

Abstract
Recent studies have reported that TBI is an independent risk factor for subsequent stroke. Here, we tested the hypothesis that TBI would exacerbate experimental stroke outcomes via alternations in neuroimmune and neurometabolic function. We performed a mild closed-head TBI and then one week later induced an experimental stroke in adult male mice. Mice that had previously experienced TBI exhibited larger infarcts, greater functional deficits, and more pronounced neuroinflammatory responses to stroke. We hypothesized that impairments in central metabolic physiology mediated poorer outcomes after TBI. To test this, we treated mice with the insulin sensitizing drug pioglitazone (Pio) after TBI. Pio prevented the exacerbation of ischemic outcomes induced by TBI and also blocked the induction of insulin insensitivity by TBI. However, tissue respiratory function was not improved by Pio. Finally, TBI altered microvascular responses including promoting vascular accumulation of serum proteins and significantly impairing blood flow during the reperfusion period after stroke, both of which were reversed by treatment with Pio. Thus, TBI appears to exacerbate ischemic outcomes by impairing metabolic and microvascular physiology. These data have important implications because TBI patients experience strokes at greater rates than individuals without a history of head injury, but these data suggest that those strokes may also cause greater tissue damage and functional impairments in that population.
Keywords: Traumatic brain injury, stroke, metabolism, vasculature, inflammation, reperfusion
Introduction
Several recent studies have reported that traumatic brain injury (TBI) is an independent risk factor for subsequent stroke (Albrecht et al., 2015; Burke et al., 2013; Chen et al., 2011; Liao et al., 2014). Even the mildest injuries (i.e., concussion) produce stroke hazard ratios of 1.27–1.74 compared to individuals without prior TBI (Burke et al., 2013; Liao et al., 2014; Liu et al., 2017). Importantly, this variable interacts with traditional risk factors including being male, hypertension, and hyperlipidemia (Morris et al., 2017). Potential links between TBI and stroke have been postulated, but only explored epidemiologically so no causative factor has been established, though there is mounting evidence that impairments in vascular function and coagulation abnormalities may be involved (Frith et al., 2012; Schwarzmaier et al., 2016). Moreover, clinically, the relative risks of treating with anticoagulants after brain injury to reduce the risk of stroke must be balanced against the increased likelihood of intracerebral hemorrhage (Albrecht et al., 2014; Zhang et al., 2012). Recreational and prescription drug use also may increase stroke incidence among TBI patients (Gill et al., 2005; Glenn, 2010).
In addition to the increased incidence of stroke, mortality is greater in stroke patients that have suffered a TBI (Liao et al., 2014). Men 20–69 years of age who suffered a stroke within 24 months of a TBI were nearly twice as likely to die as patients that had not suffered a TBI. It is also possible that strokes in the TBI population are more likely to be reported because they produce more significant signs, symptoms, and damage. Thus, the epidemiological data do not fully indicate whether TBI is increasing stroke incidence and/or exacerbating the damage associated with ischemia.
Clearly central to the vulnerability to cerebral ischemia is the cerebral vasculature and the neurovascular unit more broadly, which is often damaged by TBI(Sandsmark et al., 2019). A very large percentage of individuals with a history of TBI exhibit postmortem evidence of damage to both large and small vessels(Hay et al., 2015; Shlosberg et al., 2010). Severe damage can cause hemorrhages and vasospasm, but even short of those outcomes vessel damage can significantly contribute to post-injury dysfunction(Logsdon et al., 2015; Sandsmark et al., 2019). The CNS is highly dependent on the coordination among multiple cell types that form the neurovascular unit (endothelial cells, mural cells and glia) for the activity-dependent delivery of oxygen and glucose and the removal of waste products. Moreover, damage to the vessels has the potential to induce inflammatory events, intravascular coagulation and microthrombi that can reduce tissue oxygenation and contribute to vasogenic edema(Bartnik-Olson et al., 2014) as well as narrowing the vascular lumen. A reduction in vascular reactivity associated with narrowing vascular lumen size and coagulopathies is likely to greatly increase the risk of stroke (Ostergaard et al., 2016) and potentially impair the ability of the vasculature to reperfuse after stroke leading to much greater tissue loss and functional deficits.
Recovery from TBI is a protracted process; not only does TBI directly damage the nervous system, but it also renders the brain more vulnerable to subsequent insults. For instance, following a TBI, the functional consequences of repeated injuries are much more severe, particularly when the injuries occur close together in time (McCrory et al., 2013; Vagnozzi et al., 2008). The TBI-induced physiological changes that render the injured brain more vulnerable to subsequent insults are not fully understood. However, it is clear that recovery from brain injury is metabolically demanding and occurs during a time in which supplying energy to the brain and the removal of potentially toxic metabolic byproducts is impaired by vascular dysfunction, increased intracranial pressure, and cell intrinsic impairments in substrate metabolism (Abdul-Muneer et al., 2015; Giza and Hovda, 2014; Sandsmark et al., 2019). Here we tested the hypothesis that experimental TBI would exacerbate ischemic outcomes via alterations in metabolic and cerebrovascular function.
Materials and Methods
Data resulting from these experiments are available upon request.
Animals.
Swiss Webster mice were purchased from Charles River (Wilmington, MA) and bred at OSU and WVU. Swiss Webster mice were used because their skulls are much less likely to fracture with our injury device than other strains. Pups were weaned at 21 days of age and were housed in a 14:10 light cycle with ad libitum access to food (Harlan-Teklad #8640) and filtered tap water. All procedures were approved by the OSU and WVU Institutional Animal Care and Use Committees and were conducted in accordance with NIH guidelines. In all cases, animals were randomly assigned to groups (injury, drug, and ischemia) and all observations were conducted by researchers blind to experimental conditions.
Surgical Procedures.
A single closed-head mild TBI was conducted on male mice (6–8 weeks of age) as previously reported (Karelina et al., 2017) (see below). Given the significant sex difference reported for the incidence of TBI in young adult males, and increased mortality in post-TBI stroke in males, we elected to conduct these experiments in young male mice. (Faul et al., 2010; Liao et al., 2014).
Traumatic brain injury.
Mice were randomly assigned to an experimental group and then placed into a stereotaxic frame under inhaled isoflurane anesthesia (3% induction, 1.5% maintenance). The injury was induced with an Impact One device (Leica Biosystems, Buffalo Grove, IL). A 3mm diameter impactor tip was retracted and driven into the skull along the midline (−2mm AP relative to bregma) to a depth of 1.2 mm at a rate of 3mm/sec (dwell time: 30 msec). The skin was sutured with 6/0 nylon suture and mice were returned to their cages. The control procedure was performed identically but the impactor was not driven into the skull (just placed on the surface and retracted). A subset of mice were treated intraperitoneally (IP) with 10mg/kg Pioglitazone (Abcam) or the vehicle (5% DMSO in olive oil). Mice were treated 15 minutes after injury and again 24 hours later.
Middle cerebral artery occlusion.
Briefly, following a 7-day recovery period after TBI, mice were re-anesthetized with inhaled isoflurane (3% induction, 1.5% maintenance) and a unilateral middle cerebral artery occlusion (MCAO) was achieved by insertion of a 0.23mm occluder (Doccol) into the right middle cerebral artery via the internal carotid artery and extending 6mm beyond the internal carotid-pterygopalatine artery bifurcation. The occluder was secured and the mouse was returned to its home cage for 1 hour. Following the 1-hour occlusion period, the mouse was re-anesthetized, and reperfusion was initiated by removal of the occluder. Sham surgeries included all the surgical preparation up to the exposure of the internal carotid artery but omitted occlusion, all other aspects of the surgery were identical. All animals were treated with the analgesic buprenorphine (0.1mg/kg). MCAO mice were included in the study if a measurable infarct was present.
Determination of infarct volume.
Mice were euthanized by rapid decapitation; brains were sectioned into 4 2mm-thick coronal sections through the forebrain and incubated in 1% TTC (made in saline) for 10 minutes at 37°C. TTC produces a pink formazan product in the presence of live mitochondria, thus ischemic lesions are visualized as unstained white tissue. Slices were post-fixed in 4% paraformaldehyde overnight, photographed, and infarct area was outlined on both sides of each slice using Fiji imaging software(Schindelin et al., 2012). Infarct size was determined after correcting for edema and reported as percent of the contralateral hemisphere using the formula: (1-(total ipsilateral hemisphere – infarct)/total contralateral hemisphere) *100. Following tissue imaging for TTC, the paraformaldehyde-fixed tissue slices were cryopreserved, frozen, and further sectioned coronally on a cryostat at 40μm throughout the forebrain. The thin-sliced tissue was then used for free-floating immunohistochemistry and silver staining. Histochemistry was conducted as previously described (Karelina et al., 2017). Axon degeneration was assessed via silver staining (FD NeuroSilver) per manufacturer’s instructions. Of note, TTC-stained tissue has successfully been used for further protein analysis (i.e. immunohistochemistry) as a means of reducing the total number of animals needed for experiments, as well as providing maximal converging information regarding ischemia-related pathology and behavior from each animal (Kramer et al., 2010; Sun et al., 2012).
A separate subset of animals was transcardially perfused seven days post-TBI and tissue was cut on a cryostat and processed for immunochemical labelling of IgG, CD31 and GFAP.
Behavioral testing.
Motor function was assessed in the corner test by placing a mouse facing into the corner of two Plexiglas boards (attached at a 30° angle) to the depth at which both sides of the vibrissae were touching each board. The direction the mouse turned to exit the corner was recorded, the mouse was replaced in its home cage for 30 seconds, and the trial was repeated for a total of 10 times. The percent time turning toward the non-impaired side (right) was calculated for each mouse by a blinded observer.
Histological analysis
Following tissue imaging for TTC, the paraformaldehyde-fixed tissue slices were cryopreserved, frozen, and further sectioned coronally on a cryostat at 40μm throughout the forebrain. The thin-sliced tissue was then used for free-floating silver staining (NeuroSilver, FD Neurotechnologies, Columbia, MD) per the manufacturer’s instructions.
A separate subset of animals was transcardially perfused seven days post-TBI (or the control procedure) and the brains were postfixed, cryoprotected and frozen before being cut on a cryostat at 40 microns. Sections were then blocked in 1% normal horse serum containing 0.01% Triton-X and incubated overnight in primary antibodies CD-31 (RNDSystems, Minneapolis, MN, 1:200) and GFAP (Agilent, Santa Clara, CA 1:250) at room temperature. Tissue was then incubated in secondary antibodies (rabbit-anti mouse 546, donkey anti-goat 647, and IgG labelling was assessed with goat anti-rabbit IgG 488).
Imaging and Analysis
Axonal degeneration was assessed live, by an investigator blinded to the experimental conditions on a Leica microscope. Sections were imaged initially at low magnification (4x) and areas of degeneration were examined at higher power (20x). Experimenters made qualitative analyses of silver staining using a 4-point scale (0 = no silver staining, 3 = dense axon degeneration in white matter tracks throughout the forebrain) as previously reported (Weil et al., 2014). IgG/CD31/GFAP immunohistochemistry of a section (between bregma −1.5 and −2.0) was imaged and stitched on an Olympus VS-120 microscope with a 10X Plan S Apo/0.40 NA objective using a Monochrome XM10 camera (1376 × 1032 imaging array, 6.45 × 6.45-micron pixel size, and 14-bit digitization). Resulting images of the entire section were assessed via qualitative analysis using a 5-point scale (0 = no staining, 1 = sparse vascular staining, 2 = moderate vascular staining limited to the injury site, 3 = extensive vascular staining throughout the injury site, 4 = extensive vascular staining evident within and beyond the injury site).
Real time PCR.
Fresh brains were removed, placed in RNAlater and cortex and striatum were dissected out of the ischemic hemisphere. RNA was extracted using Trizol reagent (Life Technologies), and reverse transcribed with M-MLV. Primer and probe sets were purchased from Applied Biosystems (CD68: Mm03047343_m1, TNF-α: Mm00443258_m1, and Il1β: Mm00434228_m1) and a Taqman 18S ribosomal RNA primer/probe set was used as an internal control (Applied Biosystems #4319413E). Universal RT-PCR cycling conditions were performed on an ABI 7900HT Fast Real-Time PCR System using the TaqMan Fast Advanced Master Mix (#4444963). Gene expression is normalized to 18S rRNA and expressed as a relative quantity based on the relative standard curve method.
Ex vivo insulin stimulation.
Mice were rapidly decapitated and fresh brains were collected and sliced through the entire forebrain at 0.4 mm intervals using a McIlwain tissue chopper. Slices from each brain were evenly split among two wells containing aCSF bubbled with carbogen (127 mM NaCl, 25 mM NaHCO3, 1.2 mM NaH2PO4, 2.5 mM KCl, 1 mM MgCl2, 25 mM glucose, 2 mM CaCl2), one well was incubated with 10nM insulin (Sigma, St. Louis MO) and the other well with only aCSF for 10 minutes at 37°C (Karelina et al., 2016). Tissue was then lysed in RIPA buffer containing phosphatase and protease inhibitors (Invitrogen), centrifuged, and protein concentration was assessed with a BCA protein assay (Bio-Rad).
Western blotting.
Protein lysates were subjected to SDS-PAGE as previously reported (Karelina et al., 2016). Protein (30μg) was electrophoresed into a precast gel (Bio-Rad, 4–15%) and transferred onto a PVDF membrane. Membranes were washed with TBS containing 0.01% tween, blocked in 5% BSA and incubated overnight with goat anti-rabbit phosphorylated Akt Ser473 (1:3,000 Cell Signaling), anti-rabbit pan Akt (1:2,000 Cell Signaling), or GAPDH (1:2000 Cell Signaling) at 4°C. Following secondary antibody incubation (HRP-conjugated anti-rabbit IgG, R&D), the blots were visualized with an ECL substrate (Invitrogen). Protein expression was determined by a densitometry calculation performed in Fiji (Schindelin et al., 2012). Phosphorylated Akt expression was first determined relative to pan Akt (pAkt/Akt), then reported as percent change pAkt expression [(pAkt/Akt insulin – pAkt/Akt aCSF)/pAkt/Akt insulin X 100)].
Seahorse XFe24 Microplate-Based Respirometry
Brains were rapidly dissected following decapitation and 250-μm slices were made using a McIlwain tissue chopper. The slices were transferred to artificial cerebral spinal fluid (aCSF, pH 7.4) consisting of 120 mM NaCl, 3.5 mM KCl, 1.3 mM CaCl2, 0.4 mM KH2PO4, 1 mM MgCl2, 5 mM HEPES, 10 mM D-Glucose, plus 4 mg/mL BSA. Tissue punches (2 per animal) of the corticostriatal interface (spanning approximately .75 mm to 1.00 mm anterior to bregma) were obtained measuring 1 mm in diameter. Tissue punches were loaded into Seahorse XFe24 Islet Capture Microplate (1 punch/well) containing 525 μL aCSF per well. Islet capture screens were secured over each tissue punch, and the plated was floated in a 37°C water bath in between animals.
Methods were adapted from (Fried et a1., 2014; Schuh et al., 2011) Briefly, the drug-filled sensor cartridge was hydrated overnight with XF Calibrant (1mL/well). The day of the assay the drug-filled sensor cartridge was loaded with 10X, 11X, 12X, and 13X of the desired final concentrations of pyruvate, oligomycin, FCCP, and rotenone plus antimycin-A in aCSF (75 μL) in ports A, B, C, and D. The final concentrations were: 1 mM, 10 μM, 10 μM, and 4 μM plus 4 μM. The assay protocol consisted of the following cycle: 3-min mix, 2-min wait, 3-min measure. The protocol specified for 5 cycles before the first injection and 5 cycles following each injection. Raw data were processed with the XFe24 Analyzer Software Program (Wave 2.6.0) using the Seahorse XF Cell Mito Stress Test Report Generator (Report Generator Version 4.0). The respiratory parameters obtained from the Report Generator for each sample (in duplicate) were averaged to determine the respiratory parameters for each sample.
Cerebral blood flow assessment.
Mice were randomly assigned to receive either a TBI or control procedure and then were injected with pioglitazone or vehicle. Seven day later mice a 1.5 cm incision was made along the surface of the skull and cerebral blood flow was assessed with a MoorFLPI laser speckle contrast imager (Moor Instruments, Devon UK). Three images were collected over the course of two minutes with an exposure time of 200 msec. All animals underwent a 60-minute MCAO procedure, the occluder was withdrawn and 60 minutes into reperfusion the animals were reimaged. Tissue incisions were closed with wound clips. Change in blood flow was expressed by relativizing the ipsi- and contralateral hemispheres during reperfusion to their baseline measurements.
Statistical analysis.
Sample sizes were formulated from power analyses based on the magnitude of differences detected in preliminary studies and from previous literature. Effect sizes were >0.45 with biochemical and histological changes being larger than behavioral effects.
Thus, we powered the study to detect differences with a β>0.8. Behavioral outcomes, infarct size, PCR, and western blotting results were assessed via a two-way ANOVA (injury condition X ischemia condition, or injury condition X treatment). All significant overall results (p < 0.05) were followed up with a Tukey HSD post hoc analysis. Qualitative silver staining and microglial morphology data were analyzed using the non-parametric Kruskal-Wallis H test. Significant overall Kruskal-Wallis results were followed up by pairwise comparisons.
Results
Traumatic brain injury exacerbates ischemic infarct, functional deficits, and neuropathology.
In order to determine whether a mild TBI increases vulnerability to cerebral ischemia, mice underwent a mild closed head TBI (or control injury; CON) and were returned to their home cages for a 1-week recovery period. Cerebral ischemia was conducted via middle cerebral artery occlusion (MCAO; or sham injury) one week after brain injury. The following groups were examined: CON/sham (n = 7), TBI/sham (n = 7), CON/MCAO (n = 9), TBI/MCAO (n = 9) after excluding 3 mice (2 CON/MCAO and 1 TBI/MCAO) without measurable infarcts. Behavioral testing for functional deficits, and tissue collection for infarct analysis and immunohistochemistry occurred 24 hours after MCAO or Sham. Infarct size was significantly impacted by prior TBI such that a mild TBI induced one week prior to MCAO more than tripled infarct size relative to MCAO without prior TBI (F1,17 = 20.458, p < 0.001; Figure 1A–B), as well as significantly increased cerebral edema (F1,16 = 13.076, p = 0.003; Figure 1C). The TBI/MCAO group also exhibited a greater degree of functional deficits in the corner test (F3,28 = 3.424, p = 0.033; Figure 1D). A post hoc analysis revealed that motor asymmetry was not significantly induced in the CON/MCAO group. In addition, mild TBI alone did not produce histological evidence of cell death or motor asymmetry without MCAO.
Figure 1. TBI prior to cerebral ischemia exacerbates infarct volume, edema, and functional deficits.
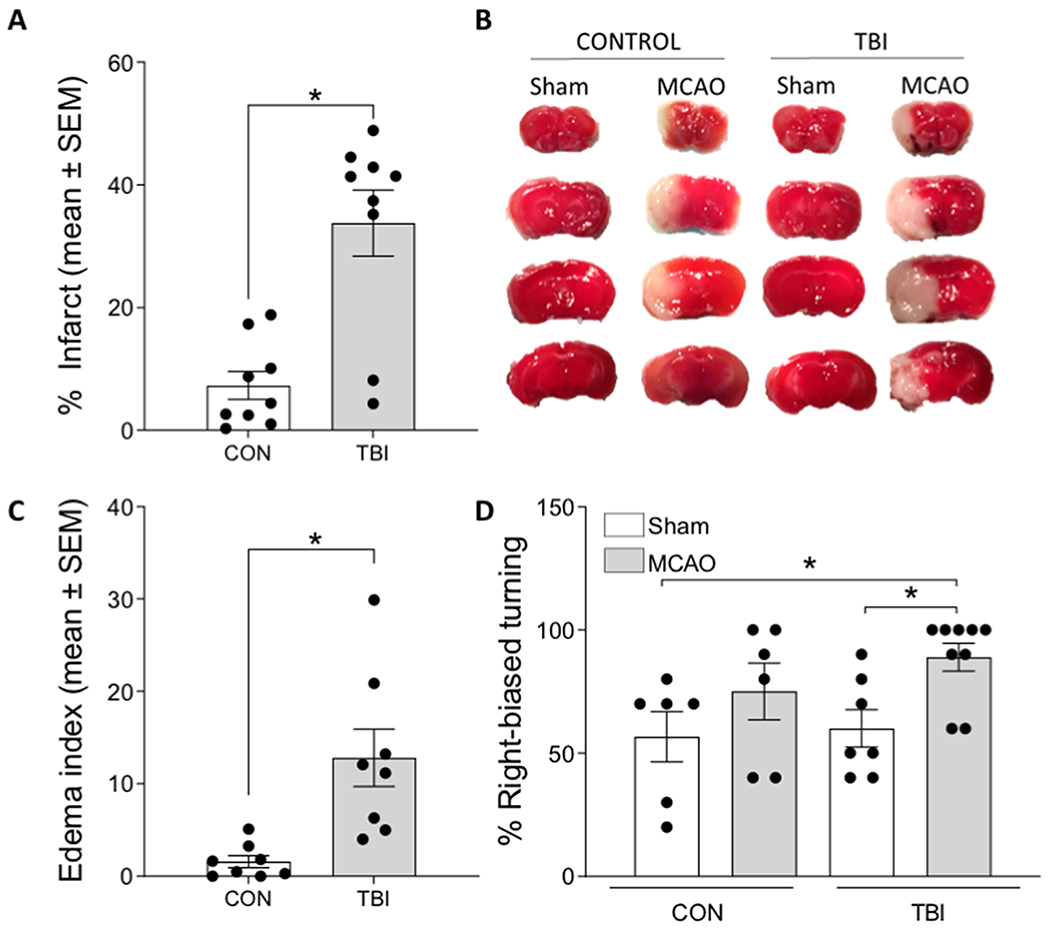
A) Mice that underwent a TBI 7 days prior to MCAO exhibited a significantly greater infarct volume, compared to those that had a control (CON) procedure prior to MCAO. B) Representative TTC stained tissue throughout the forebrain indicating healthy (red) and infarcted (white) tissue in the cortex and striatum. C) Mice that underwent a TBI prior to MCAO also exhibited significantly greater edema. D) Right-biased turning in the corner test revealed significant deficits in mice that underwent both a TBI and MCAO. Data are presented as mean +/− SEM. An asterisk (*) indicates significance at p < 0.05.
Analysis of the neuropathology revealed a statistically significant overall difference in axon degeneration between the groups as assessed with silver staining (H(3) = 15.687, p = 0.001), and a follow-up pairwise comparison revealed greater axonal degeneration in the TBI/MCAO group (Figure 2A). Given that TBI increased brain tissue vulnerability to MCAO-induced cell death (infarcted tissue), edema, and axon degeneration, we conducted an analysis of proinflammatory gene expression an identical cohort of mice (n = 5–7 per group) was examined for pro-inflammatory gene expression 24 hours after MCAO/Sham. Tissue punches from the ischemic hemisphere were collected through the cortex and striatum and relative gene expression was measured for CD68 (expressed by activated microglia) and the pro-inflammatory cytokines tumor necrosis factor (TNF) and interleukin 1-beta (IL1β). Significant group differences were determined for all the examined genes in the cortex (CD68 F3,24 = 7.614, p = 0.001; TNF F3,24 = 3.576, p = 0.032; Il1;β F3,23 = 5.244, p = 0.008). A follow-up Tukey post hoc analysis revealed the TBI/MCAO group to have significantly more CD68 and TNF gene expression relative to the CON/sham and TBI/sham groups, as well as significantly more IL1β gene expression relative to all other groups (Figure 2B). Similarly, significant group differences were determined for these genes in the striatum (CD68 F3,23 = 12.945, p < 0.001; TNF F3,24 = 6.440, p = 0.003; Il1β F3,24 = 3.716, p = 0.028). Post hoc analysis revealed the TBI/MCAO group to have significantly more CD68 gene expression relative to all other groups, as well as significantly more TNF and IL1β gene expression relative to the CON/sham and TBI/sham groups (Figure 2C).
Figure 2. Axon degeneration and pro-inflammatory gene expression following TBI and cerebral ischemia.
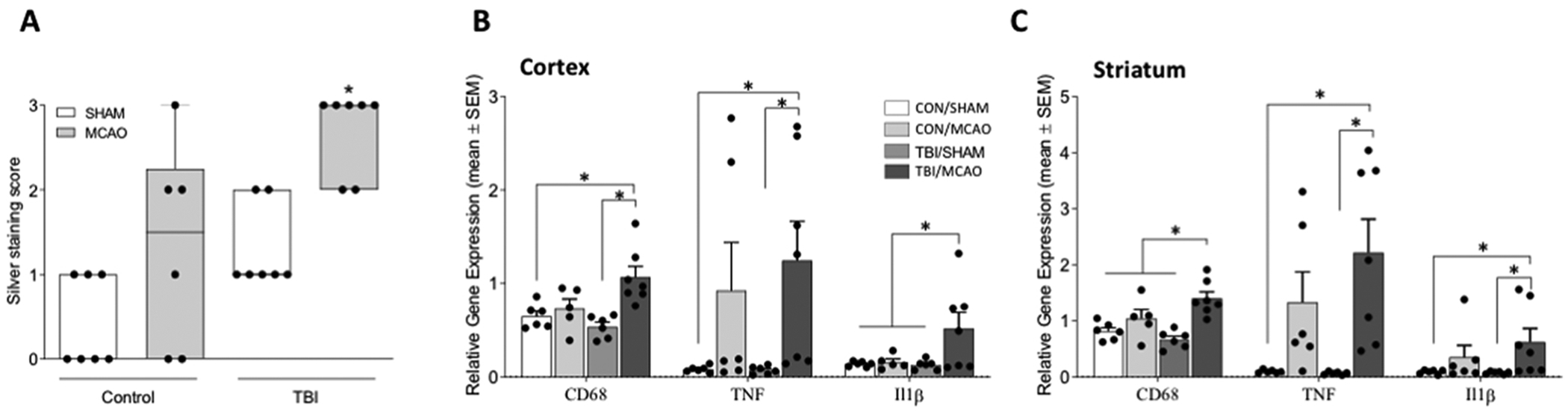
A) Mice that underwent a TBI 7 days prior to MCAO exhibited more white matter degeneration than those that had either TBI or MCAO alone. Proinflammatory cytokine gene expression is shown relative to 18S for CD68, TNF, and IL1β in the ipsilateral cortex B) and striatum C). All data are considered significant at p < 0.05. An asterisk (*) indicates significance at p < 0.05.
Pioglitazone treatment reduces vulnerability to cerebral ischemia after TBI.
TBI increases tissue vulnerability to subsequent insult in part by impairing brain metabolism, rendering the central nervous system temporarily more vulnerable to damage (Prins et al., 2010; Vagnozzi et al., 2008). Thus, additional damage to the brain during a period of central metabolic dysfunction is more severe and longer lasting. We previously reported that TBI acutely reduces central insulin sensitivity (Karelina et al., 2016). Here, we tested the effect of the insulin sensitizing drug, pioglitazone (Pio) on enhanced vulnerability to cerebral ischemia after TBI. Mice underwent TBI or control injury followed by MCAO one week later. All mice were treated with either Pio or vehicle after TBI as above and all groups underwent an MCAO 1 week after injury, resulting in the following MCAO groups: CON/Veh (n = 6), CON/Pio (n = 6), TBI/Veh (n = 6), TBI/Pio (n = 7), after excluding 7 mice from analysis due to a lack of a measurable infarct (3 CON/Pio, 3 TBI/Veh, and 1 TBI/PIO). Five days after MCAO, mice were assessed for functional deficits and infarct volume. Overall ANOVA results indicate significant group differences in infarct volume (F3,20 = 3.899, p = 0.029). Post hoc analysis revealed that among vehicle treated animals, mice that had a prior TBI had a significantly larger ischemic infarct compared to those that did not have a TBI; however, treatment with Pio significantly reduced infarct size (Figure 3A–B). Functional deficits were significantly increased after MCAO in the TBI/Veh group (F1,9 = 7.402, p = 0.03), compared to CON/Veh, but treatment with Pio after the TBI prevented the worsening of functional deficits (Figure 3C).
Figure 3. Pioglitazone reduces brain vulnerability to ischemia after TBI.
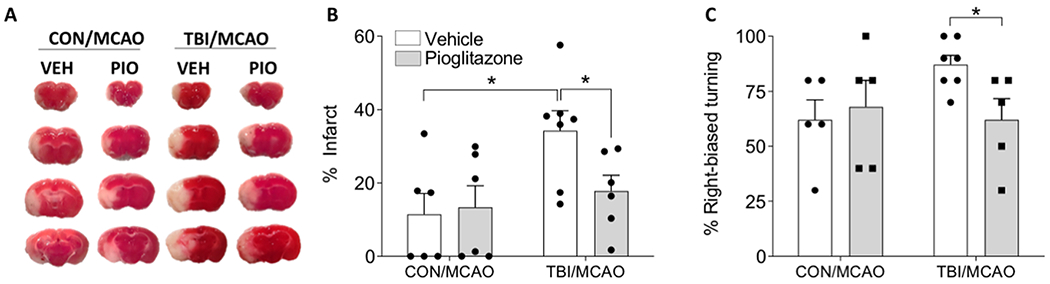
A) Representative TTC staining after MCAO from mice that first underwent control or TBI and were treated with pioglitazone (Pio; 10 mg/kg) or vehicle (Veh). B) Mice that were treated with Pio after TBI has infarct volumes similar to mice that did not have a TBI prior to MCAO. C) Treatment with PIO prevented the TBI exacerbation of right-biased turning in the corner test. An asterisk (*) indicates significance at p < 0.05.
In order to assess whether the protective effect of Pio was mediated by altered metabolic physiology an additional cohort of animals was treated with Pio (10mg/kg) (or Veh) after injury, producing the following groups: CON/Veh (n = 5), TBI/Veh (n = 5), CON/Pio (n = 5), TBI/Pio (n = 5). Forty-eight hours after injury we tested brain tissue sensitivity to insulin by measuring phosphorylated Akt after ex vivo incubation with insulin. As previously reported, TBI impairs Akt signaling in response to insulin, however treatment with Pio reversed the impairment resulting in similar insulin sensitivity between the CON/Pio and TBI/Pio groups (F3,15 = 3.530, p = 0.05; Figure 4 A–B).
Figure 4. Treatment with pioglitazone improves central insulin signaling but not respiratory parameters after TBI.
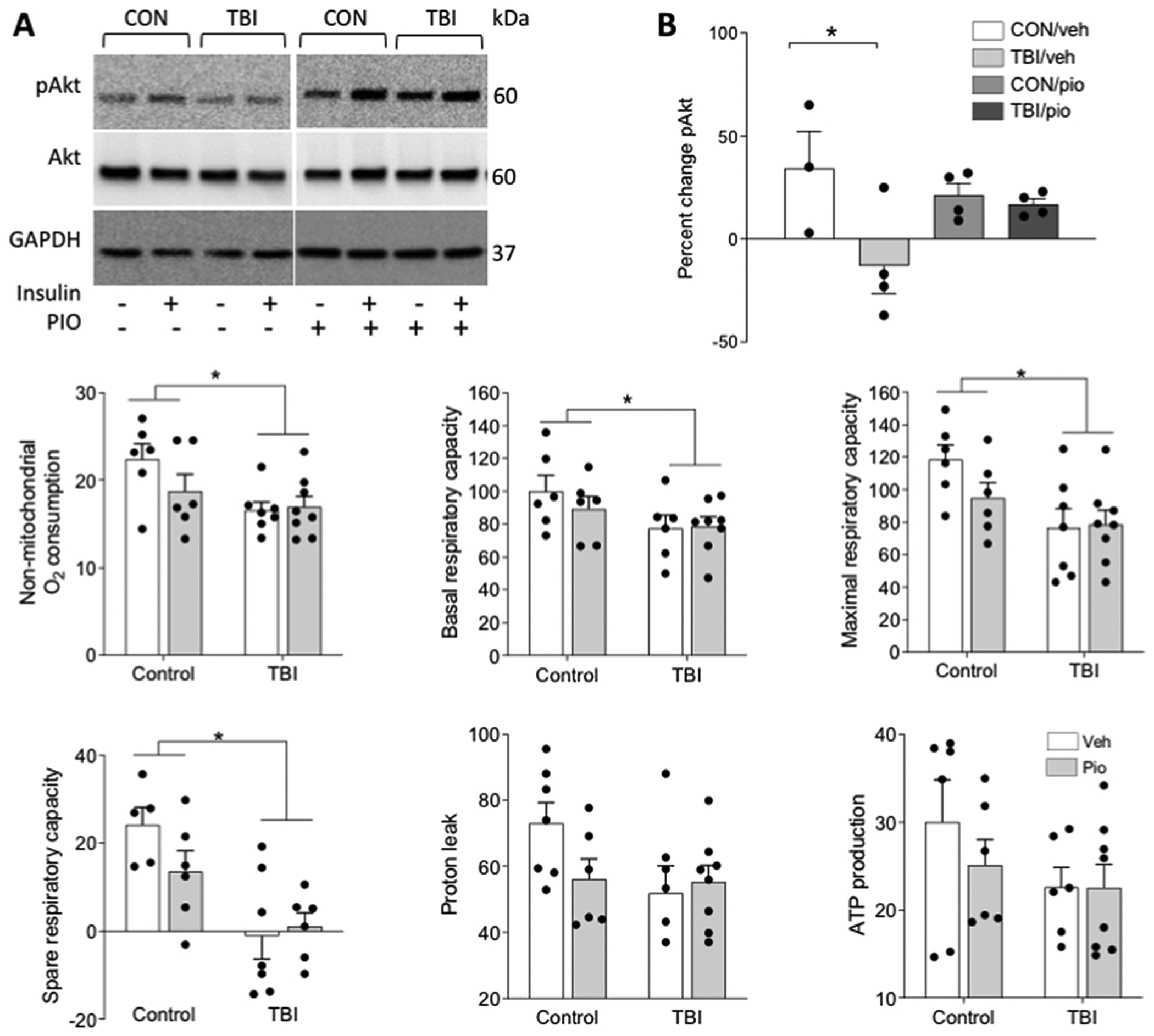
Brain tissue from control (CON) and TBI mice was split into two wells and incubated ex vivo with aCSF alone or containing 10nm insulin. A) Representative western blot bands are shown for phosphorylated Akt (Ser473), total Akt, and GAPDH. B) Data are shown as percent change pAkt expression in response to insulin. Separate animals underwent TBI or control procedure and were treated with Pio or vehicle, and seven days later one mm tissue punches were collected from 250-micron sections through the forebrain and analyzed with Seahorse respirometry. TBI reduced C) non-mitochondrial O2 production, D) basal respiratory capacity, D) maximal respiratory capacity, and E) spare respiratory capacity. There were no TBI effects on either G) proton leak or H) ATP production. There were no effects of Pio on any parameter. Data are presented as mean +/− SEM. An asterisk (*) indicates significance at p < 0.05.
Traumatic brain injuries impair central respiratory function an effect not rescued by pioglitazone treatment.
Pioglitazone administered directly after TBI was sufficient to block the induction of central insulin resistance. However, whether this manipulation would also prevent impairments in respiratory function remained unspecified. To test this, we induced TBI and treated the mice with pio or the vehicle [CON/Veh (n = 6), TBI/Veh (n = 7), CON/Pio (n = 6), TBI/Pio (n = 8)] and then collected micro-punches from the striato-cortical region seven days after injury before testing them on with a Seahorse mitostress test. Tissue punches from animals that had previously experienced a TBI exhibited reductions in non-mitochondrial O2 production (F2,22=6.354, p=0.019; Figure 4C), basal respiration (F2,22=4.535, p=0.049; Figure 4D), maximal respiration (F2,22=8.481, p=0.008; Figure 4E), and spare respiratory capacity (F2,22=8.649, p=0.008; Figure 4F). Neither proton leak (F2,22=2.26, p=0.147; Figure 4G) nor ATP production (F2,22=2.381, p=0.137; Figure 4H) were altered by injury. However, there were no effects of Pio or interactions between drug and injury in any respiratory parameter (p>0.05 in all cases).
Injury and pioglitazone alter vascular function
To generate hypotheses regarding the mechanisms of Pio-mediated protection we injured a group of animals (n=3/group) and treated them with Pio or the vehicle and collected hemispheres for the Nanostring analysis of neurological disease-related genes. Although the magnitude of the changes in gene expression was small there were a number of genes that were differentially expressed one week after injury (Figure S1A). Focusing on the key comparison between TBI-Veh and TBI-Pio, six genes were downregulated by Pio and an additional ten genes were upregulated by drug treatment (Figure S1B). This set of genes included growth factors (IGF1 and NGF) and antioxidant genes (GSR) upregulated by Pio and, several genes related to innate immune regulation and control of complement and coagulation processes (C1Qc, Casp8, TLR2, F2) that were also differentially expressed.
Thus, to determine the role of TBI-induced disruption of vascular function as a contributing factor to stroke vulnerability, animals underwent a TBI or control procedure and then one week later we administered a MCAO. Blood flow was assessed throughout the stroke procedure via laser doppler flowmetry. TBI did not alter blood flow prior to or during ischemia nor at reperfusion (p>0.05 in all cases; data not shown). Nevertheless, in order to determine whether alterations in vascular responses to injury were involved in vulnerability to TBI we injured animals and treated them with Pio (or the appropriate controls) and collected the tissue one week after brain injury. Animals that experienced TBI exhibited significant accumulation of IgG in CD31 positive vessels. There was also evidence of IgG extravasation into parenchyma. Treatment with Pio markedly attenuated this IgG accumulation both intra- and extravascularly (Figure 5; U3,26=9.66, p=0.022).
Figure 5. TBI induces vascular pathology that is attenuated by pioglitazone.
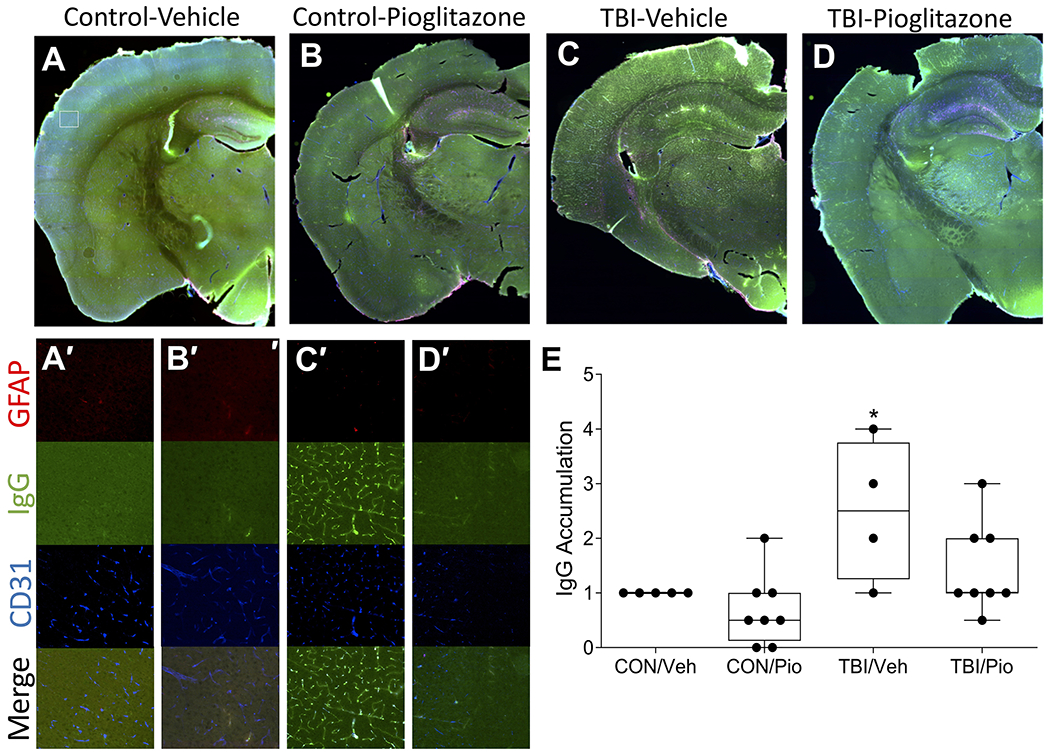
A-D) Representative sections indicate TBI induces accumulation of intravascular IgG (green) as well as mild extravasation that colocalizes with CD31 positive endothelial cells (blue) this is attenuated by pioglitazone. GFAP positive astrocytes (red) are also in close contact with IgG positive vessels. A’-D’) Corresponding insets of cortical staining. Size of insets indicated by white box in A). E) Among vehicle-treated mice TBI significantly increased IgG accumulation relative to CON, but Pio treatment reduced IgG accumulation to CON levels. An asterisk (*) indicates significance at p < 0.05.
One potential pathway through which vascular dysfunction could impair stroke outcomes is via impairments in blood flow during the reperfusion period. In order to assess this possibility, we prepared mice that had either a TBI or the control procedure and were treated with Pio or vehicle (CON/Veh (n = 13), CON/Pio (n = 10), TBI/Veh (n = 14), TBI/Pio (n = 9). We then assessed cerebral blood flow both prior to stroke and 60 minutes after removal of the ischemic occluder with laser speckle imaging. Pioglitazone significantly improved cerebral blood flow in the ischemic hemisphere (Figure 6; F1,37=6.66, p=0.014), an effect that was nearly entirely mediated by reversing the impairment in blood flow among TBI animals (F1,37=5.74, p=0.022). There were no drug or TBI effects on blood flow in the contralateral hemisphere (S2).
Figure 6. TBI impairs reperfusion blood flow, reversal by pioglitazone.
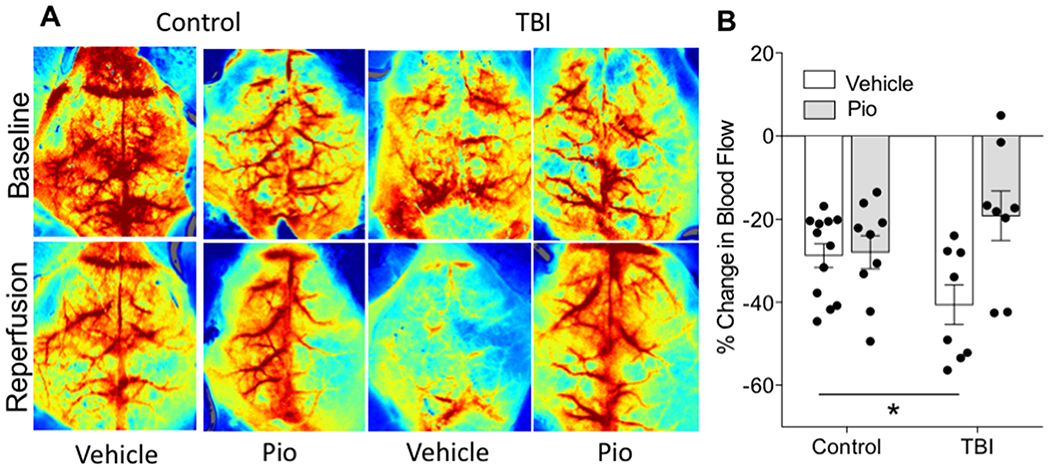
Baseline and reperfusion (60 minutes after removal of the occluder) blood flow measurements assessed with laser speckle imaging. Warmer colors indicate greater blood flow. B) Assessment of relative change in blood flow from baseline to reperfusion in the ipsilateral hemisphere. Data are presented as mean +/− SEM. An asterisk (*) indicates significance at p < 0.05.
Discussion
TBI is increasingly recognized as part of a disease process, rather than a single self-limiting event (Vespa, 2017). There is mounting epidemiological evidence that TBIs are a risk factor for both greater incidence and severity of neurological insults in the acute period after the initial injury (Chen et al., 2011; Vagnozzi et al., 2008). The TBI model procedure utilized here recapitulates that vulnerability to subsequent injury; the mild closed-head TBI produces widely distributed, but sparse, axonal degeneration without inducing a focal lesion. While the TBI alone, which is performed along the midline, does not produce appreciable behavioral deficits or functional lateralization, it does set the stage for the exacerbation of subsequent ischemic outcomes. Specifically, mice that had previously experienced a TBI had a nearly four-fold increase in lesion volume, greater edema in the ischemic hemisphere and functional impairments after MCAO. TBI also significantly upregulated neuroinflammation in both the ischemic cortex and striatum compared to mice that underwent ischemia without a history of TBI. Previously, we have reported that TBI transiently reduces insulin receptor sensitivity in the brain, a phenomenon with important implications for the regulation of both inflammatory and metabolic physiology (Karelina et al., 2016). Here, we report that TBI significantly reduced insulin sensitivity, as measured by AKT phosphorylation in a slice culture preparation; however, administration of the insulin sensitizing drug Pio immediately after TBI prevented the TBI-induced reduction in insulin receptor sensitivity. Critically, Pio administered immediately after TBI also prevented the increase in deleterious stroke outcomes including infarct development, and behavioral deficits. When we assessed tissue respiratory function, a TBI-mediated impairment was apparent one week after injury, however, these parameters were not rescued by treatment with Pio. However, Pio did significantly alter the gene expression profile after TBI, markedly reduced the vascular accumulation of serum proteins and prevented poor reperfusion blood flow after TBI.
Shortly after a TBI, the brain enters a brief period of hyperglycolysis, in which glucose utilization increases to meet the metabolic demands associated with recovery from the insult. This initial phase is followed by a prolonged period of metabolic depression that can last several weeks to months (Bergsneider et al., 1997; Oddo et al., 2008). A stroke occurs when energetic demands of tissue outstrip the metabolic fuels necessary to maintain cellular homeostasis (Smith, 2004). Most, if not all, of the ischemic cascade in stroke is initiated by depletion of substrates necessary for maintaining cellular homeostasis (George and Steinberg, 2015). Therefore, if cellular energy metabolism is already disrupted by TBI, cells in the infarct penumbral region where blood flow is reduced (Durukan and Tatlisumak, 2007) may be rendered less likely to survive and may instead be incorporated into the infarct core.
The data presented here indicate that mild TBI induces both vascular dysfunction and impairments in central metabolic signaling and respiratory function. Therefore, it seems likely that the metabolic dysfunction induced by TBI prevents the brain from mobilizing the energetically intensive processes needed for optimal recovery from an injury like cerebral ischemia. We have previously demonstrated that TBI significantly, but transiently, impairs central insulin receptor sensitivity as assessed by activation of AKT after ex vivo stimulation with insulin (Karelina et al., 2016). Although glucose metabolism in neurons and glia is not absolutely dependent on insulin signaling, there is a general enhancement of glucose uptake and utilization following insulin treatment and insulin can reduce the impairments in energy metabolism induced by oxidative stress (Blazquez et al., 2014; Duarte et al., 2006). Moreover, insulin receptor signaling in neurons and other CNS cell types is generally anti-inflammatory, anti-apoptotic and generally opposes the deleterious effects of ischemia (Garg et al., 2006; Shuaib et al., 1995). The reduction in central insulin sensitivity induced by TBI, therefore, may contribute to and/or exacerbate metabolic dysfunction and deprive the injured brain of the potential protective effects of insulin signaling. To test this hypothesis, we used the peroxisome-proliferator activated receptor (PPAR) agonist Pio and we administered it immediately after TBI but more than six days before the ischemic stroke. Critically, Pio prevented both the reduction in insulin sensitivity and the increase in infarct size in mice that had a prior TBI. Although Pio is used clinically as an insulin sensitizer for patients with type 2 diabetes, it has relatively broad neuroprotective effects that likely extend beyond its role in increasing insulin receptor sensitivity. However, as the half-life of Pio is short (approximately 9 hours in rodent serum (Hanefeld, 2001)), administration of Pio one week prior to MCAO is unlikely to have resulted in direct neuroprotection. Moreover, there was no reduction in ischemic lesion volume in Pio-treated mice that did not receive a TBI, thus we propose that the reduction of ischemic lesions in Pio-treated TBI mice is a function of reduced insulin resistance induced by TBI.
Although this Pio treatment paradigm was sufficient to restore central insulin receptor sensitivity and prevent the exacerbation in stroke damage after TBI, it did not alter the respiratory function of tissue punches collected from the injured hemisphere. TBI (regardless of drug treatment) reduced both the basal and maximal respiratory capacity of the tissue punches and also reduced the spare respiratory capacity. The injured brain thus respires at a reduced rate and also exhibits a smaller capacity for increasing respiratory output in the case of subsequent challenges. As this assay primarily measures mitochondria-mediated respiratory function, impairments in mitochondrial energy production as well as biogenesis are likely contributing to this phenotype (Fried et al., 2014; Underwood et al., 2020). Gene expression analysis one week after injury indicated a simultaneous upregulation of mitofusin-2 and down regulation of OPA-1 by injury, two genes for proteins that regulate mitochondrial fusion. However, as Pio prevented the TBI-induced increase in ischemic cell death without altering respiratory function, two critical questions remain unanswered, specifically, 1) whether TBI-induced impairments in respiratory function contribute to the increased vulnerability to injury and 2) how Pio improves stroke outcomes after TBI.
The TBI-induced impairments in respiratory function were assessed 7 days after injury (the same time course in which MCAO was induced in other cohorts). It seems unlikely that significant metabolic impairments, as assessed by Seahorse respirometry and insulin receptor sensitivity assays, would not exacerbate the outcomes after a severe metabolic challenge such as ischemic stroke. However, treatment with Pio reduced insulin receptor sensitivity and the enhanced ischemic damage after TBI but did not rescue the respiratory phenotype. Thus, the observed impairments in respiratory function are insufficient alone to exacerbate stroke damage after TBI. The current study does not rule out the possibility that respiratory deficits contribute to the increased vulnerability to ischemic damage but does suggest that other factors are involved.
One potential mechanism for the protective effect of Pio is as an anti-inflammatory agent (Deng et al., 2020). TBI prior to an ischemic injury greatly exacerbates the inflammatory responses to subsequent injury as indicated by increased expression of proinflammatory genes in the ischemic hemisphere. For instance, the cytokine gene expression for IL-1β (assessed 24 hours post-reperfusion) was significantly elevated in mice that had a TBI prior to stroke, relative to all other experimental groups. Metabolic and inflammatory signaling pathways are both interconnected and bidirectional (van Horssen and Witte, 2013). For example, the energy sensing adenosine monophosphate protein kinase (AMPK) is a sensor of energy balance, and a potential exacerbating element in stroke outcome. AMPK can also be activated by elements of the inflammatory cascade such as TNFα, and in turn regulate inflammatory signaling(Ashabi et al., 2015; Ronnett et al., 2009). Finally, impairments in mitochondrial function can result in increased reactive oxygen production and the release of damage-associated molecular patterns such as mitochondrial DNA, which can drive inflammatory signaling(Lassmann and van Horssen, 2016). Inversely, proinflammatory cytokines and other ligands of toll-like receptors can reduce both central and peripheral insulin sensitivity, impair mitochondrial energy production and survival, as well as negatively impact mitochondrial oxidative phosphorylation(Lee and Huttemann, 2014; Samavati et al., 2008). Insulin signaling is potently anti-inflammatory in the CNS, and thus loss of insulin sensitivity can cause a net increase in inflammatory tone(Dandona et al., 2007; Sun et al., 2014). Ergo, impairments in metabolic signaling are both caused by and exacerbate inflammation following CNS injury(Abcouwer et al., 2008).
An additional potential mechanism through which Pio may have provided neuroprotection from TBI-induced exacerbation of ischemic outcomes is via improving the function of the cerebral vasculature(Badhwar et al., 2017; Nicolakakis et al., 2008). Severe damage to vessels can cause hemorrhages and vasospasm, but even short of those outcomes’ vessel damage can significantly contribute to post-injury dysfunction(Logsdon et al., 2015; Sandsmark et al., 2019). The CNS is highly dependent on the coordination among multiple cell types that form the neurovascular unit for the activity-dependent delivery of oxygen and glucose and the removal of waste products. Additionally, the vasculature forms an integral component of the blood-brain-barrier (BBB) which is tasked with chemically isolating the brain from many large or charged molecules. Damage to the cerebral vasculature, which is an extremely common histological finding in head trauma patients, has the potential to significantly disrupt these critical processes(Hay et al., 2015; Shlosberg et al., 2010). Moreover, damage to the vessels has the potential to induce intravascular coagulation and microthrombi that can reduce tissue oxygenation and contribute to vasogenic edema (Bartnik-Olson et al., 2014). Astrocytes are central players in metabolic physiology of the nervous system and as structural components of the blood brain barrier. Moreover, they are critical regulators of activity-dependent blood flow. Swelling of astrocytes and in particular their end feet can both reduce BBB function and reduce the size of the vascular lumen. Although we did not detect large gene expression changes or histological evidence of astrocyte hypertrophy prior to stroke it remains important to understand the way in which TBI may alter the astrocytic response to ischemia.
Here we report that mild TBI leads to the accumulation of intravascular IgG deposits and extravasation and significant upregulation of prothrombin gene expression all of which were blocked by treatment with Pio. Changes in other genes associated with ongoing hypoxia, vascular remodeling and integrity including HIf1a, Notch1 and 3, angiopoietin 2, as well MMP19 by injury were apparent. Thus, this TBI model seems to produce a relatively mild injury to the cerebral vasculature along with some moderate breakdown of the blood brain barrier. The deposition of serum proteins like IgG into vascular walls can serve to both narrow the luminal space and form a seed for thrombus formation(Grinberg and Thal, 2010; Schreiber et al., 2012). The accumulation of intravascular serum proteins like IgG is not a common finding in the TBI literature where most moderate-severe experimental injuries produce BBB breakdown and the spilling of large molecules into the parenchyma(Pop and Badaut, 2011). This pattern of vascular histology most closely resembles cerebral small vessel disease, a set of diseases associated with white matter damage, lacunar strokes and dementia(Joutel et al., 2010).
Critically there is a strong and bidirectional link between microvasculature dysfunction and insulin resistance(Rhea and Banks, 2019). Insulin resistance in the NVU in general, and in endothelial cells particularly, can impair microvascular function and lead to reduced functional perfusion under basal and hyperemic conditions(Muniyappa and Sowers, 2013). Insulin signaling under physiological conditions can serve to balance PI3K-AKT-nitric oxide-mediated vasodilation signals with MAP kinase endothelin-mediated vasoconstriction(Vicent et al., 2003). However, insulin resistant states are commonly associated with endothelial dysfunction and a shift in the balance toward vasoconstriction and can serve as major risk factors for cardio- and cerebrovascular disorders(Katakam et al., 2009; Muniyappa and Sowers, 2013). Further, insulin can also serve to oppose the inflammatory and oxidative signaling in the NVU cells that both impair function and reinforce insulin resistance and vascular dysfunction(Kobayashi and Puro, 2007). Pioglitazone was sufficient to block the induction of insulin resistance in slices prepared from TBI animals and future studies will address whether improvements in insulin sensitivity in the cerebral vasculature itself mediate improved outcomes from stroke.
The primary strategy for the minimization of ischemic damage is to restore blood flow to the ischemic territory, clinically this can be accomplished with pharmacological or mechanical clot removers. However, it is becoming increasingly clear that reperfusion of the infarcted tissue and not recanalization of the blocked vessel is the primary predictor of ischemic outcomes(Soares Bruno et al., 2010). If the tissue does not (fully) reperfuse after the occlusion is removed outcomes will be relatively poor as the tissue is not supplied with the necessary metabolic substrates and potentially toxic waste products are not removed(Arbel-Ornath et al., 2013; Bai and Lyden, 2015; El Amki et al., 2020). The causes of incomplete reperfusion are not fully understood but likely involve vascular inflammation (with associated vascular accumulation of serum proteins and immune cells), intravascular coagulation and endothelial dysfunction(Dalkara and Arsava, 2012; El Amki et al., 2020). Here we report that TBI induces a significant impairment in reperfusion blood flow a phenomenon that is highly likely to contribute to the much larger infarct volumes among mice in those groups.
Several important limitations of this study should be pointed out. First, the TBI paradigm used here produces repeatable patterns of axonal degeneration, but we did not assess force and displacement of the actual skull which may introduce some variability. Secondly, this study was conducted in male animals. We elected to study males for two reasons. First, at the age that we are interested in, late adolescence-young adulthood, there is a significant sex difference in TBI incidence (Faul et al., 2010). Secondly, we focused on males because the single paper that has demonstrated an increase in mortality following stroke (after TBI) did so in males (Liao et al., 2014). Follow-up studies are under way to investigate the role of TBI in post-stroke mortality in females. Finally, the seven day post-TBI injury time point is relatively short and does not fully replicate the persistent increase in stroke risk that lasts many months in humans. This work is currently being extended to detect the limits of the enhancement of stroke outcomes.
There is convincing evidence that TBI increases the incidence of ischemic stroke and some epidemiological evidence that stroke outcomes are worse in patients with a history of TBI. Given the very large number of TBIs that occur annually it is critically important that we determine why this population is at a greater risk for both more strokes and worse outcomes. Here, we demonstrate that TBI exacerbates stroke outcomes, and that tissue damage and functional impairments are much worse in animals that experienced TBI several days prior to stroke. We also provide the first evidence that metabolic dysfunction in general, and central insulin resistance in particular, strongly correlates with this phenomenon. Treatment with the insulin sensitizer Pio rescued insulin resistance, inflammatory signaling, vascular dysfunction, and enhanced vulnerability to ischemia but did not alter respiratory parameters suggesting that a constellation of pathophysiological processes mediate enhanced vulnerability after injury. Stroke prophylaxis therapies, (including anti-hypertensives, blood thinners and potentially insulin sensitizing drugs) are not without risk, particularly in the TBI patient population, but the data presented here raise the possibility that assessment of central metabolic physiology could be used to predict which TBI patients (and for how long) might be at risk for greater damage if a stroke does occur, thus allowing clinicians to direct treatment at those at the greatest risk. TBI patients, by definition, have already suffered damage to their brains and thus any intervention that can reduce the likelihood or severity of subsequent injuries should be considered.
Supplementary Material
Highlights.
Traumatic brain injury increases the risk and severity of ischemic stroke
Mice with traumatic brain injuries have larger experimental strokes
Mice with traumatic brain injuries have impaired metabolic and vascular function
Vascular dysfunction after TBI may exacerbate stroke outcome by impairing reperfusion
Acknowledgments
This work supported by grants from American Heart Association, NIH P20GM109098 (to ZMW) and R01-HL-128485 (to JMH) and additional support was provided by Community Foundation for the Ohio Valley Whipkey Trust and the West Virginia University for Clinical and Translational Science Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest
None
supplementary data
References
- Abcouwer SF, Shanmugam S, Gomez PF, Shushanov S, Barber AJ, Lanoue KF, Quinn PG, Kester M, Gardner TW, 2008. Effect of IL-1beta on survival and energy metabolism of R28 and RGC-5 retinal neurons. Invest Ophthalmol Vis Sci 49, 5581–5592. [DOI] [PubMed] [Google Scholar]
- Abdul-Muneer PM, Chandra N, Haorah J, 2015. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol 51, 966–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht JS, Liu X, Baumgarten M, Langenberg P, Rattinger GB, Smith GS, Gambert SR, Gottlieb SS, Zuckerman IH, 2014. Benefits and risks of anticoagulation resumption following traumatic brain injury. JAMA Intern Med 174, 1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht JS, Liu X, Smith GS, Baumgarten M, Rattinger GB, Gambert SR, Langenberg P, Zuckerman IH, 2015. Stroke incidence following traumatic brain injury in older adults. J Head Trauma Rehabil 30, E62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbel-Ornath M, Hudry E, Eikermann-Haerter K, Hou S, Gregory JL, Zhao L, Betensky RA, Frosch MP, Greenberg SM, Bacskai BJ, 2013. Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol 126, 353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashabi G, Khalaj L, Khodagholi F, Goudarzvand M, Sarkaki A, 2015. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab Brain Dis 30, 747–754. [DOI] [PubMed] [Google Scholar]
- Badhwar A, Brown R, Stanimirovic DB, Haqqani AS, Hamel E, 2017. Proteomic differences in brain vessels of Alzheimer’s disease mice: Normalization by PPARgamma agonist pioglitazone. J Cereb Blood Flow Metab 37, 1120–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Lyden PD, 2015. Revisiting cerebral postischemic reperfusion injury: new insights in understanding reperfusion failure, hemorrhage, and edema. Int J Stroke 10, 143–152. [DOI] [PubMed] [Google Scholar]
- Bartnik-Olson BL, Holshouser B, Wang H, Grube M, Tong K, Wong V, Ashwal S, 2014. Impaired neurovascular unit function contributes to persistent symptoms after concussion: a pilot study. J Neurotrauma 31, 1497–1506. [DOI] [PubMed] [Google Scholar]
- Bergsneider M, Hovda DA, Shalmon E, Kelly DF, Vespa PM, Martin NA, Phelps ME, McArthur DL, Caron MJ, Kraus JF, 1997. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J. Neurosurg. 86, 241–251. [DOI] [PubMed] [Google Scholar]
- Blazquez E, Velazquez E, Hurtado-Carneiro V, Ruiz-Albusac JM, 2014. Insulin in the brain: its pathophysiological implications for states related with central insulin resistance, type 2 diabetes and alzheimer’s disease. Front Endocrinol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JF, Stulc JL, Skolarus LE, Sears ED, Zahuranec DB, Morgenstern LB, 2013. Traumatic brain injury may be an independent risk factor for stroke. Neurology 81, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Kang JH, Lin HC, 2011. Patients with traumatic brain injury: population-based study suggests increased risk of stroke. Stroke 42, 2733–2739. [DOI] [PubMed] [Google Scholar]
- Dalkara T, Arsava EM, 2012. Can Restoring Incomplete Microcirculatory Reperfusion Improve Stroke Outcome after Thrombolysis? Journal of Cerebral Blood Flow & Metabolism 32, 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandona P, Chaudhuri A, Mohanty P, Ghanim H, 2007. Anti-inflammatory effects of insulin. Curr Opin Clin Nutr Metab Care 10, 511–517. [DOI] [PubMed] [Google Scholar]
- Deng Y, Jiang X, Deng X, Chen H, Xu J, Zhang Z, Liu G, Yong Z, Yuan C, Sun X, Wang C, 2020. Pioglitazone ameliorates neuronal damage after traumatic brain injury via the PPARgamma/NF-kappaB/IL-6 signaling pathway. Genes Dis 7, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte AI, Proenca T, Oliveira CR, Santos MS, Rego AC, 2006. Insulin restores metabolic function in cultured cortical neurons subjected to oxidative stress. Diabetes 55, 2863–2870. [DOI] [PubMed] [Google Scholar]
- Durukan A, Tatlisumak T, 2007. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav 87, 179–197. [DOI] [PubMed] [Google Scholar]
- El Amki M, Glück C, Binder N, Middleham W, Wyss MT, Weiss T, Meister H, Luft A, Weller M, Weber B, Wegener S, 2020. Neutrophils Obstructing Brain Capillaries Are a Major Cause of No-Reflow in Ischemic Stroke. Cell Reports 33, 108260. [DOI] [PubMed] [Google Scholar]
- Faul MD, Xu L, Wald MM, Coronado VG, 2010. Traumatic brain injury in the United States: Emergency department visits, hospitalizations, and deaths, 2002–2006 , in: Centers for Disease Control and Prevention National Center for Injury Prevention and Control; (Ed.), Atlanta, GA. [Google Scholar]
- Fried NT, Moffat C, Seifert EL, Oshinsky ML, 2014. Functional mitochondrial analysis in acute brain sections from adult rats reveals mitochondrial dysfunction in a rat model of migraine. Am J Physiol Cell Physiol 307, C1017–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith D, Davenport R, Brohi K, 2012. Acute traumatic coagulopathy. Curr Opin Anaesthesiol 25, 229–234. [DOI] [PubMed] [Google Scholar]
- Garg R, Chaudhuri A, Munschauer F, Dandona P, 2006. Hyperglycemia, insulin, and acute ischemic stroke - A mechanistic justification for a trial of insulin infusion therapy. Stroke 37, 267–273. [DOI] [PubMed] [Google Scholar]
- George PM, Steinberg GK, 2015. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron 87, 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SS, Rochon PA, Herrmann N, Lee PE, Sykora K, Gunraj N, Normand SL, Gurwitz JH, Marras C, Wodchis WP, Mamdani M, 2005. Atypical antipsychotic drugs and risk of ischaemic stroke: population based retrospective cohort study. BMJ 330, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giza CC, Hovda DA, 2014. The new neurometabolic cascade of concussion. Neurosurgery 75 Suppl 4, S24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn MB, 2010. Sudden cardiac death and stroke with the use of antipsychotic medications: implications for clinicians treating individuals with traumatic brain injury. J Head Trauma Rehabil 25, 68–70. [DOI] [PubMed] [Google Scholar]
- Grinberg LT, Thal DR, 2010. Vascular pathology in the aged human brain. Acta Neuropathol 119, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanefeld M, 2001. Pharmacokinetics and clinical efficacy of pioglitazone. Int J Clin Pract Suppl, 19–25. [PubMed] [Google Scholar]
- Hay JR, Johnson VE, Young AMH, Smith DH, Stewart W, 2015. Blood-Brain Barrier Disruption Is an Early Event That May Persist for Many Years After Traumatic Brain Injury in Humans. J Neuropath Exp Neur 74, 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joutel A, Monet-Lepretre M, Gosele C, Baron-Menguy C, Hammes A, Schmidt S, Lemaire-Carrette B, Domenga V, Schedl A, Lacombe P, Hubner N, 2010. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest 120, 433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karelina K, Gaier KR, Weil ZM, 2017. Traumatic brain injuries during development disrupt dopaminergic signaling. Exp. Neurol. 297, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karelina K, Sarac B, Freeman LM, Gaier KR, Weil ZM, 2016. Traumatic brain injury and obesity induce persistent central insulin resistance. Eur. J. Neurosci. 43, 1034–1043. [DOI] [PubMed] [Google Scholar]
- Katakam PV, Domoki F, Lenti L, Gaspar T, Institoris A, Snipes JA, Busija DW, 2009. Cerebrovascular responses to insulin in rats. J Cereb Blood Flow Metab 29, 1955–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Puro DG, 2007. Loss of insulin-mediated vasoprotection: early effect of diabetes on pericyte-containing microvessels of the retina. Invest Ophthalmol Vis Sci 48, 2350–2355. [DOI] [PubMed] [Google Scholar]
- Kramer M, Dang J, Baertling F, Denecke B, Clarner T, Kirsch C, Beyer C, Kipp M, 2010. TTC staining of damaged brain areas after MCA occlusion in the rat does not constrict quantitative gene and protein analyses. J Neurosci Methods 187, 84–89. [DOI] [PubMed] [Google Scholar]
- Lassmann H, van Horssen J, 2016. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Bba-Mol Basis Dis 1862, 506–510. [DOI] [PubMed] [Google Scholar]
- Lee I, Huttemann M, 2014. Energy crisis: the role of oxidative phosphorylation in acute inflammation and sepsis. Biochim Biophys Acta 1842, 1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CC, Chou YC, Yeh CC, Hu CJ, Chiu WT, Chen TL, 2014. Stroke risk and outcomes in patients with traumatic brain injury: 2 nationwide studies. Mayo Clin. Proc. 89, 163–172. [DOI] [PubMed] [Google Scholar]
- Liu SW, Huang LC, Chung WF, Chang HK, Wu JC, Chen LF, Chen YC, Huang WC, Cheng H, Lo SS, 2017. Increased Risk of Stroke in Patients of Concussion: A Nationwide Cohort Study. Int J Environ Res Public Health 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon AF, Lucke-Wold BP, Turner RC, Huber JD, Rosen CL, Simpkins JW, 2015. Role of Microvascular Disruption in Brain Damage from Traumatic Brain Injury. Compr Physiol 5, 1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrory P, Meeuwisse W, Aubry M, Cantu B, Dvorak J, Echemendia R, Engebretsen L, Johnston K, Kutcher J, Raftery M, Sills A, Benson B, Davis G, Ellenbogen R, Guskiewicz K, Herring SA, Iverson G, Jordan B, Kissick J, McCrea M, McIntosh A, Maddocks D, Makdissi M, Purcell L, Putukian M, Schneider K, Tator C, Turner M, 2013. Consensus statement on Concussion in Sport - The 4th International Conference on Concussion in Sport held in Zurich, November 2012. Phys Ther Sport 14, e1–e13. [DOI] [PubMed] [Google Scholar]
- Morris NA, Cool J, Merkler AE, Kamel H, 2017. Subarachnoid Hemorrhage and Long-Term Stroke Risk After Traumatic Brain Injury. Neurohospitalist 7, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniyappa R, Sowers JR, 2013. Role of insulin resistance in endothelial dysfunction. Rev Endocr Metab Disord 14, 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolakakis N, Aboulkassim T, Ongali B, Lecrux C, Fernandes P, Rosa-Neto P, Tong XK, Hamel E, 2008. Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci 28, 9287–9296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo M, Schmidt JM, Mayer SA, Chiolero RL, 2008. Glucose control after severe brain injury. Curr. Opin. Clin. Nutr. Metab. Care. 11, 134–139. [DOI] [PubMed] [Google Scholar]
- Ostergaard L, Engedal TS, Moreton F, Hansen MB, Wardlaw JM, Dalkara T, Markus HS, Muir KW, 2016. Cerebral small vessel disease: Capillary pathways to stroke and cognitive decline. J Cereb Blood Flow Metab 36, 302–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop V, Badaut J, 2011. A neurovascular perspective for long-term changes after brain trauma. Transl Stroke Res 2, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins M, Hales A, Reger M, Giza C, Hovda D, 2010. Repeat traumatic brain injury in the juvenile rat is associated with increased axonal injury and cognitive impairments. Dev. Neurosci. 32, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhea EM, Banks WA, 2019. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front Neurosci 13, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S, 2009. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem 109, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavati L, Lee I, Mathes I, Lottspeich F, Huttemann M, 2008. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem 283, 21134–21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandsmark DK, Bashir A, Wellington CL, Diaz-Arrastia R, 2019. Cerebral Microvascular Injury: A Potentially Treatable Endophenotype of Traumatic Brain Injury-Induced Neurodegeneration. Neuron 103, 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S, Bueche CZ, Garz C, Kropf S, Angenstein F, Goldschmidt J, Neumann J, Heinze HJ, Goertler M, Reymann KG, Braun H, 2012. The pathologic cascade of cerebrovascular lesions in SHRSP: is erythrocyte accumulation an early phase? J Cereb Blood Flow Metab 32, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh RA, Clerc P, Hwang H, Mehrabian Z, Bittman K, Chen H, Polster BM, 2011. Adaptation of microplate-based respirometry for hippocampal slices and analysis of respiratory capacity. J Neurosci Res 89, 1979–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzmaier SM, de Chaumont C, Balbi M, Terpolilli NA, Kleinschnitz C, Gruber A, Plesnila N, 2016. The Formation of Microthrombi in Parenchymal Microvessels after Traumatic Brain Injury Is Independent of Coagulation Factor XI. J Neurotrauma 33, 1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlosberg D, Benifla M, Kaufer D, Friedman A, 2010. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol 6, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuaib A, Ijaz MS, Waqar T, Voll C, Kanthan R, Miyashita H, Liu L, 1995. Insulin Elevates Hippocampal Gaba Levels during Ischemia - This Is Independent of Its Hypoglycemic Effect. Neuroscience 67, 809–814. [DOI] [PubMed] [Google Scholar]
- Smith WS, 2004. Pathophysiology of focal cerebral ischemia: a therapeutic perspective. J Vasc Interv Radiol 15, S3–12. [DOI] [PubMed] [Google Scholar]
- Soares Bruno P, Tong E, Hom J, Cheng S-C, Bredno J, Boussel L, Smith Wade S, Wintermark M, 2010. Reperfusion Is a More Accurate Predictor of Follow-Up Infarct Volume Than Recanalization. Stroke 41, e34–e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Li J, Gao F, 2014. New insights into insulin: The anti-inflammatory effect and its clinical relevance. World J Diabetes 5, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YY, Yang D, Kuan CY, 2012. Mannitol-facilitated perfusion staining with 2,3,5- triphenyltetrazolium chloride (TTC) for detection of experimental cerebral infarction and biochemical analysis. J Neurosci Methods 203, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood E, Redell JB, Zhao J, Moore AN, Dash PK, 2020. A method for assessing tissue respiration in anatomically defined brain regions. Sci Rep 10, 13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnozzi R, Signoretti S, Tavazzi B, Floris R, Ludovici A, Marziali S, Tarascio G, Amorini AM, Di Pietro V, Delfini R, Lazzarino G, 2008. Temporal window of metabolic brain vulnerability to concussion: a pilot 1H-magnetic resonance spectroscopic study in concussed athletes--part III. Neurosurgery 62, 1286–1295; discussion 1295–1286. [DOI] [PubMed] [Google Scholar]
- van Horssen J, Witte M, 2013. Mitochondrial dysfunction: a link between inflammation and tissue damage in multiple sclerosis? Mult Scler J 19, 365–365. [Google Scholar]
- Vespa P, 2017. Traumatic brain injury is a longitudinal disease process. Curr Opin Neurol 30, 563–564. [DOI] [PubMed] [Google Scholar]
- Vicent D, Ilany J, Kondo T, Naruse K, Fisher SJ, Kisanuki YY, Bursell S, Yanagisawa M, King GL, Kahn CR, 2003. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest 111, 1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil ZM, Gaier KR, Karelina K, 2014. Injury timing alters metabolic, inflammatory and functional outcomes following repeated mild traumatic brain injury. Neurobiol. Dis. 70, 108–116. [DOI] [PubMed] [Google Scholar]
- Zhang J, Jiang R, Liu L, Watkins T, Zhang F, Dong JF, 2012. Traumatic brain injury-associated coagulopathy. J Neurotrauma 29, 2597–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.