

Abstract

The angiotensin II type 2 receptor (AT2R) has attracted much attention as a potential target for the relief of neuropathic pain, which represents an area of unmet clinical need. A series of 1,2,3,4-tetrahydroisoquinolines with a benzoxazole side-chain were discovered as potent AT2R antagonists. Rational optimization resulted in compound 15, which demonstrated both excellent antagonistic activity against AT2R in vitro and analgesic efficacy in a rat chronic constriction injury model. Its favorable physicochemical properties and oral bioavailability make it a promising therapeutic candidate for neuropathic pain.
Introduction
Endogenous angiotensin II plays an important role in the renin-angiotensin system via two types of receptors, AT1R and AT2R, which only share approximately 34% sequence identity.1,2 The two receptors have similar binding affinities to angiotensin II but differ in their functions. AT1R serves as a key regulator of blood pressure and is a validated drug target with several antagonists approved as the well-known category of antihypertensives (valsartan, irbesartan etc.), while the function of AT2R has remained enigmatic. Diverse biological functions of AT2R have been reported and are often biological in context and cell- and tissue-dependent.3,4 One of the profound effects is that activation of AT2R by agonists, such as C21, demonstrated antifibrotic effects and promoted tissue protection in cardiovascular and renal diseases.5−7 On the other hand, a growing number of studies suggest that AT2R is involved in pain modulation,8 and AT2R antagonists could relieve peripheral neuropathic pain in animal models. Therefore, the AT2R has drawn considerable attention as a new therapeutic target for treatments of different diseases. Neuropathic pain is caused by a lesion or disease affecting the somatosensory nervous system, either centrally or in the periphery.9 The population prevalence of neuropathic pain is estimated to lie between 6.9 and 10%.10 Neuropathic pain is common in cancer pain, diabetic neuropathy, postherpetic neuralgia, and trigeminal neuralgia, among others.11 The pathophysiology of neuropathic pain is not well defined. The deficient mechanistic understanding of neuropathic pain, which is poorly managed by currently available drugs due to limited efficacy and tolerability, has undoubtedly impeded the discovery of effective analgesics.12−15
Recently, an oral small-molecule AT2R antagonist, EMA401 (Figure 1), has been reported to show analgesic efficacy in animal models and in a phase II clinical trial for neuropathic pain.16−21EMA401 is a potent AT2R antagonist with high selectivity over AT1R. However, it had only moderate drug exposure and oral bioavailability in rats.17 In addition, clinical pharmacokinetic (PK) data indicated that large interindividual variation in exposure existed,18 which could, in turn, lead to variation in therapeutic effects in patients. Our aim was to develop a potent AT2R antagonist with an improved pharmacokinetic profile. Herein, we report the design, synthesis, and biological activities of a novel series of benzoxazoles as potent and selective AT2R antagonists and the in vitro and in vivo evaluation of the derived analogues, including a rat chronic constriction injury (CCI) model.
Figure 1.

Structure of EMA401.
Our medicinal chemistry efforts started from EMA401. EMA401 is an oily compound with C Log P as high as 5.48, and the diphenylacetyl moiety presumably contributes a lot to the high lipophilicity of the molecule. Docking of EMA401 to the crystal structure of AT2R22 reveals that while one of the two geminal phenyl groups is well fitted into a hydrophobic pocket formed by Tyr103, Tyr108, Leu300, and Pro301, the other one is much more solvent-exposed (Figure 2), which indicates that the diphenylacetyl moiety as a whole probably is not ideal for protein–ligand interaction, and may be amenable to substitution.
Figure 2.

Docking model of EMA401 binding to AT2R (PDB, 5UNF). AT2R in ribbons with the interaction surface colored by electrostatics, some key residues shown in sticks with carbon in gray, and EMA401 in sticks with carbon in green.
Results and Discussion
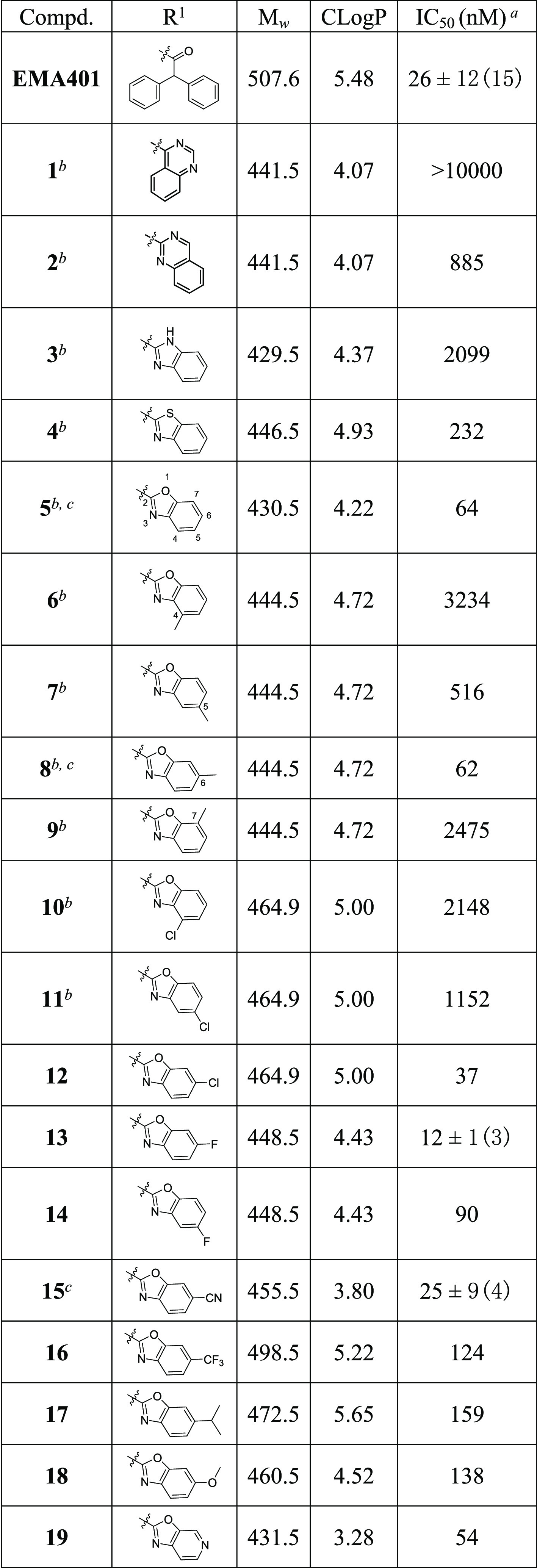
Our initial plan was to replace the diphenylacetyl of EMA401 with benzoheteroaromatic groups to maintain the possible hydrophobic interaction with AT2R and at the same time to reduce the lipophilicity of the small molecule (Table 1). Compounds 1–34 were synthesized as outlined in Schemes 1 and 2. Compounds 1–11 were readily synthesized as racemates. The isoquinoline 35(23) was assembled to give 1a–11a via SNAr reaction with heteroaryl halides or oxidative amination of benzoxazoles.24 Following saponification gave compounds 1–11. Enantiopure intermediate 41 was synthesized according to the procedures similar to others.25 Horner–Wadsworth–Emmons reaction of aldehyde 36 with phosphonate 37 followed by asymmetric catalytic hydrogenation26 produced 39 with high S-enantioselectivity (98.7% ee). Deprotection of tert-butyloxycarbonyl gave 40, which was further cyclized to 41 via a Pictet–Spengler reaction. SNAr displacement of heteroaryl halides with 41 followed by saponification afforded the desired compounds 12–19. The intermediates 14a and 15a could be further debenzylated to give phenols 42 and 43, respectively. The following alkylation with various arylmethyl halides under basic conditions or with arylmethyl alcohols under Mitsunobu reaction conditions yielded compounds 20a–34a, and subsequent saponification of the resulting esters produced the final compounds 20–34. It was worth noting that compound 15 was prepared from 36 and 37 through six steps with high yield (57% overall yield) and high enantiomeric excess (98.5% ee). Details on the synthesis of all compounds are described in the Supporting Information.
Table 1. SAR of Diphenylacetyl Replacement for hAT2R Antagonistic Activities.
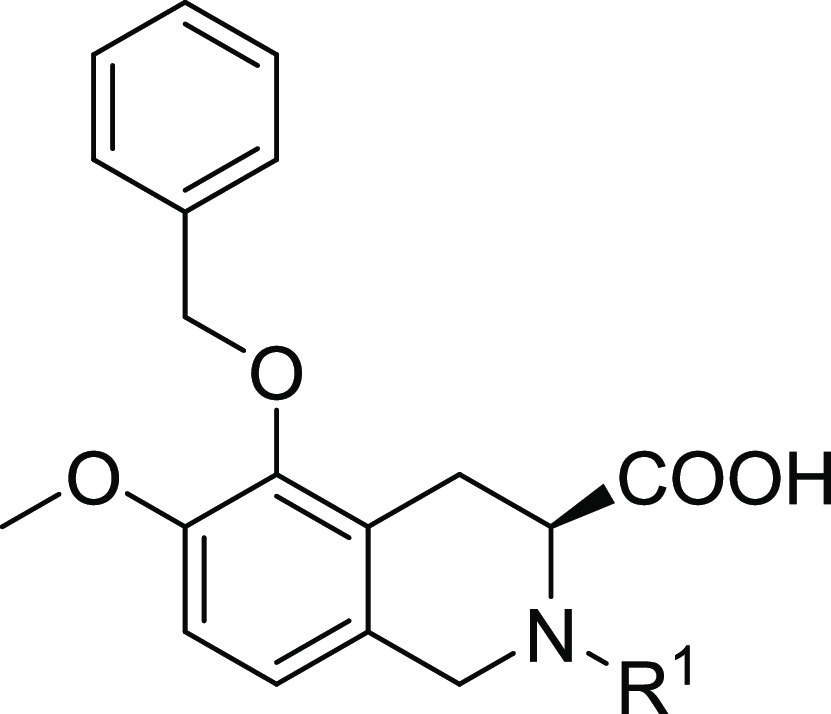
IC50 for human AT2R with standard deviation and the number of runs in brackets if applicable.
Racemate.
IC50 > 10 μM for human AT1R.
Scheme 1. Synthetic Routes of Racemic Compounds 1–11.

Reagents and conditions: (i) (a) heteroaryl halide, DIPEA, NMP, 80 °C; or (b) heteroaryl halide, DIPEA, NMP, 80 °C, and then TFA, CH2Cl2, rt; or (c) heteroaryl halide, K2CO3, DMF, rt; or (d) benzoxazole, Ag2CO3, PhCO2H, CH3CN, 60 °C; (ii) (e) NaOH, H2O, THF, MeOH, rt; or (f) LiOH, H2O, THF, rt.
Scheme 2. Synthetic Routes of Compounds 16–34.
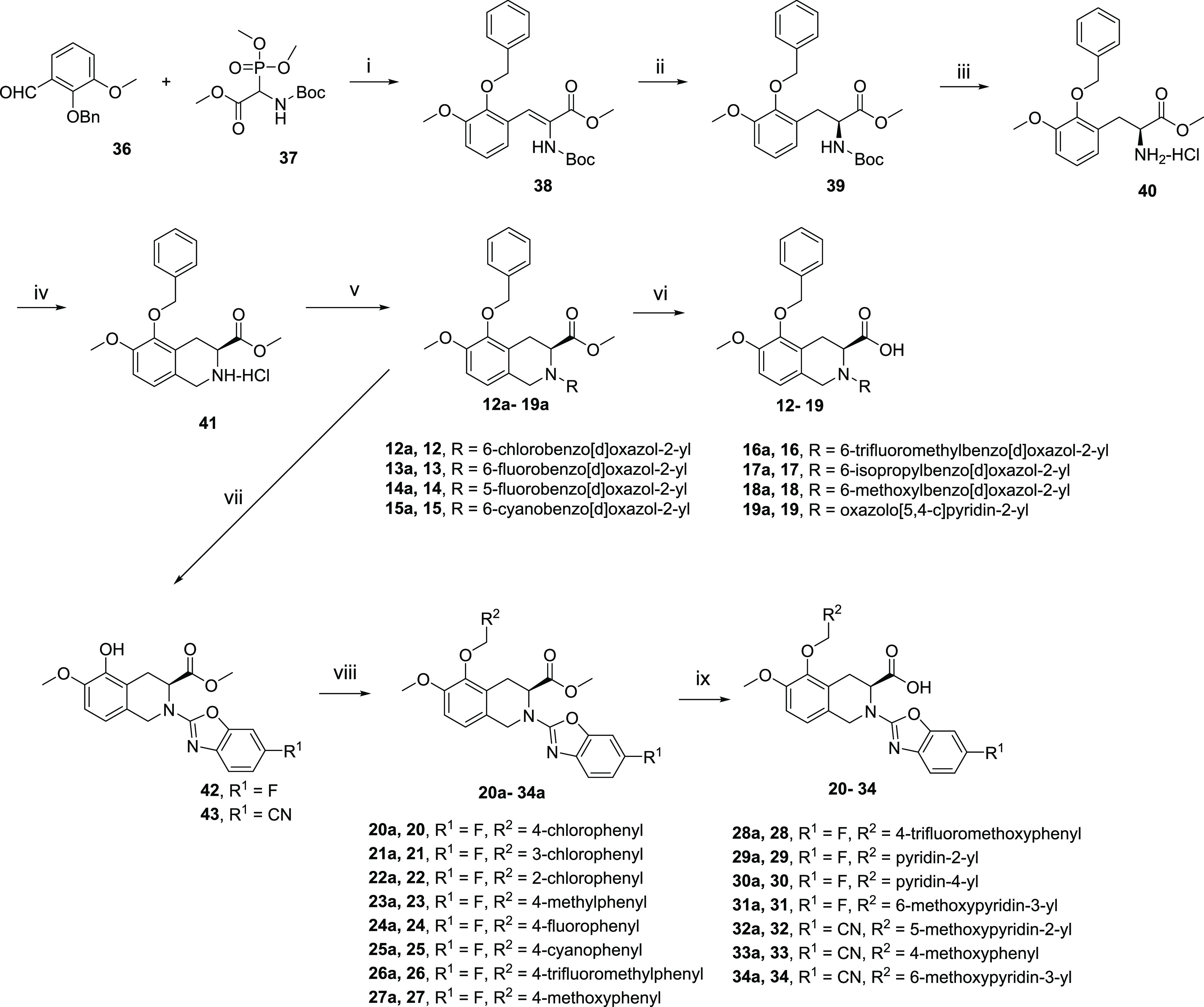
Reagents and conditions: (i) 1,1,3,3-tetramethylguanidine, THF, rt, 88%; (ii) (R)-methyl BoPhoz, [Rh(COD)2]BF4, MeOH, H2 rt; (iii) HCl, dioxane, rt, 96% for two steps; (iv) paraformaldehyde, HCl, dioxane, 70 °C, 82%; (v) (a) heteroaryl halide, Et3N, THF, 60 °C; or (b) heteroaryl halide, K2CO3, DMF, rt; (vi) (c) LiOH, H2O, THF, rt; or (d) CaCl2, NaOH, H2O, iPrOH, THF, rt; (vii) Pd/C, H2, MeOH, rt; (viii) (e) ArCH2OH, PPh3, DIAD, THF, rt; or (f) ArCH2Cl, K2CO3, NaI, DMF, rt; or (g) ArCH2Cl, K2CO3, DMF, 70 °C; (ix) (h) LiOH, H2O, THF, rt; or (j) CaCl2, NaOH, H2O, iPrOH, THF, rt.
Here, we adopted a homogeneous time-resolved fluorescence (HTRF)-based competitive binding assay (see the Supporting Information for details) to measure the inhibitory activities of compounds of interest. EMA401 had a half-maximum inhibitory concentration (IC50) of 26 nM for human AT2R (hAT2R), which was in good agreement with that of 39 nM measured via a radiolabeled ligand-binding assay.17 Replacement of the diphenylacetyl with quinazolin-4-yl led to complete activity loss (compound 1). However, replacement with quinazolin-2-yl could restore the activity to the sub-micromolar level (compound 2), which indicated that the orientation of this bicyclic aromatic ring was important. We then switched to [5,6]-bicyclic heteroaromatic rings. Benzonimidazol-2-yl (compound 3) worsened the activity when compared to compound 2. To our delight, benzothiazole-2-yl demonstrated decent activity, leading to compound 4 with an IC50 of around 200 nM. A more dramatic activity boost was seen for benzoxazole-2-yl; the resulting compound 5 showed comparable potency (IC50 = 64 nM) to EMA401 (IC50 = 26 nM) and more than one-log reduction in lipophilicity. The encouraging result promoted further structure–activity relationship (SAR) studies on the benzoxazole ring. We first did a methyl-walk on the ring. It turned out that activity was very sensitive to the substitution position. The 6-Me compound 8 kept the potency with an IC50 value of 62 nM, while 4-Me (6), 5-Me (7), and 7-Me (9) resulted in 50-, 8-, and 39-fold activity loss, respectively, when compared to compound 5. Similar trends were observed in the following cases of Cl- or F-substitution (compounds 10–12 for Cl and 13–14 for F). It was thus concluded that position-6 was the sweet spot for substitution, and the fluorine atom seemed to fit ideally to this position with compound 13 showing an even better activity than EMA401 (12 vs 26 nM). Nevertheless, substituents with different sizes or electronegativities were explored at position-6 (compounds 15–18). Size has been proven to matter more than the other factors; bulkier groups like CF3 (16), iPr (17), and OMe (18) were harmful, and more than 10-fold activity loss was observed when compared to corresponding fluorine compound 13, while the smaller cyano group was well tolerated, leading to compound 15, with an IC50 of 25 nM and C Log P below 4. Furthermore, the hydrophilic pyridine analogue (19) provided similar potency to compound 5. These results highlighted the delicate nature of the protein–ligand interaction at this region.
We next moved on to the exploration of the antagonistic ability of various arylmethyl derivatives of compounds 13 and 15 while maintaining a fluoro or a cyano substituent at the position-6 of the benzoxazole ring (Table 2). Substitution on the phenyl ring in general decreased antagonistic activities, with p-F (24, 17 nM), p-CF3 (26, 11 nM), and p-MeO (27, 5.9 nM; 33, 11 nM) as a few exceptions. It is worthy to point out that p-methoxyl substitution actually increased the activity to a single-digit nanomolar level. In addition, replacement of the phenyl ring with pyridine was tolerable, and a combination of pyridine and methoxyl gave (6-methoxypyridin-3-yl)methyl derivative 31 with a hAT2R IC50 of 6.6 nM.
Table 2. SAR of the Arylmethyl Ether Region.


IC50 for human AT2R with standard deviation and the number of runs in brackets if applicable.
IC50 > 10 μM for human AT1R.
With this new series of hAT2R inhibitors in hand, we went on to measure their selectivities over the hAT1R. Six compounds (5, 8, 15, 27, 28, and 33) with different hAT2R activities were picked for a competitive binding assay against hAT1R. Gratifyingly, none of them showed any hAT1R antagonistic activity at the highest tested concentration (10 μM).
Based on the potency and selectivity, compounds 13, 15, 27, and 31 were selected for drug metabolism and pharmacokinetic (DMPK) evaluation in the rat (Table 3), given the fact that animal efficacy studies were planned to be carried out in the rat (vide infra). EMA401 showed moderate clearance both in vitro and in vivo, moderate exposure, and low oral bioavailability in rats. In contrast, our compounds, which have smaller molecular weights and substantially lower lipophilicities, demonstrated superiority in all these DMPK parameters. Among the four compounds, compound 15 stood out. Compound 15 had the lowest in vivo clearance (1.7 mL/min/kg) and the longest half-life (4.2 h). Its exposure was excellent, and greater than 10- and 30-fold increases were realized in terms of Cmax (6657 vs 470 ng/mL for EMA401) and AUC (44,100 vs 1470 (ng·h)/mL), respectively. Furthermore, its bioavailability was also significantly improved (47 vs 12% for EMA401).
Table 3. In Vitro Metabolic Stability and PK Data of Compounds 13, 15, 22, and 31 in Ratsa.
| PK in
rats |
|||||||
|---|---|---|---|---|---|---|---|
| compd. | CLintb | Cmaxc | AUC0–24hc | T1/2d | CLd | Vdssd | F%e |
| EMA401 | 25.9 ± 1.0 | 470 ± 197 | 1470 ± 709 | 1.6 ± 0.3 | 14 ± 1 | 0.74 ± 0.1 | 12 ± 6% |
| 13 | 12.5 ± 6.7 | 5280 ± 72 | 29,367 ± 4626 | 3.5 ± 0.3 | 2.1 ± 0.3 | 0.24 ± 0.05 | 36 ± 6% |
| 15 | 8.13 ± 1.29 | 6657 ± 1745 | 44,100 ± 10,713 | 4.2 ± 1.2 | 1.7 ± 0.1 | 0.21 ± 0.06 | 47 ± 12% |
| 27 | 10.4 ± 3.0 | 2370 ± 182 | 6930 ± 1720 | 2.4 ± 0.8 | 5.3 ± 1.0 | 0.37 ± 0.17 | 22 ± 5% |
| 31 | 2.95 ± 0.18 | 5490 ± 1311 | 15,200 ± 2237 | 2.3 ± 0.4 | 5.0 ± 0.6 | 0.57 ± 0.06 | 45 ± 7% |
Units: CLint (intrinsic clearance), mL/min/kg; Cmax, ng/mL; AUC (area under the curve), (ng·h)/mL; T1/2, h; CL (clearance), mL/min/kg; Vdss (steady-state apparent volume of distribution), L/kg.
Metabolic stability in the rat liver microsome.
p.o., 10 mg/kg.
i.v., 1 mg/kg.
F%, oral bioavailability.
Compound 15 was thus further evaluated by in vitro and in vivo experiments with EMA401 as the comparator (Table 4). The metabolic stability of compound 15 was high in the human liver microsome and medium in human hepatocytes. However, EMA401 had high clearance in both the liver microsome and hepatocytes of the human. A recent paper actually reported that EMA401 was highly unstable in hepatocytes of many species, e.g., mouse, rat, dog, monkey, and human,21 and glucuronidation was one of the predominant metabolism pathways. Both compounds had high plasma protein binding with a free fraction less than 0.1% in both the rat and human. Interestingly, although compound 15 had good cell permeability with no efflux as shown in the Caco-2 cell permeability test, EMA401 had low permeability and a high efflux ratio of 7.6. Neither compound crossed the blood–brain barrier (BBB) following p.o. administration in rats, as demonstrated by their extremely low brain-to-plasma ratios. Nevertheless, the high efflux ratio of EMA401 could partially contribute to its low bioavailability in different preclinical species. Compound 15 displayed no significant inhibition of six human CYP isozymes, including CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, while EMA401 exhibited medium inhibition on CYP2C9 with an IC50 value of 7.4 μM. The drug–drug interaction assays indicated less liabilities in potential drug combination use for compound 15 than EMA401. In an in vitro cardiovascular safety evaluation, both compound 15 and EMA401 indicated no hERG potassium channel activity at concentrations up to 10 μM, avoiding potential cardiovascular liability. The single-dose PK studies in dogs were also examined. Again, compound 15 showed a superior PK profile to EMA401 with significant higher plasma exposure (>70-fold in AUC) and oral bioavailability (75 vs 39%).
Table 4. ADME, Early Safety, and Dog PK Data of Compound 15 and EMA401.
| compd. | 15 | EMA401 |
|---|---|---|
| human liver microsome stability, CLint (mL/min/kg) | 1.50 ± 1.38 | 25.0 ± 1.1 |
| human hepatocyte stability, CLint (mL/min/kg) | 26 | 66 |
| plasma protein binding (PPB, rat/human) | >99.9%/>99.9% | >99.9%/>99.9% |
| brain-to-plasma ratio in the rat | 0.012 ± 0.007 | 0.007 ± 0.001 |
| Caco-2, Papp, A-B/B-A (10–6 cm/s) | 11.3 ± 0.2/13.9 ± 0.3 | 3.3 ± 0.3/24.5 ± 1.5 |
| CYP inhibition (1A2/2C8/2C9/2C19/2D6/3A4), IC50 (μM) | >50/>50/31/>50/>50/>50 | >50/20/7.4/>50/>50/>50 |
| hERG, IC50 (μM) | >10 | >10 |
| dog PK of sodium salt at 3 mg/kg, p.o. | ||
|---|---|---|
| Cmax (ng/mL) | 19,500 ± 6366 | 2707 ± 1530 |
| AUC0–t ((ng·h)/mL) | 132,483 ± 54,459 | 1727 ± 959 |
| T1/2 (h) | 11 ± 3 | 2.3 ± 1.4 |
| bioavailability (F%) | 75 ± 31% | 39 ± 22% |
As the AT2R competitive binding assay is not enough evidence to claim agonistic or antagonistic effects of compounds, a neurite outgrowth functional assay was conducted in NG108-15 cells.27 The functional assay showed that EMA401 and compound 15 abolished neurite outgrowth induced by angiotensin II and confirmed that compound 15 had an antagonistic effect on AT2R.
Given its most balanced overall profile, compound 15 was advanced to in vivo efficacy evaluation in the chronic constriction injury (CCI) rat model of neuropathic pain (Figure 3). The analgesic efficacy of compound 15 was assessed using von Frey filaments to measure paw withdrawal thresholds (PWT) in rats after p.o. administration. Pregabalin and EMA401 were used as positive controls, and vehicle administration was used as the negative control. Mechanical allodynia was well developed in the ipsilateral hind paws of CCI rats at 13 days after CCI surgery, and the mechanical withdrawal threshold of the ipsilateral paws of CCI rats decreased to ∼6.5 g (PWT value) from a presurgery value of ∼13 g (see detailed experiments in the Supporting Information). A significantly increased paw withdrawal threshold responsive to tactile stimulation (von Frey) was observed in CCI rats orally dosed with compound 15, pregabalin, and EMA401. A clear dose-dependent antiallodynia effect was exhibited at doses of 15, 30, and 60 mg/kg for compound 15, and the duration of action at 30 and 60 mg/kg was about 2 h. Pregabalin showed a faster onset of action than compound 15 and EMA401, which probably was due to their different mechanisms of action and PK properties. Compound 15 at a dose of 30 mg/kg showed comparable efficacy to EMA401 at the same dose, which was a bit unexpected given the fact that compound 15 should have much higher systemic exposure than EMA401 at the same dosage. The difference in the tissue distribution profile or in free concentration at the action site between the two compounds may be a reason behind this discrepancy. In addition, although the two compounds showed comparable antagonistic activities toward the human AT2R, their activities toward rat AT2R were not measured due to the unavailability of a cell line expressing the rat AT2R. Thus, between-species differences may also need to be investigated and should be taken as a critical step in compound screening cascades in the future.
Figure 3.
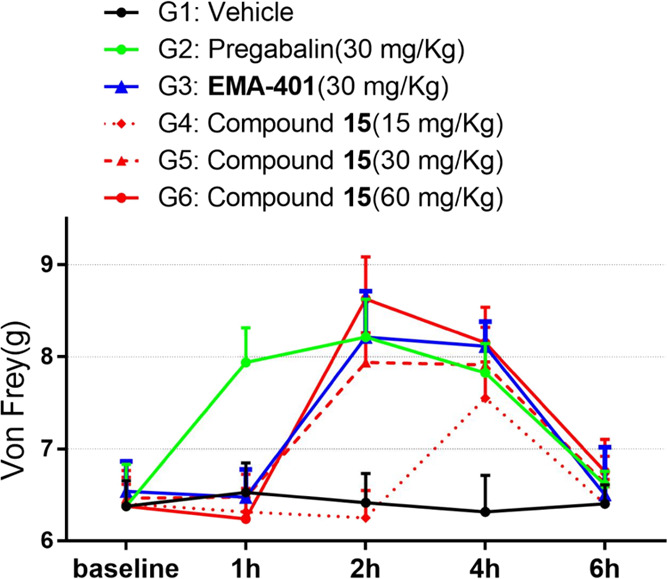
In vivo analgesic efficacy evaluation in the chronic constriction injury rat model after p.o. administration.
Finally, a non-GLP 7-day acute rat toxicity study of compound 15 and EMA401 was evaluated at multiple doses of 200, 400, and 800 mg/kg/day. Compound 15 and EMA401 were well tolerated with no serious adverse events or significant laboratory abnormalities except for salivary discharge 1 h after dosage in high- and medium-dose groups for compound 15 and in high-, medium-, and low-dose groups for EMA401. The salivation phenomenon presented a dose-dependent effect, and the high-dose group of EMA401 had the most severe salivation. There were no distinguishable gross anatomy differences in kidneys, hearts, or livers between treated groups (compound 15 and EMA401) and the vehicle group. AT2R has a vital role in cardiovascular and renal systems. Although AT2R knockout in mice increased the vasopressor response upon angiotensin II treatment and aggravated the impairment of renal function after renal injury,28,29 AT2R antagonists are unlikely to affect cardiovascular and renal functions under normal physiologic conditions. In adults, AT2R is expressed at low levels in the normal cardiovascular system and kidneys.3 It indicates that AT2R antagonists might show no potential on-target toxicity in the heart and kidneys. However, two phase II clinical trials of EMA401 have been terminated30,31 very recently due to hepatotoxicity in monkeys after 39 weeks of chronic dosing,32 which indicates that longer-term toxicity studies may be necessary for potential adverse effects.
Conclusions
In conclusion, we have discovered a series of potent AT2R antagonists with benzoxazole side-chain based on EMA401. EMA401 is metabolically highly unstable both in vitro and in vivo. Direct acylglucuronidation is the major elimination pathway, and the acyglucuronide metabolite was able to undergo acyl migration substantially under neutral conditions.21 It was well documented that acylglucuronides could form covalent bonds with proteins and other bio-macromolecules, which may have a link with adverse drug reactions of some drugs.33 However, it is too early to tell whether this metabolism pathway of EMA401 had anything to do with the termination of two recent phase II clinical trials of EMA401 due to safety concerns based on data from longer-term toxicity studies in monkeys. With the aim of improving the DMPK properties of EMA401, comprehensive rational optimization resulted in a novel series of compounds with high potencies, high selectivities over AT1R, and more importantly with distinct improvement in drug-like properties, especially the liver microsome and hepatocyte stability. Compound 15 demonstrated an excellent PK profile in rodent/nonrodent species and analgesic efficacy in the CCI rat model. Further investigations including IND enabling studies and development of compound 15 are in progress and will be reported in due course.
Experimental Section
Chemistry
All materials, such as reagents, starting materials, and solvents, were purchased from commercial suppliers and were used without further purification. Reactions were run under an argon atmosphere, unless noted otherwise. The reactions were monitored by thin-layer chromatography (TLC) and/or high-performance liquid chromatography-mass spectrometry (HPLC-MS). Analytical HPLC was performed on an Agilent 1200 (chromatographic column: Agilent Zorbax Eclipse Plus C18, 4.6 mm × 250 mm, 5 μm; T = 25 °C; λ = 245 nm; eluted with a 16 min gradient from 10 to 100% B, where A = H2O/0.05% TFA and B = ACN; F = 1.0 mL/min). For NMR measurement, samples were dissolved in deuterated solvent (CD3OD, CDCl3, or DMSO-d6), and spectra were acquired with a Bruker Avance-400 NMR spectrometer (1H: 400 MHz; 13C: 100 MHz) under standard observation conditions using tetramethylsilane (TMS) as an internal standard. The procedure for the synthesis of all compounds is given in the Supporting Information.
AT2R and AT1R Competitive Binding Assays
The AT2R competitive binding assay was conducted in the Tag-lite angiotensin AT2 cells transiently expressing the angiotensin AT2 receptor labeled with terbium (Cisbio, catalog number: C1TT1AT2). Cryopreserved Tag-lite angiotensin AT2 cells (1 mL) were thawed in a 37 °C water bath, then transferred into a conical vial containing 5 mL of 1× Tag-lite buffer, centrifuged for 5 min at 300g, and resuspended in 2.7 mL of 1× Tag-lite buffer. 10 μL of labeled cells was dispensed into each well of a 384-well plate, and then 5 μL of reference compound (angiotensin II) or 5 μL of test compound solution (10-dose with a 5-fold serial dilution starting at 10 μM) and 5 μL of Tag-lite angiotensin receptor red agonist (Cisbio, catalog number: L0007RED, 12 nM in 1× Tag-lite buffer) were added to all assay wells. The assay plate was centrifuged for 1 min at 200g and incubated at 25 °C for 1 h. The data were read and collected on an EnVision (PerkinElmer, model: 2203-1060) by the HTRF-module. IC50 was calculated by Prism 5.0 software (GraphPad). AT1R IC50 was determined by a similar assay with Tb-labeled Tag-lite AT1 stable cells (Cisbio, catalog number: C1SU1AT1), and the reference compounds for the AT1R binding assay were losartan and angiotensin II.
NG108-15 Neurite Outgrowth Assay
NG108-15 cells were purchased from the Cell Resource Center of the Institute of Basic Medical Sciences (IBMS) of the Chinese Academy of Medical Sciences and cultured in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum and 2% HAT supplement (5 mM hypoxanthine, 20 μM aminopterin, 0.8 mM thymidine). For the neurite outgrowth assay, NG108-15 cells were plated in a 6-well plate at a density of 50,000 cells per well and incubated overnight. Cells were stimulated by 0.1 μM angiotensin II or co-treated with 0.1 μM angiotensin II and 1 μM compound (applied 30 min before angiotensin II) once a day for three consecutive days. The neurite outgrowth was observed and photographed using a microscope.
Chronic Constriction Injury (CCI) Model
Male Sprague–Dawley rats were subjected to peripheral neuropathy injury by constriction of the sciatic nerve.34 Pain threshold base values were measured before and 13 days after surgery to check whether the model was successful. The rats were randomly separated into six groups (n = 8 per group). Pregabalin was used as the positive control, and the vehicle was used as the negative control. The rats were administered by oral gavage at the indicated dose of test compounds as a suspension in 0.5% CMC-Na and 0.5% Tween 80 on the 14th day. Von Frey paw withdraw thresholds (PWT) were determined in the ipsilateral hind paws at the following times post-dosing: 1, 2, 4, and 6 h.
Acknowledgments
We thank the colleagues in the analytical group, biological group, and process group for their contributions on this work.
Glossary
Abbreviations
- AT2R
angiotensin II type 2 receptor
- AT1R
angiotensin II type 1 receptor
- PK
pharmacokinetic
- CCI
chronic constriction injury
- Tyr
tyrosine
- Leu
leucine
- Pro
proline
- PDB
Protein Data Bank
- HTRF
homogeneous time-resolved fluorescence
- SNAr
nucleophilic aromatic substitution
- DIPEA
N,N-diisopropylethylamine
- NMP
N-methyl pyrrolidone
- TFA
trifluoroacetic acid
- DMF
N,N-dimethylformamide
- THF
tetrahydrofuran
- DIAD
diisopropyl azodicarboxylate
- SAR
structure–activity relationship
- DMPK
drug metabolism and pharmacokinetic
- CLint
intrinsic clearance
- CL
clearance
- AUC
area under the curve
- p.o.
per os
- i.v.
intravenous injection
- CYP
cytochrome P450 enzyme
- hERG
human ether-a-go-go related gene
- PPB
plasma protein binding
- BBB
blood–brain barrier
- Caco-2
human colorectal adenocarcinoma cell line
- PWT
paw withdrawal thresholds
- IND
investigational new drug
- TLC
thin layer chromatography
- HPLC
high-performance liquid chromatography
- MS
mass spectrometry
- ACN
acetonitrile
- NMR
nuclear magnetic resonance
- DMSO
dimethyl sulfoxide
- TMS
tetramethylsilane
- DMEM
Dulbecco’s Modified Eagle Medium
- CMC-Na
sodium carboxymethyl cellulose
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01866.
Experimental details for synthetic procedures and analytical data; in vitro and in vivo assay information (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
These studies were funded by Zhejiang Hisun Pharmaceutical Co., Ltd.
The authors declare no competing financial interest.
Supplementary Material
References
- Karnik S. S.; Unal H.; Kemp J. R.; Tirupula K. C.; Eguchi S.; Vanderheyden P. M. L.; Thomas W. G. International Union of Basic and Clinical Pharmacology XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli. Pharmacol. Rev. 2015, 67, 754–819. 10.1124/pr.114.010454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gasparo M.; Catt K. J.; Inagami T.; Wright J. W.; Unger T. International Union of Pharmacology XXIII. The Angiotensin II Receptors. Pharmacol. Rev. 2000, 52, 415–472. [PubMed] [Google Scholar]
- Juillerat-Jeanneret L. The Other Angiotensin II Receptor: AT2R as a Therapeutic Target. J. Med. Chem. 2020, 63, 1978–1995. 10.1021/acs.jmedchem.9b01780. [DOI] [PubMed] [Google Scholar]
- Pulakat L.; Sumners C. Angiotensin Type 2 Receptors: Painful, or Not. Front. Pharmacol. 2020, 11, 571994. 10.3389/fphar.2020.571994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg M.; Sumners C.; Steckelings U. M.; Hallberg A. Small-molecule AT2 Receptor Agonists. Med. Res. Rev. 2018, 38, 602–624. 10.1002/med.21449. [DOI] [PubMed] [Google Scholar]
- Isaksson R.; Lindman J.; Wannberg J.; Sallander J.; Backlund M.; Baraldi D.; Widdop R.; Hallberg M.; Åqvist J.; Gutiérrez-de-Terán H.; Gising J.; Larhed M. A Series of Analogues to the AT2R Prototype Antagonist C38 Allow Fine Tuning of the Previously Reported Antagonist Binding Mode. ChemistryOpen 2019, 8, 114–125. 10.1002/open.201800282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallander J.; Wallinder C.; Hallberg A.; Åqvist J.; Gutiérrez-de-Terán H. Structural Determinants of Subtype Selectivity and Functional Activity of Angiotensin II Receptors. Bioorg. Med. Chem. Lett. 2016, 26, 1355–1359. 10.1016/j.bmcl.2015.10.084. [DOI] [PubMed] [Google Scholar]
- Anand U.; Yiangou Y.; Sinisi M.; Fox M.; MacQuillan A.; Quick T.; Korchev Y. E.; Bountra C.; McCarthy T.; Anand P. Mechanisms Underlying Clinical Efficacy of Angiotensin II Type 2 Receptor (AT2R) Antagonist EMA401 in Neuropathic Pain: Clinical Tissue and in Vitro Studies. Mol. Pain 2015, 11, 38–49. 10.1186/s12990-015-0038-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treede R.-D.; Jensen T. S.; Campbell J. N.; Cruccu G.; Dostrovsky J. O.; Griffin J. W.; Hansson P.; Hughes R.; Nurmikko T.; Serra J. Neuropathic Pain: Redefinition and a Grading System for Clinical and Research Purposes. Neurology 2008, 70, 1630–1635. 10.1212/01.wnl.0000282763.29778.59. [DOI] [PubMed] [Google Scholar]
- van Hecke O.; Austin S. K.; Khan R. A.; Smith B. H.; Torrance N. Neuropathic Pain in the General Population: a Systematic Review of Epidemiological Studies. Pain 2014, 155, 654–662. 10.1016/j.pain.2013.11.013. [DOI] [PubMed] [Google Scholar]
- Colloca L.; Ludman T.; Bouhassira D.; Baron R.; Dickenson A. H.; Yarnitsky D.; Freeman R.; Truini A.; Attal N.; Finnerup N. B.; Eccleston C.; Kalso E.; Bennett D. L.; Dworkin R. H.; Raja S. N. Neuropathic Pain. Nat. Rev. Dis. Primers 2017, 3, 17003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham K.; Shepherd A.; Mohapatra D. P.; Haroutounian S. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr. Pain Headache Rep. 2017, 21, 28. 10.1007/s11916-017-0629-5. [DOI] [PubMed] [Google Scholar]
- Moore R. A.; Wiffen P. J.; Derry S.; Toelle T.; Rice A. S. Gabapentin for Chronic Neuropathic Pain and Fibromyalgia in Adults. Cochrane Database Syst. Rev. 2014, 4, CD007938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf C. J.; Mannion R. J. Neuropathic Pain: Aetiology, Symptoms, Mechanisms, and Management. Lancet 1999, 353, 1959–1964. 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- Mathieson S.; Maher C. G.; McLachlan A. J.; Latimer J.; Koes B. W.; Hancock M. J.; Harris I.; Day R. O.; Billot L.; Pik J.; Jan S.; Lin C.-W. C. Trial of Pregabalin for Acute and Chronic Sciatica. N. Engl. J. Med. 2017, 376, 1111–1120. 10.1056/NEJMoa1614292. [DOI] [PubMed] [Google Scholar]
- Hesselink J. K.; Schatman M. E. EMA401: an Old Antagonist of AT2R for a New Indication in Neuropathic Pain. J. Pain Res. 2017, Volume 10, 439–443. 10.2147/JPR.S128520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. T.; Wyse B. D.; Edward S. R. Small Molecule Angiotensin II Type 2 Receptor (AT2R) Antagonists as Novel Analgesics for Neuropathic Pain: Comparative Pharmacokinetic, Radioligand Binding, and Efficacy in Rats. Pain Med. 2013, 14, 692–705. 10.1111/pme.12063. [DOI] [PubMed] [Google Scholar]
- Rice A. S. C.; Dworkin R. H.; McCarthy T. D.; Anand P.; Bountra C.; McCloud P. I.; Hill J.; Cutter G.; Kitson G.; Desem N.; Raff M. EMA401, an Orally Administered Highly Selective Angiotensin II Type 2 Receptor Antagonist, as a Novel Treatment for Postherpetic Neuralgia: a Randomised, Double-Blind, Placebo-Controlled Phase 2 Clinical Trial. Lancet 2014, 383, 1637–1647. 10.1016/S0140-6736(13)62337-5. [DOI] [PubMed] [Google Scholar]
- Smith M. T.; Muralidharan A. Targeting Angiotensin II Type 2 Receptor Pathways to Treat Neuropathic Pain and Inflammatory Pain. Expert Opin. Ther. Targets 2015, 19, 25–35. 10.1517/14728222.2014.957673. [DOI] [PubMed] [Google Scholar]
- Smith M. T.; Anand P.; Rice A. S. C. Selective Small Molecule Angiotensin II Type 2 Receptor Antagonist for Neuropathic Pain: Preclinical and Clinical Studies. Pain 2016, 157, S33–S41. 10.1097/j.pain.0000000000000369. [DOI] [PubMed] [Google Scholar]
- Murgasova R.; Carreras E. T.; Suetterlin-Hachmann M.; Torrao L. R. S.; Kittelmann M.; Alexandra V.; Fredenhagen A. Non-Clinical Characterization of the Disposition of EMA401, a Novel Small Molecule Angiotensin II Type 2 Receptor (AT2R) Antagonist. Biopharm. Drug Dispos. 2020, 41, 166–183. 10.1002/bdd.2226. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Han G. W.; Batyuk A.; Ishchenko A.; White K. L.; Patel N.; Sadybekov A.; Zamlynny B.; Rudd M. T.; Hollenstein K.; Tolstikova A.; White T. A.; Hunter M. S.; Weierstall U.; Liu W.; Babaoglu K.; Moore E. L.; Katz R. D.; Shipman J. M.; Garcia-Calvo M.; Sharma S.; Sheth P.; Soisson S. M.; Stevens R. C.; Katritch V.; Cherezov V. Structural Basis for Selectivity and Diversity in Angiotensin II Receptors. Nature 2017, 544, 327–332. 10.1038/nature22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakchaure P. B.; Bremberg U.; Wannberg J.; Larhed M. Synthesis of Enantiopure Angiotensin II Type 2 Receptor [AT2R] Antagonist EMA401. Tetrahedron 2015, 71, 6881–6887. 10.1016/j.tet.2015.07.018. [DOI] [Google Scholar]
- Cho S. H.; Kim J. Y.; Lee S. Y.; Chang S. Silver-Mediated Direct Amination of Benzoxazoles: Tuning the Amino Group Source from Formamides to Parent Amines. Angew. Chem., Int. Ed. 2009, 48, 9127–9130. 10.1002/anie.200903957. [DOI] [PubMed] [Google Scholar]
- McCarthy T. D.; Naylor A.. Heterocyclic Compounds and Methods of Their Use. WO/2015/003223.
- Boaz N. W.; Mackenzie E. B.; Debenham S. D.; Large S. E.; Ponasik J. A. Synthesis and Application of Phosphinoferrocenylaminophosphine Ligands for Asymmetric Catalysis. J. Org. Chem. 2005, 70, 1872–1880. 10.1021/jo048312y. [DOI] [PubMed] [Google Scholar]
- Mahalingam A. K.; Wan Y.; Murugaiah A. M.; Wallinder C.; Wu X.; Plouffe B.; Botros M.; Nyberg F.; Hallberg A.; Gallo-Payet N.; Alterman M. Selective Angiotensin II AT2 Receptor Agonists with Reduced CYP 450 Inhibition. Bioorg. Med. Chem. 2010, 18, 4570–4590. 10.1016/j.bmc.2010.03.064. [DOI] [PubMed] [Google Scholar]
- Hein L.; Barsh G. S.; Pratt R. E.; Dzau V. J.; Kobilka B. K. Behavioural and Cardiovascular Effects of Disrupting the Angiotensin II Type-2 Receptor Gene in Mice. Nature 1995, 377, 744–747. 10.1038/377744a0. [DOI] [PubMed] [Google Scholar]
- Benndorf R. A.; Krebs C.; Hirsch-Hoffmann B.; Schwedhelm E.; Cieslar G.; Schmidt-Haupt R.; Steinmetz O. M.; Meyer-Schwesinger C.; Thaiss F.; Haddad M.; Fehr S.; Heilmann A.; Helmchen U.; Hein L.; Ehmke H.; Stahl R. A.; Böger R. H.; Wenzel U. O. Angiotensin II Type 2 Receptor Deficiency Aggravates Renal Injury and Reduces Survival in Chronic Kidney Disease in Mice. Kidney Int. 2009, 75, 1039–1049. 10.1038/ki.2009.2. [DOI] [PubMed] [Google Scholar]
- U.S. National Library of Medicine . https://clinicaltrials.gov/ct2/show/NCT03094195. [DOI] [PubMed]
- U.S. National Library of Medicine . https://clinicaltrials.gov/ct2/show/NCT03297294. [DOI] [PubMed]
- Rice A. S. C.; Dworkin R. H.; Finnerup N. B.; Attal N.; Anand P.; Freeman R.; Piaia A.; Callegari F.; Doerr C.; Mondal S.; Narayanan N.; Ecochard L.; Flossbach Y.; Pandhi S. Efficacy and Safety of EMA401 in Peripheral Neuropathic Pain: Results of Two Randomised, Double-Blind, Phase 2 Studies in Patients with Postherpetic Neuralgia and Painful Diabetic Neuropathy. Pain 2021, 10.1097/j.pain.0000000000002252. [DOI] [PubMed] [Google Scholar]
- Sawamura R.; Okudaira N.; Watanabe K.; Murai T.; Kobayashi Y.; Tachibana M.; Ohnuki T.; Masuda K.; Honma H.; Kurihara A.; Okazaki O. Predictability of Idiosyncratic Drug Toxicity Risk for Carboxylic Acid-Containing Drugs Based on the Chemical Stability of the Acyl Glucuronide. Drug Metab. Dispos. 2010, 38, 1857–1864. 10.1124/dmd.110.034173. [DOI] [PubMed] [Google Scholar]
- Austin P. J.; Wu A.; Moalem-Taylor G. Chronic Constriction of the Sciatic Nerve and Pain Hypersensitivity Testing in Rats. J. Vis. Exp. 2012, 13, e3393. 10.3791/3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.