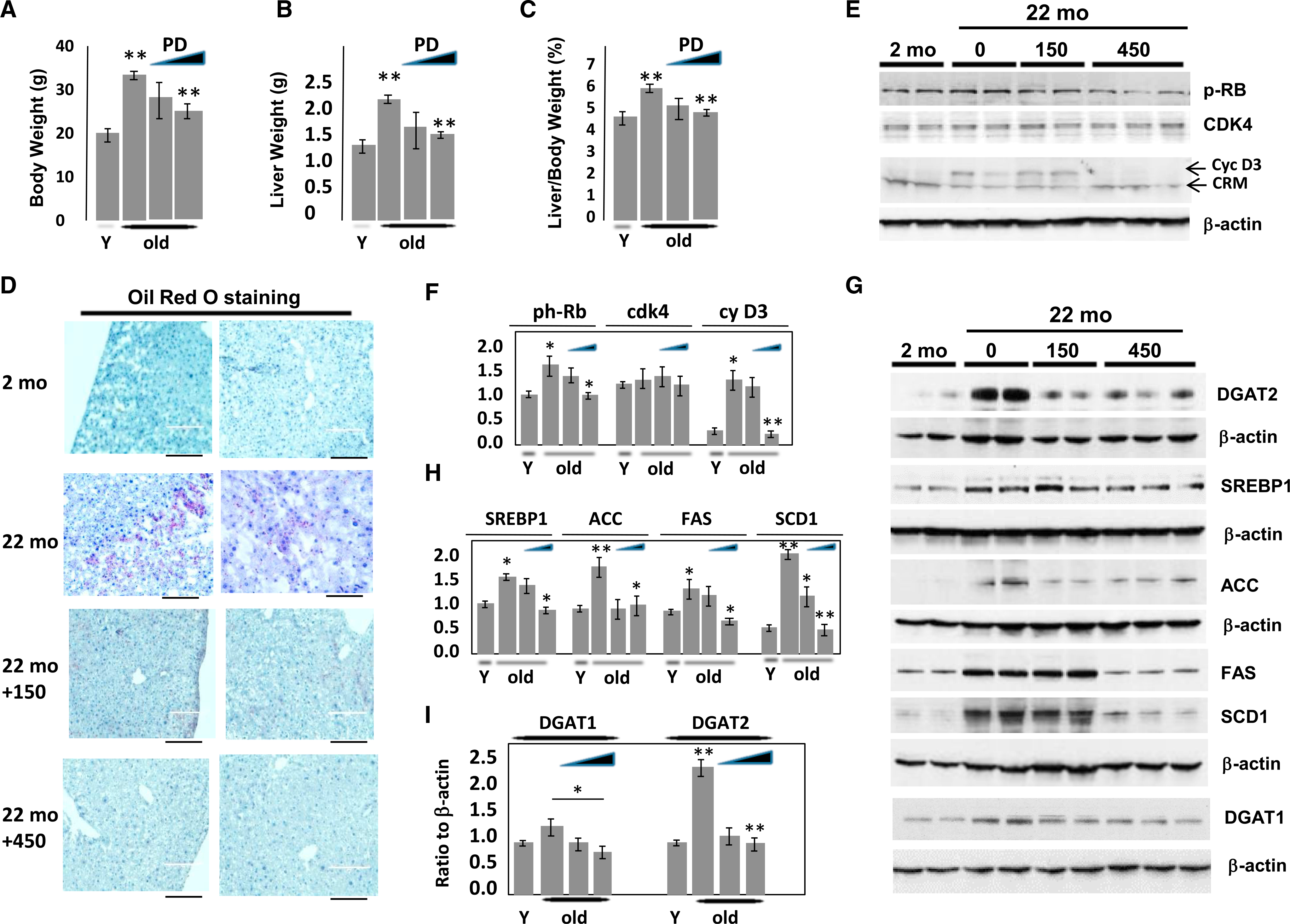
Mice were treated with PD-0332991 (150 mg/kg or 450 mg/kg with chow) for 2 weeks.
(A–C) Body weight (A), liver weight (B), and liver to body weight ratios (C) correspondingly in WT mice treated with PD-0332991. Statistically significant results are shown for comparison of untreated old mice and old mice treated with high dose of PD-0332991.
(D) A typical picture of oil red O staining of livers is shown. The scale bars represent 200 μm.
(E) Inhibition of cdk4 reduces its activity as measurement of phosphorylation of Rb substrate. Western blotting was performed with Abs to ph-S780-RB, CDK4, and cyclin D3.
(F) Levels of proteins on E were calculated as ratios to β-actin.
(G) Levels of enzymes of development of fatty liver DGAT1, DGAT2, ACC, SCD1, SREBP1, and FAS were determined by western blotting.
(H) Levels of SREBP1, ACC, FAS, and SCD1 were calculated as ratios to β-actin.
(I) Levels of DGAT1 and DGAT2 were calculated as ratios to β-actin.
In (A)-(C), (F), (H), and (I), *p < 0.05, **p < 0.01.
Oil red O staining revealed that hepatic steatosis is inhibited in old mice by the lower concentration and completely eliminated by the higher concentration of PD-0332991 (Figure 4D). We next examined whether treatments of old mice with PD-0332991 inhibits the activity of cdk4. Examination of cyclin D3 and cdk4 levels showed that cyclin D3 is elevated in livers of old mice and protein levels of cdk4 are elevated to a less degree or not changed (Figures 4E and 4F). Treatment of old mice with PD-0332991 does not change levels of cdk4 but reduces cyclin D3. Consistent with these data, phosphorylation of Rb is reduced in old mice by treatments with PD-0332991 (Figures 4E and 4F). These results and further studies of phosphorylation of C/EBPα (Figure 5) confirmed that PD-0332991 inhibits cdk4 activity. Next, we examined expression of genes that regulate the development of a fatty liver phenotype, including genes that are under the control of C/EBPα-p300 complexes. Figures 4G–4I show the results of these studies. In agreement with previous data, FAS, DGAT1, DGAT2, ACC, SCD1, and SREBP1 are elevated in old WT mice; however, all these proteins are reduced in livers of old mice treated with PD-0332991. Thus, we conclude that the inhibition of cdk4 in old WT mice eliminates hepatic steatosis.
Figure 5. Elimination of Hepatic Steatosis by Inhibition of cdl4 Involves Inhibition of C/EBPα-p300 Axis.
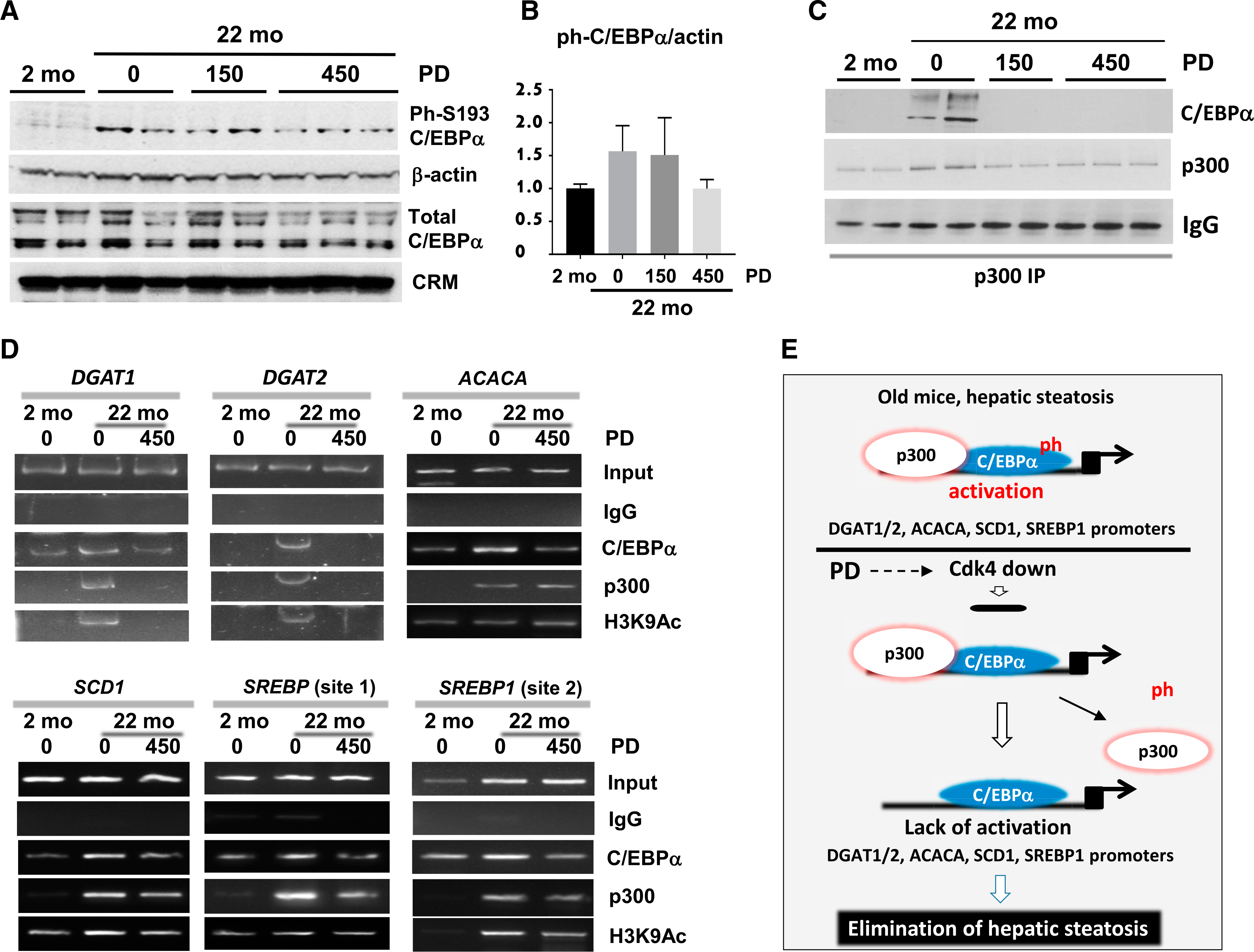
(A) Levels of ph-S193-C/EBPα and total C/EBPα were determined by western blotting.
(B) Bar graphs show a ratio of ph-Ser193-C/EBPα to β-actin.
(C) p300 was immunoprecipitated from nuclear extracts and probed with Abs to C/EBPα and to p300.
(D) ChIP assay was performed with primers covering C/EBPα site on the promoters of DGAT1, DGAT2, ACACA, SCD1, and SREBP genes. C/EBPα, p300, and H3K9Ac were immunoprecipitated from chromatin solutions and used for RT-PCR with primers covering C/EBPα binding sites. Input shows PCR products with 1/100 portion of input. *p < 0.05, **p < 0.01.
(E) A diagram showing the mechanisms by which inhibition of cdk4 eliminates hepatic steatosis through C/EBPα-p300 pathway.
Elimination of Hepatic Steatosis by PD-0332991 Strongly Correlates with the Inhibition of cdk4-C/EBPα-p300 Axis
We next examined the C/EBPα-p300 pathway in mice with inhibited cdk4. First, we examined the phosphorylation status of C/EBPα by western blotting with specific antibodies (Abs) and found that phosphorylation of C/EBPα at Ser193 is reduced by treatments with higher doses of PD-0332991 (Figures 5A and 5B). Examination of C/EBPα-p300 complexes indicated that the complexes are abundant in old WT mice but are reduced in mice treated with PD-0332991 (Figure 5C). To determine consequences of the elimination of C/EBPα-p300 complexes, we examined promoters of the enzymes that are under control of this complex. ChIP assay with mice treated by higher doses of PD-0332991 showed that inhibition of cdk4 leads to the reduction or removal of the C/EBPα-p300 complexes from all examined promoters (Figure 5D) and to partial repression of the promoters because the acetylation of histone H3 at K9 is reduced in mice with inhibited cdk4. Taken together, these studies revealed that the inhibition of cdk4 by PD-0332991 in livers of old mice eliminates hepatic steatosis and that this elimination correlates with the inhibition of cdk4-C/EBPα-p300 axis (Figure 5E).
Inhibition of cdk4 in Livers of Old Mice Causes Normalization of Multiple Pathways of Liver Biology
Examination of the cdk4-C/EBPα-p300 axis revealed that the inhibition of this pathway by PD-0332991 contributes to the elimination of hepatic steatosis. However, recent publications showed that cellular senescence (Ogrodnik et al., 2017) and E2F1-Rb pathways (Denechaud et al., 2016) are also involved in development of hepatic steatosis. Therefore, we asked whether the inhibition of cdk4 in livers of old mice might affect other pathways. Transcriptome profiling clearly demonstrated changes in gene expression in PD-0332991-treated old mice with a large number of upregulated and downregulated genes (Figure 6A). Examination of ontologies of the changes and pathways revealed that PD-0332991-mediated up- and downregulated pathways in livers of old mice belong to positive and negative regulators of fatty liver phenotype and to pathways of glucose homeostasis (Figure 6B). Interestingly, PD-0332991 also inhibited the E2F1-Rb pathway and markers of cellular senescence, such as CDKN1A (p21 protein) and ZNF385A (Hzf protein). These data show that, in addition to the inhibition of cdk4-C/EBPα-p300 axis, PD-0332991-mediated elimination of hepatic steatosis includes inhibition of other known pathways of hepatic steatosis: Rb-E2F1 and cellular senescence. Figure 6C summarizes these data and shows that the inhibition of cdk4 improves liver functions and corrects expression of genes involved in cellular senescence, pathways of proliferation, pathways regulating chromatin structure, steatosis, and C/EBPα and Rb-E2F1 pathways. Alterations of the key genes in these pathways were confirmed by qRT-PCR. Figure 6D shows an example of this verification for cell proliferation genes, markers of cellular senescence, and genes that regulate chromatin structure. In the course of examination of blood parameters, we have found that the mutation of Ser193 to Ala in C/EBPα or inhibition of cdk4 in old mice corrects cholesterol levels and partially corrects glucose levels (Figure S3). Although mechanisms of cdk4-dependent regulation of glucose in old mice are not known, levels of glucose in the cdk4-resistant S193A mice are reduced via inhibition of PEPCK and G6Pase (Jin et al., 2015), suggesting that this mechanism can also be involved in age-dependent and cdk4-dependent changes of glucose.
Figure 6. Inhibition of cdk4 in Livers of Old Mice Corrects Multiple Pathways, Including Cellular Senescence, Chromatin Structure, Steatosis, and Proliferation.
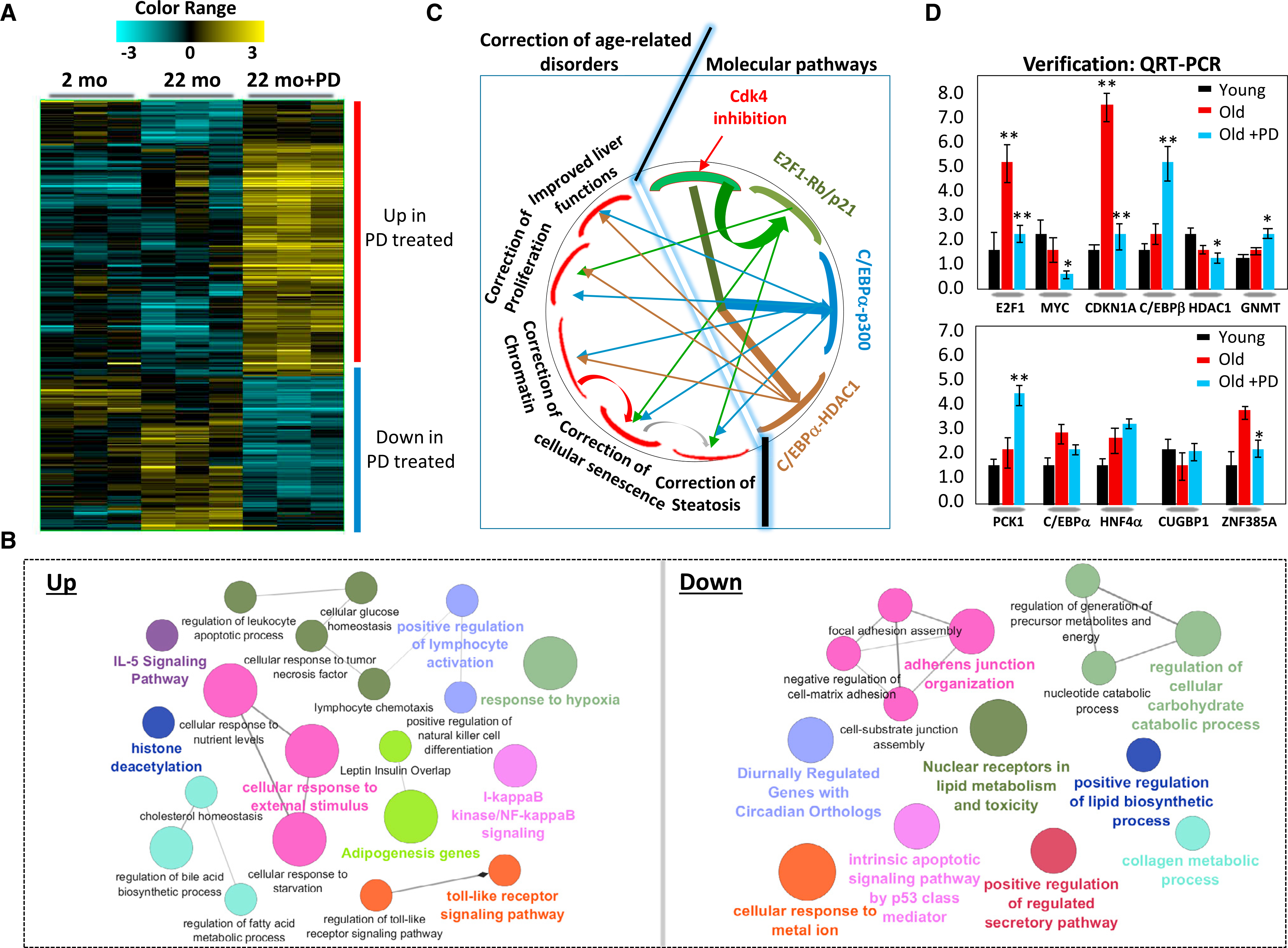
(A) Heatmap of RNA-seq assay showing clusters of up- and downregulated genes in livers of mice treated with PD-0332991.
(B) Network of up- and downregulated pathways in livers of old mice treated with PD-0332991.
(C) A diagram shows biological processes and molecular pathways that are corrected in livers of old mice by inhibition of cdk4.
(D) An example of confirmation of main changes in gene expression by qRT-PCR.
Inhibition of cdk4 in Aged Mice Eliminates Cellular Senescence Phenotype
To further examine overall healing effects of the inhibition of cdk4, we have measured additional age-related characteristics. One of the main characteristics of the aging phenotype and cellular senescence is the appearance of larger hepatocytes with increased heterochromatin regions (Jin et al., 2010; Kreiling et al., 2011). Given RNA sequencing (RNA-seq) results showing alterations in histone deacetylase HDAC1 and GMNT after inhibition of cdk4 (Figures 6B–6D), we asked whether inhibition of cdk4 might correct the chromatin structure of hepatocytes. For this goal, we stained livers with a marker of heterochromatin H3K9-trimethyI. Figures 7A and 7B show a typical result of this staining. As one can see, livers of old mice contain many massive hepatocytes with increased heterochromatin structures. Calculation of percentage of large hepatocytes with increased heterochromatin structures revealed that the livers of 22-month-old mice contain up to 25% more large hepatocytes with increased heterochromatin regions compared to less than 2% of these hepatocytes in 2-month-old mice (Figure 7C). Inhibition of cdk4 reduced the number of hepatocytes with heterochromatin enrichments.
Figure 7. Inhibition of cdk4 in Livers of Old Mice Eliminates Hepatocyte Senescence.
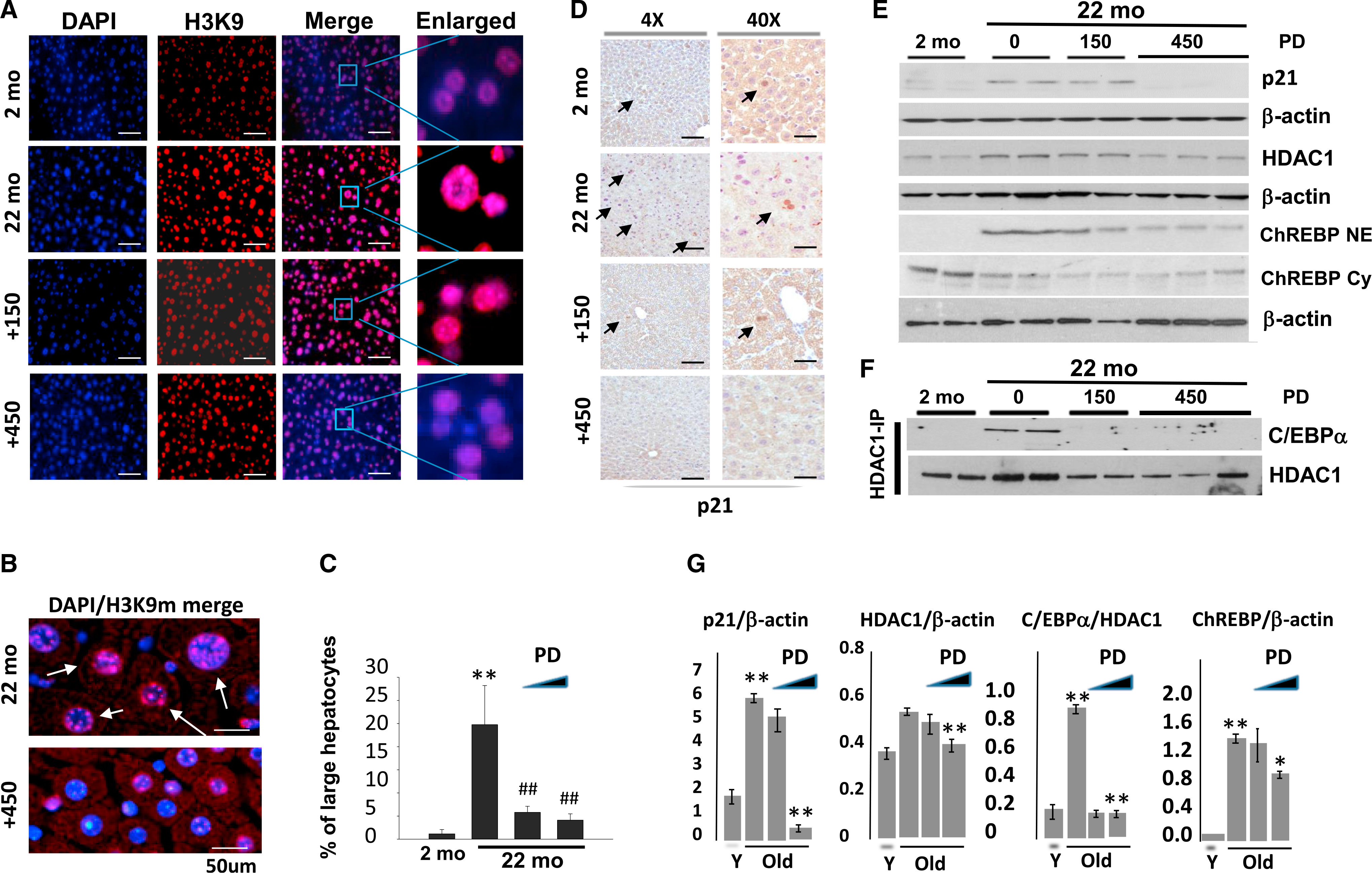
(A) Livers of young, old mice, and old mice treated with cdk4 inhibitor PD-0332991 were stained with antibodies to H3K9-trimethyl and with DAPI. Pictures were taken with 40× magnification. Scale bars represent 200 μm.
(B) A representative picture of big fields of the livers with DAPI/H3K9me (merge) staining. Arrows show gigantic hepatocytes with increased heterochromatin structures. The scale bars represent 50 μm.
(C) % of senescent hepatocytes of young, old mice, and old mice treated with cdk4 inhibitor.
(D) Immunostaining of livers with p21. Arrows show p21-positive hepatocytes. The scale bars represent 50 μm.
(E) Examination of levels of p21, HDAC1, and nuclear and cytoplasmic ChREBP by western blotting. The filters were re-probed with β-actin.
(F) Inhibition of cdk4 in livers of old mice reduces protein levels of HDAC1 and eliminates C/EBPα-HDAC1 complexes. HDAC1 was immunoprecipitated, and C/EBPα was determined by western blotting. The filter was re-probed with Abs to HDAC1.
(G) Bar graphs show calculations of protein levels of p21, HDAC1, nuclear ChREBP, and C/EBPα-HDAC1 complexes that were detected on (E) and (F).
In (C) and (G), *p < 0.05, **p or ##p < 0.01.
RNA-seq analysis and following qRT-PCR verification showed that mRNA of a marker of cellular senescence, p21, is elevated in old mice and is corrected by inhibition of cdk4 (Figure 6D). To further confirm this effect, we performed immunostaining and western blotting experiments. Figures 7D, 7E, and 7G show that p21 is elevated in livers of old mice but is reduced in mice treated with the higher dose of PD-0332991. We examined methylation of histone H3 at K9, which is also a strong marker of cellular senescence (Jin et al., 2010; Kreiling et al., 2011), and we found that PD-0332991-mediated inhibition of cdk4 reduces amounts of this indicator of cellular senescence (Figures 7A and 7B). Taken together, we conclude that the inhibition of cdk4 in old mice eliminates cellular senescence.
We next examined possible pathways by which PD-0332991 reduces heterochromatin structures in livers of old WT mice. One of the previously described pathways of the accumulation of heterochromatin foci in livers of old mice is the elevation of C/EBPα-HDAC1 complexes (Jin et al., 2010). Therefore, we suggested that this pathway might be affected by inhibition of cdk4. This hypothesis was also based on previous observations that cdk4-mediated phosphorylation of C/EBPα at Ser193 increases its interactions with HDAC1 (Jin et al., 2010). coIP studies showed that the inhibition of cdk4 leads to the disruption of C/EBPα-HDAC1 complexes (Figures 7F and 7G). These results demonstrate that the cdk4-C/EBPα pathway changes HDAC1-dependent chromatin remodeling.
Inhibition of cdk4 by PD-0332991 Changes Intracellular Localization of ChREBP Protein and Expression of Its Targets
RNA-seq approach detected alterations of mRNAs; however, aging is characterized by changes that are dependent on intracellular localization of the proteins. Therefore, we asked whether the correction of the aging phenotype in PD-0332991-treatedold mice might involve additional pathways, such as change of localization of certain proteins. It has been shown that the glucose-sensitive transcription factor ChREBP is an important regulator of fatty liver phenotype (Jois et al., 2017; Softic et al., 2017) and that it is mainly observed in the cytoplasm of young animals, but it is translocated into nucleus in livers of old animals (Salamanca et al., 2015). Therefore, we examined the levels of this protein in cytoplasmic and nuclear extracts from young, old, and old PD-0332991-treated mice. Consistent with previous reports, ChREBP is observed in cytoplasm of young mice, and it is translocated to the nucleus in livers of old mice. The inhibition of cdk4 in old mice resulted in a reduction of nuclear ChREBP, especially with higher doses of PD-0332991 (Figures 7E and 7G). Interestingly, levels of ChREBP mRNA do not differ significantly between young, old, and old PD-033291-treated mice (Figure S4A). To examine whether this reduction of nuclear ChREBP changes the expression of its targets, we analyzed known ChREBP targets using our results with RNA-seq. This bio-statistical analysis revealed that expression of a portion of ChREBP targets is altered in old mice with inhibited cdk4 (Figure S4B). Thus, these studies showed that the correction of the aging phenotype by inhibition of cdk4 includes corrections of intracellular localization of ChREBP, leading to alterations of its downstream targets. In addition to age-related disorders described above, we also analyzed insulin resistance and inflammation pathways in our RNA-seq data because these pathways are altered in livers of old mice (Oh et al., 2016; Jelenik et al., 2017). This analysis revealed a quite complex pattern of alterations in gene expression. Some of these changes suggest the correction of inflammation and insulin resistance. However, some PD-0332991-dependent alterations within these pathways are not consistent with the correction of these pathways (Figure S5). Further molecular and functional studies are required to evaluate the effects of inhibition of cdk4 on inflammation and insulin resistance.
DISCUSSION
The liver is one of the main organs that regulates body homeostasis and eliminates toxins from organism. NAFLD is a well-documented age-associated disease. It is recognized that older people develop the first step of this disease, hepatic steatosis, which in turn creates a risk for further development of NASH and HCC. In this paper, we have performed a systemic examination of the role of increase or activation of cdk4 in the development of age-associated NAFLD. We found that the degree of elevation of hepatic cdk4 in livers of patients with NAFLD correlates with the severity of NAFLD and with their age. Because the amounts of human liver biopsies are quite limited, it is not possible to examine cause or effect relationships in human samples. Therefore, we have performed mechanistic studies using young and old WT mice and recently generated cdk4-resistant C/EBPα-S193A mice. These studies demonstrated that cdk4-resistant C/EBPα-S193A mice do not develop hepatic steatosis with age due to a failure to activate cdk4-C/EBPα-p300 axis. It is interesting that promoters of key enzymes of development of fatty liver, such as enzymes of triglyceride synthesis diacylglycerol O-acyltransferase 1 (DGAT1) and diacylglycerol O-acyl-transferase 2 (DGAT2), acetyl-coenzyme A (ACACA), stearoylcoenzyme A desaturase 1 (SCD1), and sterol regulatory element binding protein 1 (SREBP1), contain binding sites for C/EBPα and are elevated in aged WT mice by the C/EBPα-p300 complex. Although, in S193A mice, the mutant C/EBPα binds to these promoters, it does not activate transcription because of a reduction or lack of interactions of C/EBPα with p300. The fact that a large number of key enzymes of fatty liver development are under the control of the cdk4-C/EBPα-p300 pathway strongly suggests that this pathway is critical for the development of age-dependent hepatic steatosis.
It has been shown that the age-associated steatosis is under control of several pathways. How does inhibition of cdk4 eliminate other known mechanisms of NAFLD, such as E2F1-Rb and cellular senescence? It has been shown that E2F1 positively contributes to the development of NAFLD under conditions of high-fat diet (Denechaudet al., 2016). In this regard, the inhibition of the phosphorylation of Rb by inhibition of cdk4 should release E2F1 and should have an opposite result. However, RNA-seq and subsequent qRT-PCR verification found that levels of E2F1 are increased in livers of old mice and that inhibition of cdk4 leads to normalization of E2F1 (Figure 6D), which likely eliminates the E2F1-dependent pathway of steatosis. Regarding the relationship between cellular senescence as the cause of hepatic steatosis and elimination of the steatosis by inhibiting cdk4, several results within our paper provide strong evidence that the inhibition of cdk4 corrects senescent hepatocytes. First, the main characteristic of hepatocyte senescence is elevation of p21, CDKNA1 gene (Aravinthan et al., 2013, 2014; Ogrodnik et al., 2017). Consistent with these reports, we found that p21 is 5- or 6-fold increased in livers of old mice and that inhibition of cdk4 normalizes levels of p21. Mechanisms of this correction are not known but might be associated with additional alterations which were found by RNA-seq analysis, which showed that the ZNF385A gene is elevated in livers of old mice and is normalized by inhibitor of cdk4 (Figure 6D). ZNF385A has been shown to upregulate p21 expression (Sugimoto et al., 2006; Zhang et al., 2012). It is possible that PD-0332991-mediated normalization of ZNF385A is involved in the reduction of p21 and in elimination of cellular senescence. Interestingly, ZNF385A also positively upregulates translation of C/EBPα (Kawagishi et al., 2008), suggesting that this activity might be also involved in elimination of cellular senescence through the inhibition of C/EBPα-p300 pathway. Another strong characteristic of the senescent hepatocytes is the increase in number of large hepatocytes with increased heterochromatin structures (Jin et al., 2010; Kreiling et al., 2011). Our results show that inhibition of cdk4 reduces the number of large hepatocytes with heterochromatin structures. Our data propose mechanisms of this correction that are associated with the reduction of C/EBPα-HDAC1 complexes. In summary, our studies suggest that the inhibition of cdk4 by US Food and Drug Administration (FDA)-approved drugs might be considered as a therapeutic approach to treat hepatic steatosis and other age-related disorders. In this regard, it is important to note that we observed accumulation of alanine transaminase (ALT)/AST (aspartate transaminase) in the blood of some old mice in experiments with higher dose of PD-0332991 (Figure 3), suggesting that further studies might be necessary for optimization of the dose of PD-0332991 for older patients.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Antibodies to C/EBPα (14AA), DGAT1 (H-255), DGAT2 (H-70), p300 (N-15 or C-20), cdk4 (C-22), and SREBP1 (C-20; sc-366) were from Santa Cruz Biotechnology. Antibodies to FASN, SCD1, and ACC were from Cell Signaling Technology. Antibodies to Ser193-ph C/EBPα were purchased from Thermo Scientific (cat. PA5–37342). HDAC1 antibody was from Upstate (cat. 05–614; CA). Acetylated histone H3K9 (H3K9Ac) antibody was purchased from Active Motif. Trimethylated histone H3K9 (H3K9m) and anti-CHREBP were from Abcam (Cambridge, UK; ab8898 and ab81958, correspondingly). Monoclonal anti-β-actin antibody was from Sigma (St. Louis, MO). Co-IP studies were performed using TrueBlot reagents as previously described (Wang et al., 2010; Jin et al., 2013, 2015).
Animal Work and Treatment of Mice with CDK4 Inhibitor PD-0332991
Experiments with animals were approved by the Institutional Animal Care and Use Committee (IACUC) at Cincinnati Children’s Hospital (protocol IACUC2014–0042). Young (2-month-old) and old (20- to 22-month-old) C57Bl6 mice (males) were purchased from The Jackson Laboratory (Bar Harbor, ME). C/EBPα-S193A mice have been generated and characterized in our lab (Jin et al., 2015, 2016). Young (2-month-old) and old (20- or 22-month-old) WT and C/EBPα-S193A mice (males and females) were used for these studies. 20-month-old C57Bl6 mice (males) were fed with chow diet (custom made by Bioserv) containing 150 mg/kg or 450 mg/kg of chow CDK4 inhibitor PD-0332991 (palbociclib HCl, Selleckchem; cat no. S1116) for 2 weeks.
Histology, Immunohistochemistry, and Oil Red O Staining
The livers were fixed overnight in buffered 10% formaldehyde, embedded in paraffin, and sectioned at a thickness of 5 μm. The sections were then stained with different antibodies against cdk4 or H3K9m. For oil red O staining of lipid droplets in frozen liver sections, the liver cryosections of 10 μm were stained with commercially available kits (oil red O color solution; cat no. 1.20419.0250; Merck, Germany).
Protein Isolation and Western Blotting
Cytoplasmic and nuclear extracts were isolated from livers as described in previous papers (Wang et al., 2010; Jin et al., 2013, 2015). Inhibitors of phosphatases were used for all samples during protein isolation process. Proteins (30–50 μg) were loaded on gradient (4%–20%) polyacrylamide gels, transferred onto membranes, and probed with antibodies against proteins of interest. To verify protein loading, each filter was re-probed with Abs to β-actin.
Transfections of Hepa-1c17 Cells with C/EBPα Constructs
WT, S193D, and S193A C/EBPα plasmids were transfected in Hepa-1c17 cells. Protein extracts were isolated 16 hr after transfection, and expression of proteins was examined by western blotting as described above.
coIP
Nuclear extracts were used for coIP. A total of 500 μg of protein extract was incubated with 2 μg of the appropriate antibody at 4° for overnight under rotation. Then, 25 μL of appropriate Ig IP Agarose Beads (Rockland) was added, followed by an hour incubation at 4° under rotation. The pellets were collected by centrifugation (6,000 rpm) and washed 3 times with IP washing buffer and then were re-suspended in 30 μL of loading buffer (with 2% of β-mercapto-ethanol). Samples were boiled for 20 min and were run in SDS-gel (4%–20% gradient gels; Bio-Rad). Western blot analyses were performed using antibody against C/EBPα. True-Blot secondary antibodies were used.
ChIP Assay
The ChIP assay was performed as described in our previous articles, using the ChIP-IT Kit (Active Motif) according to the manufacturer’s protocol. Briefly, the chromatin solutions were prepared from liver tissues, and DNA was sheared by enzymatic method. C/EBPα, p300, and H3K9Ac were immunoprecipitated from the solutions using appropriate antibodies. DNA was isolated and used for PCRs with primers covering the C/EBPα binding sites within DGAT1/2, ACACA, SCD1, and SREBP1 promoters. The sequences of the primers for these promoters are as follows: ACACA-forward: 5′-AGCCAAGCAAGCCAGTTCT TAG-3′ and ACACA-reverse: 5′-TCATGTGAAAGGCTGGTGACA-3′ (PCR: 318 bp); SCD1-forward: 5′-GAGTTGTGAGTGGCGGTAGC-3′ and SCD1-reverse: 5′-GGCTGATGGGTGACTTTTCTT-3′ (PCR: 158 bp); SREBP1-forward-site1: 5′-GGAGTAGTACGAGCCCACTG-3′; SREBP1-reverse site 1: 5′-CATGGCCCATAAGACCGTCC-3′; SREBP1-forward-site 2: 5′-TGGGTCTA GTCCTGTGGCTT-3′ (PCR: 185 bp); and SREBP1-reverse site 2: 5′-GGCCC ACGTTAAGGAAAAGT-3′ (PCR: 243 bp). PCR products were separated by 5% PAAG.
RNA-Seq Analyses
RNA-seq analysis was performed with livers of young, old, and old-PD-treated mice. RNA was isolated from 3 mice of each group. RNA sequencing libraries were prepared using Illumina TruSeq RNA preparation kit and sequenced on the Illumina HiSeq 2500, using paired-end, 100-bp reads(Illumina, San Diego, CA). Reads were aligned using mm10 annotations produced by University of California Santa Cruz (UCSC) and quantified using Kallisto, which accurately quantifies read abundances (in transcripts per million) through pseudoalignment. Statistical analysis was performed in GeneSpring 13.0. Raw counts were thresholded at 1, normalized using quantile normalization procedure, and baselined to the median of all samples (n = 25,240 transcripts). A filtration was applied to ensure analysis of reasonably expressed transcripts, requiring at least two reads in >50% of samples in at least one experimental condition (n = 12,551 transcripts). Ontological analysis of significantly differential genes was performed in the ToppGene Suite.
Statistical Analysis
All continuous values are presented as means and SD. Statistical analyses were performed using the Student’s t test. Statistical significance was accepted at either p < 0.05 (shown by one asterisk) or p < 0.01 (shown by two asterisks).
Supplementary Material
Highlights.
Elevation of cdk4 correlates with severity of NAFLD in patients
Cdk4-resistant mice do not develop age-associated hepatic steatosis
Inhibition of cdk4 in older organisms eliminates hepatic steatosis
Inhibition of cdk4 corrects many age-associated disorders
ACKNOWLEDGMENTS
This work is supported by NIH grants R01DK102597 and R01CA159942 (N.A.T.) and by Internal Development Funds from Cincinnati Children’s Hospital Medical Center (CCHMC) (SP15 to N.A.T.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA sequencing data reported in this paper is GEO: GSE104395.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and can be found with this article online at https://doi.org/10.1016Zj.celrep.2018.07.014.
Data and Software Availability
REFERENCES
- Aravinthan A, Scarpini C, Tachtatzis P, Verma S, Penrhyn-Lowe S, Harvey R, Davies SE, Allison M, Coleman N, and Alexander G (2013). Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J. Hepatol 58, 549–556. [DOI] [PubMed] [Google Scholar]
- Aravinthan A, Mells G, Allison M, Leathart J, Kotronen A, Yki-Jarvinen H, Daly AK, Day CP, Anstee QM, and Alexander G (2014). Gene polymorphisms of cellular senescence marker p21 and disease progression in non-alcohol-related fatty liver disease. Cell Cycle 13, 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SJ, and Reddy EP (2012). CDK4: a key player in the cell cycle, development, and cancer. Genes Cancer 3, 658–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursier J, and Diehl AM (2016). Nonalcoholic fatty liver disease and the gut microbiome. Clin. Liver Dis 20, 263–275. [DOI] [PubMed] [Google Scholar]
- Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, Guy CD, Seed PC, Rawls JF, David LA, et al. (2016). The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JC, Horton JD, and Hobbs HH (2011). Human fatty liver disease: old questions and new insights. Science 332, 1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denechaud PD, Lopez-Mejia IC, Giralt A, Lai Q, Blanchet E, Delacuisine B, Nicolay BN, Dyson NJ, Bonner C, Pattou F, et al. (2016). E2F1 mediates sustained lipogenesis and contributes to hepatic steatosis. J. Clin. Invest 126, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott JA, Noble ME, and Tucker JA (1999). Cyclin-dependent kinases: inhibition and substrate recognition. Curr. Opin. Struct. Biol 9, 738–744. [DOI] [PubMed] [Google Scholar]
- Engin A (2017). Non-alcoholic fatty liver disease. Adv. Exp. Med. Biol 960, 443–467. [DOI] [PubMed] [Google Scholar]
- Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ, and Schwartz GK (2012). Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin. Cancer Res 18, 568–576. [DOI] [PubMed] [Google Scholar]
- Guillory B, Jawanmardi N, Iakova P, Anderson B, Zang P, Timchenko NA, and Garcia JM (2018). Ghrelin deletion protects against age-associated hepatic steatosis by downregulating the C/EBPα-p300/DGAT1 pathway. Aging Cell 17, e12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbard L, and George J (2011). Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol 8, 35–44. [DOI] [PubMed] [Google Scholar]
- Jelenik T, Kaul K, Séquaris G, Flögel U, Phielix E, Kotzka J, Knebel B, Fahlbusch P, Hörbelt T, Lehr S, et al. (2017). Mechanisms of insulin resistance in primary and secondary nonalcoholic fatty liver. Diabetes 66, 2241–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Iakova P, Jin J, Sullivan E, Sharin V, Hong IH, Anakk S, Mayor A, Darlington G, Finegold M, et al. (2013). Farnesoid X receptor inhibits gankyrin in mouse livers and prevents development of liver cancer. Hepatology 57, 1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Wang GL, Shi X, Darlington GJ, and Timchenko NA (2009). The age-associated decline of glycogen synthase kinase 3beta plays a critical role in the inhibition of liver regeneration. Mol. Cell. Biol 29, 3867–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Wang GL, Iakova P, Shi X, Haefliger S, Finegold M, and Timchenko NA (2010). Epigenetic changes play critical role in age-associated dysfunctions of the liver. Aging Cell 9, 895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Iakova P, Breaux M, Sullivan E, Jawanmardi N, Chen D, Jiang Y, Medrano EM, and Timchenko NA (2013). Increased expression ofenzymes of triglyceride synthesis is essential for the development of hepatic steatosis. Cell Rep. 3, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Hong IH, Lewis K, Iakova P, Breaux M, Jiang Y, Sullivan E, Jawanmardi N, Timchenko L, and Timchenko NA (2015). Cooperation of C/EBP family proteins and chromatin remodeling proteins is essential for termination of liver regeneration. Hepatology 61, 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Valanejad L, Nguyen TP, Lewis K, Wright M, Cast A, Stock L, Timchenko L, and Timchenko NA (2016). Activation of CDK4 triggers development of non-alcoholic fatty liver disease. Cell Rep. 16, 744–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jois T, Chen W, Howard V, Harvey R, Youngs K, Thalmann C, Saha P, Chan L, Cowley MA, and Sleeman MW (2017). Deletion of hepatic carbohydrate response element binding protein (ChREBP) impairs glucose homeostasis and hepatic insulin sensitivity in mice. Mol. Metab 6, 1381–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagishi H, Wakoh T, Uno H, Maruyama M, Moriya A, Morikawa S, Okano H, Sherr CJ, Takagi M, and Sugimoto M (2008). Hzf regulates adipogenesis through translational control of C/EBPalpha. EMBO J. 27, 1481–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiling JA, Tamamori-Adachi M, Sexton AN, Jeyapalan JC, Munoz-Najar U, Peterson AL, Manivannan J, Rogers ES, Pchelintsev NA, Adams PD, and Sedivy JM (2011). Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell 10, 292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laish I, Mannasse-Green B, Hadary R, Biron-Shental T, Konikoff FM, Amiel A, and Kitay-Cohen Y (2016). Telomere dysfunction in nonalcoholic fatty liver disease and cryptogenic cirrhosis. Cytogenet. Genome Res 150, 93–99. [DOI] [PubMed] [Google Scholar]
- Machado MV, and Diehl AM (2016). Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 150, 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry EP, Kennedy S, Xie Y, Luo J, Jiang H, Ory DS, and Davidson NO (2015). Phenotypic divergence in two lines of L-Fabp−/− mice reflects substrain differences and environmental modifiers. Am. J. Physiol. Gastrointest. Liver Physiol 309, G648–G661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry EP, Xie Y, Kennedy SM, Graham MJ, Crooke RM, Jiang H, Chen A, Ory DS, and Davidson NO (2017). Prevention of hepatic fibrosis with liver microsomal triglyceride transfer protein deletion in liver fatty acid binding protein null mice. Hepatology 65, 836–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, et al. (2017). Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun 8, 15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh AR, Bae JS, Lee J, Shin E, Oh BC, Park SC, and Cha JY (2016). Ursodeoxycholic acid decreases age-related adiposity and inflammation in mice. BMB Rep. 49, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamanca A, Bárcena B, Arribas C, Fernández-Agulló T, Martínez C, Carrascosa JM, Ros M, Andrés A, and Gallardo N (2015). Aging impairs the hepatic subcellular distribution of ChREBP in response to fasting/feeding in rats: Implications on hepatic steatosis. Exp. Gerontol 69, 9–19. [DOI] [PubMed] [Google Scholar]
- Schmucker DL (2005). Age-related changes in liver structure and function: Implications for disease? Exp. Gerontol 40, 650–659. [DOI] [PubMed] [Google Scholar]
- Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, Willoughby J, Harbison C, Fitzgerald K, Ilkayeva O, et al. (2017). Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Invest 727, 4059–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M, Gromley A, and Sherr CJ (2006). Hzf, a p53-responsive gene, regulates maintenance of the G2 phase checkpoint induced by DNA damage. Mol. Cell. Biol 26, 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko NA (2009). Aging and liver regeneration. Trends Endocrinol. Metab 20, 171–176. [DOI] [PubMed] [Google Scholar]
- Timchenko NA (2011). Senescent liver. In Molecular Pathology of the Liver Diseases, Monga SPS (Springer Science + Business Media; ), pp. 279–290. [Google Scholar]
- Valanejad L, Lewis K,Wright M, Jiang Y, D’Souza A, Karns R, Sheridan R, Gupta A, Bove K, Witte D, et al. (2017). FXR-Gankyrin axis is involved in development of pediatric liver cancer. Carcinogenesis 38, 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti L, and Dongiovanni P (2014). CDKN1A: a double-edged sword in fatty liver? Cell Cycle 13, 1371–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdelho Machado M, and Diehl AM (2016). Role of Hedgehog signaling pathway in NASH. Int. J. Mol. Sci 17, E857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G-L, Shi X, Salisbury E, Sun Y, Albrecht JH, Smith RG, and Timchenko NA (2006). Cyclin D3 maintains growth-inhibitory activity of C/EBPalpha by stabilizing C/EBPalpha-cdk2 and C/EBPalpha-Brm complexes. Mol. Cell. Biol 26, 2570–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G-L, Shi X, Haefliger S, Jin J, Major A, lakova P, Finegold M, and Timchenko NA (2010). Elimination of C/EBPalpha through the ubiquitin-proteasome system promotes the development of liver cancer in mice. J. Clin. Invest 120, 2549–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XP, Liu F, and Wang W (2012). Regulation of the DNA damage response by p53 cofactors. Biophys. J 102, 2251–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.