

Abstract
Maintenance of genome stability suppresses cancer and other human diseases and is critical for organism survival. Inevitably, during a life span, multiple DNA lesions can arise due to the inherent instability of DNA molecules or due to endogenous or exogenous DNA damaging factors. To avoid malignant transformation of cells with damaged DNA, multiple mechanisms have evolved to repair DNA or to detect and eradicate cells accumulating unrepaired DNA damage. In this review, we discuss recent findings on the role of Sp1 (specificity factor 1) in the detection and elimination of cells accumulating persistent DNA strand breaks. We also discuss how this mechanism may contribute to the maintenance of physiological populations of healthy cells in an organism, thus preventing cancer formation, and the possible application of these findings in cancer therapy.
INTRODUCTION AND HISTORICAL PERSPECTIVE
Transcription factor Sp1 (specificity factor 1) was traditionally viewed as a basal transcription factor responsible for the regulation of housekeeping genes. However, recent data have indicated that Sp1 is an important player in the DNA damage response (DDR), controlling cellular life/death decisions in the event of DNA repair deficiency. These recent findings transform our understanding of the intracellular response to the accumulation of persistent DNA strand breaks and the role of Sp1 in this process.
Sp1, ubiquitously expressed in mammalian cells, was first isolated in 1983 by Dynan and Tjian as a eukaryotic transcription specificity factor that binds to the SV40 early promoter (1). It was later shown that Sp1 activity is modulated by three Cys-2–His-2 zinc finger motifs, which bind to a consensus 5′-GGGGCGGGG-3′ (GC box) element (2). Subsequently, Sp1 has been recognized to function as a transcription factor, required for transcription at many TATA promoters as well as regulating TATA-less promoters via protein–protein interactions or interplay with other transcription factors and components of the basal transcriptional machinery (3). Unsurprisingly, Sp1 is an essential gene and Sp1 knockout mice die at the very early stages of development, showing a broad range of abnormalities (4).
The importance of Sp1 in cellular function is not to be underestimated. Numerous studies have linked Sp1 to a multitude of cellular pathways and processes associated with the pathogenesis of a number of diseases, with cancer perhaps the best studied (5). Sp1 overexpression is seen in a number of cancer cell types, where high levels of Sp1 correlate with poorer patient prognosis [reviewed in (6)]. Downregulation of Sp1 in cancer cell lines (including breast, kidney, pancreatic, lung and colon cancers), triggered by Sp1 inhibitors or DNA damaging anticancer drugs, leads to decreased survival and the inhibition of cell growth [reviewed in (7)].
As with many other transcription factors, to accomplish its various functions, Sp1 undergoes different posttranslational modifications, including phosphorylation, O-linked glycosylation and sumoylation. Among these, phosphorylation that can lead to varying effects on Sp1 function is the best-studied modification [reviewed in (8)]. Phosphorylation by various kinases at different sites within the protein can result in inhibition or stimulation of DNA binding and transcriptional activity of Sp1, although in many cases the biological role of these modifications remains unknown (9). The first observation of the link between Sp1 phosphorylation and the DDR was published in 2007 by Oh et al. (10). They demonstrated that in normal human fibroblasts treated with hydrogen peroxide, Sp1 protein levels decreased progressively and that inhibition of ATM/ATR kinases partially prevented this downregulation. Interestingly, they also showed that the decrease in Sp1 protein level was due to its accelerated proteasomal degradation, suggesting that Sp1 level might be modulated by the DDR (10). Since this initial observation, several studies investigating phosphorylation of Sp1 in response to DNA damage have been published. For example, it was demonstrated that serine 101 (Ser-101) of Sp1 is a critical ATM phosphorylation site and that ATM-dependent phosphorylation of Sp1 modulates cellular survival in response to DNA damage (11). Moreover, it was also shown that silencing Sp1 with siRNA sensitizes cells to DNA damage, and that exogenous expression of wild-type Sp1, but not a mutant containing a Ser-101 to alanine substitution, can rescue cell survival after ionizing irradiation, confirming that phosphorylation of Ser-101 is required for the DDR (11,12). However, there were still several questions as to the exact function of Sp1 in the DDR that remained unanswered. In addition, understanding its putative role in the DDR has also been made more difficult by discrepancies between several studies, particularly with respect to Sp1 protein stability and activity. First, in studies that used cancer cells, the authors did not observe a decrease in Sp1 level after hydrogen peroxide treatment (11,12), as was shown for normal cells (10). Second, Ser-101 phosphorylation after DNA damage in cancer cells did not affect transcriptional activity from an Sp1-responsive promoter, thereby questioning whether the role of Sp1 in the DDR is related to its transcriptional activity (12). Third, cell cycle arrest is one of the major consequences of DDR activation in normal cells and DNA single-strand breaks (SSBs) produced directly by hydrogen peroxide treatment or during repair of oxidized DNA bases by base excision repair (BER) would not be converted to DNA double-strand breaks (DSBs) during DNA replication. Therefore, as ATM was considered a DSB-responsive protein (13) at the time these studies were carried out, it was not clear how hydrogen peroxide, which does not directly generate DSBs, led to ATM activation. Additionally, there are several lines of evidence to indicate that Sp1 may be involved in the transcription regulation of a large number of DNA repair genes. For example, early studies indicated that downregulation of Sp1 affects the transcription of genes involved in non-homologous end joining (NHEJ), and several studies have linked Sp1 with the expression of BER genes (14–20). This suggests that Sp1 may well regulate a large number of DNA repair genes. Regulation of BER genes through Sp1 is of particular interest as BER is the frontline pathway responsible for repairing physiologically relevant endogenous DNA lesions (21), including SSBs that occur due to chemical instability of the DNA molecule (22–24). In fact, the high number of the key BER genes (APE1, XRCC1 and DNA polymerase β) putatively regulated through Sp1 suggests that the protein may play a role in modulating gene expression across the whole BER pathway, thereby affecting BER homeostasis. It has also been proposed that Sp1 in combination with p53 might be involved in BER regulation (15,25), which would align with the hypothesis that Sp1 is involved in regulation of BER in response to DNA strand breaks. However, the observations that Sp1 participates in the regulation of DNA repair genes involved in BER has further complicated attempts to understand the role of Sp1 during the DDR. Although Sp1 is clearly required during the DDR and controls expression of DNA repair enzymes, it was not clear why the amount of Sp1 declines in response to persistent DNA damage, therefore likely reducing DNA repair capacity.
THE EMERGING ROLE OF Sp1 IN THE ELIMINATION OF CELLS CONTAINING PERSISTENT DNA DAMAGE
In light of these findings, a number of studies have begun to address the unanswered questions. In 2015, Khoronenkova and Dianov found that in normal human fibroblasts, unrepaired SSBs activate the ATM signalling pathway to ensure early detection of these lesions and coordinate the timing of their repair with DNA replication. Since it was already known that ATM phosphorylates Sp1 in response to DNA damage, these findings established an important link between BER, unrepaired SSBs, ATM and Sp1 phosphorylation (26), an axis investigated in detail in a later study. In a more recent publication, the same group demonstrated that persistent DNA strand breaks led to phosphorylation of Sp1 at Ser-101 solely by ATM and that this phosphorylation results in Sp1 proteasomal degradation (27). Using independent methods, the authors demonstrated that other major DDR kinases, including ATR, DNA-PKcs, CHK1 and CHK2, were not involved in Ser-101 phosphorylation in response to persistent DNA strand breaks. Moreover, the authors used an in vitro kinase assay to confirm that ATM phosphorylates Sp1 directly at Ser-101 and that this was the only canonical ATM site modified after DNA damage. Potentially, Sp1 may regulate expression (upregulation and downregulation) of many genes by binding to the GC box or through interaction with other transcription factors and chromatin proteins (5). In particular, it was also noted that loss of Sp1 led to upregulation of proapoptotic genes and was associated with increased sensitivity to cell elimination, either through apoptosis or by the innate immune system through natural killer cells (27). This was an important observation as although Sp1 has previously been linked to the expression of genes involved in apoptosis (28,29), the molecular mechanism was not clear. In support of these findings, a genome-wide shRNA screen integrated with transcriptomic and chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) analysis identified that Sp1 is an essential determinant of p53-induced apoptosis in human cancer cells (30).
The question, therefore, arose: ‘What is the role of Sp1 degradation in response to DNA damage?’ It was hypothesized that Sp1 is a part of the intracellular DNA damage signalling system that detects terminally damaged and potentially genetically compromised cells, to initiate their elimination in order to promote survival of the whole organism (27). The authors proposed a model whereby ATM-dependent regulation of Sp1, upon persistent DNA strand breaks, promotes the removal of DNA repair deficient and potentially precancerous cells (Figure 1). As both ATM and Sp1 phosphorylation are barely detectable under unstressed physiological conditions, the model suggests that the basal expression of BER genes and therefore BER capacity is sufficient to maintain genome stability (Figure 1, left-hand panel). In response to an accumulation of unrepaired DNA strand breaks, however, ATM phosphorylates Sp1, thereby destabilizing it. Loss of Sp1 results in downregulation of BER genes, which reduces the repair capacity of the pathway. Ultimately, this leads to a self-accelerating cycle resulting in the accumulation of more DNA strand breaks (Figure 1, right-hand panel). The authors proposed that this scenario might typify the response to excessive acute DNA damage, or to failures in DNA repair. In either case, this mechanism ensures the elimination of such DNA damage loaded, potentially precancerous cells. The possibility of switching from the repair process to apoptosis in case of excessive DNA damage was proposed earlier, although the exact mechanism was not clear (31). Interestingly, a similar system promoting selective cell elimination by downregulating DNA repair was recently proposed by Ponath and Kaina. They suggested that monocytes, which express lower levels of BER proteins, could be purposefully eliminated by macrophages, their differentiated counterpart, when these cells generate an reactive oxygen species (ROS) burst during the inflammatory response (32). Downregulation of BER via ATM phosphorylation of Sp1 is also reminiscent of the downregulation of RAD51 during apoptosis (33). It has previously been suggested that ATM may be under selective pressure for inactivation during tumorigenesis (34), and indeed, loss of ATM function has been correlated with cancer development (35,36). These observations might explain how potentially genetically unstable DNA repair deficient cells could escape DNA damage control mechanisms, promoting cancer formation. Based on this model, we propose that ATM-dependent modulation of Sp1 level plays a key role in the cellular life/death decision-making process in normal cells. Ordinarily, the level of DNA repair enzymes is able to provide an efficient repair of endogenous DNA damage and can be induced only moderately at the protein level (1.3–1.5-fold) in response to increased loads of DNA damage (37,38). We suggest that under unstressed conditions, or even after acute DNA damage, if arising DNA strand breaks are quickly repaired and do not accumulate, the amount of Sp1 and DNA repair proteins moderately fluctuates around basal levels (Figure 2). However, when the amount of DNA strand breaks exceeds DNA repair capacity (point of no return), then hyperactivation of ATM leads to Sp1 degradation and consequently to DNA repair downregulation and, ultimately, activation of cell death mechanisms (point of cell death, Figure 2).
Figure 1.
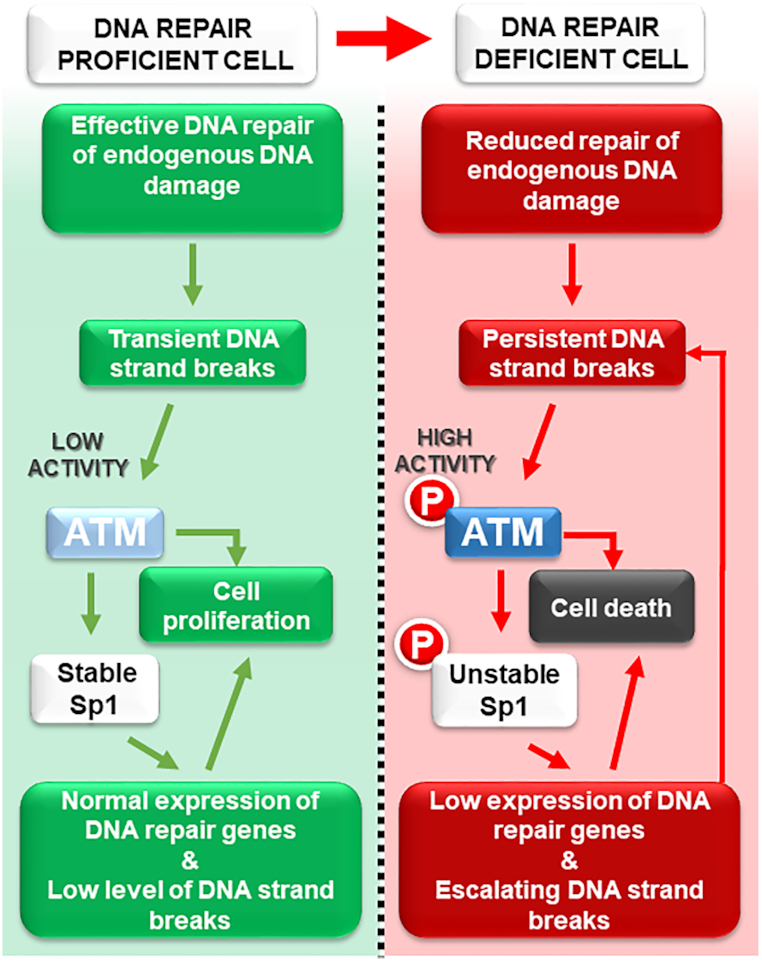
Detection and elimination of DNA repair deficient cells. Adapted from Fletcher et al. (27). Left: In a DNA repair proficient cell (normal cells), DNA damage is kept in check by sufficient DNA repair mechanisms. Low ATM activity ensures stable Sp1 protein levels, promoting the efficient expression of DNA repair genes. Right: Persistent unrepaired DNA damage can arise under conditions of excessive exogenous DNA damage, or a DNA repair deficiency (found in many cancer cells) as well as from oncogene-induced replication stress (49,50). This leads to ATM activation, which triggers Sp1 phosphorylation and subsequent degradation that in turn leads to reduced expression of DNA repair genes. Under these circumstances, the cellular load of DNA damage is worsened by the decreased expression of DNA repair genes, leading to sustained ATM phosphorylation and, ultimately, activation of a proapoptotic cascade and cell death.
Figure 2.
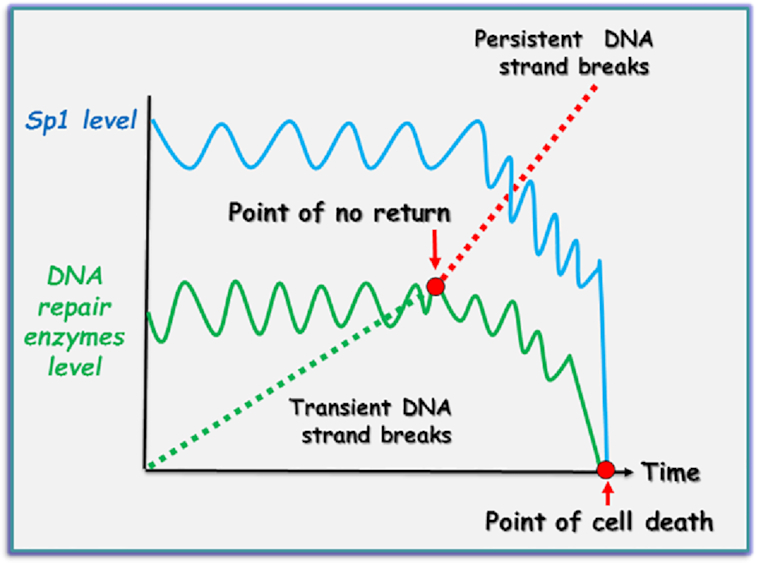
Proposed mechanism for life/death decision making in genetically compromised cells. In healthy cells, arising DNA strand breaks are quickly repaired and do not accumulate. Correspondingly, the amount of Sp1 and DNA repair proteins only moderately fluctuates around basal levels. However, when the amount of DNA strand breaks exceeds the DNA repair capacity of the cell (point of no return), hyperactivation of ATM leads to Sp1 degradation and consequently to DNA repair downregulation, resulting in activation of cell death mechanisms (point of cell death).
In cases where the Sp1-dependent mechanism fails to detect and eliminate cells containing DNA strand breaks during the G1 phase of the cell cycle, ‘damaged’ cells still can be eliminated in the G2 phase. It was recently demonstrated that unrepaired DSBs activate a DNA damage checkpoint in G2 phase to trigger an ATR-dependent cell cycle arrest to repair DSBs. However, if DNA repair cannot be accomplished, then cells containing persistent DSB permanently exit the cell cycle, entering either senescence or apoptosis (39). It is quite possible that this process is Sp1 independent as it was recently demonstrated that cells that are able to proliferate in spite of the persistent DNA SSBs (like many cancer cells do) are still able to activate Sp1-independent downregulation of NHEJ (40).
A TRANSLATIONAL PERSPECTIVE ON CANCER TREATMENT
As mentioned earlier, many cancer cells overexpress Sp1, which correlates with poor patient survival. Moreover, downregulation of Sp1 leads to decreased survival of cancer cells (7). These observations have provided a strong rationale for the development of anticancer drugs targeting Sp1. For example, mithramycin A (MTA) is a potent inhibitor of Sp1, which preferentially binds to GC-rich regions of DNA (41), thus preventing binding of Sp1 to promoter regions. Although MTA has a high killing potential for cancers cells (42–44), the major problem with this inhibitor is its high toxicity towards normal cells (45). More recently, however, promising Sp1 inhibitors that are less toxic to normal cells have been tested in both cell culture and xenograft models. These compounds demonstrated a high potential for killing cancer cells and also triggered cancer growth delay in xenograft and transgenic mouse tumour models (46–48), supporting the hypothesis that targeting Sp1 through chemical inhibition is a valid clinical approach that deserves further attention.
CONCLUSIONS AND FUTURE WORK
A better understanding of the molecular mechanisms involved in genomic integrity checkpoint may explain how physiological populations of healthy cells are maintained in an organism, by detecting and eliminating genetically compromised cells. However, the paramount task is to understand how some cancer cells may avoid this ‘genome stability checkpoint’. Understanding this process will open a new avenue for developing novel cancer therapy.
Contributor Information
Polina S Loshchenova, Department of Natural Sciences, Novosibirsk State University, Pirogova 2, Novosibirsk 630090, Russian Federation; Institute of Cytology and Genetics, Russian Academy of Sciences, Lavrentyeva 10, Novosibirsk 630090, Russian Federation.
Svetlana V Sergeeva, Department of Natural Sciences, Novosibirsk State University, Pirogova 2, Novosibirsk 630090, Russian Federation; Institute of Cytology and Genetics, Russian Academy of Sciences, Lavrentyeva 10, Novosibirsk 630090, Russian Federation.
Sally C Fletcher, Institute of Cancer and Genomic Sciences, University of Birmingham, Birmingham B15 2TT, UK.
Grigory L Dianov, Department of Natural Sciences, Novosibirsk State University, Pirogova 2, Novosibirsk 630090, Russian Federation; Institute of Cytology and Genetics, Russian Academy of Sciences, Lavrentyeva 10, Novosibirsk 630090, Russian Federation; Institute for Radiation Oncology, Department of Oncology, University of Oxford, Old Road Campus Research Building, Oxford OX3 7DQ, UK.
FUNDING
Russian Science Foundation [19-74-20069 to G.L.D.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Dynan W.S., Tjian R. Isolation of transcription factors that discriminate between different promoters recognized by RNA polymerase II. Cell. 1983; 32:669–680. [DOI] [PubMed] [Google Scholar]
- 2. Letovsky J., Dynan W.S. Measurement of the binding of transcription factor Sp1 to a single GC box recognition sequence. Nucleic Acids Res. 1989; 17:2639–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Naar A.M., Ryu S., Tjian R. Cofactor requirements for transcriptional activation by Sp1. Cold Spring Harb. Symp. Quant. Biol. 1998; 63:189–199. [DOI] [PubMed] [Google Scholar]
- 4. Marin M., Karis A., Visser P., Grosveld F., Philipsen S. Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell. 1997; 89:619–628. [DOI] [PubMed] [Google Scholar]
- 5. O’Connor L., Gilmour J., Bonifer C. The role of the ubiquitously expressed transcription factor Sp1 in tissue-specific transcriptional regulation and in disease. Yale J. Biol. Med. 2016; 89:513–525. [PMC free article] [PubMed] [Google Scholar]
- 6. Beishline K., Azizkhan-Clifford J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015; 282:224–258. [DOI] [PubMed] [Google Scholar]
- 7. Vizcaino C., Mansilla S., Portugal J. Sp1 transcription factor: a long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015; 152:111–124. [DOI] [PubMed] [Google Scholar]
- 8. Chang W.C., Hung J.J. Functional role of post-translational modifications of Sp1 in tumorigenesis. J. Biomed. Sci. 2012; 19:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tan N.Y., Khachigian L.M. Sp1 phosphorylation and its regulation of gene transcription. Mol. Cell Biol. 2009; 29:2483–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oh J.E., Han J.A., Hwang E.S. Downregulation of transcription factor, Sp1, during cellular senescence. Biochem. Biophys. Res. Commun. 2007; 353:86–91. [DOI] [PubMed] [Google Scholar]
- 11. Olofsson B.A., Kelly C.M., Kim J., Hornsby S.M., Azizkhan-Clifford J. Phosphorylation of Sp1 in response to DNA damage by ataxia telangiectasia-mutated kinase. Mol. Cancer Res. 2007; 5:1319–1330. [DOI] [PubMed] [Google Scholar]
- 12. Iwahori S., Yasui Y., Kudoh A., Sato Y., Nakayama S., Murata T., Isomura H., Tsurumi T. Identification of phosphorylation sites on transcription factor Sp1 in response to DNA damage and its accumulation at damaged sites. Cell. Signal. 2008; 20:1795–1803. [DOI] [PubMed] [Google Scholar]
- 13. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003; 3:155–168. [DOI] [PubMed] [Google Scholar]
- 14. Aasland D., Reich T.R., Tomicic M.T., Switzeny O.J., Kaina B., Christmann M. Repair gene O6-methylguanine-DNA methyltransferase is controlled by SP1 and up-regulated by glucocorticoids, but not by temozolomide and radiation. J. Neurochem. 2018; 144:139–151. [DOI] [PubMed] [Google Scholar]
- 15. Antoniali G., Marcuzzi F., Casarano E., Tell G. Cadmium treatment suppresses DNA polymerase delta catalytic subunit gene expression by acting on the p53 and Sp1 regulatory axis. DNA Repair (Amst.). 2015; 35:90–105. [DOI] [PubMed] [Google Scholar]
- 16. Bocangel D., Sengupta S., Mitra S., Bhakat K.K p53-mediated down-regulation of the human DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) via interaction with Sp1 transcription factor. Anticancer Res. 2009; 29:3741–3750. [PMC free article] [PubMed] [Google Scholar]
- 17. Christmann M., Kaina B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 2013; 41:8403–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ludwig D.L., Chen F., Peterson S.R., Nussenzweig A., Li G.C., Chen D.J. Ku80 gene expression is Sp1-dependent and sensitive to CpG methylation within a novel cis element. Gene. 1997; 199:181–194. [DOI] [PubMed] [Google Scholar]
- 19. Narayan S., Wilson S.H. Kinetic analysis of Sp1-mediated transcriptional activation of the human DNA polymerase beta promoter. Oncogene. 2000; 19:4729–4735. [DOI] [PubMed] [Google Scholar]
- 20. Nishida Y., Mizutani N., Inoue M., Omori Y., Tamiya-Koizumi K., Takagi A., Kojima T., Suzuki M., Nozawa Y., Minami Y.et al. Phosphorylated Sp1 is the regulator of DNA-PKcs and DNA ligase IV transcription of daunorubicin-resistant leukemia cell lines. Biochim. Biophys. Acta. 2014; 1839:265–274. [DOI] [PubMed] [Google Scholar]
- 21. Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993; 362:709–715. [DOI] [PubMed] [Google Scholar]
- 22. Caldecott K.W. XRCC1 protein: form and function. DNA Repair (Amst.). 2019; 81:102664. [DOI] [PubMed] [Google Scholar]
- 23. Dianov G.L., Hubscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 2013; 41:3483–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Caldecott K.W. DNA single-strand break repair. Exp. Cell Res. 2014; 329:2–8. [DOI] [PubMed] [Google Scholar]
- 25. Poletto M., Legrand A.J., Fletcher S.C., Dianov G.L. p53 coordinates base excision repair to prevent genomic instability. Nucleic Acids Res. 2016; 44:3165–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khoronenkova S.V., Dianov G.L. ATM prevents DSB formation by coordinating SSB repair and cell cycle progression. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:3997–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fletcher S.C., Grou C.P., Legrand A.J., Chen X., Soderstrom K., Poletto M., Dianov G.L. Sp1 phosphorylation by ATM downregulates BER and promotes cell elimination in response to persistent DNA damage. Nucleic Acids Res. 2018; 46:1834–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grande L., Bretones G., Rosa-Garrido M., Garrido-Martin E.M., Hernandez T., Fraile S., Botella L., de Alava E., Vidal A., Garcia del Muro X.et al. Transcription factors Sp1 and p73 control the expression of the proapoptotic protein NOXA in the response of testicular embryonal carcinoma cells to cisplatin. J. Biol. Chem. 2012; 287:26495–26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirose T., Horvitz H.R. An Sp1 transcription factor coordinates caspase-dependent and -independent apoptotic pathways. Nature. 2013; 500:354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H., Zhang Y., Strose A., Tedesco D., Gurova K., Selivanova G. Integrated high-throughput analysis identifies Sp1 as a crucial determinant of p53-mediated apoptosis. Cell Death Differ. 2014; 21:1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bernstein C., Bernstein H., Payne C.M., Garewal H. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat. Res. 2002; 511:145–178. [DOI] [PubMed] [Google Scholar]
- 32. Ponath V., Kaina B. Death of monocytes through oxidative burst of macrophages and neutrophils: killing in trans. PLoS One. 2017; 12:e0170347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flygare J., Armstrong R.C., Wennborg A., Orsan S., Hellgren D. Proteolytic cleavage of HsRad51 during apoptosis. FEBS Lett. 1998; 427:247–251. [DOI] [PubMed] [Google Scholar]
- 34. Negrini S., Gorgoulis V.G., Halazonetis T.D. Genomic instability—an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010; 11:220–228. [DOI] [PubMed] [Google Scholar]
- 35. Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A.et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Choi M., Kipps T., Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol. Cancer Ther. 2016; 15:1781–1791. [DOI] [PubMed] [Google Scholar]
- 37. Parsons J.L., Tait P.S., Finch D., Dianova II, Allinson S.L., Dianov G.L. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol. Cell. 2008; 29:477–487. [DOI] [PubMed] [Google Scholar]
- 38. Cabelof D.C., Raffoul J.J., Yanamadala S., Guo Z., Heydari A.R. Induction of DNA polymerase beta-dependent base excision repair in response to oxidative stress in vivo. Carcinogenesis. 2002; 23:1419–1425. [DOI] [PubMed] [Google Scholar]
- 39. Feringa F.M., Raaijmakers J.A., Hadders M.A., Vaarting C., Macurek L., Heitink L., Krenning L., Medema R.H. Persistent repair intermediates induce senescence. Nat. Commun. 2018; 9:3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loshchenova P.S., Sergeeva S.V., Limonov D.V., Guo Z., Dianov G.L. Sp1-independent downregulation of NHEJ in response to BER deficiency. DNA Repair (Amst.). 2019; 86:102740. [DOI] [PubMed] [Google Scholar]
- 41. Van Dyke M.W., Dervan P.B. Chromomycin, mithramycin, and olivomycin binding sites on heterogeneous deoxyribonucleic acid. Footprinting with (methidiumpropyl-EDTA)iron(II). Biochemistry. 1983; 22:2373–2377. [DOI] [PubMed] [Google Scholar]
- 42. Miller D.M., Polansky D.A., Thomas S.D., Ray R., Campbell V.W., Sanchez J., Koller C.A. Mithramycin selectively inhibits transcription of G-C containing DNA. Am. J. Med. Sci. 1987; 294:388–394. [DOI] [PubMed] [Google Scholar]
- 43. Blume S.W., Snyder R.C., Ray R., Thomas S., Koller C.A., Miller D.M. Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J. Clin. Invest. 1991; 88:1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scroggins B.T., Burkeen J., White A.O., Chung E.J., Wei D., Chung S.I., Valle L.F., Patil S.S., McKay-Corkum G., Hudak K.E.et al. Mithramycin A enhances tumor sensitivity to mitotic catastrophe resulting from DNA damage. Int. J. Radiat. Oncol. Biol. Phys. 2018; 100:344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kennedy B.J. Metabolic and toxic effects of mithramycin during tumor therapy. Am. J. Med. 1970; 49:494–503. [DOI] [PubMed] [Google Scholar]
- 46. Previdi S., Malek A., Albertini V., Riva C., Capella C., Broggini M., Carbone G.M., Rohr J., Catapano C.V. Inhibition of Sp1-dependent transcription and antitumor activity of the new aureolic acid analogues mithramycin SDK and SK in human ovarian cancer xenografts. Gynecol. Oncol. 2010; 118:182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shinde D., Albino D., Zoma M., Mutti A., Mapelli S.N., Civenni G., Kokanovic A., Merulla J., Perez-Escuredo J., Costales P.et al. Transcriptional reprogramming and inhibition of tumor-propagating stem-like cells by EC-8042 in ERG-positive prostate cancer. Eur. Urol. Oncol. 2019; 2:415–424. [DOI] [PubMed] [Google Scholar]
- 48. Scott D., Chen J.M., Bae Y., Rohr J. Semi-synthetic mithramycin SA derivatives with improved anticancer activity. Chem. Biol. Drug Des. 2013; 81:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bartkova J., Horejsi Z., Koed K., Kramer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C.et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005; 434:864–870. [DOI] [PubMed] [Google Scholar]
- 50. Kotsantis P., Petermann E., Boulton S.J. Mechanisms of oncogene-induced replication stress: jigsaw falling into place. Cancer Discov. 2018; 8:537–555. [DOI] [PMC free article] [PubMed] [Google Scholar]