

Abstract
The non-nucleoside analog gemcitabine has been the standard of care for treating pancreatic cancer. The drug shows good potency in pancreatic cancer cells in vitro but, due to poor bioavailability, requires administration in large doses by infusion and this systemic exposure results in significant toxicity for the patient. Genes have been identified that, when silenced by siRNA, synergize with gemcitabine treatment and offer a means of reducing the gemcitabine dosage required for efficacy. However, benefiting from the synergism between the two agents requires that the gemcitabine and siRNA penetrate the same cells. To ensure co-delivery, we incorporated gemcitabine covalently within siRNAs against targets synergistic with gemcitabine (CHK1 or RAD17). We demonstrated that specific bases within an siRNA can be replaced with gemcitabine to increase efficacy. The result is a single drug molecule that simultaneously co-delivers gemcitabine and a synergistic siRNA. The siRNA–gemcitabine constructs demonstrate a 5–30-fold improvement in potency compared with gemcitabine alone. Co-delivering a CHK1 siRNA–gemcitabine construct together with a WEE1 siRNA resulted in a 10-fold improvement in IC50 compared with gemcitabine alone. These constructs demonstrate efficacy across a wide array of pancreatic tumor cells and may represent a novel therapeutic approach for treating pancreatic cancer.
INTRODUCTION
Gemcitabine is the primary therapeutic in treating pancreatic cancer (1) but is also used in the treatment of a number of other cancers, including cholangiocarcinoma (2), non-small cell lung cancer (3), ovarian cancer (4) and breast cancer (5). Gemcitabine (2′,2′-difluoro-2′-deoxycytidine) is a pyrimidine-based nucleoside analog that, when administered systemically, is taken up by nucleoside transporters, activated by triphosphorylation by deoxycytidine kinase and can then be incorporated into either RNA or DNA. It replaces the nucleic acid cytidine during DNA replication and can inhibit tumor growth since new nucleosides cannot be attached to this nucleoside mimic, resulting in apoptosis of the cells (1).
Improved therapies are desperately needed to combat the poor prognosis of pancreatic cancer. Targeted therapies have been actively sought to potentiate the efficacy of gemcitabine in killing tumor cells and therefore allow a reduction of the dose required for therapeutic efficacy. In this quest, one significant approach has been the use of siRNAs to silence specific genes that results in an increase in potency and/or efficacy of gemcitabine.
Azorsa et al. (6) identified siRNAs against the gene CHK1 that potentiate the effect of gemcitabine on pancreatic cancer cells in culture. A subsequent manuscript by Fredebohm et al. (7) validated CHK1 as a potentiator of gemcitabine toxicity in pancreatic tumor cells but also identified several additional targets that improved the activity of gemcitabine when silenced. One of these targets was RAD17 (7), which demonstrated a profound improvement in the action of gemcitabine in reducing cell viability when silenced by shRNAs. Ma et al. have demonstrated that gemcitabine can be synthesized as a polymer by combining amidites of the nucleoside, and that these polymers form nanogels with activity against cancer (8).
We therefore speculated that we could synthesize siRNAs with gemcitabines included within their sequence to result in co-delivery of two synergistic agents to the same cells at the same time.
We selected the 19-mer CHK1 siRNA sequence identified in the paper by Azorsa et al. (6) to modify the sequence with gemcitabine—added as an amidite during synthesis of each of the strands of the siRNA duplex. We also examined gemcitabine modifications to a 25-mer RAD17 siRNA and a 25-mer siRNA against CHK1 on viability of pancreatic tumor cells. Various modifications based on position within the siRNA or the number of gemcitabines attached were tested for their ability to improve efficacy or potency of the constructs in reducing cell viability in pancreatic cancer cells in culture.
MATERIALS AND METHODS
Cell lines
The pancreatic cancer cell lines BxPC3, MIA PaCa-2, Panc1, Capan-2 and CFPAC-1 were obtained from ATCC (Rockville, MD). Huh7 cell line was obtained from Sekisui XenoTech, LLC (Kansas City, KS). Cells were cultured in standard media supplemented with 10% FBS: BxPC3 in RPMI-1640 Medium; MIA PaCa-2, Panc1 and Huh7 in DMEM; Capan-2 in McCoy’s 5A (Modified) Medium; and CFPAC-1 in Iscove’s Modified Dulbecco’s Medium. All media were obtained from ATCC.
Cell line authentication
Cells were authenticated by ATCC (Manassas, VA) using short tandem repeat profiling and were last validated at the completion of the reported experiments.
siRNA design
The 19-mer siRNA sequence against CHK1 sequence was taken from the paper by Azorsa et al. (6). This siRNA included a dTdT overhang at the 3′ end of each strand. We also designed 25-mer blunt-ended siRNA sequences against RAD17 and CHK1 that were identical for both mouse and human genes. Sequences used in the study are shown in Table 1.
Table 1.
Structures of gemcitabine-modified siRNA sequences
| Label | Target | Sense strand | Antisense strand |
|---|---|---|---|
| NS | NS | 5′-CGAGCAGGGUAUCGACGAUUACAAA-3′ | 5′-UUUGUAAUCGUCGAUACCCUGCUCG-3′ |
| Chk1az | CHK1 | 5′-AAGAAAGAGAUCUGUAUCAAUdTdT-3′ | 5′-AUUGAUACAGAUCUCUUUCUUdTdT-3′ |
| Chk1az-3′s2G | CHK1 | 5′-AAGAAAGAGAUCUGUAUCAAU[GEM][GEM]dTdT-3′ | 5′-AUUGAUACAGAUCUCUUUCUUdTdT-3′ |
| Chk1az-3′s4G | CHK1 | 5′-AAGAAAGAGAUCUGUAUCAAU[GEM][GEM][GEM][GEM]dTdT-3′ | 5′-AUUGAUACAGAUCUCUUUCUUdTdT-3′ |
| Chk1az-3′s6G | CHK1 | 5′-AAGAAAGAGAUCUGUAUCAAU[GEM][GEM][GEM][GEM][GEM][GEM]dTdT-3′ | 5′-AUUGAUACAGAUCUCUUUCUUdTdT-3′ |
| NS-a2G | NS | 5′-CGAGCAGGGUAUCGACGAUUACAAA-3′ | 5′-UUUGUAAUCGU[GEM]GAUACCCUG[GEM]UCG-3′ |
| NS-s2G | NS | 5′-CGAGCAGGGUAU[GEM]GACGAUUA[GEM]AAA-3′ | 5′-UUUGUAAUCGUCGAUACCCUGCUCG-3′ |
| NS-s2a2G | NS | 5′-CGAGCAGGGUAU[GEM]GACGAUUA[GEM]AAA-3′ | 5′-UUUGUAAUCGU[GEM]GAUACCCUG[GEM]UCG-3′ |
| Chk1 | CHK1 | 5′-CCUGUGGAAUAGUACUUACUGCAAU-3′ | 5′-AUUGCAGUAAGUACUAUUCCACAGG-3′ |
| Chk1-3′s2G | CHK1 | 5′-CCUGUGGAAUAGUACUUACUGCAAU[GEM] [GEM]-3′ | 5′-AUUGCAGUAAGUACUAUUCCACAGG-3′ |
| Chk1-s3G_short | CHK1 | 5′-__UGUGGAAUAGUA[GEM]UUA [GEM]UG [GEM]AAU-3′ | 5′-AUUGCAGUAAGUACUAUUCCACAGG-3′ |
| Chk1-3′s2G_short | CHK1 | 5′-__UGUGGAAUAGUACUUACUGCAAU[GEM][GEM]-3′ | 5′-AUUGCAGUAAGUACUAUUCCACAGG-3′ |
| Chk1-5′s2_3′s2G | CHK1 | 5′-[GEM][GEM]UGUGGAAUAGUACUUA[GEM]UG [GEM]AAU-3′ | 5′-AUUGCAGUAAGUACUAUUCCACAGG-3′ |
| Chk1az-a2G | CHK1 | 5′-AAGAAAGAGAUCUGUAUCAAUdTdT-3′ | 5′-AUUGAUACAGAUCU[GEM]UUU[GEM]UUdTdT-3′ |
| Chk1az-s2G | CHK1 | 5′-AAGAAAGAGAU[GEM]UGUAU[GEM]AAUdTdT-3′ | 5′-AUUGAUACAGAUCUCUUUCUUdTdT-3′ |
| Chk1az-a2s2G | CHK1 | 5′-AAGAAAGAGAU[GEM]UGUAU[GEM]AAUdTdT-3′ | 5′-AUUGAUACAGAUCU[GEM]UUU[GEM]UUdTdT-3′ |
| RAD17 | RAD17 | 5′-CCAACAAUUAUGAUGAAAUUUCUUA-3′ | 5′-UAAGAAAUUUCAUCAUAAUUGUUGG-3′ |
| RAD17-s2G | RAD17 | 5′-CCAACAAUUAUGAUGAAAUUUCUUA[GEM][GEM]-3′ | 5′-UAAGAAAUUUCAUCAUAAUUGUUGG-3′ |
The sequences of the sense and antisense strands for the gemcitabine–siRNA constructs used within the siRNAs are shown. The sequences shown were used to anneal to generate duplex siRNAs. NS: non-silencing control sequence; Chk1az: 19-mer derived from Azorsa et al. (6).
siRNA synthesis
Oligonucleotides were synthesized using an ABI 394 automated DNA/RNA synthesizer. Syntheses were conducted at 0.2 μmol scale using either a controlled pore glass support (1000 Å) bearing the 3′ RNA base of the intended sequence or a polystyrene support coupled with a universal linker (USIII-PS, Glen Research, LLC, Loudoun, VA).
TBDMS-protected RNA phosphoramidites and supports (Glen Research, LLC, Sterling, VA) along with N-benzoyl-2-deoxy-5-O-DMT-2′,2′-difluorocytidine 3-CE (gemcitabine) phosphoramidite (Advent Bio, Elk Grove, IL) were coupled using standard phosphoramidite synthesis procedures including 5-ethylthio-1H-tetrazole activator. The 5′-dimethoxytrityl (DMT) group was retained for use in downstream purification.
Cleavage from the solid support for oligonucleotides containing gemcitabine was achieved using 2 M ammonia solution in methanol (Acros Organics, Morris Plains, NJ) followed by addition of 1 ml of concentrated ammonium hydroxide prior to deprotection overnight at 40oC. Oligonucleotides lacking gemcitabine phosphoramidite were cleaved and deprotected using 1:1 ammonium hydroxide/40% methylamine.
Removal of the 2′-TBDMS protection was completed using dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO) and triethylamine trihydrofluoride solution (Sigma-Aldrich, St. Louis, MO) containing triethylamine (Sigma-Aldrich, St. Louis, MO).
The deprotection reaction was quenched with RNA quenching buffer (Glen Research, LLC, Sterling, VA) and immediately purified using a Glen-Pak RNA cartridge (Glen Research, LLC, Sterling, VA). Purified products were dried down and converted to the sodium salt form via ethanol precipitation.
Compounds were reconstituted in RNase-free water, quantified and characterized by reversed-phase high-performance liquid chromatography prior to annealing and use.
siRNA validation of target silencing
Cells were seeded in 12-well plates at a density of 2.5 × 105 cells/well and allowed to attach overnight. On the next day, cells were transfected with different variants of gemcitabine-modified Chk1 or non-silencing (NS) siRNA using Lipofectamine RNAiMAX (Life Technologies, Carlsbad, CA) according to the manufacturer’s recommendations.
Twenty-four to forty-eight hours after transfection, total RNA was extracted using RNeasy Plus Mini Kit (QIAGEN), following the protocol suggested by the manufacturer. cDNA synthesis was performed using Maxima First Strand cDNA Kit (Life Technologies). Relative RNA level was determined by SYBR Green RT-qPCR. The sequences of primers were as follows: for CHK1 5′-TTGTGGAAGACTGGGACTTG-3′ (forward) and 5′-ATTTTCTGGACAGTCTACGGC-3′ (reverse), for RAD17 5′-TTTTCCTGACTTCTGCCTACC-3′ (forward) and 5′-TCTTCCAAAGTGTCGCTTCAG-3′ (reverse), for RRM1 5′-ACCGCCCACAACTTTCTAG-3′ (forward) and 5′-CCAGTAGCCCGAATACAACTC-3′ (reverse), for RRM2 5′-AAGGACATTCAGCACTGGG-3′ (forward) and 5′-AGCGGGCTTCTGTAATCTG-3′ (reverse), and for β-actin 5′-ACCTTCTACAATGAGCTGCG-3′ (forward) and 5′-CCTGGATAGCAACGTACATGG-3′ (reverse). Amplification conditions were set at 50°C for 5 min, 95°C for 20 s, and included 40 cycles of 95°C for 15 s and 60°C for 1 min. mRNA level in all samples was calculated relative to RNA level of a non-treated control. β-Actin was used as an internal standard for all samples.
Western blot
Cells were plated in a six-well plate at 2 × 105 cells/well overnight. Cells were transfected with 5 nM corresponding siRNA for 72 h. Cells were then lysed in M-PER (Thermo Scientific) buffer containing a protease inhibitor cocktail (Sigma-Aldrich) and a phosphatase inhibitor cocktail (Sigma-Aldrich).
Forty micrograms of protein were subject to immunoblotting analysis. Proteins were separated by NuPAGE 4–12% Bis-Tris gel (Invitrogen) and transferred to PVDF membrane by iBlot 2 Transfer Stacks (Thermo Fisher). Anti-CHK1 (1:5000 dilution, Abcam), anti-RRM1, anti-RRM2, Phospho-Histone H2AX (1:1000 dilution, Cell Signaling) and anti-GAPDH (1:1000 dilution, Santa Cruz) antibodies were used to detect proteins. Proteins were visualized with Alexa Fluor 647 or 488 conjugated secondary antibodies (Invitrogen) using an Azure c400 Imaging System (Azure Biosystems).
Cell cytotoxicity assays
Cells were seeded in 384-well plates at a density of 0.5 × 103 to 1 × 103 cells/well. On the next day, cells were treated with serially diluted gemcitabine or siRNAs delivered by Lipofectamine RNAiMAX (Life Technologies, Carlsbad, CA). Forty-eight to one hundred twenty hours after addition of gemcitabine or transfection, the number of viable cells was determined with CellTiter-Glo® 2.0 reagent (Promega, Madison, WI) by measuring luminescent signal using a Cytation 5 plate reader (BioTek Inc., Winooski, VT). All values were compared to values generated for cells treated with non-silencing (NS) siRNA or to non-treated control and reported as the percentage cell viability. Values represent the mean ± standard deviation (SD) (n = 4).
Calculation of IC50 values
The IC50 values of the gemcitabine, or gemcitabine-modified siRNAs (concentration of drug needed to inhibit 50% of growth), were derived from a sigmoidal dose response (variable slope) curve using GraphPad Prism 8.4.2 software (GraphPad Software, San Diego, CA).
Presentation of results
Unless stated in the figures, all experiments were performed using four replicates at each data point within the experiment and the experiments were repeated at least twice. Error bars in the figures represent the mean ± SD of the data.
RESULTS
Adding gemcitabine to sense strand of CHK1 siRNA increases cell killing
We compared an unmodified 19-mer siRNA (Chk1az) with the same sequence where two (Chk1az-3′s2G), four (Chk1az-3′s4G) or six gemcitabines (Chk1az-3′s6G) were added at the 3′ end of the sense strand on cell viability in BxPC3 cells. These sequences (shown in Table 1) were compared with gemcitabine alone (Gem; Figure 1A). Gemcitabine itself produced a full dose response in this cell line and at this incubation time (120 h) produced a 3nM IC50 with 100% reduction in cell viability at concentrations above 50nM. Unmodified 19-mer siRNA (Chk1az) gave an IC50 of 0.16nM. However, the efficacy maxed out at ∼70% inhibition of cell viability, even at this prolonged exposure time. The expectation that more gemcitabines would improve the potency of the siRNAs was not met and a sequence with two gemcitabines was more potent than either the four- or six-gemcitabine constructs. The two-gemcitabine construct showed an ∼115-fold improvement in IC50 (0.026nM) relative to gemcitabine alone (3nM).
Figure 1.
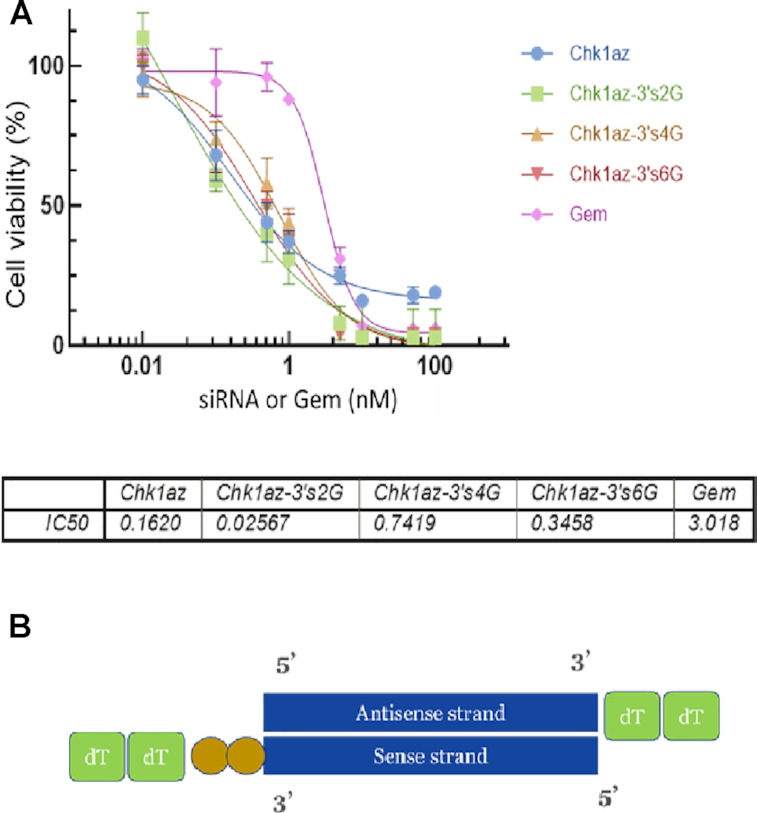
Effect of adding additional gemcitabines on the 3′ end of the sense strand on efficacy and potency. BxPC3 cells seeded in 384-well plates at 1000 cells/well were transfected with various concentrations of siRNA constructs or exposed to gemcitabine for 120h. (A) Dose curve of Chk1az siRNA, gemcitabine alone and various forms of siRNA–gemcitabine constructs. Cell viability was measured by CellTiter-Glo® 2.0 reagent. Chk1az: the unmodified 19-mer siRNA against CHK1; Chk1za-3′s2G, Chk1az-3′s4G and Chk1az-3′s6G: the same sequences of the Chk1az siRNA but with two (Chk1az-3′s2G), four (Chk1az-3′s4G) and six (Chk1az-3′s6G) gemcitabine nucleotides added at the 3′ end of the sense strand prior to annealing with the same antisense strand; Gem: gemcitabine alone. The data are presented as mean ± SD from a single experiment with four replicates for each setting. The experiment was repeated twice with similar results. (B) Diagram of Chk1az-3′s2G. Two gemcitabines (gold circles) are shown attached to the 3′ end of the sense strand ahead of the dTdT group.
We tested whether the result observed with the gemcitabine-modified siRNAs in BxPC3 cells was dependent on the duration of exposure to these agents (Figure 2). At each time point, the unlabeled siRNA (Chk1az) showed a weaker effect at the highest concentration (100nM) than the gemcitabine-modified siRNAs (containing two, four or six gemcitabines). While there was not much difference in potency or efficacy between the siRNA containing two gemcitabines (Chk1az-3′s2G) compared with four (Chk1az-3′s4G) or six gemcitabines (Chk1az-3′s6G), the maximum degree of cell killing by each of the constructs (at 100 nM) increased with exposure duration [from ∼40% at 48 h (Figure 2A) to ∼70% at 72 h (Figure 2B) and 100% at 120 h (Figure 2C)].
Figure 2.
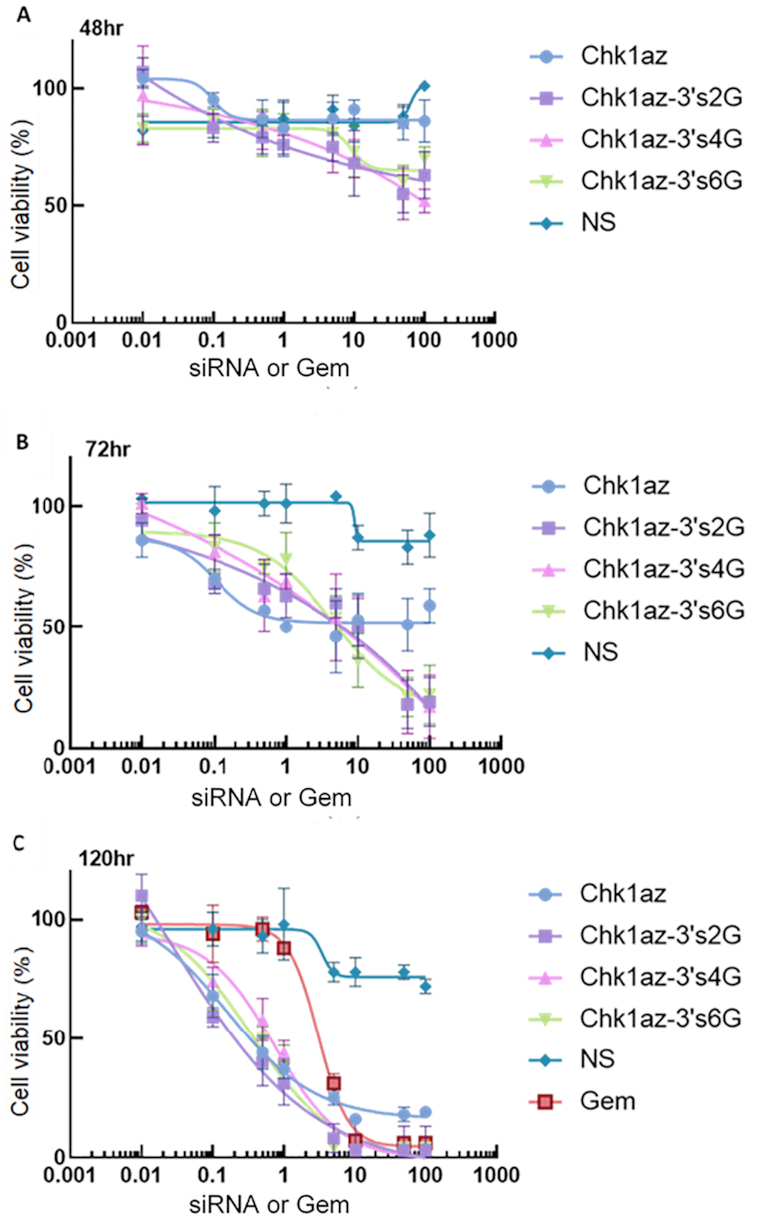
Time dependence of efficacy of siRNA and siRNA–gemcitabine constructs on cell viability. BxPC3 cells seeded in 384-well plates at 1 × 103 cells/well were transfected with siRNA constructs or exposed to gemcitabine at the concentration indicated below. The cell viability was measured with CellTiter-Glo® 2.0 reagent at (A) 48h, (B) 72h and (C) 120h. NS: non-silencing siRNA control; Gem: gemcitabine alone; Chk1Az: 19-mer siRNA against CHK1 unmodified; Chk1az-3′s2G, Chk1az-3′s4G and Chk1az-3′s6G: Chk1az siRNA modified with two, four and six gemcitabine moieties on the 3′ end of the sense strand. The data are presented as mean ± SD from a single representative experiment in quadruplicates. The experiment was repeated twice with similar results.
We also identified a potent blunt-ended 25-mer siRNA against CHK1 and compared the effect of silencing using this siRNA with the 19-mer siRNA (6) on viability of BxPC3 cells (Figure 3A). The blunt-ended 25-mer siRNA against CHK1 demonstrated greater efficacy than the Azorsa CHK1 siRNA sequence with ∼80% decrease in cell viability at 100nM compared with 50% reduction for the latter. Interestingly, all three siRNAs incorporating two gemcitabines per molecule, either on the 3′ ends (Chk1az-3′s2G or Chk1-3′s2G) or in place of cytidines within the sense strand sequence (Chk1az-s2G), gave a similar dose response with improvement in maximal efficacy—killing almost 100% of cells at a concentration of 30nM.
Figure 3.
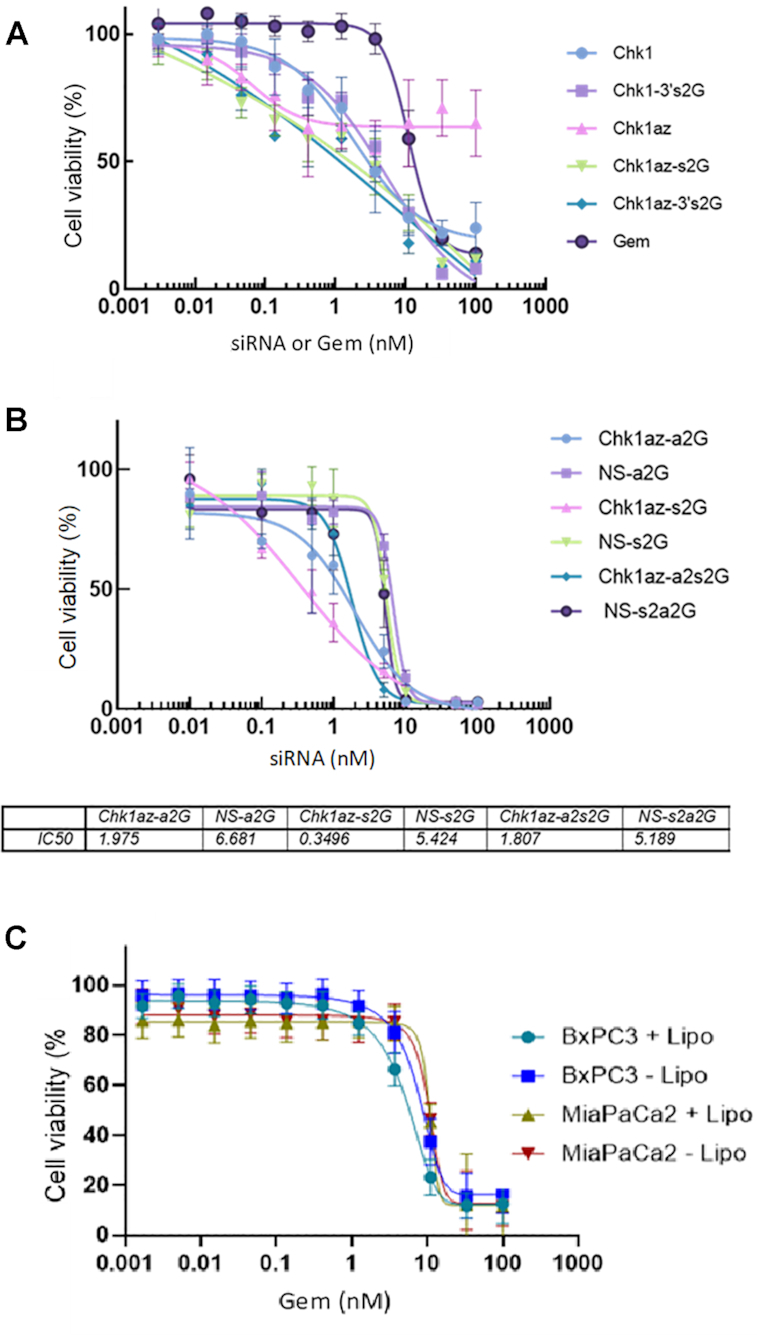
Potency of other forms of modified Chk1 siRNA in killing pancreatic cells. BxPC3 cells plated at 1000 cells/well in a 384-well plate were transfected the next day and incubated for 96h prior to measuring cell viability using CellTiter-Glo® 2.0 reagent. siRNA data were normalized to NS siRNA. The data are presented as mean ± SD from a single experiment with four replicates per data point. The experiment was repeated three times with similar results. (A) Effect of adding two gemcitabine nucleotides to the sense strand of each siRNA on viability of treated pancreatic cells. (B) Effect of inclusion of gemcitabines in place of cytidines in the antisense strand on cell viability. (C) Effect of transfection reagent on gemcitabine efficacy.
Replacing cytidine nucleotides with gemcitabines in antisense strand enhances potency of CHK1 siRNA
We further determined whether the gemcitabine nucleotide could be used to replace the cytidine nucleotides within the siRNA in the antisense strand of the siRNA (Figure 3B).
Incorporating two gemcitabines into the sense strand of the siRNA targeting CHK1 (Chk1az-s2G), we get the combined effect of silencing this gene and the effect of releasing gemcitabine within the cells—producing a shift in the IC50 (0.35 nM) while also maintaining full efficacy (Figure 3B). When two gemcitabines are inserted into the sense strand of the NS siRNA, we see only the effect of gemcitabine release on the apparent IC50 (5.4nM). Therefore, just adding two gemcitabines to the CHK1 siRNA resulted in ∼15-fold improvement over having two gemcitabines in the sense strand of an NS siRNA.
Adding two gemcitabines in the antisense strand of CHK1 and NS siRNAs, we see that the Chk1–gemcitabine IC50 (Chk1az-a2G) was reduced to 1.9nM compared with the NS siRNA (NS-a2G) at 6.68nM (Figure 3B). The IC50 for the combination of siRNA strands where both the CHK1 antisense and sense strands each have two gemcitabines incorporated (Chk1az-a2s2G; 1.8nM) is very close to the IC50 for the siRNA incorporating the two gemcitabines only in the antisense strand (Chk1az-a2G; 1.98nM) suggesting that there is little benefit in adding the two additional gemcitabines.
The curves for NS-s2G, NS-a2G and NS-s2a2G all overlap and have a much higher IC50 than those where CHK1 was silenced. The former siRNAs have no gene silencing and therefore all their effect is solely due to release of the two gemcitabines present on the sense strand during cleavage—providing similar IC50 values and effects comparable to the dose response for gemcitabine alone (Figure 3B).
There was concern that lipofectamine may alter the sensitivity of the cells to gemcitabine, so we analyzed the effect of lipofectamine on the dose response effect of gemcitabine in two of the cell lines (BxPC3 and MIA PaCa-2 cells; Figure 3C). No change in IC50 was noted between the two treatments (gemcitabine ± lipofectamine) in either cell line.
Validation of gene silencing by gemcitabine-modified siRNAs
To validate that the modified siRNAs could still silence their respective gene target, we examined MIA PaCa-2 cells transfected with different variants of CHK1/gemcitabine siRNA or RAD17/gemcitabine constructs (each at 10 nM; Figure 4A and B, respectively). Twenty-four hours after transfection, the relative level of CHK1 and RAD17 RNA was determined by SYBR Green RT-qPCR (Figure 4A and B). The expression level of β-actin was used as a normalizer. Under these conditions, placing two gemcitabines on the NS siRNA had no effect on gene silencing. However, adding two gemcitabines to the sense strand of CHK1 siRNA (Chk1-3′s2G) or on RAD17 (Rad17-s2G) resulted in silencing of the relevant gene by >90% in each case. The degree of silencing was equivalent between the gemcitabine-modified and the unmodified siRNA sequences (Figure 4A and B). The silencing by CHK1 siRNA translated into a reduction of the protein as shown in the western blot (Figure 4C). No silencing of CHK1 or RAD17 was observed with NS siRNA (modified with gemcitabine or not; Figure 4A); consequently, there was no change in protein level in CHK1 in the western blot (Figure 4C). CHK1 siRNA alone (Chk1) and in combination with gemcitabines (Chk1-3′s2G) produced a marked increase in phospho-gamma-H2AX levels when measured at 40nM and exposed for 24 h (Figure 4C). Gemcitabine produced an increase in phospho-gamma-H2AX only at 80nM and 48 h exposure (data not shown). In order to validate that the response we were seeing with the gemcitabine constructs was being mediated at least in part by the gemcitabine moieties being released inside the cells, we examined the effect of gemcitabine on expression level of RRM1 and RRM2. Gemcitabine at increasing concentrations (5–80nM) produced an increase in expression of RRM1 and RRM2 in a Western blot (Figure 4D). Gemcitabine-containing constructs mimicked this effect, whereas siRNAs lacking gemcitabines did not show this increase (Figure 4E). We further validated that this response could be mimicked by the constructs using RT-qPCR (Figure 4F and G). RRM1 was upregulated ∼3-fold in cells exposed to gemcitabine alone (Gem) but was reduced ∼40% in response to CHK1 siRNA (Chk1; Figure 4F). Adding gemcitabine to the NS siRNA (NS-s2G) produced a similar increase as mediated by gemcitabine alone (3.3-fold). However, addition of gemcitabine to CHK1 siRNA (Chk1-3′s2G) produced a slightly smaller effect on RRM1 induction (2.8-fold). Gemcitabine alone produced a much greater increase in RRM2 level (7-fold) as measured using RT-qPCR (Figure 4G). A similar response was observed with the NS siRNA containing two gemcitabines. CHK1 siRNA alone also produced a small increase in the RRM2 signal (1.9-fold) and this was reflected by a larger increase in the RRM2 signal for the Chk1-3′s2G (10.6-fold) than for NS-2G (7.5-fold)—presumably due to the additive effect of gemcitabine and CHK1 silencing on the signal.
Figure 4.
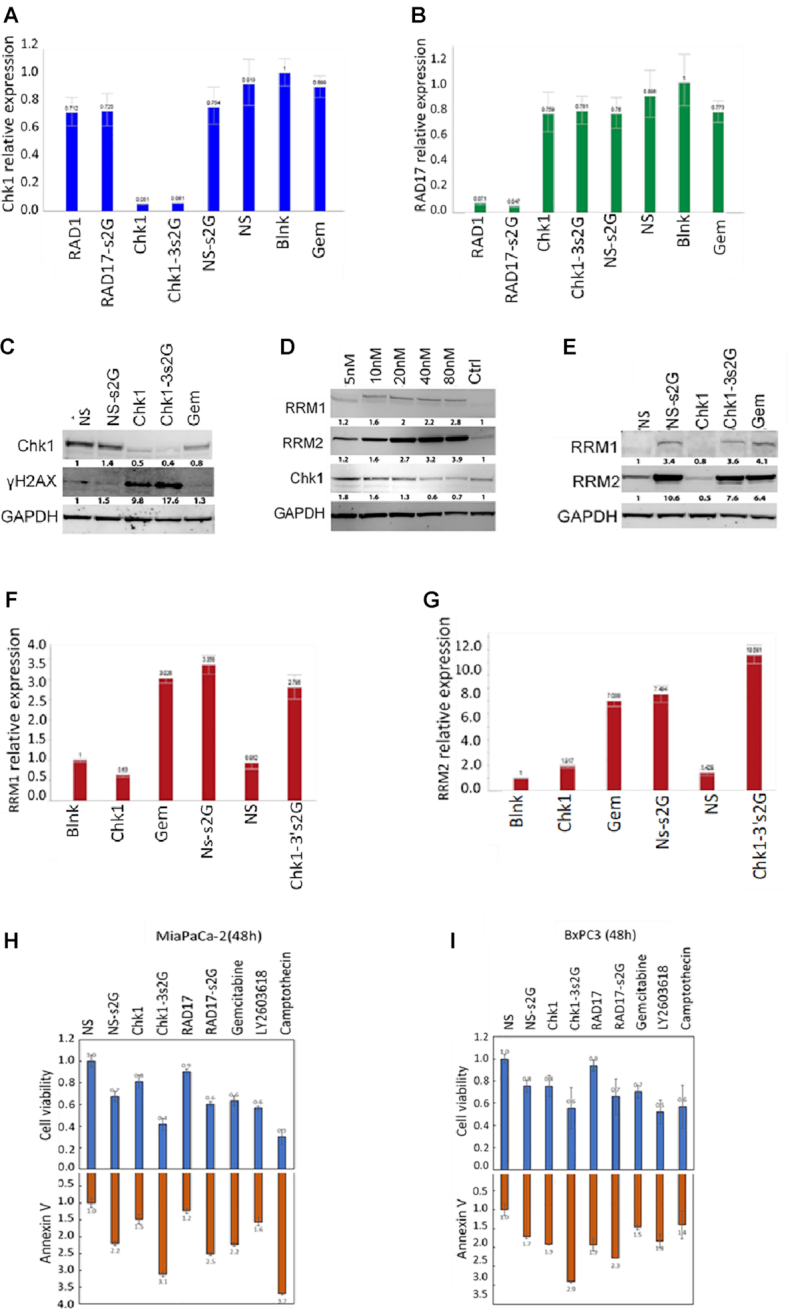
Effect of modified and unmodified siRNA on gene expression and apoptosis induction. (A, B) CHK1 and RAD17 gene expression levels of cells transfected with siRNA containing gemcitabines inserted within the sense/antisense strands. MIA PaCa-2 cells (2.5 × 105 cells/well in a 12-well plate) were transfected with gemcitabine-modified siRNAs (Chk1-3′s2G, Rad17-s2G and NS-s2G) and non-modified siRNAs (Chk1, Rad17 and NS) at a concentration of 10nM for 24h. The gene expression level of CHK1 and RAD17 RNA was determined by SYBR Green RT-PCR and normalized to β-actin. (C) Western blot of CHK1 and gamma-H2AX levels in MIA PaCa-2 cells exposed to 40nM of test agents after 24h exposure. (D, E) Western blot of RRM1 and RRM2 levels in MIA PaCa-2 cells exposed to increasing concentrations of gemcitabine for 48h and exposed to 40nM of test agents for 48h. (F, G) RT-qPCR analysis of expression of RRM1 and RRM2. MIA PaCa-2 cells were exposed to 40nM of each reagent indicated for 48h and then cells were harvested and used to assess expression of RRM1 and RRM2 using RT-qPCR. (H, I) MIA PaCa-2 and BxPC3 cells were transfected with either 100nM of gemcitabine-modified or non-modified siRNAs or exposed to gemcitabine at a concentration of 100nM. LY2603618 and camptothecin were added at a concentration of 2μM. Annexin V level (lower panels) was determined after a 48h incubation by using RealTime-Glo Annexin V Apoptosis and Necrosis Assay (Promega, Madison, WI, cat. # JA1011). Cell viability (upper panels) was assessed with RealTime-Glo MT Cell Viability Assay (Promega, Madison, WI, cat. # G97111). All values were compared to values generated for cells treated with NS siRNA and reported as the fold change in cell viability or annexin V level. Values represent the mean ± SD (n = 4) from a single experiment repeated twice.
To ensure that the data for cell viability corresponded with apoptosis of the cells, we used the RealTime-Glo MT Cell Viability Assay (Promega) and the RealTime-Glo Annexin V Apoptosis and Necrosis Assay (Promega) to measure effects of treatment with the constructs on cell viability and apoptosis in MIA PaCa-2 cells (Figure 4H) and BxPC3 cells (Figure 4I). Results were normalized to the effect of NS siRNA. For all treatments, there was a correlation between effect on cell viability and effect on apoptosis—with decreasing viability resulting from an increase in apoptosis.
Effect of number of gemcitabine modifications and location on efficacy
Using the sequences for the sense strand and antisense strand modifications shown in Table 1, we examined the effect on cell viability of transfecting these reagents into BxPC3 (Figure 5A) and MIA PaCa-2 cells (Figure 5B) and evaluated specificity by comparing effects in a liver cancer cell line Huh7 (Figure 5C).
Figure 5.
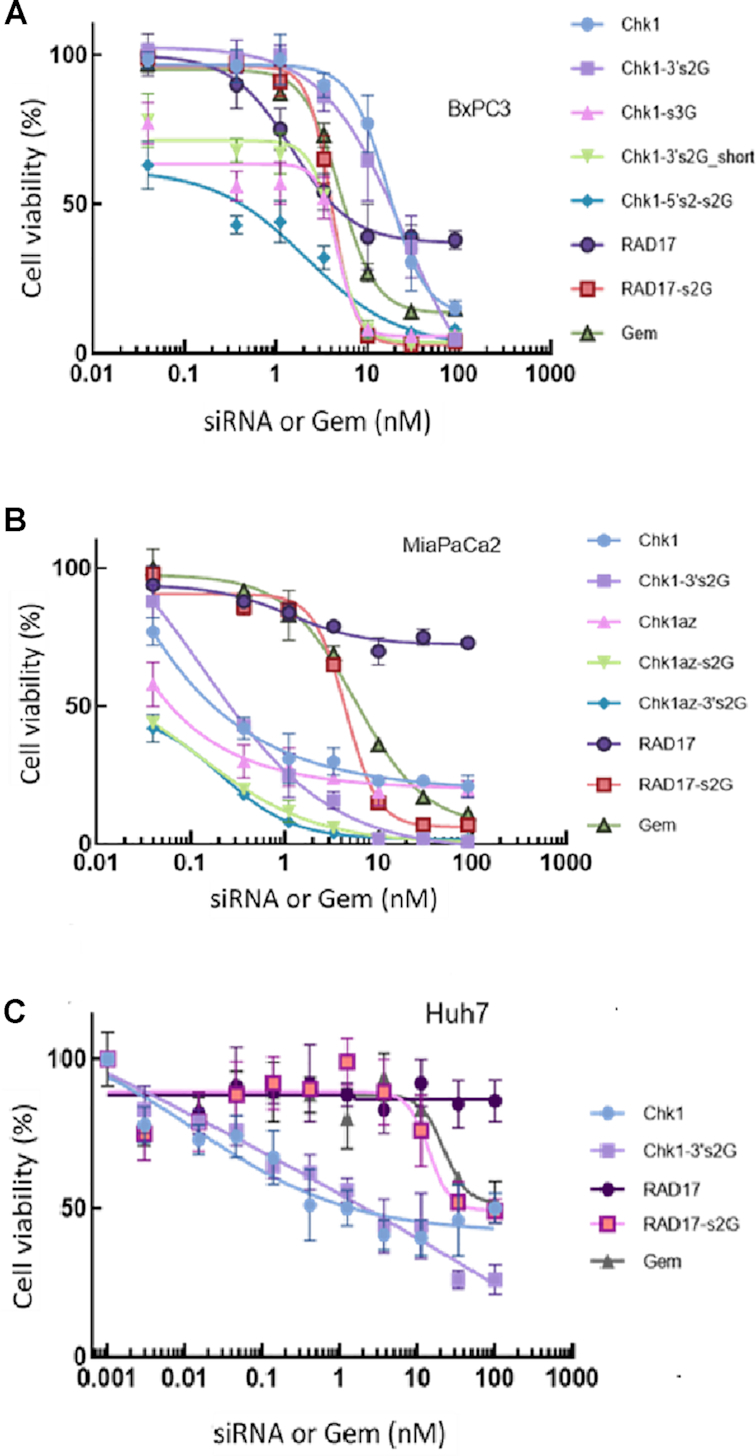
Effect of gemcitabine position and number of modifications in the sense strand of the siRNA on efficacy and potency of the siRNA/gemcitabine combination. Various unmodified/gemcitabine-modified siRNAs (Table 1) were transfected into BxPC3 (A), MIA PaCa-2 (B) or Huh7 cells (C) at a density of 1000 cells/well in 384-well plates and incubated for 96h. Cell viability was measured using CellTiter-Glo® 2.0 reagent. Data are shown as the mean ± SD for four replicates for each data point. The experiment was replicated three times with similar results. All data are normalized to unmodified NS siRNA treatment (data not shown).
In BxPC3 cells (Figure 5A), unmodified siRNA against RAD17 (labeled RAD17) exhibited a dose-dependent decrease in cell viability with an IC50 of ∼4nM and a maximal efficacy of only 60% reduction in cell viability. When this siRNA had two gemcitabine moieties added at the 3′ end of the sense strand, we observed a dramatic improvement in efficacy (100% cell killing at 30nM) with no change in the potency (IC50 ∼ 4nM).
A 25-mer blunt-ended siRNA sequence against CHK1 was demonstrated to have a very potent effect on cell viability when used without gemcitabine modification (Chk1; IC50 0.25nM and efficacy 90% at 100nM). Like RAD17, adding two gemcitabines to the 3′ end of the sense strand resulted in a product (Chk1-3′s2G) with a similar IC50 value but greater efficacy (∼100% killing at 30nM in MIA PaCa-2 cells; Figure 5B).
Shortening the 5′ end of the sense strand of the Chk1 siRNA containing either two gemcitabines (Chk1-3′s2G_short) or three gemcitabines (Chk1-3′s3G) resulted in the same effect (IC50 ∼ 3nM; maximal efficacy of 83–97% at 30nM; Figure 5A).
Using the same sense strand sequence with four gemcitabines replacing the cytidines within the siRNA sequence (Chk1-5′s2_s2G) gave similar IC50 and efficacy values to the sequence containing just two gemcitabines (Chk1-3′s2G). This result suggests that the gemcitabine released from the sense strand containing either two or four gemcitabines produces the same improvement in efficacy—suggesting a saturation effect or a lack of efficacy in releasing or processing the gemcitabine moieties. We thought that the exonucleases may not be able to cleave and release a second gemcitabine when it was directly attached to another gemcitabine in the same sequence (e.g. in Chk1-3′s2G in Table 1). However, even when we separated the gemcitabines by nuclease-sensitive nucleotides (e.g. in Chk1-3′s3G or Chk1-5′s2_3′s2G), we did not observe any further improvement in potency or efficacy (Figure 5A). These results suggest that release of one or two gemcitabines from each siRNA may provide the effects seen.
We studied the effect of gemcitabine and unmodified siRNAs (with the same 96 h treatment duration as used against BxPC3 cells) on MIA PaCa-2 cells (Figure 5B), another pancreatic cancer cell line. The 19-mer siRNA (Chk1az) and the 25-mer blunt-ended siRNA designed against CHK1 (Chk1) both showed similar efficacy (maximal inhibition ∼80%). The RAD17 siRNA, however, showed greatly reduced efficacy when used alone against the MiaPaCa2 cells (maximum inhibition of only ∼30%) than against BxPC3 cells (maximal inhibition of ∼60%).
Addition of two gemcitabines to the CHK1-Az 19-mer sequence, either on the 3′ end of the sense strand (Chk1az-3′s2G) or by replacement of the cytidines within the sense strand (Chk1az-s2G), resulted in a significant improvement in efficacy of the siRNA in killing MIA PaCa-2 cells (maximal inhibition ∼100% at 10nM; Figure 5B). The same was true for the addition of two gemcitabines on the 3′ end of the sense strand of the 25-mer siRNA (Chk1-3′s2G), which also resulted in maximal efficacy at 10nM with ∼100% cell killing versus MIA PaCa-2 cells. While the unmodified 25-mer siRNA targeting RAD17 (RAD17) showed almost no efficacy against MIA PaCa-2 cell viability (maximal ∼25%), addition of two gemcitabines to the 3′ end of the sense strand of this siRNA (RAD17-s2G) dramatically increased the efficacy of this construct (inhibition of 93% at 30nM). All modified siRNAs now demonstrated a maximal efficacy beyond that achievable using gemcitabine alone (maximal inhibition of 90% at 100nM).
Examining the effect of gemcitabine and siRNA constructs on the liver cancer cell line Huh7 (Figure 5C), we observed that gemcitabine alone has limited efficacy—producing a maximal cell killing of only 40% at the highest concentration tested (100nM). Unmodified siRNA against RAD17 at the same concentration demonstrated no effect on cell viability. RAD17-s2G only produced a 40% reduction in cell viability at 100nM. CHK1 siRNA alone showed some inhibitory activity against the cell line and this was augmented by inclusion of gemcitabines (Chk1-3′s2G). However, the number of cells killed by the modified CHK1 siRNA in Huh7 cells was lower than that observed for pancreatic cells at 100nM [∼65% in Huh7 (Figure 5C) compared with 100% killing in the pancreatic cells BxPC3 and MIA PaCa-2 (Figure 5A and B)].
Gemcitabine-modified siRNAs demonstrate efficacy across a number of pancreatic tumor cells
We tested the unmodified 25-mer siRNA against CHK1 (Chk1), the gemcitabine-modified sequence (Chk1-3′s2G) and gemcitabine alone (Gem) for efficacy and potency against a number of pancreatic tumor types (Figure 6). The gemcitabine-modified siRNA against CHK1 showed a dramatic improvement in efficacy when used in each of the five cell lines (Capan-1, MIA PaCa-2, CFPAC-1, Panc1 and BxPC3 cells). The gemcitabine-modified siRNA (33nM) produced >80% killing in all of these cell lines.
Figure 6.

Efficacy against pancreatic tumor cell types. One thousand cells per well in 384-well plates were transfected with the siRNAs or treated with gemcitabine for 96h. Cell viability was measured using CellTiter-Glo® 2.0 reagent. Dose curves in (A) BxPC3, (B) CFPAC-1, (C) Panc1, (D) Capan-2 and (E) MIA PaCa-2. Chk1: 25-mer siRNA against CHK1 gene; Chk1-3′s2G: CHK1 modified with two gemcitabines at the 3′ end of the sense strand; Gem: gemcitabine alone. Data are shown as the mean ± SD for four replicates for each data point. The experiment was replicated three times with similar results. All data are normalized to unmodified NS siRNA treatment (data not shown).
Co-transfection with WEE1 siRNA improves efficacy and potency of CHK1–gemcitabine construct
Inclusion of WEE1 siRNA or the small molecule WEE1 inhibitor (MK1775) together with CHK1–gemcitabine siRNA (Chk1-3′s2G) produces a dramatic shift in the IC50 value compared with CHK1–gemcitabine alone (Chk1-3′s2G) in MIA PaCa-2 and BxPC3 cells (Figure 7). The IC50 for CHK1–gemcitabine (Chk1-3′s2G) + WEE1 siRNA (at 0.1nM) was reduced to <0.9nM in MIA PaCa-2 cells compared with CHK1–gemcitabine (Chk1-3′s2G) alone at ∼3nM and gemcitabine alone at ∼10nM. This represents an improvement of ∼10-fold. Maximal efficacy of the combination with WEE1 siRNA or the inhibitor was equivalent to CHK1–gemcitabine alone (Chk1-3′s2G) with a maximal inhibition of cell viability in MIAPaCa-2 cells of 100% occurring with both treatments at ∼6nM.
Figure 7.
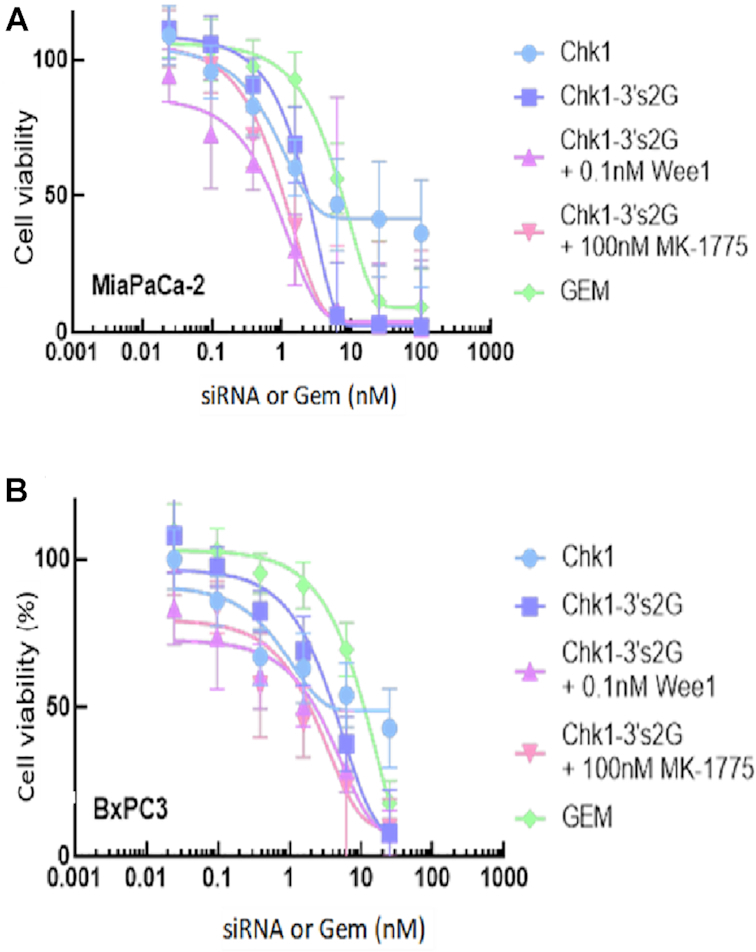
Enhanced efficacy in the presence of WEE1 siRNA and WEE1 inhibitor. MIA PaCa-2 cells (A) or BxPC3 cells (B) seeded at a density of 1000cells/well in a 384-well plate were treated with gemcitabine or transfected with various gemcitabine-modified siRNAs for 96h. Cell viability was measured by CellTiter-Glo® 2.0 reagent. Chk1: unmodified CHK1 siRNA; Chk1-3′s2G: CHK1 siRNA with two gemcitabines on the 3′ end of the sense strand; Chk1-3′s2G + 0.1nM WEE1: Chk1-3′s2G + 0.1nM WEE1 siRNA; Chk1-3′s2G + 100nM MK-1775: Chk1-3′s2G + 100nM MK-1775 (small molecule inhibitor of WEE1). All data are normalized to unmodified NS siRNA treatment (data not shown). Curves are generated using asymmetric sigmoidal regressions (5PL) conducted in GraphPad Prism. The data are presented as the mean ± SD from a single experiment with four replicates. The experiment was repeated twice with similar results.
DISCUSSION
We rationalized that we could use gemcitabine amidites to synthesize a hybrid siRNA–gemcitabine molecule that could then be delivered as a single agent—ensuring co-delivery of gemcitabine and siRNA to the same cell. We further speculated that it should be possible to include gemcitabines within an siRNA duplex sequence designed to target a gene whose suppression would augment the action of the gemcitabine in killing tumor cells—reducing the dosage required to observe a therapeutic effect and decreasing the potential for toxic side effects observed when gemcitabine alone is administered to patients.
In the current studies, we used the previously published 19-mer siRNA sequence against CHK1 (6) that had demonstrated potentiation of gemcitabine action when gemcitabine was administered separately from the siRNA. CHK1 is required for homologous recombination in mammalian cells (9). When we appended four or six gemcitabines on the 3′ end of the sense strand, we observed no additive effect greater than adding two gemcitabines and there was no increase in potency or efficacy (Figure 1). Adding two, four or six gemcitabine moieties only resulted in the potency expected to correspond to the release of one gemcitabine from each sequence. The degree of effect observed using the modified oligos was dependent on the time of exposure of the cells to the compounds (Figure 2). This may be as a result of exo- or endonucleases not being able to cleave the gemcitabines or it may be as a result of very slow release and subsequent phosphorylation to activate the gemcitabine moieties (10) that are typically taken up by nucleoside transporters (11).
Gemcitabine is an analog of the nucleotide cytidine and so can be incorporated directly into an siRNA sequence in place of the cytidines. In order to determine whether gemcitabines would be better processed if separated by native nucleotides, we identified two cytidines in the Azorsa siRNA sequence (6) that could be replaced on the sense strand and four on the antisense strands, respectively. We observed that insertion of gemcitabines in place of cytidines in the sense strand resulted in a product (Chk1-3′s2G) with improved IC50 and efficacy (IC50 = 0.38nM; Figure 3A) compared with unmodified siRNA (Chk1) incubated for the same time (IC50 = 3nM; Figure 3A).
Adding gemcitabines to the antisense strand in place of cytidines had the potential to interfere with the loading of the siRNA into the RISC complex (12,13) or the subsequent recognition of the cognate mRNA and cleavage by Dicer since, on interaction with the siRNA duplex, Ago2 cleaves the sense strand between nucleotides 10 and 11 from the 5′ end of the antisense strand (12). Inclusion of gemcitabines in and around this location had the potential to inhibit the cleavage. However, we also saw that adding two gemcitabines to the antisense strand (e.g. Chk1az-a2G; Figure 3B) improved potency and efficacy (IC50 1.8nM) but not as marked as for the sense strand (Chk1az-s2G; Figure 3B). Furthermore, combining the two strands [now containing four gemcitabines total (Chk1az-a2s2G)], we did not see a further improvement in potency (1.98nM) that might be expected if the principal contribution was from release of gemcitabines from both strands (Figure 3B).
RNAi approaches are prone to off-target effects and manipulating the seed sequence might change the target spectrum of the siRNA and might decrease knockdown of the target while also increasing the potency for off-targets. However, the gemcitabines were added at the 3′ end of the sense strand—and this is not the seed region recognition site for siRNAs (the seed region is defined as a six-nucleotide stretch corresponding to positions 2–7 on the antisense siRNA strand) (14). We purposefully avoided replacing cytidines with gemcitabines at these positions in the antisense strand to avoid this potential issue. Furthermore, inclusion of two gemcitabines at non-seed regions in the CHK1 sequence (e.g. in Chk1az-s2G) produced an identical degree of inhibition (as in Chk1az-3′s2G; see Figure 3A).
Gemcitabine addition as a control for effects of the constructs was typically performed without the use of lipofectamine as a delivery agent. However, there was concern that lipofectamine may alter the sensitivity of the cells to gemcitabine in the constructs, so we analyzed the effect of lipofectamine on the dose response effect of gemcitabine in two of the cell lines (BxPC3 and MIAPaCa-2 cells; Figure 3C). No significant difference was observed in the IC50 values for gemcitabine added with or without lipofectamine—alleviating this concern.
RAD17 was previously identified as another target that demonstrated a dramatic improvement in the action of gemcitabine in reducing cell viability when silenced by siRNAs (7). RAD17 acts upstream of CHK1 and is involved in detecting gemcitabine-induced stalled replication forks (7). We therefore designed an siRNA against RAD17 and modified this sequence with gemcitabines as for the CHK1 sequence.
The efficacy of the modified and unmodified siRNAs was confirmed in RT-qPCR analysis examining the silencing induced by the CHK1 and RAD17 constructs (Figure 4A and B). We observed target-specific gene silencing by the siRNAs—either unmodified siRNA or siRNA modified with gemcitabine produced equivalent silencing against CHK1 or RAD17, respectively, with >90% silencing by each species (Figure 4A and B).
Both CHK1 25-mer siRNA (Chk1) and gemcitabine-modified 25-mer siRNA (Chk1-3′s2G) demonstrated a reduction of the target protein (CHK1) in western blot analysis, whereas an NS siRNA or a gemcitabine-modified NS siRNA (NS-s2G) had no effect on protein levels (Figure 4C), suggesting that the siRNA functions independently, irrespective of the presence of the gemcitabines. Furthermore, we observed an increase in phospho-gamma-H2AX levels induced by Chk1 alone or Chk1 modified with gemcitabines (Chk1-3′s2G; Figure 4C). The study was performed using 40nM of the constructs and, at this concentration, gemcitabine had little effect on phospho-gamma-H2AX levels itself (we observed an effect of gemcitabine on gamma-H2AX induction only at 80nM and at 48h incubation; data not shown). Consequently, the change in gamma-H2AX was due primarily to the Chk1 silencing—in agreement with previous publications (15) showing that depleting CHK1 results in an increase in phospho-gamma-H2AX signal. The gamma-H2AX signal was dramatically increased in the gemcitabine-containing siRNA construct (Chk1-3′s2G; Figure 4C)—suggesting some additive effect of gemcitabine on the induction caused by Chk1 silencing.
To determine whether the response observed with the gemcitabine construct was a result of release of gemcitabine, we examined the effect of gemcitabine on several key proteins. Gemcitabine has previously been demonstrated to result in an upregulation of RRM1 and RRM2 in western blots after a 48h exposure to 40nM gemcitabine in MIA PaCa2 cells (16). In a dose response assay using western blot analysis to measure RRM1 and RRM2 levels in response to gemcitabine (Figure 4D), we observed an increase in RRM1 and a greater increase in RRM2 (compared to control) equivalent to that observed previously by Liang et al. (16) using the same concentration of gemcitabine at the same time point. Moreover, the siRNA constructs containing gemcitabine modifications mimicked the effect of gemcitabine alone in increasing levels of RRM1 and RRM2 as evidenced by the western blot (Figure 4E).
Furthermore, using RT-qPCR to measure the transcript for RRM1 and RRM2 at 24h, we see that gemcitabine alone mimics the response seen in the western blots (increasing RRM1 and RRM2 expression; Figure 4F and G). We also see increases in RRM1 equivalent to that induced by gemcitabine alone by the siRNA constructs containing gemcitabine (either NS siRNA: NS-s2G or Chk1: Chk1-3′s2G). The unmodified siRNAs (NS or CHK1, respectively) did not show an increase in RRM1 signal—suggesting that the increase is mediated by the gemcitabines present on the siRNA constructs. Chk1-3′s2G produced a larger increase in RRM2 levels than either gemcitabine alone or the NS siRNA gemcitabine construct (NS-s2G; Figure 4G). These data further confirm the mechanism of action of the siRNA construct by release of gemcitabine to produce these effects as shown previously (16).
We examined whether the data for cell viability corresponded with apoptosis of the cells by comparing constructs in assays using RealTime-Glo Cell Viability Assay (Promega) with the RealTime-Glo Annexin V Apoptosis and Necrosis Assay (Promega) to measure cell viability and apoptosis in BxPC3 and MIA PaCa-2 cells (Figure 4H and I). Results were normalized to the effect of NS siRNA. For all treatments, there was a correlation between effect on cell viability and effect on apoptosis—with decreasing viability resulting from an increase in apoptosis. Camptothecin and the CHK1 inhibitor LY2603618 were used as positive controls in the assay. As shown in Figure 4H and I, there was a direct correlation in the signal observed for cell viability with the signal for apoptosis where an increase in the observed apoptosis signal resulted in a decrease in cell viability.
As in (7), RAD17 siRNA alone had almost no effect on cell viability in MIA PaCa-2 cells and only produced ∼60% reduction in cell viability in BxPC3 cells—even at the highest concentrations tested (100nM). However, appending two gemcitabine moieties onto the 3′ end of the sense strand of this siRNA resulted in >90% inhibition at 30nM—demonstrating the synergism between the siRNA and appended gemcitabine (Figure 5).
To examine specificity of the constructs for pancreatic cancer cells, we also explored the effect of gemcitabine, and unmodified and gemcitabine-modified siRNAs against CHK1 and RAD17 on the liver cancer cell line Huh7 (Figure 5C) that, like all of the pancreatic tumor cell lines tested, also carries mutations in p53 (17). The cell line showed no response to unmodified RAD17 siRNA and only limited sensitivity to gemcitabine alone (∼40% inhibition at the highest concentration tested, 100nM). The effect of RAD17-s2G also showed a limited response (equivalent to gemcitabine alone). Chk1 siRNA alone yielded ∼40% inhibition at the highest concentration tested and this was marginally increased in the gemcitabine construct (Chk1-3′s2G; Figure 5C). The effect of RAD17-s2G and Chk1-3′s2G in this cell line was much lower than the effect observed in BxPC3 and MiaPaca2 cells (Figure 5A and B). Huh7 cell line is still a tumor cell line that divides at a faster rate than many of the pancreatic cancer cells tested (doubling time ∼36h compared with Panc1 ∼52h, MiaPaCa2 ∼40h, BxPC3 ∼48–60h, CFPAC-1 cells ∼31h and Capan-2 ∼96h). Consequently, we may expect some sensitivity to gemcitabine mechanism of action to inhibit cell growth, but the lower efficacy of the constructs in this cell line suggests some degree of specificity toward pancreatic tumor cells.
Chk1 siRNAs modified with gemcitabines showed their relevance for use as a potential therapeutic to treat pancreatic cancer since they demonstrated efficacy against a number of pancreatic cancer cells where gemcitabine had little effect on cell viability (Figure 6). The greatest improvement in efficacy was observed in Capan-2 cells, followed by Panc1 and then CFPAC-1; MIA PaCa-2 and BxPC3 both demonstrated the smallest improvement in efficacy. However, in all cells tested, Chk1-3′s2G produced >80% inhibition of cell viability at 30nM.
Previous publications determined that WEE1 inhibitors can synergize with CHK1 inhibitors to reduce proliferation in various cell lines (18–20). It was further suggested that WEE1 and CHK1 inhibition could augment activity of gemcitabine (21). WEE1 is a tyrosine kinase and one of the key proteins regulating the G2 checkpoint (22). The tumor suppressor protein p53 is a key regulator of cell cycle arrest in response to DNA damage (23). The synthetic lethal effect of CHK1 and WEE1 inhibition in combination with DNA-damaging agents has been shown to be dependent on p53 deficiency (22,24–26). Since MIA PaCa-2 cell lines carry deleterious missense mutations in the DNA binding domain of p53 (17,27), we utilized these cells to evaluate the effect of Chk1–gemcitabine siRNA and WEE1 siRNA as well as the wee1 inhibitor (MK1775) on cell viability (Figure 7A). Silencing WEE1 definitely augmented the activity of CHK1 siRNA modified with gemcitabine (Chk1-3′s2G) in MIA PaCa-2 cells (Figure 7A) with a shift in the dose response curve from an IC50 of ∼3nM (for Chk1-3′s2G alone) to <1nM in the presence of 0.1nM WEE1 siRNA. This compared with gemcitabine alone at ∼10nM IC50. The combination therefore improved the IC50 compared to gemcitabine alone by >10-fold and may provide additional benefit in a potential therapeutic where both molecules are delivered concomitantly to cancer cells. A similar response was also identified using the small molecule wee1 inhibitor MK1775—suggesting that this is consistent with inhibition of Wee1 signaling by the two distinct mechanisms. Similar responses were also observed in BxPC3 cells (Figure 7B).
Combined WEE1 and CHK1 inhibition forces mitotic entry from S phase in the absence of chemotherapy (24) and these data demonstrate that there is also a significant synergism with gemcitabine.
While small molecules such as MK1775 (also known as AZD1775, a WEE1 inhibitor) have demonstrated sufficient safety to be included with gemcitabine in clinical studies (28), the hematological toxicity observed may be avoided by using a nanoparticle-formulated siRNA cocktail delivered to the tumor site.
We have previously demonstrated the utility of a histidine–lysine branched peptide nanoparticle (HKP) carrying siRNA on viability of tumors in vivo (29). HKP has proven to be safe and effective at siRNA delivery to xenograft tumors in vivo and can be formulated into nanoparticles of ∼100 nm diameter by direct mixing with the siRNAs. HKP can carry multiple siRNAs (30) and consequently can be used to deliver Chk1–gemcitabine siRNA and Wee1 siRNA for treating tumors in vivo. It will be important to validate the siRNA–gemcitabine combinations work in vivo and we are currently pursuing these experiments.
In summary, we have demonstrated that incorporation of gemcitabines into siRNA sequences that target genes whose suppression synergizes with gemcitabine provides a pronounced reduction in viability across multiple pancreatic tumor cell lines as compared to treatment with either agent alone. Additionally, combining the gemcitabine-modified siRNA against CHK1 with an siRNA against WEE1 may further augment activity. These gemcitabine-modified siRNAs, with or without additional potentiating agents, may represent a novel therapeutic option for treating pancreatic and other cancers.
Contributor Information
Vera Simonenko, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
Xiaoyong Lu, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
Eric Roesch, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
Daniel Mutisya, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
Chunbo Shao, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
Qian Sun, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
Athéna Patterson-Orazem, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
Marcus McNair, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
Aranganathan Shanmuganathan, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
Patrick Lu, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
David M Evans, Sirnaomics Inc., Suite 280, 401 Professional Drive, Gaithersburg, MD 20879, USA.
FUNDING
All funding for the published work was provided by Sirnaomics Inc.
Conflict of interest statement. None declared.
REFERENCES
- 1. Burris H.A. 3rd, Moore M.J., Andersen J., Green M.R., Rothenberg M.L., Modiano M.R., Cripps M.C., Portenoy R.K., Storniolo A.M., Tarassoff P.et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 1997; 15:2403–2413. [DOI] [PubMed] [Google Scholar]
- 2. Jo J.H., Song S.Y. Abdeldayem H. Chemotherapy of cholangiocarcinoma: current management and future directions. Topics in the Surgery of the Biliary Tree. 2018; London: IntechOpen; 35–52. [Google Scholar]
- 3. Muggia F., Diaz I., Peters G.J. Nucleoside and nucleobase analogs in cancer treatment: not only sapacitabine, but also gemcitabine. Expert Opin. Investig. Drugs. 2012; 21:403–408. [DOI] [PubMed] [Google Scholar]
- 4. Le T.N., Harvey R.E., Kim C.K., Brown J., Coleman R.L., Smith JA. A retrospective evaluation of activity of gemcitabine/platinum regimens in the treatment of recurrent ovarian cancer. Gynaecol. Oncol. Res. Pract. 2017; 4:16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xie Z., Zhang Y., Jin C., Fu D. Gemcitabine-based chemotherapy as a viable option for treatment of advanced breast cancer patients: a meta-analysis and literature review. Oncotarget. 2018; 9:7148–7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Azorsa D.O., Gonzales I.M., Basu G.D., Choudhary A., Arora S., Bisanz K.M., Kiefer J.A., Henderson M.C., Trent J.M., Von Hoff D.D.et al. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J. Transl. Med. 2009; 7:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fredebohm J., Wolf J., Hoheisel J.D., Boettcher M. Depletion of RAD17 sensitizes pancreatic cancer cells to gemcitabine. J. Cell Sci. 2013; 126:3380–3389. [DOI] [PubMed] [Google Scholar]
- 8. Ma Y., Mou Q., Zhu L., Su Y., Jin X., Feng J., Yan D., Zhu X., Zhang C. Polygemcitabine nanogels with accelerated drug activation for cancer therapy. Chem. Commun. 2019; 55:6603–6606. [DOI] [PubMed] [Google Scholar]
- 9. Sørensen C.S., Hansen L.T., Dziegielewski J., Syljuåsen R.G., Lundin C., Bartek J., Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005; 7:195–121. [DOI] [PubMed] [Google Scholar]
- 10. Plunkett W., Huang P., Searcy C.E., Gandhi V. Gemcitabine: preclinical pharmacology and mechanisms of action. Semin. Oncol. 1996; 23:3–15. [PubMed] [Google Scholar]
- 11. Zhang J., Visser F., King K.M., Baldwin S.A., Young J.D., Cass C.E. The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev. 2007; 26:85–110. [DOI] [PubMed] [Google Scholar]
- 12. Carthew R.W., Sontheimer E.J. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009; 136:642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Filipowicz W. RNAi: the nuts and bolts of the RISC machine. Cell. 2005; 122:17–20. [DOI] [PubMed] [Google Scholar]
- 14. Jackson A.L., Bartz S.R., Schelter J., Kobayashi S.V., Burchard J., Mao M., Li B., Cavet G., Linsley P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003; 21:635–637. [DOI] [PubMed] [Google Scholar]
- 15. Gagou M.E., Zuazua-Villar P., Meuth M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol. Biol. Cell. 2010; 21:739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liang M., Zhao T., Ma L., Guo Y. CHK1 inhibition sensitizes pancreatic cancer cells to gemcitabine via promoting CDK-dependent DNA damage and ribonucleotide reductase downregulation. Oncol. Rep. 2018; 39:1322–1330. [DOI] [PubMed] [Google Scholar]
- 17. Bouaoun L., Sonkin D., Ardin M., Hollstein M., Byrnes G., Zavadil J., Olivier M. TP53 variations in human cancers: new lessons from the IARC TP53 Database and genomics data. Hum. Mutat. 2016; 37:865–876. [DOI] [PubMed] [Google Scholar]
- 18. Guertin AD., Martin MM., Roberts B., Hurd M., Qu X., Miselis N.R., Liu Y., Li J., Feldman I., Benita Y., Bloecher A.et al. Unique functions of CHK1 and WEE1 underlie synergistic anti-tumor activity upon pharmacologic inhibition. Cancer Cell Int. 2012; 12:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davies K.D., Cable P.L., Garrus J.E., Sullivan F.X., von Carlowitz I., Huerou Y.L., Wallace E., Woessner R.D., Gross S. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol. Ther. 2011; 12:788–796. [DOI] [PubMed] [Google Scholar]
- 20. Carrassa L., Chila R., Lupi M., Ricci F., Celenza C., Mazzoletti M., Broggini M., Damia G. Combined inhibition of Chk1 and Wee1 in vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle. 2012; 11:2507–2517. [DOI] [PubMed] [Google Scholar]
- 21. Koh S.B., Wallez Y., Dunlop C.R., Bernaldo de Quirós Fernández S., Bapiro T.E., Richards F.M., Jodrell D.I. Mechanistic distinctions between CHK1 and WEE1 inhibition guide the scheduling of triple therapy with gemcitabine. Cancer Res. 2018; 78:3054–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen Z., Xiao Z., Gu W.Z., Xue J., Bui M.H., Kovar P., Li G., Wang G., Tao Z.F., Tong Y.et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int. J. Cancer. 2006; 119:2784–2794. [DOI] [PubMed] [Google Scholar]
- 23. Alberts B. Molecular Biology of the Cell. 2002; NY: Garland Science. [Google Scholar]
- 24. Aarts M., Sharpe R., Garcia-Murillas I., Gevensleben H., Hurd M.S., Shumway S.D., Toniatti C., Ashworth A., Turner N.C. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012; 2:524–539. [DOI] [PubMed] [Google Scholar]
- 25. Landau H.J., McNeely S.C., Nair J.S., Comenzo R.L., Asai T., Friedman H., Jhanwar S.C., Nimer S.D., Schwartz G.K. The checkpoint kinase inhibitor AZD7762 potentiates chemotherapy-induced apoptosis of p53-mutated multiple myeloma cells. Mol. Cancer Ther. 2012; 11:1781–1788. [DOI] [PubMed] [Google Scholar]
- 26. Ma Z., Yao G., Zhou B., Fan Y., Gao S., Feng X. The Chk1 inhibitor AZD7762 sensitises p53 mutant breast cancer cells to radiation in vitro and in vivo. Mol. Med. Rep. 2012; 6:897–903. [DOI] [PubMed] [Google Scholar]
- 27. Petitjean A., Mathe E., Kato S., Ishioka C., Tavtigian S.V., Hainaut P., Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007; 28:622–629. [DOI] [PubMed] [Google Scholar]
- 28. Leijen S., van Geel R.M.J.M., Pavlick A.C., Tibes R., Rosen L., Razak A.R.A, Lam R., Demuth T., Rose S., Lee MA.et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J. Clin. Oncol. 2016; 34:4371–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan Z., Zou H., Tian F., Grandis J.R., Mixson A.J., Lu P.Y., Li L.Y. Human rhomboid family-1 gene silencing causes apoptosis or autophagy to epithelial cancer cells and inhibits xenograft tumor growth. Mol. Cancer Ther. 2008; 7:1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou J., Zhao Y., Simonenko V., Xu J.J., Liu K., Wang D., Shi J., Zhong T., Zhang L., Zeng L.et al. Simultaneous silencing of TGF-β1 and COX-2 reduces human skin hypertrophic scar through activation of fibroblast apoptosis. Oncotarget. 2017; 8:80651–80665. [DOI] [PMC free article] [PubMed] [Google Scholar]