

Abstract
MicroRNAs (miRNAs) are small non-coding RNAs with diverse functions in post-transcriptional regulation of gene expression. Sequence and length variants of miRNAs are called isomiRs and can exert different functions compared to their canonical counterparts. The Cancer Genome Atlas (TCGA) provides isomiR-level expression data for patients of various cancer entities collected in a multi-center approach over several years. However, the impact of batch effects within individual cohorts has not been systematically investigated and corrected for before. Therefore, the aim of this study was to identify relevant cohort-specific batch variables and generate batch-corrected isomiR expression data for 16 TCGA cohorts. The main batch variables included sequencing platform, plate, sample purity and sequencing depth. Platform bias was related to certain length and sequence features of individual recurrently affected isomiRs. Furthermore, significant downregulation of reported tumor suppressive isomiRs in lung tumor tissue compared to normal samples was only observed after batch correction, highlighting the importance of working with corrected data. Batch-corrected datasets for all cohorts including quality control are provided as supplement. In summary, this study reveals that batch effects present in the TCGA dataset might mask biologically relevant effects and provides a valuable resource for research on isomiRs in cancer (accessible through GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164767).
Graphical Abstract
Graphical Abstract.
On the impact of batch effect correction in TCGA isomiR expression data.
INTRODUCTION
MicroRNAs (miRNAs) are endogenous, single-stranded RNA molecules that function as guides in RNA silencing processes and thereby conduct post-transcriptional inhibition of roughly one third of all genes (1,2). In cancer, aberrant miRNA expression evokes dysregulated gene expression. By either acting on tumor suppressor- or oncogenes, miRNAs themselves can act as tumor suppressive miRNAs or oncomiRs. Furthermore, the expression of some miRNAs is regulated by tumor-suppressor- or oncogenes (3).
One precursor miRNA molecule is not only the origin of one specific mature miRNA, long discussed as the ‘one arm–one miRNA’ assumption. During maturation, a great variety of miRNA isoforms (isomiRs) is generated and can be detected using next generation sequencing technologies (4,5). Compared to their canonical counterpart, these isomiRs differ in length at their 3′ or 5′ end and/or in their internal sequence. A combination of several alteration types to the canonical isomiR sequence is possible.
The isomiR expression between different organs and tissue types is highly variable (6). Even though the specific role of most non-canonical isomiRs is unknown, biological relevance has been characterized for some examples. For instance, by targeting functionally related genes, they can work as cooperative partners to their corresponding canonical isomiR (7). For 5′ isomiRs in particular, relevance regarding the evolution of miRNA genes is assumed due to altering target sequences (8). Even a difference of just one nucleotide at the 5′-end of isomiRs can lead to a substantial difference in their target mRNA spectrum (9,10).
In recent studies, isomiR expression levels were used as biomarkers to develop diagnosis and classification models for different cancer types as well as for pan-cancer multiclass predictions (11–15). Even though in some of these models, predictors could be narrowed down to only a few isomiRs, the mechanistic role of isomiRs, with few exceptions, remains unknown. Inconsistencies in isomiR annotation, data pre-processing and handling of batch effects further complicate the biological interpretation of such models.
In case of lung cancer, the potential role of certain miRNAs has been previously reviewed by Ebrahimi and Sadroddiny (16) and Lin et al. (17), followed up by several studies highlighting the differential expression of specific miRNAs and promoting the biomarker potential of miRNAs due to presence and stability in blood and urine (18–20). As an example, miR-30c-5p has been described as tumor suppressor in lung cancer. Specifically, its expression is downregulated in tumors compared to adjacent normal tissue and its suppression is associated with more aggressive phenotypes in vitro (21). While functional relevance has been established for a variety of miRNAs, large-scale studies on the expression and function on isomiR-level in lung cancer have not been reported on so far.
The Cancer Genome Atlas (TCGA) provides an unprecedented amount of isomiR and miRNA expression data from cancer and normal samples along with clinical information of the patients. Thereby, it is a valuable resource for computational studies of the unknown mechanisms of isomiR processing and function in different cancer types (22). The sample-linked Omics datasets collected and pre-processed by TCGA enable to connect information about clinical parameters, genetic status, epigenetic marks, transcriptome and (iso)miRNAome of individual cancer patients. As for today, data from more than 30 000 patients covering 33 different tumor types are publicly available (23). Out of these, isomiR quantification data are available for 11 022 samples corresponding to 10 250 cases (24).
A general challenge for multi-centered studies is the presence of confounding variables negatively influencing data consistency. In the case of TCGA, data generation, sample harvesting, processing and sequencing was not only spread along a time period of more than one decade but also took place in different collaborating institutions (25). In such high-throughput sequencing processes, batch effects are unavoidable (26). In addition to this, reproducibility of miRNA sequencing itself is still subject to continuous improvement, as even current standard protocols, sequencing platforms and sequencing depth do not always allow to directly compare sequencing results (27).
Prior to moving the data to the NCI Genomic Data Commons (GDC), technical biases and batch effects were described for most datasets (28). Although the MD Anderson Cancer Center of the university of Texas hosts a web page with solely computationally batch corrected TCGA data (https://bioinformatics.mdanderson.org/public-software/tcga-batch-effects/), a systematic analysis of the batch effects and technical biases in the harmonized TCGA isomiR and miRNA expression quantification data specifically, is missing so far (27). MiRNAs were sequenced with two different Illumina platforms and the libraries were prepared using two different protocols throughout data generation. Those two batch effects have been described and removed using a batch correction method based on Empirical Bayes (29). For TCGA isomiR expression quantification data, to our knowledge, batch effects have not yet been described in the literature.
Here, we show that multiple batch effects are not only present across different TCGA projects as described before, but also affect the data within individual isomiR expression quantification datasets. Before batch correction, filtering steps were applied both on sample and isomiR level to enhance data quality and reduce manifestation of batch effects. We here performed and benchmarked isomiR annotation correction as well as batch effect removal for 16 TCGA cohorts with isomiR expression data. Specifically, we compared two different algorithms, limma and ComBat, based on linear and empirical Bayes models, respectively, and identified ideal parameter combinations for batch correction of each data set. Batch corrected data along with substantial quality control is provided as a supplement to this article for further use in the research community. Using the TCGA-LUSC dataset as an example, we characterize the effect of potential confounding variables, demonstrate the efficient and specific correction of the resulting batch effects and show that biologically relevant differences are potentially masked due to the batch effects. In addition, we provide evidence that the detection bias related to the use of different sequencing platforms is partly associated with specific sequence features of the respective isomiRs. These features include read length, GC content and first nucleotide of the isomiR. Of note, we provide evidence that deep sequencing is required to ensure high quality data especially for rare isomiRs.
MATERIALS AND METHODS
Data download and pre-processing
Our results are based on the isoform expression quantification data generated by TCGA Research Network (https://www.cancer.gov/tcga). The open access, GRCH38 build (hg38) aligned data were downloaded from the GDC harmonized database using the Bioconductor R package TCGAbiolinks (version 2.12.6) (30). Sixteen TCGA projects, including at least 290 samples each, were examined for confounding effects (Table 1). The data comprise different sample types: primary solid tumor, recurrent solid tumor, additional primary tumor, metastatic tumor as well as solid normal tissue. The primary solid tumor and recurrent solid tumor data types were combined as ‘tumor sample’ during the analysis since only few samples were available for tumor sample types other than primary solid tumor. Due to limited sample size, metastatic tumor samples were excluded from the analysis (Table 1). Analyses were performed for tumor samples separately as well as tumor and normal samples combined if the respective data was available. To improve data quality for batch correction, samples with low coverage were excluded from the analysis, as described by Chu et al. (22). Therefore, unless the description of the data regarding sequencing depth before batch correction was in scope, a filtering step of at least 1 million mapped reads per sample was introduced.
Table 1.
Isoform expression quantification data from 16 different TCGA projects
| Project | Abbrev. | Primary solid tumor | Recurrent/addit. tumor | Metastatic tumor | Solid tissue normal | Solid tissue normal (same plate as matched tumor sample) | Batch cor. tumor samples | Batch cor. tumor + normal samples | Expression comparison between sequencing platforms |
|---|---|---|---|---|---|---|---|---|---|
| Urothelial bladder carcinoma | BLCA | 417 | 0 | 1 | 19 | 19 | x | x | |
| Breast invasive carcinoma | BRCA | 1096 (1051) | 0 | 7 | 104 (103) | 86 | x | x | x |
| Cervical squamous cell carcinoma and endocervical adenocarcinoma | CESC | 307 | 0 | 2 | 3 | 3 | x | ||
| Colon adenocarcinoma | COAD | 455 (404) | 1 | 1 | 8 (0) | 0 | x | x | |
| Head-neck squamous cell carcinoma | HNSC | 523 (517) | 0 | 2 | 44 | 42 | x | x | x |
| Kidney renal clear cell carcinoma | KIRC | 544 (535) | 1 | 0 | 71 | 68 | x | x | x |
| Cervical kidney renal papillary cell carcinoma | KIRP | 291 | 0 | 0 | 34 | 33 | x | x | |
| Low grade glioma | LGG | 512 (510) | 18 | 0 | 0 | 0 | x | ||
| Liver hepatocellular carcinoma | LIHC | 372 (371) | 3 | 0 | 50 | 48 | x | x | |
| Lung adenocarcinoma | LUAD | 519 (506) | 2 | 0 | 46 (40) | 26 | x | x | x |
| Lung squamous cell carcinoma | LUSC | 478 (456) | 0 | 0 | 45 | 36 | x | x | x |
| Ovarian cancer | OV | 491 (452) | 8 (7) | 0 | 0 | 0 | x | ||
| Prostate adenocarcinoma | PRAD | 498 | 0 | 1 | 52 | 51 | x | x | |
| Stomach adenocarcinoma | STAD | 446 (432) | 0 | 0 | 45 | 41 | x | x | x |
| Thyroid cancer | THCA | 506 (504) | 0 | 8 | 59 | 59 | x | x | |
| Uterine corpus endometrial carcinoma | UCEC | 545 (512) | 1 | 0 | 33 | 18 | x | x | x |
The number of samples per corresponding sample type is listed per project. In parenthesis, the number of samples is displayed after excluding samples with a total number of mapped reads below 1 million. Further, it is indicated for how many normal tissue samples and patient-matched tumor samples library preparation was conducted on the same plate. If at least 18 normal samples were available, batch correction was performed not only including tumor samples but also with both tumor and normal samples. IsomiR expression between the two sequencing platforms was conducted for eight cohorts that utilized both the Illumina GA and HiSeq sequencing platform.
The GDC miRNA analysis workflow is based on the British Columbia Genome Sciences Centre miRNA Profiling Pipeline, as described by Chu et al. (22). Three different data files from miRNA sequencing are stored in the harmonized portal from the NCI GDC: aligned reads, miRNA expression quantification that associates miRNAs with the read count and the normalized read count as reads-per-million-miRNA-mapped (RPM) and the isoform expression quantification data with additional isoform coordinates (https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/miRNA_Pipeline/). However, the end position of the isoform coordinates is labeled exclusively in the isoform expression quantification file, thus with an offset of one compared to standard annotation formats. This finding has been added to the GDC ‘Release Notes—Known Issues and Workarounds’ as well as to the BCGSC miRNA Profiling Pipeline website after contacting the authors (https://docs.gdc.cancer.gov/Data/Release_Notes/Data_Release_Notes/#data-release-220, https://www.bcgsc.ca/resources/software/mirna-profiling). The offset was corrected by subtracting one from the end position.
Genomic locations of mature miRNAs contained in miRBase version 22.1 (http://www.mirbase.org/) were extracted. These positions were flanked by 3 nt on each side. This resulted in the annotation of 49 isoforms per mature miRNA, from |-3|-3| to |3|3| with all possible combinations of 5′ and 3′ ends (Supplementary Table S1).
Gathering information about potential confounders
Different potential confounders and their values were pre-defined and derived from individual data sources. The TCGA barcode itself provides information on multiple variables such as the plate (of library preparation) and the sample type. Information on patient related data such as sex, year of birth, vital status and tumor stage were provided in clinical files by the GDC harmonized portal. The tumor purity was derived from the TCGAbiolinks R package using the TCGAtumor_purity function. As this function aims to filter out samples with tumor purity below a certain threshold, purity was derived in steps of 10% by adjusting the threshold by 0.1. The cpe variable, a consensus measurement from several purity estimation methods, served as a threshold. For the TCGA-STAD project, no tumor purity information was available. The tumor subtype was derived from the TCGAbiolinks R package using the PanCancerAtlas_subtypes function, focusing on the Selected_Subtype variable (30). The number of total mapped reads, the sequencing depth per sample, was derived from the isoform expression quantification data files by calculating the total sum of read counts. The Illumina sequencing platform details were mentioned in the supplement by Thorsson et al. (31) and were downloaded from the GDC website (https://gdc.cancer.gov/about-data/publications/panimmune). According to TCGA information, library preparation protocols did not differ within the 16 TCGA-projects and were therefore not included as potential confounders. Confounding factors are stored in Supplementary Table S2.
Assessment of confounding effects
Assessment of confounding effects and removal of batch effects was performed separately for each TCGA project. To improve data quality, an arbitrary median expression filter of 15 RPM was introduced, representing a trade-off between sensitivity and specificity, and confounding effects were assessed with both the reduced and full dataset. The log2 transformed expression matrices were used as the basis for a principal component analysis (PCA). For a confounder analysis, PCA scatterplots with PC1-10 were generated and potential confounders were identified. Furthermore, boxplots comparing PC1-10 per batch label were used to compare confounding effects before and after batch effect correction, as described by Goh, Wang and Wong (32). In addition to the visualization of confounding effects, the difference of PC values between the batch labels was statistically assessed. Categorical variables with more than two labels (plate, subtype and tumor stage) were tested with the Kruskal Wallis test, categorical variables with two labels (platform and gender) with a Wilcoxon Rank Sum test. The association between the PCs and the sequencing depth as a continuous variable was tested using a Spearman correlation. For the ordinal variable tumor purity, the Kendall Tau Rank correlation was calculated. The P-values were false discovery rate (FDR) adjusted using the p.adjust function from the stats R package. The association between potential confounder and PC was significant in case of a q-value < 0.01. For correlation testing, an additional threshold for the minimal absolute correlation coefficient of 0.25 was used. On an individual isomiR level, the expression difference between samples from different plates was tested using ANOVA. Statistical analyses were conducted using the stats R package (version 3.6.0) (33). q-values were visualized in forest and bubble plots using the ggplot2 R package (version 3.3.2) (34).
Confounding effects were assessed before and after batch effect removal in order to evaluate the ideal parameter combination for the batch effect removal. Additionally, t-SNE projections were generated for the uncorrected and final batch corrected data for different confounders using the Rtsne package which computes the Barnes-Hut implementation of t-Distributed Stochastic Neighbors (version 0.15) (35,36) setting perplexity to 25 and using 1000 iterations.
Batch effect correction
Batch effects were removed from log2-transformed and RPM-normalized expression data using different batch effect correction strategies. The removeBatchEffects function from the limma R package is based on a linear model that is fit to the data in order to calculate and remove the given batch effects (version 3.40.6) (37). As a second approach, the ComBat function from the sva R package was applied (version 3.32.1) (38). To this end, the data are adjusted for batch effects by using an empirical Bayes model, as described by Johnson, Li and Rabinovic (39). To remove two different batch effects, the functions were applied sequentially. The sample type was used as a variable of interest during batch effect removal to conserve the biological difference between normal and tumor samples.
Assessment of systematic sequencing bias introduced by different sequencing platforms
Heatmaps for the comparison of isomiR expression between samples sequenced on the two different platforms Genome Analyzer (GA) and HiSeq were generated calculating z-scores on the log2 transformed expression values for each isomiR with a median expression of >15 RPM within the TCGA-LUSC cohort. To avoid additional confounders, normal samples were excluded from this analysis. All GA sequenced samples and the same number of randomly chosen HiSeq-sequenced samples were used to overcome imbalance between the groups. The heatmap.2 function from the gplots package in R was used to draw the heatmaps (version 3.0.4) (40).
To assess the effect of the batch correction on differential expression between the platforms statistically, two-sided t-tests were performed comparing log2 transformed isomiR expression values for each isomiR on the two platforms. P-values were FDR corrected for multiple testing.
To identify recurrently affected isomiRs, these t-tests were repeated for the other TCGA datasets which contained samples sequenced on the GA, i.e. BRCA, COAD, HNSC, KIRC, LUAD, STAD and UCEC, together with LUSC resulting in eight analyzed datasets. The results were filtered for isomiRs with an absolute log2 fold change between median expression detected by GA and HiSeq of at least 0.5 and a q-value < 0.05 in six out of eight entities to obtain a dataset with isomiRs highly affected by platform related batch effects.
For each isomiR expressed > 15 RPM in at least six out of the eight projects, the sequence information was retrieved from miRbase and length, GC content and position as well as percentage of individual nucleotides were determined. The outlier isomiR list was further separated in isomiRs ‘under-represented’ or ‘over-represented’ in patients sequenced by GA compared to HiSeq and a ‘non-affected’ list containing all isomiRs that were neither over- nor under-represented. These sublists were then used for statistical comparison of all isomiRs expressed above 15 RPM. Differences in the distribution of read lengths and GC content were analyzed by two-sided student's t-test and visualized as violin plots. Furthermore, the frequency of individual nucleotides and the GC content at the first position were compared between the ‘union’ list and the ‘over-represented’ and ‘under-represented’ isomiRs by two-tailed Fisher's exact test. Relative nucleotide distribution and GC content at the first position were visualized as heatmaps or barplots, respectively.
Effects of the batch correction on biological messages
Differential expression between tumor and normal samples of the TCGA-LUSC cohort was investigated for all isomiRs expressed at above 15 RPM before and after batch correction using a t-test with Welch modification and subsequent FDR correction for multiple testing.
Programming languages and computing resources
All analyses were performed using the computational infrastructure of the German Cancer Research Center (DKFZ), Heidelberg. High performance computing clusters were used with portable batch systems (PBS) and Load Sharing Facility (LSF) platforms, running on CentOS Linux 7. Scripts were run using R 3.6.0.
RESULTS
Adjustment of isomiR annotation is a crucial step prior to working with the TCGA data
Various studies have utilized isomiR quantification data provided by TCGA to investigate the potential of isomiR-resolution small RNA expression data for identification of novel biomarkers or to generate hypotheses on isomiR function. To our knowledge, these studies frequently used the data as provided by TCGA or GDC, respectively. However, being aware of the potential substantial bias batch effects can cause especially in large-scale multi-centered studies, we aimed here at their characterization and correction to enable more robust research on isomiRs in cancer in the future.
The GDC isoform expression quantification data is provided as data tables including the read count, normalized read count per million mapped reads and miRNA, as well as information about the miRNA region and the isoform coordinates, therefore the start and end position. To map the isoform coordinates to the corresponding isomiR, we first generated a mapping file. Briefly, human mature miRNAs annotated in miRBase version 22.1 were defined as canonical isomiR (|0|0|) for each miRNA stem. These coordinates were shifted by a maximum of 3 nt at the 3′ and 5′ end, resulting in a total of 49 isoforms and their coordinates were defined per miRNA stem (Supplementary Table S1). Their expression associated through the isoform coordinates of the GDC isoform expression quantification data. The notation of isoforms derived from the same arm of a stem loop can be visualized in a matrix-like structure (Figure 1A and B).
Figure 1.

Characterization of the TCGA-LUSC isoform quantification data. (A) Different isomiRs are derived from the pre-miRNA sequence, with varying 3′ and 5′ ends. In this example, the canonical isoform |0|0| (dark blue) and the isoforms |0|1| (dark green), |1|1| (light green), |1|0| (light blue) and |-1|-1| (orange) are depicted. (B) The matrix serves as a representation of isoforms and their respective notation. Color code matches panel (A). (C) Matrix representation of median expression of isoforms derived from the hsa-let-7a-5p stem loop before and after annotation correction of the end position in the TCGA-LUSC data set, colored by log2 median expression. The hsa-let-7a-5p isomiRs are derived from three genetic loci on chromosome 9 (plus strand), 11 (minus strand) and 22 (plus strand). (D) Distribution of canonical isomiRs among all isomiRs for each miRNA arm in percent before (left panel) and after (right panel) annotation correction. Red color marks the area in which canonical isomiRs account for <50% of the reads, i.e. more than 50% of reads coming from this arm are non-canonical.
In the following, we use the LUSC cohort as an example to illustrate our observations. This cohort is highly affected by multiple batch affects and includes both tumor and normal samples. In addition, we provide a summary for the other cancer entities in the supplementary information. When first examining the expression matrices derived from this annotation process, we observed an obvious discrepancy for sequence-identical isomiRs derived from different genomic locations such as hsa-let-7a-5p (Figure 1C, upper panel). Specifically, isomiRs attributed to the plus strand of the genome exhibited a systematic shift in the 3′ annotation, whereas isomiRs expressed from the minus strand were systematically shifted at the 5′ end. Reads which cannot be mapped unambiguously to a genomic origin had been randomly assigned to one of the possible origins by the GDC pipeline (21). Therefore, sequence-identical miRNAs are expected to exhibit similar isomiR expression profiles. This analysis showed that the TCGA miRNA profiling pipeline annotated the chromosomal end position of each isomiR feature exclusively. This annotation is not supported by the common data file formats such as *.bed or *.gff3 resulting in a mis-annotation of isomiRs.
The left panels of Figure 1C sum up the expression values attributed to different genomic loci yielding the final expression values for the respective isomiRs. Comparing those, it becomes apparent that this mis-annotation can affect not only the isomiR annotation an expression value is assigned to, but also distort isomiR expression patterns, both leading to potential misinterpretation of the derived data. After adjusting the isomiR annotation based on this observation, the expected coherent expression values for all sequence-identical isomiRs could be observed (Figure 1C, lower panel).
We next sought to broadly characterize the isomiR expression dataset. The TCGA-LUSC cohort contains isomiR expression data for 478 tumor samples, 456 of them were sequenced with a sequencing depth of at least 1 000 000 mapped reads. Reads were mapped to 1816 canonical isomiRs, 6773 3′ isomiRs, 3824 5′ isomiRs and 12 971 mixed isomiRs derived from 2118 distinct miRNA arms. To further analyse the effects of the annotation correction, we compared the ratio of reads from the canonical isomiR to the reads of all non-canonical isomiRs for each miRNA arm. Annotation correction leads to a shift in distribution of the percentage of isomiRs that are canonical and cases in which the canonical version accounts for <50% of the isomiR reads. The percentage falls from 94% before correction to 68% after correction (Figure 1D). The majority of the other isoforms were only mapped to a few reads indicating that the provided sequencing depth might be insufficient to ensure robust sequencing of rare isoforms (Supplementary Figure S1).
Multiple batch effects are present in the TCGA-LUSC isoform expression quantification dataset
After annotation adjustment, we next aimed to identify potential confounding variables in the TCGA-LUSC isomiR expression dataset accounting for potential batch effects. For that purpose, PCA was performed on pre-filtered data with 660 isomiRs with a median expression above 15 RPM, and the association of potential confounders with the principal components (PC) 1–10 was investigated. The variables gender, plate (during library preparation), tumor purity, sequencing depth (total number of mapped reads), sample type, year of birth, vital status and tumor stage were analyzed as potential confounders.
Furthermore, as two different sequencing platforms were used to sequence the samples, Illumina Genome Analyzer II (GA) and Illumina HiSeq (HiSeq), the platform variable was included as well. Significant associations were mainly found between the PCs and the batch variables plate, platform, purity and sequencing depth (Supplementary Figure S2a).
Batch correction was compared using two different methods: the removeBatchEffect function of the limma R package is based on a linear model that is fit to the data, including the batch effect, which is then removed. In contrast, the ComBat function of the sva R package is based on an empirical Bayes model. Batch correction was performed with different batch variable combinations, consisting of plate, platform and purity. Since the sequencing depth is a continuous variable, it could not serve as a batch variable for correction. Up to two batch variables were included in sequential order during batch effect removal. While limma failed to correct efficiently for the prominent batch variable plate in case of the LUSC cohort, the ComBat function with plate as first batch variable and tumor purity as second batch variable efficiently removed all batch effects (Supplementary Figure S2).
First, we focused on the batch bias associated with the plate variable. Here, a significant association was detected in PC 1–3, 5, 7, 9 and 10 when analyzing the first 10 PCs, making it the most prominent batch variable for the LUSC cohort. The PCA plot of PC1 and PC2 demonstrates a clear batch bias before batch correction that is removed by the ComBat function as indicated by the colored centroids of each plate (Figure 2A and B).
Figure 2.

Batch effects dominate the isoform expression quantification data. (A) PCA plots colored by plate. The mean PC1 and PC2 values were calculated per plate to visualize each centroid in color, actual data points are added in gray. PCA was based on the log2 TCGA-LUSC isoform quantification data before batch correction. The percentage values in parenthesis indicate the variance explained by the corresponding principal component. (B) PCA plots colored by plate as described in (A), after sequential batch correction with plate as first batch variable and purity as second. (C) The association between sequencing platforms and PCs was determined using Wilcoxon Rank Sum tests. Log10 of the resulting q-value is shown before and after batch correction. (D) PC2 of TCGA-LUSC tumor samples with different tumor purity estimates before batch correction, displaying an ordinal association between tumor purity and PC2. Purity estimation of tumor samples was performed using the TCGAbiolinks function TCGAtumor_purity. (E) Box plots of PC2 distribution for TCGA-LUSC samples with differing tumor purity as described in (D) after batch correction. (F) Association between the total number of reads and PC 1–10. Log10 of the q-value of the correlation is shown for each PC. (G) The distribution of sequencing depth (number of total mapped reads) of the TCGA-LUSC isoform expression quantification data per plate, colored by sequencing platform. (H) Spearman correlation between number of isomiRs with non-zero expression and number of mapped reads (sequencing depth) in the TCGA-LUSC cohort. Median expression of the detected isomiRs in RPM is color coded as indicated. (I) Relative isomiR detection per TCGA-LUSC tumor sample for each isomiR type. Canonical isomiRs (|0|0|) as well as 3′ isomiRs (|0|X|), 5′ isomiRs (|X|0|) and mixed isomiRs (|X|Y|) with X and Y unequal 0 and between -3 and 3 were annotated to the provided isoform expression quantification data. In total, 1816 canonical isomiRs from miRBase were detected along with 6759 3′ isomiRs, 3822 5′ isomiRs and 12 916 mixed isomiRs, summing up to 25 313 total isomiRs detected in at least one patient.
We next investigated the impact of the batch variable tumor purity where statistical analyses had shown a significant association with PC2. This association was observed to be ordinal before and completely absent after batch correction (Figure 2D and E). No significant associations between tumor purity and any of the first 10 principle components was observed after batch correction (Supplementary Figure S2). Further, a batch bias was detected in the sequencing depth variable in PC1-3; which was also successfully removed upon batch correction (Figure 2F). Of note, correction for the batch variables plate and purity also efficiently removed the pronounced association of the variable platform (Figure 2C).
These observations let us further investigate the interconnection between the batch variables sequencing depth, plate and platform. In general, we observed that library preparations from the same plate were primarily sequenced on the same platform and that sequencing by Illumina GA generally resulted in a lower sequencing depth (Figure 2G) (24). The successful elimination of batch effects associated with the platform variable by only correcting for plate and tumor purity indicates that our approach was not over-correcting the data as this would likely not abolish interconnected batch effects so efficiently.
In general, we observed that batch effects were less pronounced when limiting the dataset to the 660 isomiRs with a median expression above 15 RPM as compared to including all isomiRs with non-zero expression in any patient (n = 25 400) (Supplementary Figure S3). Of note, especially the batch variable sequencing depth was corrected for less efficiently in the dataset comprising all detected isomiRs. This raised the question if variation introduced into the data by the varying sequencing depth present in the dataset would be a consequence of unsaturated detection of isomiR species. Indeed, the number of total mapped reads and the number of isomiRs detected in a given sample are highly correlated (R = 0.77) (Figure 2H). Saturation in the number of isomiRs detected is not even reached in samples sequenced with a relatively high coverage of 6 000 000 mapped reads. This observation was confirmed for each isomiR type independently (canonical form, 3′, 5′ and mixed isomiRs) (Figure 2I). Even in the upper quartile of sequencing depth, for each individual patient only about 35% of canonical isomiRs measured in at least one patient of the cohort were detected. This number drops gradually with sequencing depth to around 25% for patients sequenced with a sequencing depth corresponding to the lower quartile. A similar bias can be observed for the less abundant classes as well, just at a much lower level of relative detection in individual patients. In contrast, limiting the analysis to the 660 isomiRs with a median expression above 15 RPM, most isomiRs (646–651) are detected in all patients, irrespective of sequencing depth further emphasizing that a focus on more abundant isomiR species substantially reduces the impact of sequencing depth on data quality (Supplementary Figure S4a).
Along this line, we next investigated the impact of confounding effects on the TCGA-LUSC miRNA expression quantification dataset including all miRNAs with non-zero expression in at least one patient (n = 1580). Since all read counts mapped to different isomiRs derived from the miRNA arm are summed up to the final miRNA read count in this dataset, fewer individual species are detected, but in turn at a higher median expression. Nevertheless, there is high correlation (r = 0.81, P = 2.2e-16) between the number of miRNAs detected and the number of total mapped reads per sample (Supplementary Figure S4b). Therefore, not only for isomiR detection, but for miRNA sequencing in general, higher sequencing depth would be required to obtain more robust data with more information content. In general, the miRNA data are less affected by batch effects, especially in PC1-3 that explain most of the variance in the data (Supplementary Figure S5).
In summary, we successfully corrected for substantial batch effects in the TCGA-LUSC isomiR expression quantification data set by sequential batch correction using ComBat with plate as first and tumor purity as second batch variable (Supplementary Figure S2). PCA with batch corrected data shows that the batch effects were a large source of variation within the original dataset potentially interfering with the biologically relevant content of the data.
Platform-dependent expression differences on individual isomiR level enable identification of recurrent outlier isomiRs
After identification, characterization and removal of batch effects from the TCGA-LUCS isomiR quantification dataset, we next sought to further investigate the impact of the described batch effects on an individual isomiR level. Here, we decided to focus on the more robust subset of isomiRs with a median expression >15 RPM to avoid overestimation of differences due to low expression in combination with low sequencing depth. For that purpose, we focused on the binary batch variable platform and first visualized expression difference between the two different platforms GA and HiSeq as heatmaps showing the mean of log2-transformed, z-scaled expression values for tumor tissue samples. Most isomiRs showed pronounced differences in mean expression values between the two different platforms which could be mostly abolished by batch correction (Figure 3A and B). There is no indication of a trend in expression differences between GA and HiSeq sequenced samples. Results from FDR corrected t-tests confirmed that differences were significant for most isomiRs (494 out of 660) before, but not after batch correction (1 out of 660) (Figure 3C). Similar to the platform batch variable, the plate batch effects were largely removed on individual isomiR level (Supplementary Figure S6).
Figure 3.

Outlier isomiRs can be identified by comparing the same isomiR sequenced on different platforms. (A) Heatmaps showing means of log2 transformed, z-scored values for tumor isomiR expression of patients’ tumor material analyzed on different sequencing platforms. A total of 115 samples sequenced on HiSeq were randomly chosen to equal the number of samples sequenced on GA. (B) After batch correction, variance is reduced. (C) Differences between expression values derived from different platforms are non-significant after batch correction. Association between log2 transformed expression values of the two different sequencing platforms for each isomiR. Student's t-test followed by FDR correction for multiple testing before and after batch correction (green and orange, respectively). (D andE) left panel: Means of log2 transformed isomiR expression values for patient tumor material sequenced on the GA (n = 131) or the HiSeq (n = 336) platform before and after batch correction. Outlier isomiRs show significantly different sequencing results on the different platforms (FDR corrected Student's t-test, padj < 0.05, log2 fold change difference >|0.5|) only before correction. Middle/right panels: Density plots for two exemplary isomiR expression values sequenced on either GA or HiSeq before and after batch correction.
Next, we aimed to investigate which isomiRs were especially affected by platform batch effects. For that purpose, we first investigated the overall correlation between isomiR expression levels detected by either sequencing platform (Figure 3D, left panel). While we globally observed a good correlation of log2 mean expression (Spearman r = 0.89, P = 2.2e-16), consistent with our findings in the heatmap, there was a substantial number of outlier isomiRs defined by a q-value below 0.05 and an absolute log2 fold change expression difference > 1.5. Density plots of two exemplary outlier isomiRs, hsa-miR-143-3p|0|0| and hsa-miR-22-3p|2|0| show the systematic expression difference prior to batch correction (Figure 3D, middle and right panel). Identical analyses of the batch-corrected data reveal an almost perfect correlation between the platforms on a global isomiR level (r = 0.999, P = 2.2e-16) and no residual outlier isomiRs once again highlighting the success of our batch correction approach (Figure 3E, left panel). Furthermore, this could also be confirmed on an individual isomiR level with the same examples analysed before batch correction (Figure 3E, middle and right panel).
IsomiRs which are recurrently represented differently between sequencing platforms share distinct length and sequence parameters
Utilizing the TCGA isomiR expression quantification data from eight different projects where both sequencing platforms were used (BRCA, COAD, HNSC, KIRC, LUAD, LUSC, STAD and UCEC), we next hypothesized that the isomiRs affected by platform bias might be recurrent between different datasets. This would indicate a systematic bias in sequencing results obtained from both platforms on an isomiR level and a list of such recurrent outlier isomiRs might be a helpful resource for future work with the TCGA isomiR expression quantification data as well as with future isomiR sequencing experiments in general.
To address this question, we first defined a list of recurrent isomiRs with a median expression >15 RPM in at least six out of the eight included tumor entities (n = 546, Supplementary Table S3). Within this list, we next identified recurrent outlier isomiRs which were either significantly over-represented (n = 69) or under-represented (n = 51) in patients sequenced by GA compared to HiSeq, respectively with a FDR < 0.05 and an absolute log2 fold change > 0.5 in six out of the eight included tumor entities (referred to as ‘over-represented’ and ‘under-represented’ isomiR list; Supplementary Table S4). As reference, we defined the residual 426 isomiRs as ‘non-affected’ isomiR list. Reasoning that these isomiR lists might differ from each other in certain sequence features, we analysed GC content in general, occurrence of certain nucleotides, especially at the first position within the detected sequence and read length.
To investigate a systematic bias introduced by the average GC content, we first compared this parameter between the three isomiR lists (Figure 4A). For isomiRs in the ‘non-affected’ list, the average GC content was 46.1%. While ‘over-represented’ isomiRs also had a GC content of 46.1% and were not significantly different from ‘non-affected’ isomiRs (P = 0.97), ‘under-represented’ isomiRs exhibited a significantly higher GC content of 50.8% (P = 0.003).
Figure 4.

IsomiRs recurrently expressed differentially between the two sequencing platforms in at least six different cancer entities (‘non-affected’/‘non-aff.’) were analysed with regard to their sequence divergence when over-or under-represented (‘over-rep.’, ‘under-rep.’) in the GA sequencing results compared to the HiSeq results, respectively. (A) GC content of the isomiR sequences in the three different categories. Statistics: Student's t-test, n.s. = non-significant, **: P < 0.01. (B) Comparison of base in the first position of the isomiRs. Statistics: see Supplementary Table S5. (C) Comparison of the GC/ AT content at the first positions of the isomiRs. Statistics: Fisher's exact test, *: P < 0.05, **: P < 0.01. (D) Comparison of the length of the isomiRs of the three groups. Statistics: Student's t-test, **: P < 0.01, ****: P < 0.0001.
Along this line, we next analysed the distribution of nucleotides at the first position in reads. Remarkably, the ‘over-represented’ isomiR list showed significantly more G nucleotides in the first position and significantly fewer A nucleotides whereas the ‘under-represented’ isomiR list contained fewer C nucleotides in the first position when compared to the ‘non-affected’ list (Figure 4B and Supplementary Table S5). Assuming that GC or AT content at the first position of an isomiR potentially affects adapter ligation during library preparation, we combined these nucleotides for our analysis, respectively (Figure 4C). Indeed, we observed a significant bias toward a higher overall GC content of 43.5% in ‘over-represented’ isomiRs compared to 26.1% in the ‘non-affected’ list of isomiRs (P = 0.003) whereas ‘under-represented’ isomiRs exhibited a significantly lower GC content than ‘non-affected’ ones (9.8% GC, P = 0.009).
Lastly, we investigated if there is an association between sequencing bias and isomiR read length: ‘Over-represented’ isomiRs were significantly shorter with 20.9 nt on average (P = 0.001), while ‘under-represented’ isomiRs were significantly longer with an average length of 22.5 nt compared to the ‘non-affected’ list with 21.5 nt (P = 1.19 e-9) (Figure 4D).
In summary, these results indicate a systematic bias towards shorter reads with an overall unchanged GC content, but a higher GC content at the first position in the reads on the GA platform in comparison to the HiSeq platform. This might either arise from systematic differences between the two platforms or slight deviations in library preparation protocols throughout the project as samples sequenced by GA were generally prepared earlier than the samples sequenced on the HiSeq platform. Furthermore, these insights might serve as a valuable resource for the analysis of other isomiR or miRNA sequencing datasets as it points towards systematic misinterpretation of relative isomiR abundance depending on library preparation protocols and sequencing platforms.
Different combination of parameters combinations for batch correction provide optimal cohort-specific results
After characterizing isomiRs systematically affected by the sequencing platform, we next aimed to investigate, characterize and correct batch effects with the approach described for the LUSC dataset above for 15 additional projects containing isomiR quantification data for at least 290 patients per cohort (Table 1). Indeed, we observed a similar manifestation of batch effects for all these projects using again the datasets reduced to isomiRs with a median expression above 15 RPM (Supplementary Figures S7–21). For each individual cohort, batch correction was performed with different batch variable combinations, consisting of plate, platform and purity. Since the sequencing depth is a continuous variable, it could not serve as a batch variable with the algorithms used. Up to two batch variables were included in sequential order during batch effect removal. For each cohort, the parameter combination with least remaining batch effects was chosen as preferred combination (summarized in Supplementary Table S6). Interestingly, the four cohorts for which limma batch correction performed better than combat (BLCA, COAD, KIRC and LGG) had a significantly higher number of samples sequenced with the Illumina GA than the HiSeq platform (Chi-Square test, P < 2.2 e-16) going hand in hand with a lower sequencing depth (t-test, P = 4.2 e-6) (Table 1).
As shown in Figure 5, plate batch effects were identified for all cancer entities, and sequencing platform batch effects were observed for all projects consisting of patient samples sequenced with both Illumina GA and Illumina HiSeq. Tumor purity was another batch effect found in most projects apart from CESC, LUAD and STAD. Information on different tumor subtypes was provided by the R package TCGAbiolinks for the TCGA-BLCA, TCGA-BRCA, TCGA-LIHC, TCGA-PRAD and TCGA-THCA projects. Interestingly, in these cohorts several PCs were significantly associated with those subtypes. Besides the projects TCGA-LUAD and TCGA-THCA, a highly significant correlation was found between the sequencing depth, and different PCs. Thus, the variance in the dataset can partly be explained by altering sequencing depth between the samples.
Figure 5.

Association between potentially confounding variables and principal components before and after batch correction in 16 TCGA isomiR expression quantification datasets. PCA was performed using the log2 expression values of isomiRs with a median threshold > 15 RPM. For each variable, association was statistically tested as follows: plate and subtype variable using a Kruskal Wallis test, platform variable using a Wilcoxon Rank Sum test, for the ordinal purity variable a Kendall Tau Rank correlation and for the sequencing depth variable a Spearman correlation. Colors indicate -log10 of the resulting q-values, the red scale represents values above the significance threshold (q-value < 0.01), the blue color scale non-significant q-values. Sizes of the dots indicate the percentage of variance that can be explained by the corresponding PC. Blue arrows indicate the batch variable 1 that was chosen for batch correction for the respective TCGA project in the same row, the yellow arrow the batch variable 2. The BLCA, COAD, KIRC and LGG datasets were batch corrected using the limma algorithm, whereas the other 12 cohorts showed better correction results using ComBat.
The plate and platform batch effects were successfully removed from all 16 projects. However, especially the tumor purity batch effect is still present in 9 of the 16 datasets. Also, the correlation between PCs and sequencing depth is still partly present in 8 of the 16 datasets. Only for TCGA-LIHC, TCGA-LUSC, TCGA-OV, TCGA-STAD and TCGA-UCEC, all significant batch effects were completely removed. Since different isomiR expression levels are expected between different tumor subtypes, batch correction should not reduce variance and significance in the subtype variable. Indeed, after batch effect removal, the PCs are still significantly different between the subtypes. These findings indicate that biological variance in the data set is not removed during batch correction.
Variance between normal and tumor samples is sustained by batch correction
To further focus on the maintenance of biological variance in the data sets, batch correction was applied to the 12 TCGA isomiR expression quantification datasets including at least 18 patient-matched normal samples (Table 1). To ensure biologically meaningful results and at the same time reduce the impact of limited sequencing depth, all subsequent analyses were performed on the more robust subset of isomiRs sequenced with at least 15 RPM on average in the respective cohort and the quality of batch correction examined (Supplementary Figure S22). In general, batch effects introduced by the variables plate and platform were less efficiently removed when normal samples were included. This might be partly explained by unequal distribution of normal samples across plates and platforms. In contrast, residual batch effects associated with sequencing depth and purity were similar between both approaches. Of note, potentially biologically relevant variance due to different disease subtypes was similarly retained in both settings.
To further investigate the impact of batch correction on biologically relevant variance in the data, we generated t-SNE projections of the log2 transformed expression data before batch correction and after batch correction. As shown in Figure 6A for the uncorrected TCGA-LUSC dataset, similar to the results obtained by PCA, t-SNE projections revealed strong clustering of patients based on the batch variables platform, sequencing depth and plate. Of note, normal samples clustered distinct from tumor samples already before batch correction. Consistent with our previous results, all batch effects were removed efficiently in the TCGA-LUSC data set. At the same time, biologically meaningful effects were not impaired by batch correction and tumor and normal samples clearly cluster separately from each other (Figure 6B), emphasizing that exclusion of poorly sequenced samples alone cannot replace batch correction. Similar results were achieved for the additional 11 TCGA projects (Supplementary Figures S23–32).
Figure 6.

Strong biological effects remain after batch correction. tSNE plots of (A) non-corrected data containing all samples containing read counts greater zero, (B) non-corrected data containing only samples with an overall expression of at least 1 million mapped reads and a median expression of at least 15 rpm and (C) batch effect corrected isomiR expression data for the different patients. Platform (left panel), sequencing depth (middle panel) and sample type (tumor/normal, right panel) are colored.
For relative comparison of isomiR expression in patient-matched tumor and normal samples, batch effect correction may seem avoidable. However, as clustering of patients based on the log2 fold change in expression between tumor and normal samples from the same and different plates show, this only applies for samples that had their libraries prepared on the same plate and were sequenced with the same platform (Supplementary Figure S33 and Table 1).
Batch effects mask biologically meaningful isomiRs in the LUSC dataset
After confirming that global differences in isomiR expression between tumor and normal samples are maintained during batch correction, we next aimed to examine on an individual isomiR level whether biological effects are revealed upon batch correction, again using the TCGA-LUSC data set as an example. For this purpose, we identified isomiRs with different expression levels in tumor and normal tissue by t-test with subsequent FDR correction. For most isomiRs, the significance of the expression difference between tumor and normal samples did not change. However, for a subset of 19 of 656 isomiRs, significant differences in expression were revealed after batch correction (in the corrected samples absolute log2 fold change of 0.4 between tumor and normal samples, q-value < 0.05 and q-value > 0.05 in the uncorrected samples), while a larger subset of 25 isomiRs are not differentially expressed any more (Figure 7A). These subsets were not associated with expression levels of the respective isomiRs (Figure 7B). Therefore, we conclude that batch correction did affect rare and abundant isomiRs similarly.
Figure 7.
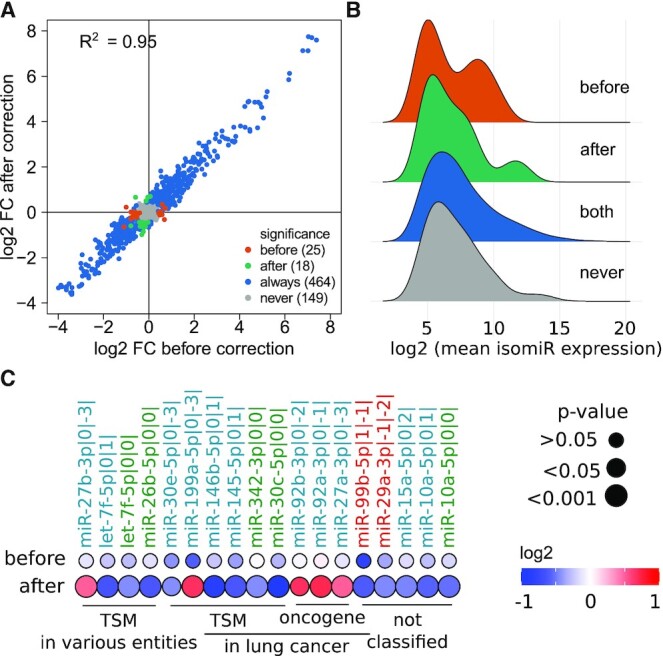
Significant differential expression in tumor versus normal tissue of a new candidate isomiR set after batch correction. (A) Comparison of fold changes of expression in tumor versus normal before and after batch correction leads to a subset of isomiRs whose significant expression can only be observed after batch correction. (significance: log2 FC difference between tumor and normal amples > |0.4|, q-value < 0.05) (B) IsomiRs which are significantly differentially expressed either only before or only after batch correction show an expression spectrum well spread within all other isomiRs. For more information on significance see (A). (C) Roles in cancer of the canonical versions of the 19 isomiRs which are only significant after correction. If roles were controversial between tumor types, we focused on the role in lung cancer.As 5′ isomiRs have altered target spectra to the canonical versions, they were generally defined as ‘not classified’. 3′ isomiR names are colored in blue, 5′ in red and canonical isomiR names in green. TSM = tumor suppressor isomiR.
Looking more carefully at a subset of 21 isomiRs with a significant unadjusted P-value only after correction of batch effects and an absolute log2 fold change of 0.4 between tumor and normal samples, we next investigated their published roles in cancer (Figure 7C). Here, we assumed that 3′ isomiRs (blue) generally exert similar functions as their canonical counterparts (green) whereas the functions of 5′ isomiRs (red) are generally less studied and harder to predict due to shifted seed sequences and altered target spectra. Therefore, we defined them to have a not classified function, just as three isomiRs with canonical seed sequences but controversial reports in literature (41–43).
Importantly, the roles of isomiRs with canonical seed sequences (i.e. canonical forms and 3′ isomiRs) as tumor suppressive or oncogenic miRNAs were largely in accordance with the significant differences in expression between tumor and normal samples unmasked upon batch correction. For instance, the canonical forms of hsa-miR-30c-5p, hsa-miR-342-3p and hsa-let-7f-5p were detected as significantly downregulated in tumors compared to normal tissues only in the batch corrected data and have described tumor suppressive functions in lung cancer and also partly in other cancer entities (21,44–47). A tumor suppressive function for hsa-let-7f-5p specifically in lung cancer has not been shown yet, however, various members of the let-7 miRNA family have been found to be tumor suppressive in almost all cancer entities (48). Unexpectedly, hsa-miR-27a-3p and hsa-miR-27b-3p were detected as significantly upregulated in tumor samples after batch correction whereas literature describes an antitumoral role of these two isomiRs in lung cancer (49,50), however no study could be found investigating their specific role in lung squamous cell carcinoma which might differ from those in other lung cancer types. Of note, hsa-miR-199a-5p has previously been described as tumor suppressor in lung cancer despite its independently observed upregulation in lung cancer samples compared to healthy tissues which is in line with our observation (51).
DISCUSSION
In this study, we identify and remove annotation errors and batch effects occurring in TCGA isoform expression quantification data and thereby provide an important new resource for downstream analysis of isomiR expression in cancer patients. Specifically, we could illustrate that misinterpretation of isomiR annotation can lead to strong misconception of the resulting dataset (Figure 1C) and this might have already affected previous studies based on the isomiR expression quantification dataset from TCGA. Therefore, we strongly recommend using our correctly annotated data in the future. Similarly, the recently proposed mirGFF3 format can be applied, also to other small RNA datasets independent from TCGA (52). In addition, data distortion by factors such as sequencing platform, plate, sequencing depth and tumor purity were identified within isomiR expression quantification datasets from individual cancer entities and can be found across all 16 TCGA datasets which were investigated here (Figure 5 and Supplementary Figures S23–32). Variation in sample processing over a long time period and between different collaborating institutes is a known confounder in TCGA pan-cancer experiments (25) and with a sequencing span of 10 years (2008–2018) for the TCGA miRNA data (24), batch effects are unavoidable (26). Comparing different combinations of batch variables (plate, platform and tumor purity) and using two different batch effect removal algorithms on the expression quantification data (limma and ComBat), we identified the best correction options i.e. the one with the least remaining batch effects for each dataset. Thereby, we could completely remove plate and platform effects from all datasets. Effects from batch variables such as purity and sequencing depth could not completely be removed in some cohorts but were clearly reduced (Figure 5). Hence, when analyzing isomiR expression using our provided batch corrected data within individual cohorts, it is strongly advisable to control for residual batch effects specifically for isomiRs under investigation (Figure 4 and Supplementary Table S2) to ensure robust and reliable results. Further, even if no significant effects are detected after batch correction, we cannot exclude the possibility that unknown or insignificant associations might still impact the biological interpretation of the data.
Batch effect removal was performed without and with normal samples where applicable (Table 1). Even though correction without normal samples improved batch correction results (Figure 5 and Supplementary Figure S22), we are aware that normal samples are of importance for many biological questions. Indeed, we confirmed that differences between tumor and normal samples were retained after successful batch correction in the TCGA-LUSC dataset (Figure 6) and other relevant tumor entities (Supplementary Figures S23–32). Furthermore, comparative expression analysis on the level of individual isomiRs between tumor and normal samples showed that some previously published biological differences were indeed masked by batch bias before correction and only became apparent after batch correction in the TCGA-LUSC dataset (Figure 6). We assume similar phenomena also in other relevant cancer entities. Concomitantly, isomiRs which showed significant differences in expression between tumors and normal samples before batch correction were not re-identified among the significantly differentially expressed ones after correction pointing to the necessity of batch correction to obtain reliable results.
As batch correction comes with potential side effects such as e.g. unknown or overestimated effects leading to false negative or positive results, or incomplete removal of batch effects, avoiding the necessity of batch correction completely, is desirable (32,53). Importantly, removal of batch effects is only required for downstream analysis such as PCA, clustering and data visualization, or for direct expression comparison between patients. For other statistical analyses, for instance differential expression analysis, modeling of the batch variables as covariates is preferable (32). To this end, our study provides valuable insights to which variable should be included as covariate in which TCGA cohort. Batch effects identified in this study are mostly related directly or indirectly to sequencing depth, in part due to use of different platforms with varying capabilities. IsomiR expression quantification is even more delicate than miRNA expression analysis as all isoforms of one miRNA arms are summed up in miRNA datasets. Due to the substantially higher complexity and resulting lower expression levels of individual isomiRs species (Figure 2H and I; Supplementary Figure S4), a high and equal sequencing depth for isomiR sequencing is vital to obtain comparable results and also to comprise isomiRs apart from the usually highest expressed canonical form (Supplementary Figure S1) (54). Therefore, sequencing depth as central issue for batch effect generation should get taken in more consideration for experimental design. In addition, we highly recommend careful and substantial quality control especially when analyzing expression patterns of individual isomiRs provided as supplement to this study to ensure robust and biologically meaningful interpretations.
Another potential source for biases in sequencing results derives from usage of different platforms for sequencing. In fact, we find significant, platform dependent differences in isomiR length, general GC content and GC content at the first position and provide a list of recurrently affected isomiRs that should be treated with care when used for further research. The usage of different sequencing platforms frequently comes with use of different library preparation protocols, another factor which plays an important role regarding batch biases. Protocols have different sequence biases (e.g. due to adapter ligation, reverse transcription and amplification) leading to major differences in small RNA sequencing outcome and batch-bias: while each mRNA consists of various possible fragments with different GC contents, each isomiR is only represented by one sequence with a defined GC content leading to more significant sequencing biases (27,55,56). To this end, clear labeling of the platform and library preparation protocol facilitates identification of potential batch effects and usage of this parameter for batch correction. While this information is provided in the GDC legacy portal, it is not available in the harmonized data portal reducing awareness for this substantial batch variable. Hence, development of a standardized protocol for small RNA sequencing including standards for preparation protocol, sequencing depth and appropriate platform would likely allow better comparability of results and reduce batch effects and thereby provide valuable new data to the research community and should be developed and used for future studies.
Together, our study highlights the importance of annotation and batch correction as well as careful quality control when working with the TCGA isomiR quantification datasets. We provide both a mapping gff3 file for automated annotations of isomiRs shifted by up to 3 nt at either end compared to the canonical isomiR as well as substantial benchmarking of confounding variables in 16 large TCGA cohorts (Supplementary Table S1, GEO series GSE164767). Given the success of batch correction especially with respect to the batch variables plate and platform, the resulting corrected expression matrices for these cohorts will be a valuable resource for the research community.
DATA AVAILABILITY
All data are publicly available and accessible as described in the ‘Data download and pre-processing’ section. The batch corrected data have been deposited in NCBI’s Gene Expression Omnibus (57) and are accessible through GEO Series accession number GSE164767 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164767). R code for data download, pre-processing, annotation and batch correction as well as for the implementation of described analyses and main figures are publicly available on GitHub (https://github.com/susibing/BatchCor_TCGAisomiR).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Yoann Pageaud for the thorough and helpful code review.
Author contributions: S.I. optimized and performed batch correction, analyzed the data, generated figures, participated in study design and writing of the manuscript. B.E.M. analyzed the data, generated figures and participated in writing of the manuscript. M.M. performed the analysis of sequence bias of batch effects. H.R.M. participated in downloading and pre-processing of the data. L.F. and C.K. conceived the study and participated in writing the manuscript.
Notes
Present address: Susanne Ibing, Digital Health Center, Hasso Plattner Institute, University of Potsdam, Prof.-Dr-Helmert-Strasse 2-3, 14482 Potsdam, Germany.
Contributor Information
Susanne Ibing, Division of Applied Bioinformatics, German Cancer Research Center (DKFZ), Berliner Straße 41, 69120 Heidelberg, Germany.
Birgitta E Michels, Division of Molecular Genome Analysis, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 580, 69120 Heidelberg, Germany.
Moritz Mosdzien, Division of Molecular Genome Analysis, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 580, 69120 Heidelberg, Germany.
Helen R Meyer, Division of Molecular Genome Analysis, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 580, 69120 Heidelberg, Germany.
Lars Feuerbach, Division of Applied Bioinformatics, German Cancer Research Center (DKFZ), Berliner Straße 41, 69120 Heidelberg, Germany.
Cindy Körner, Division of Molecular Genome Analysis, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 580, 69120 Heidelberg, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) [437616695 to B.E.M.]; Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) [Research Consortium FOR 2674 to L.F.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bartel D.P. MicroRNA target recognition and regulatory functions. Cell. 2009; 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hammond S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015; 87:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou K., Liu M., Cao Y. New insight into microRNA functions in cancer: oncogene-microRNA-tumor suppressor gene network. Front. Mol. Biosci. 2017; 4:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Desvignes T., Batzel P., Berezikov E., Eilbeck K., Eppig J.T., McAndrews M.S., Singer A., Postlethwait J.H. microRNA nomenclature: a view incorporating genetic origins, biosynthetic pathways, and sequence variants. Trends Genet. 2015; 31:613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Telonis A.G., Loher P., Jing Y., Londin E., Rigoutsos I. Beyond the one-locus-one-miRNA paradigm: microRNA isoforms enable deeper insights into breast cancer heterogeneity. Nucleic Acids Res. 2015; 43:9158–9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ludwig N., Leidinger P., Becker K., Backes C., Fehlmann T., Pallasch C., Rheinheimer S., Meder B., Stähler C., Meese E. et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016; 44:3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cloonan N., Wani S., Xu Q., Gu J., Lea K., Heater S., Barbacioru C., Steptoe A.L., Martin H.C., Nourbakhsh E. et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011; 12:R126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan G.C., Chan E., Molnar A., Sarkar R., Alexieva D., Isa I.M., Robinson S., Zhang S., Ellis P., Langford C.F. et al. 5′ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 2014; 42:9424–9435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manzano M., Forte E., Raja A.N., Schipma M.J., Gottwein E. Divergent target recognition by coexpressed 5′-isomiRs of miR-142-3p and selective viral mimicry. RNA. 2015; 21:1606–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salem O., Erdem N., Jung J., Münstermann E., Wörner A., Wilhelm H., Wiemann S., Körner C. The highly expressed 5′isomiR of hsa-miR-140-3p contributes to the tumor-suppressive effects of miR-140 by reducing breast cancer proliferation and migration. BMC Genomics. 2016; 17:566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ibuki Y., Nishiyama Y., Tsutani Y., Emi M., Hamai Y., Okada M., Tahara H. Circulating microRNA/isomiRs as novel biomarkers of esophageal squamous cell carcinoma. PLoS One. 2020; 15:e0231116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lan C., Peng H., McGowan E.M., Hutvagner G., Li J. An isomiR expression panel based novel breast cancer classification approach using improved mutual information. BMC Med. Genomics. 2018; 11:73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liao Z., Li D., Wang X., Li L., Zou Q. Cancer diagnosis through isomiR expression with machine learning method. Curr. Bioinform. 2016; 13:57–63. [Google Scholar]
- 14. Telonis A.G., Magee R., Loher P., Chervoneva I., Londin E., Rigoutsos I. Knowledge about the presence or absence of miRNA isoforms (isomiRs) can successfully discriminate amongst 32 TCGA cancer types. Nucleic Acids Res. 2017; 45:2973–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang S., Zheng Z., Chen P., Wu M. Tumor classification and biomarker discovery based on the 5′isomiR expression level. BMC Cancer. 2019; 19:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ebrahimi A., Sadroddiny E. MicroRNAs in lung diseases: recent findings and their pathophysiological implications. Pulm. Pharmacol. Ther. 2015; 34:55–63. [DOI] [PubMed] [Google Scholar]
- 17. Lin P.Y., Yu S.L., Yang P.C. MicroRNA in lung cancer. Br. J. Cancer. 2010; 103:1144–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zaporozhchenko I.A., Morozkin E.S., Ponomaryova A.A., Elena Y., Cherdyntseva N. V, Zheravin A.A., Pashkovskaya O.A. Profiling of 179 miRNA expression in blood plasma of lung cancer patients and Cancer-Free individuals. Sci. Rep. 2018; 8:6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luo J., Shi K., Yin S., Tang R., Chen W., Huang L., Gan T., Cai Z., Chen G. Clinical value of miR-182-5p in lung squamous cell carcinoma: a study combining data from TCGA, GEO, and RT-qPCR validation. World J. Surg. Oncol. 2018; 16:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koppers-Lalic D., Hackenberg M., de Menezes R., Misovic B., Wachalska M., Geldof A., Zini N., de Reijke T., Wurdinger T., Vis A. et al. Non–invasive prostate cancer detection by measuring miRNA variants (isomiRs) in urine extracellular vesicles. Oncotarget. 2016; 7:22566–22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xia Y., Chen Q., Zhong Z., Xu C., Wu C., Liu B., Chen Y. Down-regulation of MIR-30C promotes the invasion of non-small cell lung cancer by targeting MTA1. Cell. Physiol. Biochem. 2013; 32:476–485. [DOI] [PubMed] [Google Scholar]
- 22. Chu A., Robertson G., Brooks D., Mungall A.J., Birol I., Coope R., Ma Y., Jones S., Marra M.A. Large-scale profiling of microRNAs for the Cancer Genome Atlas. Nucleic Acids Res. 2016; 44:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Camacho D.M., Collins K.M., Powers R.K., Costello J.C., Collins J.J. Next-generation machine learning for biological networks. Cell. 2018; 173:1581–1592. [DOI] [PubMed] [Google Scholar]
- 24. Gao G.F., Parker J.S., Reynolds S.M., Silva T.C., Wang L.B., Zhou W., Akbani R., Bailey M., Balu S., Berman B.P. et al. Before and after: comparison of legacy and harmonized TCGA genomic data commons’ data. Cell Syst. 2019; 9:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tomczak K., Czerwińska P., Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp. Oncol. 2015; 1A:A68–A77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leek J.T., Scharpf R.B., Bravo H.C., Simcha D., Langmead B., Johnson W.E., Geman D., Baggerly K., Irizarry R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010; 11:733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giraldez M.D., Spengler R.M., Etheridge A., Godoy P.M., Barczak A.J., Srinivasan S., De Hoff P.L., Tanriverdi K., Courtright A., Lu S. et al. Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat. Biotechnol. 2018; 36:746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lauss M., Visne I., Kriegner A., Ringnér M., Jönsson G., Höglund M. Monitoring of technical variation in quantitative high-throughput datasets. Cancer Inform. 2013; 12:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hoadley K.A., Yau C., Hinoue T., Wolf D.M., Lazar A.J., Drill E., Shen R., Taylor A.M., Cherniack A.D., Thorsson V. et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018; 173:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Colaprico A., Silva T.C., Olsen C., Garofano L., Cava C., Garolini D., Sabedot T.S., Malta T.M., Pagnotta S.M., Castiglioni I. et al. TCGAbiolinks: an R/bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016; 44:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A. et al. The immune landscape of cancer. Immunity. 2018; 48:812–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goh W.W. Bin, Wang W., Wong L Why batch effects matter in Omics Data, and how to avoid them. Trends Biotechnol. 2017; 35:498–507. [DOI] [PubMed] [Google Scholar]
- 33. R Core Team R: a Language and Environment for Statistical Computing. 2019; Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 34. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2016; NY: Springer. [Google Scholar]
- 35. Van Der Maaten L., Courville A., Fergus R., Manning C. Accelerating t-SNE using Tree-Based Algorithms. J. Mach. Learn. Res. 2014; 15:3221–3245. [Google Scholar]
- 36. Van der Maaten L., Hinton G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008; 9:2579–2605. [Google Scholar]
- 37. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leek J.T., Johnson W.E., Parker H.S., Fertig E.J., Jaffe A.E., Storey J.D., Zhang Y., Torres L.C. sva: Surrogate Variable Analysis. 2019; R package version 3.32.1; https://bioconductor.org/packages/release/bioc/html/sva.html. [Google Scholar]
- 39. Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007; 8:118–127. [DOI] [PubMed] [Google Scholar]
- 40. Warnes G.R., Bolker B., Bonebakker L., Gentleman R., Liaw W.H.A., Lumley T., Maechler M., Magnusson A., Moeller S., Schwartz M. gplots: Various R Programming Tools for Plotting Data. 2020; R package version 3.1.0; https://cran.r-project.org/package=gplots. [Google Scholar]
- 41. Agirre X., Jiménez-Velasco A., San José-Enériz E., Garate L., Bandrés E., Cordeu L., Aparicio O., Saez B., Navarro G., Vilas-Zornoza A. et al. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol. Cancer Res. 2008; 6:1830–1840. [DOI] [PubMed] [Google Scholar]
- 42. Ran J., Li Y., Liu L., Zhu Y., Ni Y., Huang H., Liu Z., Miao Z., Zhang L. Apelin enhances biological functions in lung cancer A549 cells by downregulating exosomal miR-15a-5p. Carcinogenesis. 2020; 42:243–253. [DOI] [PubMed] [Google Scholar]
- 43. Li Y., Zhang H., Dong Y., Fan Y., Li Y., Zhao C., wang C., Liu J., Li X., Dong M. et al. MiR-146b-5p functions as a suppressor miRNA and prognosis predictor in non-small cell lung cancer. J. Cancer. 2017; 8:1704–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tai M.C., Kajino T., Nakatochi M., Arima C., Shimada Y., Suzuki M., Miyoshi H., Yatabe Y., Yanagisawa K., Takahashi T. miR-342-3p regulates MYC transcriptional activity via direct repression of E2F1 in human lung cancer. Carcinogenesis. 2015; 36:1464–1473. [DOI] [PubMed] [Google Scholar]
- 45. Li Y., Jiang Q., Xia N., Yang H., Hu C. Decreased expression of microRNA-375 in nonsmall cell lung cancer and its clinical significance. J. Int. Med. Res. 2012; 40:1662–1669. [DOI] [PubMed] [Google Scholar]
- 46. Xie X., Liu H., Wang M., Ding F., Xiao H., Hu F., Hu R., Mei J. miR-342-3p targets RAP2B to suppress proliferation and invasion of non-small cell lung cancer cells. Tumor Biol. 2015; 36:5031–5038. [DOI] [PubMed] [Google Scholar]
- 47. Yan J.W., Lin J.S., He X.X. The emerging role of miR-375 in cancer. Int. J. Cancer. 2014; 135:1011–1018. [DOI] [PubMed] [Google Scholar]
- 48. Chirshev E., Oberg K.C., Ioffe Y.J., Unternaehrer J.J. Let-7 as biomarker, prognostic indicator, and therapy for precision medicine in cancer. Clin. Transl. Med. 2019; 8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sun Y., Xu T., Cao Y.W., Ding X.Q. Antitumor effect of miR-27b-3p on lung cancer cells via targeting Fzd7. Eur. Rev. Med. Pharmacol. Sci. 2017; 21:4113–4123. [PubMed] [Google Scholar]
- 50. Yan X., Yu H., Liu Y., Hou J., Yang Q., Zhao Y. miR-27a-3p functions as a tumor suppressor and regulates non-small cell lung cancer cell proliferation via targeting HOXB8. Technol. Cancer Res. Treat. 2019; 18:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51. Wang Q., Ye B., Wang P., Yao F., Zhang C., Yu G. Overview of microRNA-199a regulation in cancer. Cancer Manag. Res. 2019; 11:10327–10335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Desvignes T., Loher P., Eilbeck K., Ma J., Urgese G., Fromm B., Sydes J., Aparicio-Puerta E., Barrera V., Espín R. et al. Unification of miRNA and isomiR research: the mirGFF3 format and the mirtop API. Bioinformatics. 2020; 36:698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nygaard V., Rødland E.A., Hovig E. Methods that remove batch effects while retaining group differences may lead to exaggerated confidence in downstream analyses. Biostatistics. 2016; 17:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Loher P., Londin E.R., Rigoutsos I. IsomiR expression profiles in human lymphoblastoid cell lines exhibit population and gender dependencies. Oncotarget. 2014; 5:8790–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wright C., Rajpurohit A., Burke E.E., Williams C., Collado-Torres L., Kimos M., Brandon N.J., Cross A.J., Jaffe A.E., Weinberger D.R. et al. Comprehensive assessment of multiple biases in small RNA sequencing reveals significant differences in the performance of widely used methods. BMC Genomics. 2019; 20:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dard-Dascot C., Naquin D., d’Aubenton-Carafa Y., Alix K., Thermes C., van Dijk E. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genomics. 2018; 19:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are publicly available and accessible as described in the ‘Data download and pre-processing’ section. The batch corrected data have been deposited in NCBI’s Gene Expression Omnibus (57) and are accessible through GEO Series accession number GSE164767 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164767). R code for data download, pre-processing, annotation and batch correction as well as for the implementation of described analyses and main figures are publicly available on GitHub (https://github.com/susibing/BatchCor_TCGAisomiR).