

Abstract

In this paper, we present an additional, new cage-GABA compound, called 4-amino-1-(4′-dimethylaminoisopropoxy-5′,7′-dinitro-2′,3′-dihydro-indol-1-yl)-1-oxobutane-γ-aminobutyric acid (iDMPO-DNI-GABA), and currently, this compound is the only photoreagent, which can be applied for GABA uncaging without experimental compromises. By a systematic theoretical design and successful synthesis of several compounds, the best reagent exhibits a high two-photon efficiency within the 700–760 nm range with excellent pharmacological behavior, which proved to be suitable for a complex epileptic study. Quantum chemical design showed that the optimal length of the cationic side chain enhances the two-photon absorption by 1 order of magnitude due to the cooperating internal hydrogen bonding to the extra nitro group on the core. This feature increased solubility while suppressing membrane permeability. The efficiency was demonstrated in a systematic, wide range of in vitro single-cell neurophysiological experiments by electrophysiological as well as calcium imaging techniques. Scalable inhibitory ion currents were elicited by iDMPO-DNI-GABA with appropriate spatial–temporal precision, blocking both spontaneous and evoked cell activity with excellent efficiency. Additionally, to demonstrate its applicability in a real neurobiological study, we could smoothly and selectively modulate neuronal activities during artificial epileptic rhythms first time in a neural network of GCaMP6f transgenic mouse brain slices.
Introduction
Caged compounds are excellent tools to simulate and modulate neuronal activity patterns from the subcellular to network level.1 Although highly efficient excitatory caged molecules have already been synthesized,2−4 the development of inhibitory caged molecules for two-photon (2P) microscopy has proven to be a more difficult task.5,6 The previously reported photo-activable caged-GABA compounds are just at the limit of their usability, and they have exhibited several drawbacks in their stability, solubility, 2P uncaging efficiency, or antagonistic effect on GABAA receptors.6−9 During complex physiological brain activity patterns, excitatory and inhibitory processes appear in a delicate balance, which is upset during pathological progressions, such as epileptic activities. The 2P uncaging technique provides an opportunity for rapid, light-initiated release of neurotransmitters.5,10−14
The precise, spatially and temporally controlled in situ release of neurotransmitters (mainly glutamate)15,16 is a major benefit of the 2P uncaging technique, which enabled numerous studies of ion channel kinetics,17,18 receptor distributions,19−21 synaptic transmission,21−24 synaptic integration,25−27 postsynaptic mechanisms,28,29 and network circuitry.30−33 Caged glutamate is the most widely used caged neurotransmitter for possibly two main reasons. One is the obvious central role of glutamate in neural signaling, which renders it as the most needed tool. The second is the properties of the available caged glutamate compounds that permit precise research to be performed. These properties are the high uncaging efficiency, the low effect on target receptors in the absence of illumination, and a suitable pharmacological profile, all stemming from the structure of the caged molecule. So far, numerous cage scaffolds were developed simultaneously,3 such as nitrobenzyl,34−36 coumarine,37−40 pyridine,5 β-carboline,41 and quinolone,7 but nitroindoline-based (NI)9 compounds are the most widely used. The nitroindilines mononitroindolyl glutamate (MNI-Glu)8,10,42,43 and the improved dinitroindolyl glutamate (DNI-Glu)44,45 show good pharmacological character, and they have been used successfully previously.8,15,27,43,45−47
Compared to other photostimulation procedures (e.g., optogenetics), the advantage of 2P uncaging is the rapid, repeatable stimulation within a micron volume on a specific segment of the selected cell. Furthermore, it does not require altering the genetic code and protein composition of cells. While cage molecules carrying the excitatory glutamate neurotransmitter have been developed for 2P experiments (e.g., DNI-Glu15,48 and MNI-Glu),8,10,42,43 relatively little attention and “success” have been achieved in the development of a cage compound carrying the inhibitory GABA molecule.
Although the focus has been on glutamate, GABA is also a prime candidate for a caged compound as it is fundamental in inhibitory–excitatory balance as the primary inhibitory neurotransmitter. Several caged GABA compounds have been reported previously in studies of GABA receptor kinetics and distribution in neural and glial compartments.9,34−36,38−40 However, these compounds exhibited a relatively low 2P uncaging efficiency and limited concentrations that can be employed without a substantial effect on GABAA receptors in the absence of illumination and suboptimal pharmacological properties.6,8 Our ultimate goal is to design a photoreagent, which is able to modulate simulated and real activity patterns too. On one hand, it allows a better understanding of their structure–activity relationship. On the other hand, it opens new therapeutic possibilities for the elimination or correction of abnormal brain activity patterns such as epilepsy. Although optogenetic neuronal modulation of epileptic activity is increasingly used,49,50 to understand how neurons are capable of classifying the excitatory and inhibitory inputs to their dendrites, both in health and disease, we need to use appropriate techniques. In the present work, we aimed to develop a novel caged GABA compound, with high uncaging efficacy and a reliable pharmacological profile besides the minimal off-target effect. For this reason, we have carried out a complex chemical and physiological validation process, including computational design, chemical synthesis, structure–activity relationship, in vitro neurophysiological testing, and their functional application in an epileptic study. To be concise, we focused only on a few derivatives. For the physiological studies, we applied four modern methods: 2P imaging, GABA uncaging, electrophysiology, and the transgenic mouse model, which was made in-house. As a result, we proved that 4-amino-1-(4′-dimethylaminoisopropoxy-5′,7′-dinitro-2′,3′-dihydro-indol-1-yl)-1-oxobutane-γ-aminobutyric acid (iDMPO-DNI-GABA) is the most effective compound among the designed and prepared molecules. Finally, using iDMPO-DNI-GABA, we have shown the selective blockade of neuronal activity associated to epileptic rhythms.
Results and Discussion
Our neurobiological goal was to selectively modulate the activity of neurons in a network during in vitro epileptic rhythms. Preliminary neurobiological studies and quantum chemical computations have shown only a moderate quantum yield on the previously reported caged GABA compounds (CDNI-GABA,40 DPNI GABA49), and they suffered by a number of side effects. Consequently, to realize our goal, we have carried out a structure–activity development on the caged scaffold by quantum mechanics (QM) methods in order to explore the best photoreagents, having high 2P absorption (TPA).
To meet the rigorous demands of 2P uncaging experiments, first we have designed several novel concept caged GABA compounds,51 and their TPAs as well as photoreactivities were computed, with the aim to improve the uncaging efficacy. Second, among many models, the four relevant derivatives were selected for synthesis and the subsequent multilevel neuropharmacological studies (1, 2a, 2b, 3). Our original starting point was the chemical structure of DNI-Glu, which was confirmed to be an outstanding caged glutamate compound reported in several publications by both experimental and QM methods.15,52 Therefore, the first and directly derived GABA compound is DNI GABA (1, Figure 1, step I), which is used primarily as the reference point. According to our hypothesis, the TPA can be originated by the quadrupole moment of a molecule, which is in contrast with the one-photon transition, which is depending on the dipole moment. To increase the TPA, we extended the size of the molecule in all dimensions in space and charge on the side chain (Figure 1, step II). An analogous strategy was attempted previously in the case of CDNI-GABA,8,47 extending the side chain of the indoline core; however, the incorporated negative side chain has yielded only little benefit. Contrary to the net neutral CDNI-GABA, we plan to include additional positively charged and branched side chains at position 4 (Figure 1, step II), with different lengths for compounds 2a and 2b. This resulted in the net double positive charge of these molecules, which effectively inhibits their penetration through the cell membrane and simultaneously increases its water solubility. For the fourth compound (3), we plan to block chemically the GABA amino group, which is assumed to be responsible for the undesired GABAA receptor activity. In this concept, we prepared a reversely bound nitroindolyl-caged-GABA with an introduced carbamide linker, DNI-CO-GABA (3, Figure 1, step III).
Figure 1.

Conceptual strategy of the present work. The present work is originated from DNI-Glu15,48 retaining the effective dinitro substitution as DNI-GABA (1, in step I). Subsequently, we introduced the novel cationic side-chain conception as iDMPO-DNI-GABA (2a) and iDMBO-DNI-GABA (2b, step II) in contrast to the anionic ones.9,53 Finally, we reverse the binding type of GABA as DNI-CO-GABA (3) in step (III).
Quantum Chemical Modeling
Preparatory uncaging tests showed that GABA is successfully released from 1, 2a, and 2b but not from 3, regardless of illumination strength. We hypothesized that the highly stable urea-type linker in 3 prevents the photochemical reaction, so this compound was excluded from further experiments.
In order to describe the remaining compounds 1, 2a, and 2b, we carried out a detailed theoretical study involving 2P excitation (Dalton 2020.alpha)54 and mechanistic study (Gaussian 16)55 at the B3LYP/6-31G(d,p)//PCM(water) level of theory. Quantum chemical modeling has confirmed that the optimal length of the cationic side chain enhances TPA by 1 order of magnitude due to an internal hydrogen bonding (HB).
Conformational Study
Caged GABA derivatives 2a and 2b have numerous conformers at the GABA side chain; however, for the sake of simplicity, we selected only its linear GABA conformer, which represents the lowest energy arrangement. Apart from this, the amino alkyl side chains can maintain two relevant conformers, a linear arrangement (I) and a ring arrangement (II), as shown in Scheme 1. The Gibbs-free-energy difference of the two conformers was corrected by an explicit solvent model, resulting from the HB of water and the NO2 group. It is noteworthy that for 2a, the energetically preferred conformer is undoubtedly the cyclic 2a-II. For 2b, instead of the cyclic 2b-II, the linear 2b-I conformer appears to be the more relevant conformer. This energy difference can be explained by the much stronger HBs in 2a-II, in contrast to the solo HB in 2b-II. In 2a-II, the more optimal arrangement of the 9-member ring is beneficial, compared to the 10-member variant in 2b. The weak HB for 2b-II is overcome by the HB effect of H2O to NO2, which was estimated as ca. 3 kJ mol–1 by the same method (see the Supporting Information).
Scheme 1. Energetic Comparison of the Two Relevant Conformers of 2a and 2b; With Respect to the Linear Form (I), the ΔG Values Are Corrected by the Explicit Effect of the Aqueous Solvation, Indicated by the Arrows (ΔG*).

HB = hydrogen bonding.
Quantum Chemical Modeling of TPA
We computed the TPA cross-section (σ) of DNI-GABA (1) and the most relevant conformers of iDMPO-DNI-GABA (2a-II) as well as iDMBO-DNI-GABA (2b-I) and compared to the recomputed values of MNI-Glu and DNI-Glu using the Dalton 2020.alpha program in the gas phase at the B3LYP/6-31G(d,p) level of theory.54 The 2P transition moment tensor components (S) were calculated and analyzed (Table 1).
Table 1. Computed 2P Transition Tensor Components (S), 2P Transition Energy (E in eV), Probabilities (Df, Dg and D in a.u.) Cross-Section Values (σ in a.u.) for the S0–S1 Transition of MNI-Glu, DNI-Glu, DNI-GABA (1), and the Relevant Conformers of iDMPO-DNI-GABA (2a-II) and iDMBO-DNI-GABA (2b-I) Computed at the B3LYP/6-31G(d,p) Level of Theory by Using Dalton 2020.alpha, Assuming a Monochromatic Light Source with Linear Polarization.
| compounds | Sxx | Syy | Szz | Sxy | Sxz | Syz | E (eV) | Df (a.u.) | Dg (a.u.) | D (a.u.) | σ (a.u.) | σ (GM) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MNI-Glu | 3.0 | 5.9 | –0.4 | –7.2 | –1.4 | 1.2 | 3.63 | 2.40 | 5.11 | 25.24 | 0.244 | 0.46 | |
| DNI-Glu | 39.9 | 0.4 | –0.4 | 1.9 | –5.3 | 0.4 | 3.59 | 53.00 | 55.30 | 327.2 | 0.309 | 0.59 | |
| DNI-GABA | 1 | 3.0 | –1.5 | –0.2 | 1.9 | –0.5 | 0.6 | 3.50 | 0.06 | 0.67 | 2.8 | 0.025 | 0.05 |
| iDMPO-DNI-GABA | 2a-II | –1.8 | 26.8 | 2.3 | 6.0 | –2.3 | 5.4 | 3.48 | 25.00 | 29.00 | 166.0 | 1.470 | 2.79 |
| iDMBO-DNI-GABA | 2b-I | –8.7 | –3.0 | 0.1 | –6.3 | 1.4 | 0.5 | 3.57 | 4.54 | 5.57 | 31.4 | 0.293 | 0.56 |
| ratio of 1/DNI-Glu | 0.001 | 0.012 | 0.009 | 0.081 | 0.081 | ||||||||
| ratio of 1/2a-II | 416.7 | 43.3 | 59.3 | 58.8 | 58.8 | ||||||||
| ratio of 1/2b-I | 75.7 | 8.3 | 11.2 | 11.7 | 11.7 | ||||||||
The two diagonal tensor components Sxx and Syy represent only high values for 1, while other tensor components proved to be almost negligible, which corresponds to its rather flat chromophore. It appears that a charge transfer exists from OMe to NO2 along the direction x and it behaves as an antenna. For compounds 2a-II and 2b-I, more off-diagonal tensor components play important roles with higher values, referring to the more three-dimensional-shaped chromophores, compared to 1 and DNI-Glu, which contribute to the larger net transition probability values for these molecules.
The TPA cross-section values (σ) calculated from linear transition probability (D) and the excitation energy (E) (Table 2) are based on a method published earlier.56−59 Here, we focused only on the dominant conformers of compounds 2a-II and 2b-I. The results showed about 58.8 times larger σ value for iDMPO-DNI-GABA (2a-II) and 11.7 times larger σ value for iDMBO-DNI-GABA (2b-I) than for DNI-GABA (1). These results predict a 1 order of magnitude increase in TPA for the S0–S1 excitations of 2a and 2b, which influences the overall photochemical process positively.
Table 2. Comparison of Biological Efficiency in IPSC Maximum Amplitude and Area (Relative to DNI-GABA, 1).
| iDMPO-DNI-GABA, 2a |
iDMBO-DNI-GABA, 2b |
||
|---|---|---|---|
| IPSC max | IPSC area | IPSC max | IPSC area |
| 4.78 | 5.88 | 1.44 | 1.12 |
Quantum Chemical Modeling of the Photochemical Uncaging Process Versus the Ground-State Hydrolysis Mechanism
The reaction mechanism of the photochemical release of GABA from 1 and the two forms of 2a and 2b were modeled and compared by quantum chemical tools (Figure 2, Table 1) based on an earlier method.15 The explored photochemical mechanisms for these GABA derivatives were analogous to those of the previously published DNI-Glu. All the caged GABA derivatives (1, 2a, and 2b) followed the same pathways with close enthalpy levels according to the substituents and the conformers. The initial state A(S0) represents the ground state, and after the excitation, it follows the following sequence of states: A(S0) → B(S1) → C(S1) → D(T1) → TS1 → E(T1) → TS2 → F(T1) → TS3 → G(T1) → H(S0). This process involves the excitation [A(S0) → B(S1)], followed by the geometrical relaxation [B(S1) → C(S1)]. The high-energy excited singlet state C(S1) tends to transform to the energetically close triplet state D(T1) via intersystem crossing (ISC). In this triplet state, an acyl transfer reaction can occur preferably in two low-energy elementary steps [TS1 → E(T1) → TS2 → F(T1)], while the acyl group migrates from the indoline nitrogen to the nitro oxygen. The E(T1) minima were also confirmed in an earlier publication.52 The forming F(T1) triplet state is considerably stable, lying close to its ground-state analogue [I(S0)], which allows the spontaneous de-excitation via a second ISC. This F(T1) state also leads to the triplet product state [G(T1)] through TS3, resulting in the ground-state zwitterionic free GABA by the N–O bond cleavage. G(T1) finally de-excites to the ground state [H(S0)]. The reactive species I(S0) can undergo a ground-state rearrangement of the O-acyl form to the N-acyl, reaching back to the starting position A(S0) via TS4. The process of A → C → D → F → I → A can be considered as an undesired “short circuit” of the uncaging process, which only emits and dissipates the laser energy to heat, recovering the starting state.
Figure 2.

Left: Summary of the schematic potential enthalpy (ΔH) profile of the photochemical (read and blue) and ground-state (black) mechanism for compounds 1, 2a, and 2b at the B3LYP/6-31G(d,p)//PCM(water) level of theory. Right: Computed enthalpy values (ΔH, in kJ mol–1) of different states (A–I, TS1–TS4) for compounds 1, 2a, and 2b, relative to state A(S0). ISC is estimated from the scanning of the reagent and reactant. (ainitial state; b,cestimated by scanning along the reaction coordinate).
In conclusion of the photochemical pattern, no significant differences were found between the enthalpy profiles and their values of 1, 2a, and 2b. However, the predicted TPA differed significantly; it proved to be 58.8 and 11.7 times larger values for 2a and 2b, respectively, as compared to 1. Based on these findings, three compounds were selected and subjected to biological testing, with predicted low (1), medium (2b), and high (2a) photoactivity.
Chemical Synthesis
The synthesis and the purification of DNI-GABA (1) were carried out according to a previously published pathway with a medium overall yield (35%, for details, see the Materials and Methods section).44 The synthesis of 2a and 2b was started on an analogous synthetic route (Scheme 2), building up the molecule by alkylation of the 4-hydroxy-indole (7) using 1-chloro-N,N-dimethylpropan-2-amine or 3-chloro-N,N-2-trimethylpropan-1-amine in the presence of NaH. Then, indole derivatives were reacted with NaBH3CN, leading to the corresponding indoline derivatives 8a,b. The isolated 8a contains minor (27%) regioisomer also, which is different on its side chain due to the formation of the asymmetric aziridine cation from 1-chloro-N,N-dimethylpropan-2-amine. In the subsequent step, they were acylated by N-Boc-GABA, resulting in the last intermediates (6a,b), respectively. Before the final dinitration step, the Boc protecting group was removed by trifluoro acetate (TFA), and then, the intermediates were nitrated by 3 equiv of NO2·BF4 in dry dichloromethane (DCM) at a low temperature (0–5 °C), yielding the desired products 2a and 2b. After purification by preparative high-performance liquid chromatography (HPLC) (eluent A: 0.1%TFA–water; eluent B: MeCN), the overall yields were between 20 and 25% with an excellent chemical purity (>99%) as a TFA salt. The free GABA content was appropriately low (<1 ppm), confirmed by an HPLC method developed earlier.60 The minor and major isomers of 2a were separated by preparative HPLC and tested in uncaging experiments. As the isomers yielded equivalent results, 2a was subsequently used in biological experiments as an isomeric mixture. The synthesis of compound 3 followed a similar synthetic strategy, starting from the known 4-OMe-indoline 7 by reacting 1,1′-carbonyldiimidazole (CDI) and then GABA-OtBu. The resulting intermediate 13 was dinitrated by NO2·BF4 in dry DCM, yielding the final product. In all the cases, the last steps and the subsequent purification by preparative HPLC as well as lyophilization were carried out in darkness to avoid unwanted photochemical degradation that could lead to the appearance of free GABA. According to our accumulated experience,51 the TFA salt proved to be more stable and more resistant toward hygroscopic degradation in the solid form.
Scheme 2. Synthesis of the Three Cage-GABA Derivatives, iDMPO-GABA (2a), iDMBO-GABA (2b), and DNI-CO-GABA (3).
In Vitro Characterization of the Selected Caged GABA Compounds (1, 2a, and 2b)
Neurophysiological experiments were carried out in three stages. In the primary assay, we compared the inhibitory postsynaptic currents (IPSCs) evoked with the three compounds (1, 2a, and 2b) under the same experimental conditions, which resulted in iDMPO-DNI-GABA (2a) as the most active candidate. In the secondary assay (the side-activity and applicability test), we examined the effect of 2a on intrinsic cellular properties and evaluated its ability to modulate neuronal input and output. Finally, we used iDMPO-DNI-GABA (2a) to modulate the participation of single neurons in a rhythmic, epileptic population activity in genetically modified, GCaMP6f mouse brain slices for the first time.
Primary Assay and the Activity Measurement: iDMPO-DNI-GABA (2a) Uncaging Evokes Significantly Larger IPSCs Compared to the Other Compounds
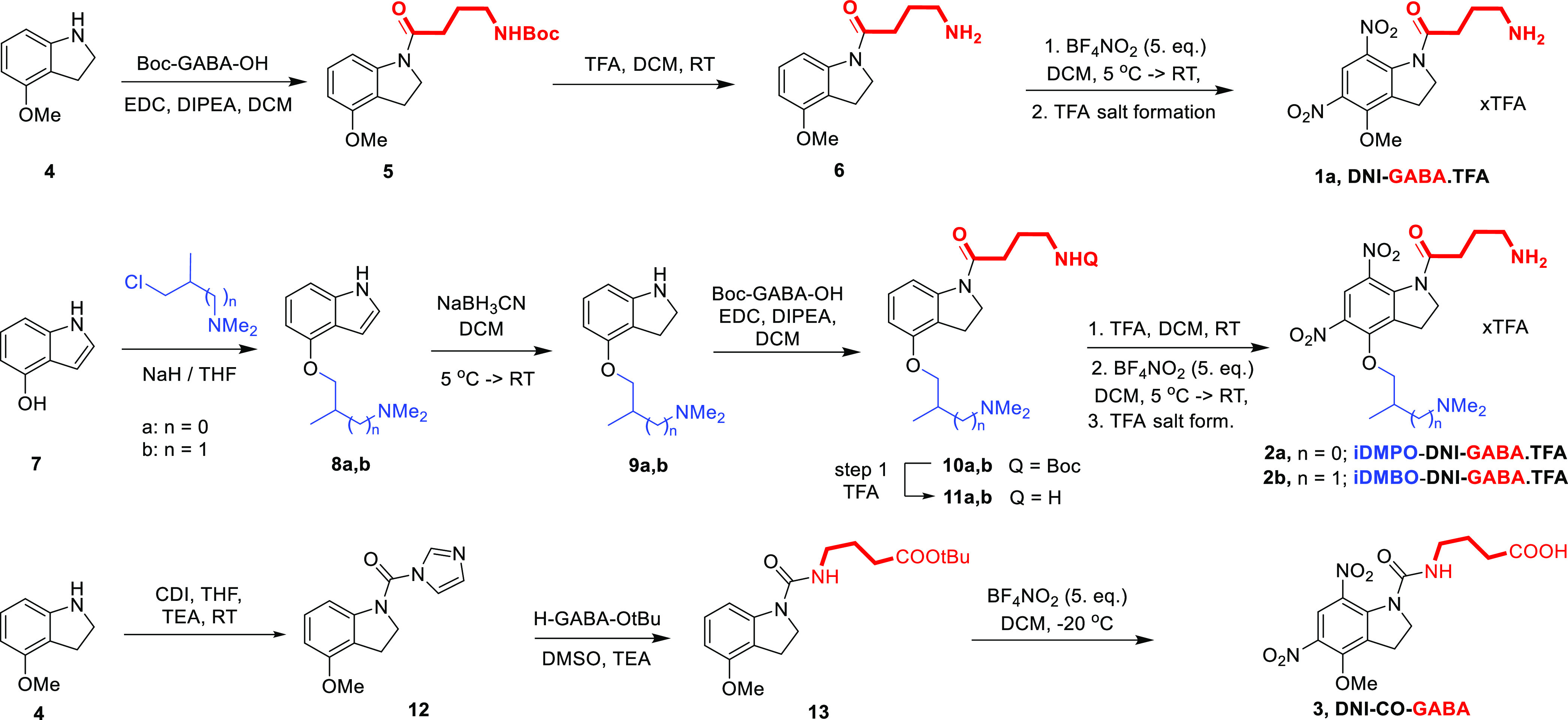
To select the most effective caged GABA compound for neurophysiological experiments, we compared IPSC amplitudes evoked with the three compounds on cortical pyramidal neurons (Figure 3) systematically under the standard conditions. Pairwise comparisons were made on the same neurons by wash-in and wash-out of the compounds (all at a 2.5 mM concentration) to get unquestionably comparable values. The spatial location and pattern of uncaging sites and the temporal order of their activation as well as all other parameters (e.g., laser intensity and pixel dwell time) were unchanged. Under these well-defined conditions, we found that uncaging of iDMPO-DNI-GABA (2a) evoked approximately 5-fold larger IPSCs on target neurons compared to the ones evoked by uncaging of DNI-GABA (1). This difference largely remained fixed when we compared iDMPO-DNI-GABA (2a) to iDMBO-DNI-GABA (2b), while uncaging of iDMBO-DNI-GABA (2b) evoked only marginally larger IPSCs compared to DNI-GABA (1, Figure 3D–F, in Table 2). Therefore, we selected iDMPO-DNI-GABA (2a) as the best candidate and limited further experiments on it exclusively. Moreover, the membrane permeability of the compound is very low.
Figure 3.
(A) Maximal intensity z-projection image of a cortical pyramidal neuron filled with Alexa 594; the yellow dot indicates the location of the 2P GABA uncaging site in the presence of DNI-GABA (1) and iDMPO-DNI-GABA (2a); in 2.5 mM. (B) Representative example showing 2P uncaging (740 nm)-evoked somatic IPSCs of the same neuron in the presence of DNI-GABA (1) and iDMPO-DNI-GABA (2a) cage compounds, respectively. (C) Comparison of the uncaging-evoked IPSCs of iDMPO-DNI-GABA (2a) to that of DNI-GABA (1) and iDMBO-DNI-GABA (2b) compounds [n = 4, mean ± standard error of the mean (SEM), respectively]. The green line represents the uncaging time. (D,E) Same data as in (C). Bar graphs of IPSC amplitude (left) and amplitude area (right), respectively. (F) Data of (D,E) are normalized to the IPSC amplitudes of iDMPO-DNI-GABA (2a). Normalized to iDMPO-DNI-GABA (2a): iDMBO-DNI-GABA (2b) and DNI-GABA (1) max: 0.31 ± 0.11 and 0.22 ± 0.02 area: 0.19 ± 0.09 and 0.16 ± 0.02. Asterisks indicate significance (*p < 0.05, ***p < 0.001).
Secondary Assay and the Compatibility and Applicability Test: iDMPO-DNI-GABA (2a) Effectively Modulates Neuronal Input and Output Signals without Significant Toxic Effects on the Basic Physiological Properties
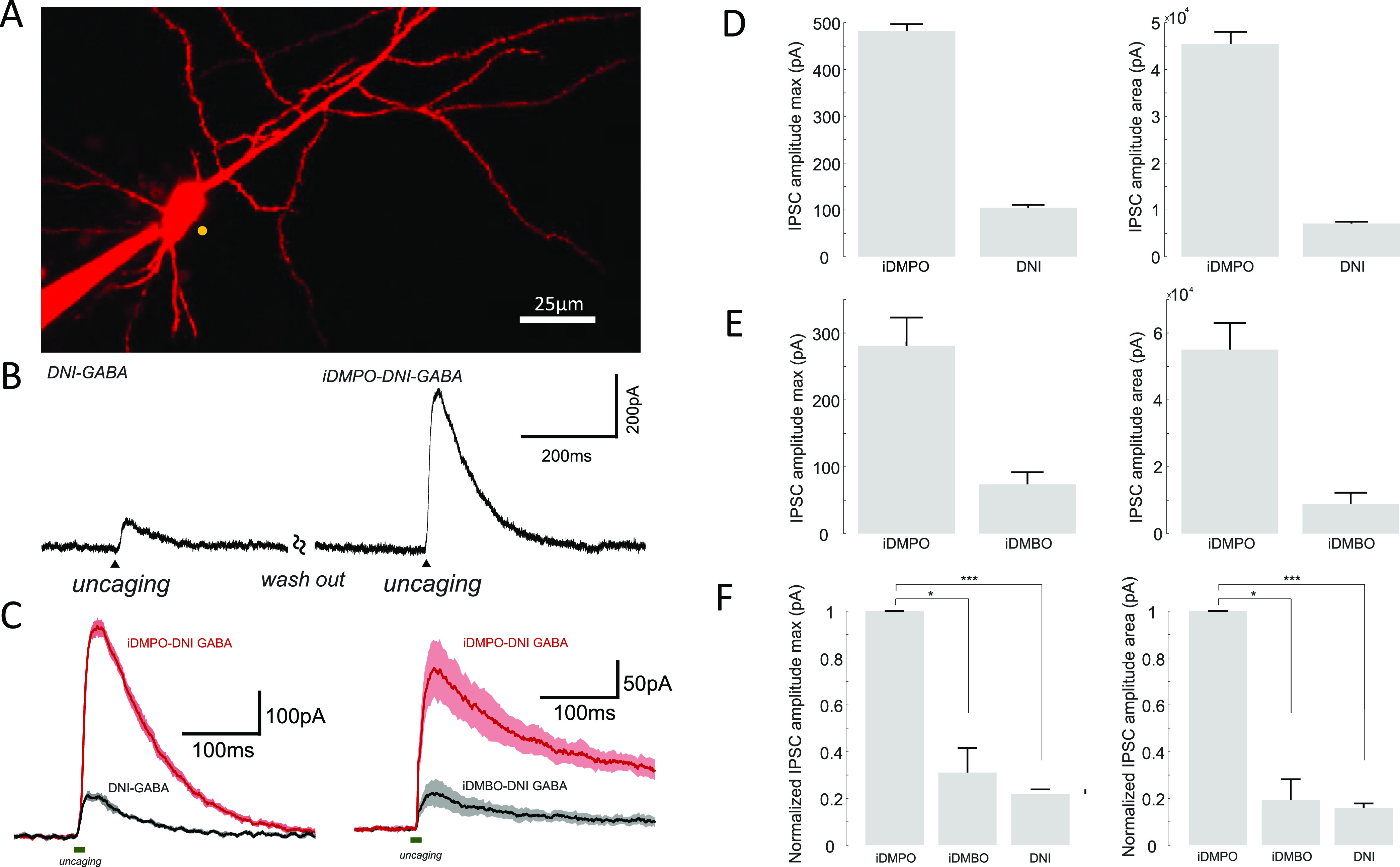
In this stage, we tested extensively and thoroughly the effects and effectivity of iDMPO-DNI-GABA (2a) and characterized its ability to modulate neuronal input and output signals, the basic physiological properties, excitation wavelength dependence, laser power, reversal of the uncaging current, and the spatial precision on the soma and the dendrite. First, we tested how the bath application of 2a could affect the basic physiological properties of pyramidal neurons (Figure S1). We patched and filled pyramidal cells with Fluo-4 Ca2+-sensitive and Alexa 594 anatomical dyes and measured the fluorescence and membrane potential changes to somatically injected current ramps (ramp test). We did not find any significant difference in the resting membrane potential of patched neurons neither in the shape, frequency, half width, amplitude, rise time, and decay time of action potentials in the presence of 2a compared to control periods before wash-in (Figure S1B, Table S1). The Ca2+ fluorescence signals of evoked backpropagating action potentials also showed no significant differences in amplitude (52 ± 0.4 vs 47 ± 0.2%, n = 4 cells, p = 0.3) or area (111.66 ± 6.92 vs 102.96 ± 2.89, n = 4 cells, p = 0.3) before and after application of iDMPO-DNI-GABA (2a; Figure S1C,D). Next, we validated that the effects of 2a uncaging were indeed mediated by released GABA. Irradiation at 740 nm for 1 ms near the cell body of a patched pyramidal neuron produced a robust outward current (Figure 4), which was abolished by bath application of 100 μM gabazine, a specific blocker of GABAA receptors61 (control: 48.72 ± 16.977 pA vs gabazine: 3.77 ± 1.92 pA, n = 4, p = 0.05) (Figure 4B).
Figure 4.
(A) Image of the measured pyramidal neuron filled with Fluo-4 and Alexa 594. The yellow dot indicates the location of the 2P GABA uncaging site in the presence iDMPO-DNI-GABA (2a) (2.5 mM). (B) Photolysis of iDMPO-DNI-GABA (2a) with 740 nm laser light (arrowhead) evokes IPSCs in the pyramidal neuron that is blocked by bath-applied gabazine (100 μM); example traces (left), average responses from four cells (right) (n = 4, mean ± SEM). Somatically evoked action potential (Ap) block: action potential amplitude before and after 2a uncage: 76.50 ± 4.88 and 31.24 ± 0.63 mV, respectively (n = 3 cells), p = 0.0096. (C) Maximal intensity z-projection image of a cortical pyramidal neuron filled with Fluo-4 and Alexa 594. The yellow dot represents the location of 2P uncaging, and the yellow line indicates the 2P imaging site along the dendrite. Somatically evoked Ap-associated backpropagated Ca2+ transients before and after 2a uncage: 1.24 ± 0.13 and 0.03 ± 0.07 (in ΔF/F0), respectively (n = 3 cells), p = 0.00504. (D) Current injection induces somatically recorded single backpropagation action potential and triggers calcium transients at the proximal dendritic segment (left and right black traces, respectively). Uncaging of iDMPO-DNI GABA (2a) blocks somatically evoked backpropagation action potentials and the corresponding Ca2+ signal (left and right red traces). (E) Somatically recorded spontaneous backpropagation action potentials can be blocked by single (left) or repetitive GABA uncaging (right).
Measuring the uncaging evoked IPSCs while holding the patched cells at different membrane potential values (10 mV intervals from 0 to −60 mV) showed that the reversal of the uncaging evoked currents are close to the reported chloride reversal potential62 (−60.77 ± 4.87 mV, n = 12 cells) (Figure S2). We also explored the wavelength dependence of photolysis-evoked IPSCs (Figure S3). Varying excitation wavelength between 700 and 820 nm showed that the amplitudes of IPSCs were the largest at 740 nm, revealing the 2P cross-section peak of iDMPO-DNI-GABA (2a) (Figure S3C). IPSC amplitudes also showed a linear relationship with applied laser power at a given wavelength (Figure S4). Next, we measured the spatial accuracy of the 2P uncaging of 2a (Figure S5). We denoted a series of points in a straight line at different distances from the soma (Figure S5C,D) or dendrite (Figure S6A,B) of pyramidal neurons and measured the IPSCs evoked by uncaging at these points one at a time. Uncaging closer to the soma evoked larger-amplitude IPSCs, and the IPSC amplitude approached 0 at a distance of 9.23 ± 1.85 μm from the membrane of the soma (n = 6 cells). Apart from these, a further important question is whether iDMPO-DNI-GABA (2a) can block effectively neuronal output signals. To test this, we somatically evoked single bAp-s and measured the associated calcium transients in the proximal dendritic segment (Figure 4D). Moreover, in another aspect, we raised the resting membrane potential of the pyramidal neurons, eliciting bursting, and then performed single or repetitive uncaging near the soma to prevent neuronal firing. We found that 2P uncaging of iDMPO-DNI-GABA (2a) is able to generate sufficient neuronal hyperpolarization to block single action potentials or even action potential burst (Figure 4D,E). Furthermore, dendritic Ca2+ signals corresponding to blocked action potentials were also diminished (Figure 4D right). According to the literature, we compared antagonist behavior of iDMPO-DNI-GABA (2a) and the well-known CDNI-GABA molecule.9,40 Both CDNI-GABA and iDMPO-DNI-GABA decreased the evoked IPSC amplitude and area significantly (Figure S8C–I). We found that the rate of changes in these self-controlled experiments was bigger in CDNI-GABA than in iDMPO-DNI-GABA. Besides this, we concluded that there is no significant (n.s.) difference between the two GABA cage compounds when compared to each other by their normalized data (Figure S8J). As we found that uncaging of iDMPO-DNI-GABA (2a) can evoke IPSCs repetitively with high spatial and temporal precision and could effectively block neuronal activity, which are necessary for selective and specific uncaging experiments, we moved on to test the compound in a functional experiment.
Tertiary Assay and the Direct Application: Rhythmic Epileptic Activity Can Be Selectively Modulated by iDMPO-DNI-GABA (2a)
In the final stage, we have shown that even during large-scale pathological network activities, such as in vitro epileptic events, rapid and precise uncaging of GABA with iDMPO-DNI-GABA (2a) can modulate the participation of neurons in the ongoing network activity. Monitoring the activity of hundreds of neurons requires widespread labeling and fast sampling of individual neurons. Therefore, we used transgenic mice expressing GCaMP6f under the Thy1 promoter (Figure S7), which predominantly labels pyramidal cells,63 and multiple line scanning with 2P calcium imaging, combined with 2P uncaging. To generate population activity in brain slices, we washed in 4-aminopyridine (in 50 μM), which triggered epileptic-like events involving most of the neuronal network (Figure 5). These rhythmic events were followed by LFP electrodes placed in close proximity to the imaging site. After the pathological population activity was established, we selected a single, rhythmically active neuron and uncaged iDMPO-DNI-GABA (2a) near its soma locally, discretely, and repetitively. We found that uncaging suppressed the Ca2+ responses [before: 0.46 ± 0.04 Hz (mean ± SEM), during: 0.03 ± 0.02 Hz (mean ± SEM), and after: 0.45 ± 0.05 Hz (mean ± SEM), n = 12 cells] of the targeted neuron reliably and its participation in the ongoing population activity was entirely blocked (Figure 5). This effect persisted through multiple successive uncaging periods, while previous activity resumed unchanged between suppressions. Other measured neurons were unaffected by this manipulation and maintained their activity during the uncaging periods. The selective modulation of cells was also sustainable trace by trace without any negative effect of their normal physiological conditions even during longer experiments (Figure 5E–H).
Figure 5.

(A) Maximal intensity z-projection of the cortical region in Thy1-GCaMP6f transgenic mice for pyramidal cell population imaging in vitro. Yellow circles indicate measured pyramidal neurons. The red circle indicates the selectively modulated cell. (B) Representative fluorescence Ca2+ responses recorded by multiple line scanning of pyramidal neurons during evoked epileptic events. The image shows the activity pattern of the simultaneously measured 18 pyramidal neurons in (A). Triangles indicate repetitive uncaging on the modulated cell (#3). (C) Ca2+ transients from (B) with associated LFP recording (green). The red transient indicates the Ca2+ signal of the cell modulated by iDMPO-DNI-GABA (2a), while the other neurons’ activity remained intact (black). The colored bar represents the time interval of repetitive GABA uncaging. (D) Enlarged view from (C) shows the activities of cells #2, #3 (modulated), and #9. (E) Same maximal intensity z-projection image as in (A) shows another manipulated pyramidal cell, #5. Dashed circles indicate uncaging locations at the soma and at the edge of the field of view as a reference [red (p1) and yellow (p2), respectively]. (F) Trace-by-trace modulation of the pyramidal cell activities by iDMPO-DNI-GABA (2a). The colored bar represents the time interval of uncaging. (G) Activities of neurons before, during, and after iDMPO-DNI-GABA uncaging (before: 0.46 ± 0.04 Hz, during: 0.03 ± 0.02 Hz, and after: 0.45 ± 0.05 Hz, n = 12, p < 0.001). (H) Representative traces show cell #5 activities when the uncaging location was set at the cell [red; p1 on (E)] and at the reference uncaging location without a cell [yellow on (E); p2].
In summary, our novel caged GABA compound enables the reproduction of somatic or dendritic inhibitory inputs selectively and exclusively on individual cells due to the precise spatial and temporal control of 2P uncaging. As a result of thorough chemical development and biological testing, iDMPO-DNI-GABA (2a) is a well-tuned and potent GABA cage molecule.
Conclusions
Our designed and synthetized cage GABA compound, iDMPO-DNI-GABA (2a), equipped with a novel cationic side chain, proved to be highly effective for the selective modulation of single or multiple neurons in physiological and epileptic networks. Quantum chemical study indicated that this compound could undergo the same photochemical mechanism upon excitation as DNI-Glu; however, its theoretical 2P cross-section increased by 1 order of magnitude for the optimal side-chain length. This benefit can be attributed to the cooperating internal HB.
An effective five-step synthesis was elaborated with a good overall yield, and the measured 2P efficiency of iDMPO-DNI-GABA (2a) proved to be very effective. The cationic side chain also increased its water solubility (>2.5 M in the buffer), while membrane permeability could be suppressing. The neurobiological efficiency of the compound was demonstrated in a wide range of in vitro single-cell neurophysiological experiments by electrophysiological and calcium imaging. Scalable and stable inhibitory ion currents were elicited by iDMPO-DNI-GABA with appropriate spatial–temporal precision, blocking spontaneous and evoked cell activity with a high efficiency, with no significant side effects. With this structurally fine-tuned cage-GABA photoreagent, neuronal activities could be modulated selectively in GCaMP6f transgenic mouse brain slices during epileptic rhythms for the first time, allowing novel in vivo neuroscientific applications and therapeutic methods in the future.
Materials and Methods
Chemical Synthesis and Analytical Methods
Amino acid derivatives were obtained from Bachem, and other chemicals were purchased from Sigma-Aldrich. Reagents were of the highest purity available. 1H, 13C, and NMR spectra were recorded in dimethyl sulfoxide-d6 (DMSO-d6) with a Bruker Avance III spectrometer operating at 500 and 125 MHz. High-resolution mass spectrometry (HRMS) spectra were recorded using an Agilent 6230 TOF LC/MS spectrometer. In some cases, preparative HPLC was applied, Agilent Prep HPLC, on a Gemini 250 × 50.00 mm; 10 μm, C18, 110A column in 0.2% TFA or ammonium carbonate in water (eluent A) and the acetonitrile (eluent B) liquid phase using the gradient method.
Preparation of DNI-GABA (1)
Synthesis of tert-Butyl (4-(4-Methoxyindolin-1-yl)-4-oxobutyl)carbamate (5)
Synthesis of 4 has been previously reported.1 4-(tert-Butoxycarbonylamino)butyric acid, N-Boc-GABA (12.46 g, 61.3 mmol, 1.0 equiv), was dissolved in EtOAc (480 mL). N,N-Diisopropylethylamine (11 mL, 63.1 mmol, 1.2 equiv) was added. The mixture was cooled to 0 °C; 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 10.1 mL, 57.1 mmol, 0.95 equiv) was added, and the solution was stirred at room temperature for 10 min. 4-Methoxyindoline (4, 13.7 g, 92.0 mmol, 1.5 equiv) was added, and the mixture was stirred for 60 min. The mixture was cooled to 0 °C; 1 M HCl was added (pH = 3); the phases were separated; and the organic phase was washed carefully with saturated aq NaHCO3 solution (200 mL), distilled water (2 × 200 mL), and brine (200 mL). The organic phase was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. This residue (12.10 g) was purified by flash chromatography (eluent: DCM and ethanol from 0 to 20% using gradient elution) to give 11.75 g (57%) of 6 as a colorless oil.
1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.70 (1H, d, J = 8.0 Hz, C[7]–H), 7.12 (1H, t, J = 8.1 Hz, C[6]–H), 6.83 (1H, br s, NH), 6.65 (1H, d, J = 8.2 Hz, C[5]–H), 4.05 (2H, t, J = 8.4 Hz, 2 × C[1]–H), 3.78 (3H, s, 3 × C[9]–H), 3.02–2.96 (4H, m, 2 × C[2]–H, 2 × C[11]–H), 2.40 (2H, t, J = 7.0 Hz, 2 × C[13]–H), 1.68 (2H, m, 2 × C[12]–H), 1.37 (9H, s, 9 × C[16]–H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) 170.5 (C[10]), 155.6 (C[14]), 155.5 (C[4]), 144.3 (C[8]), 128.5 (C[6]), 118.0 (C[3]), 109.0 (C[7]), 105.9 (C[5]), 77.4 (C[15]), 55.1 (C[9]), 47.7 (C[1]), 39.9 (C[11]), 32.2 (C[2]), 28.2 (C[16]), 25.0 (C[12]), 24.6 (C[13]). HRMS (ES+): found 335.19627, C18H27O4N2 ([M + H]+) requires 335.19653.
Synthesis of 4-Amino-1-(4-methoxyindolin-1-yl)butan-1-one (6)
tert-Butyl (4-(4-methoxyindolin-1-yl)-4-oxobutyl)carbamate (5, 10.1 g, 30.0 mmol) was dissolved in TFA (30 mL), and the solution was stirred at room temperature for 60 min. Methanol (100 mL) was added, and the solvent was evaporated under reduced pressure. Methanol (100 mL) was added again, and the solvent was evaporated. Diethyl ether (50 mL) was added, and the solid precipitate was filtered, washed with diethyl ether, and dried. This residue (TFA salt of 7, a colorless solid, 9.67 g, 92%) was used without further purification.
1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.87 (3H, br s, NH3), 7.70 (1H, d, J = 8.2 Hz, C[7]–H), 7.13 (1H, t, J = 8.2 Hz, C[6]–H), 6.67 (1H, d, J = 8.2 Hz, C[5]–H), 4.06 (2H, t, J = 8.6 Hz, 2 × C[1]–H), 3.78 (3H, s, 3 × C[9]–H), 3.01 (2H, t, J = 8.6 Hz, 2 × C[2]–H), 2.88 (2H, m, 2 × C[11]–H), 2.55 (2H, t, J = 7.0 Hz, 2 × C[13]–H), 1.85 (2H, m, 2 × C[12]–H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) 169.9 (C[10]), 155.5 (C[4]), 144.1 (C[8]), 128.6 (C[6]), 118.1 (C[3]), 109.0 (C[7]), 106.1 (C[5]), 55.2 (C[9]), 47.6 (C[1]), 38.5 (C[11]), 31.6 (C[2]), 24.6 (C[13]), 22.0 (C[12]). HRMS (ES+): found 235.14392, C13H19O2N2 ([M + H]+) requires 235.14410.
Synthesis of 4-Amino-1-(4-methoxy-5,7-dinitroindolin-1-yl)butan-1-one, DNI-GABA (1)
The solution of compound 6 (0.5 g; 2.1 mmol) in acetonitrile (6 mL) was cooled to 10 °C, and nitronium tetra-fluoroborate (1.11 g; 8.4 mmol, 4 equiv) was added portionwise. The reaction mixture was stirred at room temperature for 60 min. After the reaction was completed, NaHCO3 (1.50 g) was added and stirred for 30 min. The mixture was filtered and evaporated. After evaporation, the residue was purified by preparative HPLC (water–acetonitrile–0.1% TFA, using the gradient method). After purification, the fractions were lyophilized. DNI-GABA·TFA 1 was isolated as a yellow powder (0.14 g, yield 20%).
1H NMR (500 MHz, DMSO-d6): δ (ppm) 8.35 (1H, s, C[6]–H), 7.78 (3H, br s, 3 × NH), 4.31 (2H, t, J = 8.2 Hz, 2 × C[1]–H), 4.02 (3H, s, 3 × C[9]–H), 3.40 (2H, t, J = 8.2 Hz, 2 × C[1]–H), 2.83 (2H, m, 2 × C[11]–H), 2.69 (2H, t, J = 7.0 Hz, 2 × C[13]–H), 1.83 (2H, p, J = 7.5 Hz, 2 × C[12]–H). 13C NMR (126 MHz, DMSO-d6): δ (ppm) 170.9 (C[10]), 151.9 (C[4]), 139.7 (C[7]), 137.5 (C5), 133.8 (C[8]), 129.4 (C[6]), 121.5 (C[3]), 61.0 (C[9]), 49.8 (C[1]), 38.2 (C[11]), 31.5 (C[12]), 26.8 (C[13]), 22.2 (C[2]). HRMS (ES+): found 325.11357, C13H17O6N4 ([M + H]+) requires 325.11426.
Synthesis of iDMPO-DNI-GABA (2a)
Synthesis of 1-((1H-Indol-4-yl)oxy)-N,N-dimethylpropan-2-amine (8a)
NaOMe (2.0 g, 41.25 mmol, 1.1 equiv) was dissolved in anhydrous MeOH (15 mL) and toluene (50 mL), and 4-hydroxyindole (7; 5.0 g, 3.75 mol, 1.0 equiv) in toluene (50 mL) was added. The mixture was stirred at reflux temperature for 60 min. 1-Chloro-N,N-dimethylpropan-2-amine (4.56 g, 3.75 mmol, 1.0 equiv) in toluene (3 mL) was added. The reaction mixture was stirred at reflux temperature for 2.5 h and cooled to room temperature. 100 mL of ethyl acetate was added, and the mixture was filtered through a perlite layer. The organic solution was washed with saturated aqueous sodium carbonate solution (50 mL) and water (3 × 50 mL) and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. This residue (3.0 g, 37%) was used without further purification. Pure 8a was isolated by preparative HPLC (eluent water–acetonitrile–0.1% TFA, gradient method).
1H NMR (500 MHz, DMSO-d6): δ (ppm) 11.18 (1H, br s, indole NH), 10.06 (1H, br s, aliphatic NH), 7.26–7.24 (1H, m, C[6]–H), 7.07 (1H, d, J = 8.2 Hz, C[7]–H), 7.03–7.00 (1H, m, C[1]–H), 6.56–6.55 (2H, m, C[2]–H, C[5]–H), 4.37 (1H, dd, J = 11.2, 3.9 Hz, C[9]–H), 4.29 (1H, dd, J = 11.2, 6.5 Hz, C[9]–H), 2.87 (6H, m, 6 × C[12]–H), 2.84–2.81 (1H, m, C[10]–H), 1.40 (3H, d, J = 6.9 Hz, 3 × C[11]–H). 13C NMR (126 MHz, DMSO-d6): δ (ppm) 150.9, 137.4, 123.8, 121.6, 118.3, 105.7, 100.3, 98.4, 66.3, 59.7, 40.1, 10.9. HRMS (ES+): found 219.14882, C13H19ON2 ([M + H]+) requires 219.14919.
Synthesis of 1-(Indolin-4-yloxy)-N,N-dimethylpropan-2-amine (9a)
1-((1H-Indol-4-yl)oxy)-N,N-dimethylpropan-2-amine (8a, 3.00 g, 13.7 mmol, 1 equiv) was dissolved in acetic acid (15 mL), and the mixture was cooled down to 0 °C. Sodium cyanoborohydride (0.86 g, 13.7 mmol, 1 equiv) was added, and the mixture was stirred at room temperature for 60 min. The mixture was cooled to 0 °C, and aqueous sodium hydroxide (20 m/m %, ca. 35 mL) was added (pH = 9). The organic compounds were extracted by DCM (4 × 40 mL), the combined organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. This residue (9a, 3.0 g, 99%) was used without further purification. HRMS (ES+): found 221.16445, C13H21ON2 ([M + H]+) requires 221.16484.
Synthesis of tert-Butyl (4-(4-(2-(Dimethylamino)propoxy)indolin-1-yl)-4-oxobutyl)carbamate (10a)
4-(tert-Butoxycarbonylamino)butyric acid, N-Boc-GABA (3.99 g, 19.6 mmol, 1.4 equiv), was dissolved in EtOAc (60 mL). N,N-Diisopropylethylamine (5.7 mL, 32.7 mmol, 2.35 equiv) was added. The mixture was cooled to 0 °C; EDC (5.7 mL, 32.4 mmol, 2.35 equiv) was added, and the solution was stirred at room temperature for 10 min. 9a indoline (3.0 g, 13.6 mmol, 1.0 equiv) in EtOAc (80 mL) was added dropwise, and the mixture was stirred for 60 min. The mixture was cooled to 0 °C; 1 M HCl was added (pH = 3); the phases were separated; and the organic phase was washed carefully with saturated aq NaHCO3 solution (40 mL), distilled water (3 × 40 mL), and brine (50 mL). The organic phase was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. This residue (4.87 g) was used without further purification. Pure 10a was isolated by preparative HPLC (eluent water–4 g NH4HCO3/10 L water–acetonitrile, gradient method).
1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.69 (1H, d, J = 7.8 Hz, Ar-H), 7.10 (1H, t, J = 8.1 Hz, Ar-H), 6.84 (1H, br s, NH), 6.67 (1H, d, J = 8.2 Hz, Ar-H), 4.12–4.00 (3H, m, C[1]–H, 2 × C[9]–H), 3.86 (1H, dd, J = 9.7, 6.3 Hz, C[1]–H), 3.07–2.93 (4H, m, 2 × C[14]–H, 2 × C[16]–H), 2.90 (1H, q, J = 6.2 Hz, C[10]–H), 2.41 (2H, t, J = 6.9 Hz, 2 × C[2]–H), 2.25 (6H, s, 6 × C[12]–H), 1.68 (2H, q, J = 6.9 Hz, 2 × C[15]–H), 1.38 (9H, s, 9 × C[19]–H), 1.05 (3H, d, J = 6.8 Hz, 3 × C[11]–H). 13C NMR (75 MHz, DMSO-d6): δ (ppm) 170.4, 155.6, 154.8, 144.3, 128.5, 118.4, 109.0, 106.9, 77.4, 69.7, 57.5, 47.6, 41.1, 32.2, 28.2, 24.7, 24.4, 12.3. HRMS (ES+): found 406.26911, C22H36O4N3 ([M + H]+) requires 406.27003.
Synthesis of 4-Amino-1-(4-(2-(dimethylamino)propoxy)indolin-1-yl)butan-1-one (11a)
10a (4.87 g, 12.0 mmol) was dissolved carefully in TFA (15 mL) at 0 °C, and the solution was stirred at room temperature for 60 min. Methanol (20 mL) was added, and the solvent was evaporated under reduced pressure. Methanol (20 mL) was added again, and the solvent was evaporated. The TFA salt of 11a, a brownish oil (3.52 g, 55%), was used without further purification. Pure 11a was isolated by preparative HPLC (water–4 g NH4HCO3/10 L water–acetonitrile, gradient method).
1H NMR (300 MHz, DMSO-d6): δ (ppm) 10.26 (1H, br s, NH), 7.93 (2N, br s, NH2), 7.74 (1H, d, J = 7.8 Hz, Ar-H), 7.15 (1H, t, J = 8.1 Hz, Ar-H), 6.72 (1H, d, J = 8.2 Hz, Ar-H), 4.41–4.17 (10H, m, 2 × C[9]–H), 4.08 (2H, t, J = 8.3 Hz, 2 × C[1]–H), 3.80 (1H, q, J = 4.7 Hz, C[10]–H), 3.09 (2H, t, J = 8.3 Hz, 2 × C[16]–H), 2.96–2.79 (8H, m, 2 × C[14]–H, 6 × C[12]–H), 2.56 (2H, t, J = 6.8 Hz, 2 × C[2]–H), 1.86 (2H, q, J = 6.9 Hz, 2 × C[15]–H), 1.36 (3H, d, J = 6.9 Hz, 3 × C[11]–H). 13C NMR (75 MHz, DMSO-d6): δ (ppm) 170.06, 153.87, 144.36, 128.67, 118.88, 109.80, 107.25, 66.35, 59.61, 47.64, 38.50, 31.75, 24.59, 22.04, 10.91. HRMS (ES+): found 306.21753, C17H28O2N3 ([M + H]+) requires 306.21760.
Synthesis of 4-Amino-1-(4-(2-(dimethylamino)propoxy)-5,7-dinitroindolin-1-yl)butan-1-one (2a)
The solution of compound 11a (8.0 g; 26.1 mmol) in acetonitrile (80 mL) was cooled to 10 °C, and nitronium tetra-fluoroborate (6.90 g; 52.2 mmol, 2 equiv) was added portionwise. The reaction mixture was stirred at room temperature for 4 h. After the reaction was completed, THF (70 mL) and NaHCO3 (17 g) were added and stirred for 30 min. The mixture was filtered, dried over MgSO4, and evaporated. The isomer mixture of 2a and 2a_2 (3.50 g, 34%) was obtained as a brownish oil. Pure 2a and 2a_2 were separated by preparative HPLC (eluent water–4 g NH4HCO3/10 L water–acetonitrile, gradient method).
For 2a: 1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.95 (1H, br s, NH), 8.41 (1H, s, C[6]–H), 7.81 (3H, br s, 3 × NH3), 5.04–5.00 (1H, m, C[10]–H), 4.39–4.30 (2H, m, 2 × C[9]–H), 3.45–3.39 (2H, m, 2 × C[1]–H), 3.38–3.34 (2H, m, 2 × C[16]–H), 2.90 (6H, s, 6 × C[12]–H), 2.86 (2H, br s, 2 × C[15]–H), 2.70 (2H, t, J = 7.0 Hz, 2 × C[2]–H), 1.89–1.77 (2H, m, 2 × C[14]–H), 1.19 (3H, d, J = 6.4 Hz, 3 × C[11]–H). 13C NMR (101 MHz, DMSO-d6): δ (ppm) 171.07, 147.70, 139.67, 138.36, 134.32, 130.82, 121.97, 73.64, 61.05, 49.80, 39.4, 38.23, 31.64, 27.15, 22.18, 17.09. HRMS (ES+): found 396.18743, C17H26N5O4+ ([M + H]+) requires 396.18776.
For 2a_2: 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.01 (1H, br s, NH), 8.42 (1H, s, C[6]–H), 7.83 (3H, br s, 3 × NH3), 4.50–4.42 (1H, m, C[9]–H), 4.34 (2H, t, J = 8.0 Hz, 2 × C[10]–H), 3.45–3.39 (2H, m, 2 × C[1]–H), 3.43 (2H, t, J = 8.0 Hz, 2 × C[16]–H), 2.90–2.80 (8H, m, 6 × C[12]–H, 2 × C[15]–H), 2.71 (2H, t, J = 7.8 Hz, 2 × C[2]–H), 1.89–1.77 (2H, m, 2 × C[14]–H), 1.35 (3H, d, J = 6.8 Hz, 3 × C[11]–H). 13C NMR (101 MHz, DMSO-d6): δ (ppm) 171.07, 150.00, 140.95, 137.23, 134.26, 130.44, 122.13, 71.31, 60.01, 49.83, 39.40, 38.18, 31.58, 26.76, 22.13, 10.30. HRMS (ES+): found 396.18830, C17H26N5O4+ ([M + H]+) requires 396.18776.
Synthesis of iDMBO-DNI-GABA (2b)
Synthesis of 3-((1H-Indol-4-yl)oxy)-N,N,2-trimethylpropan-1-amine (8b)
NaOMe (8.0 g, 165 mmol, 1.1 equiv) was dissolved in anhydrous MeOH (80 mL) and toluene (80 mL), and 4-hydroxyindole (20.0 g, 150 mol, 1.0 equiv) in toluene (100 mL) and anhydrous MeOH (80 mL) were added. The mixture was stirred at reflux temperature for 60 min. 3-Chloro-N,N,2-trimethylpropan-1-amine (57.3 g, 37.5 mmol, 2.5 equiv) in xylene (50 mL) was added. The reaction mixture was stirred at reflux temperature for 6 h and cooled to room temperature. 100 mL of ethyl acetate was added, and the mixture was filtered through a perlite layer. The organic solution was washed with saturated aqueous sodium carbonate solution (200 mL) and water (3 × 100 mL) and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. Hexane (70 mL) was added, and the solid precipitate was filtrated and dried. This residue (8b, 20.0 g, 57%) was used without further purification. Pure S5 was isolated by preparative HPLC (eluent water–acetonitrile–0.1% TFA, using the gradient method).
1H NMR (600 MHz, DMSO-d6): δ (ppm) 11.15 (1H, br s, NH), 9.73 (1H, br s, NH), 7.23 (1H, t, J = 2.6 Hz, Ar-H), 7.03 (1H, d, J = 8.1 Hz, Ar-H), 6.99 (1H, t, J = 7.8 Hz, Ar-H), 6.50–6.47 (2H, m, 2 × Ar-H), 4.03–3.98 (2H, m, 2 × C[9]–H), 3.31–3.26 (1H, m, C[12]–H), 3.16–3.10 (1H, m, C[12]–H), 2.90 (3H, d, J = 4.2 Hz, 3 × C[13]–H), 2.84 (3H, d, J = 4.2 Hz, 3 × C[13]–H), 2.48 (1H, m, C[10]–H), 1.13 (3H, d, J = 6.8 Hz, 3 × C[11]–H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) 158.6 (q, J = 34.3 Hz), 151.7, 137.4, 123.6, 121.7, 118.4 (q, J = 294 Hz), 116.3, 105.2, 100.0, 98.4, 69.9, 60.5, 43.5, 42.7, 29.3, 15.0. HRMS (ES+): found 233.16449, C14H21ON2+ ([M + H]+) requires 233.16484.
Synthesis of 3-(Indolin-4-yloxy)-N,N,2-trimethylpropan-1-amine (9b)
3-((1H-Indol-4-yl)oxy)-N,N,2-trimethylpropan-1-amine (8b, 19.61 g, 89 mmol, 1 equiv) was dissolved in acetic acid (62 mL), and the mixture was cooled down to 0 °C. Sodium cyanoborohydride (5.58 g, 89 mmol, 1 equiv) was added, and the mixture was stirred at room temperature for 60 min. The mixture was cooled to 0 °C, and aqueous sodium hydroxide (20 m/m %) was added (pH = 10). The organic compounds were extracted by ethyl acetate (3 × 100 mL); the combined organic layers were dried over anhydrous magnesium sulfate and filtered, and the solvent was evaporated under reduced pressure. The crude product was recrystallized from ethyl acetate and yielded 9b (19.5 g, 99%) as a yellowish oil.
1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.65 (1H, br s, NH), 7.20 (1H, t, J = 8.0 Hz, Ar-H), 6.78 (1H, d, J = 7.8 Hz, Ar-H), 6.75 (1H, d, J = 8.2 Hz, Ar-H), 3.96 (2H, d, J = 5.7 Hz, 2 × C[9]–H), 3.66 (2H, t, J = 8.0 Hz, 2 × C[1]–H), 3.21 (1H, dd, J = 12.8, 6.6 Hz, C[12]–H), 3.11–3.01 (3H, m, 2 × C[2]–H, C[12]–H), 2.87 (3H, s, 3 × C[13]–H, NMe), 2.81 (3H, s, 3 × C[13]–H, NMe), 2.43 (1H, dq, J = 12.7, 6.4 Hz, C[10]–H), 1.13 (3H, d, J = 6.8 Hz, 3 × C[11]–H, C–Me). 13C NMR (151 MHz, DMSO-d6): δ (ppm) 158.5 (q, J = 33.7 Hz), 155.15, 143.32, 129.24, 120.63, 116.6 (q, J = 294 Hz), 108.61, 108.49, 70.28, 60.25, 45.50, 43.07, 42.95, 29.07, 26.33, 14.86. HRMS (ES+): found 235.18014, C14H23ON2+ ([M + H]+) requires 235.18049.
Synthesis of tert-Butyl (4-(4-(3-(Dimethylamino)-2-methylpropoxy)indolin-1-yl)-4-oxobutyl)carbamate (10b)
4-(tert-Butoxycarbonylamino)butyric acid, N-Boc-GABA (10.31 g, 34 mmol, 1.13 equiv), was dissolved in EtOAc (80 mL). N,N-Diisopropylethylamine (6.6 mL, 37.9 mmol, 1.25 equiv) was added. The mixture was cooled to 0 °C; EDC (5.62, 5.7 mL, 32 mmol, 1.06 equiv) was added, and the solution was stirred at room temperature for 10 min. 9b indoline (7.0 g, 30.2 mmol, 1.0 equiv) in EtOAc (80 mL) was added dropwise, and the mixture was stirred for 60 min. The mixture was cooled to 0 °C; 1 M HCl was added (pH = 3); the phases were separated; and the organic phase was washed carefully with saturated aq Na2CO3 solution (100 mL), distilled water (3 × 50 mL), and brine (50 mL). The organic phase was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The 10b product (5.31 g, 41%) was used without further purification. Pure 10b was isolated by preparative HPLC (eluent water–acetonitrile–0.1% TFA, using the gradient method).
1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.69 (1H, br s, NH), 7.71 (1H, d, J = 8.1 Hz, Ar-H), 7.12 (1H, t, J = 8.1 Hz, Ar-H), 6.84 (1H, br s, NH), 6.64 (1H, d, J = 8.2 Hz, Ar-H), 4.08 (2H, t, J = 8.5 Hz, 2 × C[17]–H), 3.93 (2H, d, J = 5.7 Hz, 2 × C[9]–H), 3.24–3.18 (1H, m, C[1]–H), 3.11–3.02 (3H, m, C[1]–H, 2 × C[12]–H), 2.98 (2H, dd, J = 6.8, 6.0 Hz, 2 × C[15]–H), 2.87 (3H, d, J = 4.0 Hz, 3 × C[13]–H), 2.81 (3H, d, J = 4.0 Hz, 3 × C[13]–H), 2.48–2.34 (3H, m, 2 × C[2]–H, C[10]–H), 1.68 (2H, q, J = 7.1 Hz, 2 × C[16]–H), 1.37 (9H, s, 9 × C[20]–H), 1.08 (3H, d, J = 6.8 Hz, 3 × C[11]–H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) 170.53, 158.3 (q, J = 33.7 Hz), 155.68, 154.49, 144.35, 128.50, 118.54, 116.6 (q, J = 294 Hz), 109.33, 106.87, 77.41, 70.08, 60.30, 47.67, 43.19, 42.87, 39.41, 32.24, 29.13, 28.25, 24.51, 24.47, 14.88. HRMS (ES+): found 420.28489, C23H38O4N3+ ([M + H]+) requires 420.28568.
Synthesis of 4-Amino-1-(4-(3-(dimethylamino)-2-methylpropoxy)indolin-1-yl)butan-1-one (11b)
10b (5.1 g, 12.2 mmol, 1 equiv) was dissolved in TFA (10 mL) at −10 °C and then stirred at room temperature for 2 h. After completion (followed by HPLC), 15 mL of MeOH was added, the mixture was concentrated under reduced pressure, 15 mL of MeOH was added again, and the mixture was concentrated under reduced pressure to give the crude product (4.6 g), which was used without further purification. The pure product was isolated by preparative HPLC (0.1% TFA in water–acetonitrile, using the gradient method), and then, the collected fractions were lyophilized. 11b was obtained as a yellowish solid.
1H NMR (500 MHz, DMSO-d6): δ (ppm) 9.66 (1H, br s, NH), 7.88 (3H, br s, 3 × NH), 7.71 (1H, d, J = 8.1 Hz, Ar-H), 7.13 (1H, t, J = 8.1 Hz, Ar-H), 6.66 (1H, d, J = 8.2 Hz, Ar-H), 4.08 (2H, t, J = 8.5 Hz, 2 × C[17]–H), 3.94 (2H, d, J = 5.7 Hz, 2 × C[9]–H), 3.25–3.18 (1H, m, C[1]–H), 3.11–3.02 (3H, m, C[1]–H, 2 × C[12]–H), 2.92–2.84 (5H, m, 3 × C[13]–H, 2 × C[15]–H), 2.81 (3H, d, J = 4.0 Hz, 3 × C[13]–H), 2.56 (2H, t, J = 6.9 Hz, 2 × C[2]–H), 2.42 (1H, td, J = 7.1, 6.5, 6.3 Hz, C[10]–H), 1.86 (2H, q, J = 7.1 Hz, 2 × C[16]–H), 1.08 (3H, d, J = 6.8 Hz, 3 × C[11]–H). 13C NMR (126 MHz, DMSO-d6): δ (ppm) 169.95, 154.50, 144.17, 128.51, 118.55, 109.25, 107.03, 70.06, 60.25, 47.56, 43.19, 42.82, 38.45, 31.67, 29.11, 24.47, 21.98, 14.85. HRMS (ES+): found 320.23316, C18H30O2N3+ ([M + H]+) requires 320.23325.
Synthesis of 4-Amino-1-(4-(3-(dimethylamino)-2-methylpropoxy)-5,7-dinitroindolin-1-yl)butan-1-one (2b)
11b (305 mg, 0.96 mmol, 1 equiv) was dissolved in DCM (8 mL) and cooled to −20 °C. Nitronium tetrafluoroborate (381 mg, 2.86 mmol, 3.0 equiv) was added in five portions, and the reaction mixture was stirred at −20 °C, followed by HPLC. After completion (20–30 min), 2 g of solid NaHCO3 was added (pH > 7). The mixture was filtered, dried over MgSO4, filtered, and concentrated under reduced pressure to give the crude product, which was purified by preparative HPLC (0.1% TFA in water–acetonitrile, gradient method), and then, the collected fractions were lyophilized. 2b (98 mg, 25%) was obtained as a yellowish solid.
1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.68 (1H, br s, NH), 8.38 (1H, s, C[6]–H), 7.87 (3H, br s, 3 × NH3), 4.32 (2H, t, J = 8.1 Hz, 2 × C[1]–H), 4.20 (2H, d, J = 5.3 Hz, 2 × C[9]–H), 3.41 (2H, t, J = 8.1 Hz, 2 × C[2]–H), 3.22 (1H, dd, J = 13.0, 6.0 Hz, C[12]–H), 3.08 (1H, dd, J = 13.0, 7.4 Hz, C[12]–H), 2.89–2.80 (8H, m, 6 × C[13]–H, 2 × C[17]–H), 2.42 (1H, td, J = 12.5, 6.8 Hz, C[10]–H), 2.70 (2H, t, J = 7.2 Hz, 2 × C[16]–H), 1.84 (2H, q, J = 7.4 Hz, 2 × C[15]–H), 1.08 (3H, d, J = 6.8 Hz, 3 × C[11]–H). 13C NMR (101 MHz, DMSO-d6): δ (ppm) 170.9, 150.8, 139.8, 137.1, 133.8, 129.4, 121.8, 75.3, 59.6, 49.8, 39.4, 38.2, 31.6, 29.7, 26.9, 22.1, 14.5. HRMS (ES+): found 410.20303, C18H28O6N5+ ([M + H]+) requires 410.20341.
Synthesis of DNI-CO-GABA (3)
Preparation of (1H-Imidazol-1-yl)(4-methoxyindolin-1-yl)methanone (12)
4-Methoxyindoline (5, 4.20 g, 28.15 mmol) was dissolved in DCM (200 mL). Triethylamine (5.89 mL, 42.23 mmol, 1.5 equiv) was added. CDI (6.85 g, 42.23 mmol, 1.5 equiv) was added in six portions, and the reaction mixture was stirred at room temperature for 30 min. The organic mixture was concentrated under reduced pressure to give the crude product. Diethyl ether (15 mL) was added, and white crystals were formed which were filtered and died in a desiccator. 12 (6.84 g, 100%) was used without further purification.
1H NMR (500 MHz, DMSO-d6): δ (ppm) 8.22 (1H, s, C[11]–H), 7.66–7.64 (1H, m, C[12]–H), 7.30–7.15(2H, m, C[6]–H, C[13]–H), 7.10–7.06 (1H, m, C[7]–H), 6.77 (1H, m, C[5]–H), 3.85 (2H, t, J = 8.2 Hz, 2 × C[1]–H), 3.81 (3H, s, 3 × C[9]–H), 3.01 (2H, t, J = 8.2 Hz, 2 × C[2]–H). 13C NMR (126 MHz, DMSO-d6): δ (ppm) 155.7 (C[4]), 148.1 (C[10]), 142.9 (C[8]), 137.0 (C[11]), 128.9 (C[13]), 128.7 (C[6]), 119.5 (C[12]), 118.2 (C[3]), 109.1 (C[7]), 107.2 (C[5]), 55.4 (C[9]), 50.8 (C[1]), 25.1 (C[2]). HRMS (ES+): found 244.10760, C13H14O2N3+ ([M + H]+) requires 244.10805.
Preparation of tert-Butyl 4-(4-Methoxy-2,3-dihydro-1H-indene-1-carboxamido)butanoate (13)
12 (2.0 g, 8.22 mmol) was suspended in DMSO (10 mL), and triethylamine (2.3 mL, 16.50 mmol, 2.0 equiv) and 1.76 g (9.00 mmol, 1.1 equiv) of tert-butyl 4-aminobutanoate hydrochloride were added. The reaction mixture was stirred for 8 h at 110 °C; then, 200 mL of ethyl acetate was added, and the mixture was washed with distilled water (3 × 80 mL). The organic solution was concentrated under reduced pressure to give the crude product as a brownish oil, which was purified by recrystallization from diethyl ether. 13 (1.58 g, 57%) was obtained as a white solid.
1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.47 (1H, d, J = 8.0 Hz, C[7]–H), 7.04 (1H, t, J = 8.2 Hz, C[6]–H), 6.58 (1H, t, J = 5.4 Hz, NH), 6.50 (1H, d, J = 8.2 Hz, C[5]–H), 3.85 (2H, t, J = 8.8 Hz, 2 × C[1]–H), 3.75 (3H, s, 3 × C[9]–H), 3.11 (2H, q, J = 6.5 Hz, 2 × C[2]–H), 2.97 (2H, t, J = 8.8 Hz, 2 × C[11]–H), 2.22 (2H, t, J = 7.5 Hz, 2 × C[13]–H), 1.68 (2H, t, J = 7.2 Hz, 2 × C[12]–H), 1.40 (9H, s, 9 × C[16]–H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) 172.1 (C[14]), 155.4 (C[4]), 154.7 (C[10]), 145.6 (C[8]), 128.4 (C[6]), 116.6 (C[3]), 107.5 (C[7]), 103.9 (C[5]), 79.4 (C[15]), 55.0 (C[9]), 47.1 (C[1]), 39.0 (C[2]), 32.3 (C[12]), 27.8 (C[16]), 25.3 (C[13]), 24.4 (C[11]). HRMS (ES+): found 335.19603, C18H27O4N2+ ([M + H]+) requires 335.19653.
Synthesis of 4-(4-Methoxy-2,3-dihydro-1H-indene-1-carboxamido)butanoic Acid (3)
tert-Butyl 4-(4-methoxy-2,3-dihydro-1H-indene-1-carboxamido)butanoate (13, 800 mg, 2.39 mmol) was dissolved in acetonitrile (15 mL) and cooled to −20 °C. Nitronium tetrafluoroborate (953 mg, 7.17 mmol, 3.0 equiv) was added in five portions, and the reaction mixture was stirred at −20 °C, followed by HPLC. After completion (20–30 min), 4 g of solid NaHCO3 was added (pH > 7). The mixture was filtered, dried over MgSO4, filtered, and concentrated under reduced pressure to give the crude product, which was purified by preparative HPLC (0.1% TFA in water–acetonitrile, gradient method), and then, the collected fractions were lyophilized. 3 (334 mg, 38%) was obtained as a yellow solid.
1H NMR (500 MHz, DMSO-d6): δ (ppm) 8.30 (1H, br s, NH), 7.63 (1H, t, J = 5.6 Hz, C[6]–H), 4.17 (2H, t, J = 8.7 Hz, C[1]–H), 3.97 (3H, s, 2 × C[9]–H), 3.38 (2H, t, J = 8.6 Hz, 2 × C[1]–H), 3.11 (2H, dd, J = 12.7, 6.7 Hz, 2 × C[11]–H), 2.26 (2H, t, J = 7.4 Hz, 2 × C[13]–H), 1.68 (2H, p, J = 7.2 Hz, 2 × C[12]–H). 13C NMR (126 MHz, DMSO-d6): δ (ppm) 174.2, 154.6, 152.1, 143.3, 135.4, 131.8, 128.1, 122.2, 60.8, 51.4, 30.9, 26.2, 24.7. HRMS (ES+): found 335.19603, C18H27O4N2+ ([M + H]+) requires 335.19653.
Transgenic Mice
The Thy1 promoter construct containing the 6.5 kb Thy1.2 genomic insert with most of the coding region deleted was a gift from Joshua Sanes (Addgene plasmid # 20736). A unique XhoI cloning site was used to insert the GCamp6f complementary DNA (cDNA) from the pGP-CMV-GCaMP6f plasmid (a gift from Douglas Kim & GENIE Project, Addgene plasmid # 40755). The GCaMP6f cDNA was cut by BglII and NotI, and the fragment ends were blunted by Klenow fill-in before ligation. For pronuclear microinjection, an 8.2 kb long PvuI fragment was isolated from the construct. Genotyping was carried out by an RQ-PCR TaqMan assay designed on the eGFP (Mr00660654_cn, Thermo Fisher). Several founders were analyzed, and the 82nd line was selected for the biological experiments by the comparable expression and activity level in both the cortex and hippocampus.64,65
According to the literature, the Thy1 promoter predominantly represents a pyramidal cell type.63 With this technology, we can monitor cell activities at the cellular and subcellular levels, and we can also study the modulation of the cells during photostimulation. In the first step, we characterized the expression of the GCaMP6f sensor (Figure S7A–G) in two different brain areas of the measured animals (cortex, n = 20 animals and hippocampus, n = 20 animals). Then, the physiological patterns of the cells with whole cell recording were tested. In accordance with the literature, we described the basic properties of cells measured by electrophysiological experiments and calcium imaging (ramp test, somatic-evoked action potential and action potential sequences, characterization of evoked dendritic calcium signals) (Figure S7A–G). The obtained results prove that the GCaMP6f labeling is in conformity with the literature, that is, it has no negative effect on the physiological values and characters of the cells.
Slice Preparation and Electrophysiology
Experiments were performed in accordance with the Hungarian Act of Animal Care and Experimentation [40/2013 (II.14)]. The Animal Care and Experimentation Committee of the Institute of Experimental Medicine and the Animal Health and Food Control Station approved the experimental design. Acute hippocampal slices were prepared from 15- to 20-day-old mice and mice using isoflurane anesthesia, followed by swift decapitation. Horizontal (300 μm) brain slices were cut with a vibratome and stored at room temperature in the artificial cerebrospinal fluid (ACSF) (in mM: 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose) as previously described.26,27,66 Hippocampal neurons in the CA1 stratum radiatum near the border of the stratum lacunosum-moleculare were visualized using 900 nm infrared oblique illumination. Whole-cell recordings were made at 32 °C (MultiClamp 700B, Digidata 1440; Molecular Devices, Sunnyvale, CA, USA) with glass electrodes (current clamp: 6–9 MΩ; voltage clamp: 3–5 MΩ) filled with (in mM): 125 K-gluconate, 20 KCl, 10 N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid, 10 di-tris-salt phosphocreatine, 0.3 Na-GTP, 4 Mg-ATP, 10 NaCl, 0.1 Fluo-4, 0.1 Alexa 594, and 0.008 biocytin. Cells with a resting membrane potential more negative than −50 mV were accepted. The recorded cells were classified as hippocampal pyramidal cells according to their electrophysiological properties. GABA IPSCs were induced by focal synaptic stimulation in the presence of AP5 (60 μM) and CNQX (10 μM). Glass electrodes (6–9 MΩ) filled with ACSF were placed at a distance of 10–15 μm from the soma (stimulation: 0.1 ms, 10–50 V, a 10 ms pulse interval, 1 stimulus; BioStim, Supertech). All evoked IPSCs were verified for synaptic delay. Data acquisition was performed using either pClamp8 or pClamp10 (Molecular Devices) and manufacturing execution system (MES, Femtonics Ltd.) software.
Measurement of Free GABA Concentration
Spontaneous hydrolysis of DNI-GABA·TFA derivatives (1–3) was analyzed by the direct measurement of GABA concentration by the previously published standard method by the o-phthalaldehyde mercaptopropionic acid derivatization method.60 The cage compound was dissolved in distilled water or in 12 mL of ACSF (a final concentration 2.5 mM). The analytical setup consisted of a Waters 996 PDA detector and a Waters 474 detector, a Waters 616 controller quaternary pump, and a Waters 717 autosampler, operating with the Millennium Software. The analytical column was a Thermo Hypersil GOLD 20 cm × 4.6 mm, 5 μm with a guard column. Detections were performed simultaneously on PDA (190 and 400 nm) and Fl detectors (λEx/λEm = 337/454 nm). Eluent system: A: 0.05 M sodium acetate of pH 7.20 ± 0.05; B: 0.1 M sodium acetate–acetonitrile–methanol (46/44/10) at pH 7.20 ± 0.05. Elutions were performed in the gradient mode (at 40 °C): starting with 1% B for 1 min with a 1.3 mL/min flow rate, reaching 100% B, and a 2.0 mL/min flow rate within 7 min; afterward, 3 min isocratic elution with 100% B, and finally returning to the initial concentration (1% B) in 1 min and equilibrating for 4 min with this B content (Figure S9).
2P Imaging
2P imaging started 15–20 min after attaining the whole-cell configuration on a 2P laser-scanning system (Femto2D, Femtonics Ltd.) using femtosecond lasers (830–850 nm) (Mai Tai HP, Spectra-Physics). The multiple line scanning method26 was used to image long dendritic segments. At the end of each experiment, a series of images were taken across the depth of the volume encompassing the imaged neuron. Measurement control, real-time data acquisition, and analysis were performed with a MATLAB-based program (MES, Femtonics Ltd.) and using custom-written software.
2P Uncaging
After achieving the whole-cell mode and filling pyramidal cells or interneurons with 100 μM Fluo-4, the bath solution was changed to ACSF containing 2.5 mM DNI-GABA·TFA (1), iDMPO-DNI-GABA·TFA (2a), iDMBO-DNI-GABA·TFA (2b), or DNI-CO-GABA (3). Photolysis of caged GABA was performed with 690–830 nm ultrafast, pulsed laser light (Mai Tai HP Deep See, Spectra-Physics or Chameleon Ultra II, Coherent). The intensity of the uncaging laser beam was controlled with an electro-optical modulator (model 350-80 LA, Conoptics). Dispersion compensation was set to have maximal response at the depth of uncaging (50–80 μm from the surface). The uncaging laser beam was coupled to the imaging optical pathway with a dichroic mirror (custom laser combiner, z750bcm; Chroma Technology Corp.). Chromatic aberration was compensated for at the focal plane. Radial and axial alignment errors between the imaging and uncaging point spread functions were held with two motorized mirrors below 100 and 300 nm, respectively. Imaging was interleaved with 2P GABA uncaging periods when galvanometers jumped to the selected uncaging locations (within <60 μs jump time) and returned to the imaging trajectory thereafter. Positions of uncaging sites were finely adjusted according to background images taken. Line scan data were also used to avoid overlapping between uncaging locations and the dendrite. Photolysis of caged GABA was performed in a single or clustered pattern (a 0.8 ± 0.1 μm distance between inputs) along the dendrite or around the soma. Small drifts of the sample (approximately 0.1–0.2 μm/min) were compensated manually according to regularly taken background images and fluorescence pixel intensities in uncaging locations during photostimulation. The same uncaging pattern in the same dendritic location was used during comparison of different uncaging materials. The perfusion rate was set to 6 mL/min to increase the exchange rate of ACSF containing different uncaging materials. Unless otherwise indicated, data are presented as the mean ± SEM. Statistical comparisons were performed using Student’s paired t-test.
Statistics
We measured the overall photochemical yields of IDMPO-DNI-GABA (2a), IDMBO-DNI-GABA (2b), and DNI-CO-GABA (3) relative to the responses of DNI-GABA (1) by calculating the relative change in the second order of laser intensity (2P irradiation), which is required to generate overlap in the responses. We used unconstrained non-linear optimization to minimize the distance between the two-point sets, where the changed variable was the scale factor applied to the 2P irradiation bringing the two-point sets into overlap. The distance between the point sets was defined by the sum
where xi and xj are the laser intensity data corresponding to the two data sets, while yi and yj are the corresponding EPSP or Ca2+ transient amplitudes. S was set to 2 mV for EPSP and 2% for Ca2+ transient measurements (Figure 3D).
Computational Methods
All computations were carried out with the Gaussian 16 program package (G16)55 using convergence criteria of 3.0 × 10–4, 4.5 × 10–4, 1.2 × 10–3, and 1.8 × 10–3 for the gradients of the root-mean-square (rms) force, maximum force, rms displacement, and maximum displacement vectors, respectively. Computations were carried out at the B3LYP/6-31G(d,p) level of theory67 using the IEFPCM method with the parameters of water.68−70 The method and basis sets were chosen for their reliability in agreement with the studies established earlier.10,71 The vibrational frequencies were computed at the same levels of theory, as used for geometry optimization, in order to properly confirm that all structures reside at minima on their potential energy hypersurfaces. Thermodynamic functions U, H, G, and S were computed at 298.15 K using the quantum chemical, rather than the conventional, thermodynamic reference state.
Acknowledgments
The authors are deeply grateful to János Szabadics for his helpful discussions. The authors also thank Joshua Sanes for providing Addgene plasmid # 20736.
Glossary
Abbreviations
- DNI
5,7-dinitroindolinyl
- DPNI
1,3-bis(dihydroxyphosphoryloxy)propan-2-yloxy
- CDNI
4-carboxymethoxy-5,7-dinitroindolinyl
- ISC
intersystem crossing
- iDMBO
3-(dimethylamino)-2-methylpropyloxy
- iDMPO
2-dimethylaminopropyloxy
- IPSC
inhibitory postsynaptic current
- MNI
7-(mono)nitroindolyl
- NI
nitroindolyl
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01164.
Spectral data, NMR spectra, computed coordinates, and neurobiological investigations (PDF)
Author Contributions
B.C. and D.P. contributed equally to this work. Caged compounds were synthesized by C.L.H., E.K., and O.F.; compound characterization was made by E.K.; HRMS measurements were carried out by M.D.; in vitro measurements were performed by B.C., M. Majoros, G. Szalay, D.P., and Z.S.; transgenic mice were prepared by G. Szabó., F.E., and Z. Máté; analysis was carried out by B.C., Z. Mucsi, E.K., and D.P.; quantum chemical computation was performed by Z. Mucsi, I.G.C., and M.S.; the uncaging microscope was designed and made by B.R., G.K., and G. Szalay; software was written by G.K.; this manuscript was written by B.C., G.J., M. Madarász, E.K., Z. Mucsi, and D.P. with comments from all authors; B.C., E.K., and Z. Mucsi supervised the project.
This work was supported by the National Research, Development, and Innovation Fund of Hungary (KFI-18-2018-00097, VKE-18-2018-00032, OTKA PD 128612), the János Bolyai Research Scholarship of the Hungarian Academy of Sciences, and the European Research Council (ERC682426).
The authors declare no competing financial interest.
Notes
B.R. and G.K. are founders of Femtonics and are members of its scientific advisory board.
Supplementary Material
References
- Yu H.; Li J.; Wu D.; Qiu Z.; Zhang Y. Chemistry and Biological Applications of Photo-Labile Organic Molecules. Chem. Soc. Rev. 2010, 39, 464–473. 10.1039/B901255A. [DOI] [PubMed] [Google Scholar]
- Klán P.; Šolomek T.; Bochet C. G.; Blanc A.; Givens R.; Rubina M.; Popik V.; Kostikov A.; Wirz J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2013, 113, 119–191. 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstain R.; Slanina T.; Kand D.; Klán P. Visible-to-NIR-Light Activated Release: From Small Molecules to Nanomaterials. Chem. Rev. 2020, 120, 13135–13272. 10.1021/acs.chemrev.0c00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T.; Lu S.; Sun J.; Xu Z.; Yang X.; Wang F.; Ma Y.; Shi Y. S.; Chen X. Sanger’s Reagent Sensitized Photocleavage of Amide Bond for Constructing Photocages and Regulation of Biological Functions. J. Am. Chem. Soc. 2020, 142, 3806–3813. 10.1021/jacs.9b11357. [DOI] [PubMed] [Google Scholar]
- Korzycka K. A.; Bennett P. M.; Cueto-Diaz E. J.; Wicks G.; Drobizhev M.; Blanchard-Desce M.; Rebane A.; Anderson H. L. Two-Photon Sensitive Protecting Groups Operating via Intramolecular Electron Transfer: Uncaging of GABA and Tryptophan. Chem. Sci. 2015, 6, 2419–2426. 10.1039/C4SC03775H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi D. D.; Trigo F. F.; Semmelhack M. F.; Wang S. S.-H. Synthesis and Biological Evaluation of Bis-CNB-GABA, a Photoactivatable Neurotransmitter with Low Receptor Interference and Chemical Two-Photon Uncaging Properties. J. Am. Chem. Soc. 2014, 136, 1976–1981. 10.1021/ja411082f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran C.; Gallavardin T.; Petit M.; Slimi R.; Dhimane H.; Blanchard-Desce M.; Acher F. C.; Ogden D.; Dalko P. I. Two-Photon “Caging” Groups: Effect of Position Isomery on the Photorelease Properties of Aminoquinoline-Derived Photolabile Protecting Groups. Org. Lett. 2015, 17, 402–405. 10.1021/ol5035035. [DOI] [PubMed] [Google Scholar]
- Passlick S.; Ellis-Davies G. C. R.. Chromatically Independent, Two-Color Uncaging of Glutamate and GABA with One- or Two-Photon Excitation. In Optochemical Biology; Deiters A., Ed.; Academic Press, 2019; Vol. 624, Chapter 9, pp 167–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M.; Hayama T.; Kasai H.; Ellis-Davies G. C. R. Two-Photon Uncaging of γ-Aminobutyric Acid in Intact Brain Tissue. Nat. Chem. Biol. 2010, 6, 255–257. 10.1038/nchembio.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer G.; Heckel A. Biologically Active Molecules with a “Light Switchs”. Angew. Chem., Int. Ed. 2006, 45, 4900–4921. 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]
- Ellis-Davies G. C. R. Caged Compounds: Photorelease Technology for Control of Cellular Chemistry and Physiology. Nat. Methods 2007, 4, 619–628. 10.1038/nmeth1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal H. K.; Janicek R.; Chi S.-H.; Perry J. W.; Niggli E.; Ellis-Davies G. C. R. Calcium Uncaging with Visible Light. J. Am. Chem. Soc. 2016, 138, 3687–3693. 10.1021/jacs.5b11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembitskaya Y.; Wu Y.-W.; Semyanov A. Tonic GABAA Conductance Favors Spike-Timing-Dependent over Theta-Burst-Induced Long-Term Potentiation in the Hippocampus. J. Neurosci. 2020, 40, 4266–4276. 10.1523/JNEUROSCI.2118-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarski D.; Sugiyama A.; Breton G.; Rakers C.; Ono D.; Schulte A.; Tama F.; Itami K.; Szymanski W.; Hirota T.; Feringa B. L. Controlling the Circadian Clock with High Temporal Resolution through Photodosing. J. Am. Chem. Soc. 2019, 141, 15784–15791. 10.1021/jacs.9b05445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pálfi D.; Chiovini B.; Szalay G.; Kaszás A.; Turi G. F.; Katona G.; Ábrányi-Balogh P.; Szőri M.; Potor A.; Frigyesi O.; Lukácsné Haveland C.; Szadai Z.; Madarász M.; Vasanits-Zsigrai A.; Molnár-Perl I.; Viskolcz B.; Csizmadia I. G.; Mucsi Z.; Rózsa B. High Efficiency Two-Photon Uncaging Coupled by the Correction of Spontaneous Hydrolysis. Org. Biomol. Chem. 2018, 16, 1958–1970. 10.1039/c8ob00025e. [DOI] [PubMed] [Google Scholar]
- Olson J. P.; Kwon H.-B.; Takasaki K. T.; Chiu C. Q.; Higley M. J.; Sabatini B. L.; Ellis-Davies G. C. R. Optically Selective Two-Photon Uncaging of Glutamate at 900 Nm. J. Am. Chem. Soc. 2013, 135, 5954–5957. 10.1021/ja4019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan L.; Hess G. P. Picrotoxin Inhibition Mechanism of a γ-Aminobutyric AcidA Receptor Investigated by a Laser-Pulse Photolysis Technique. Biochemistry 2005, 44, 8523–8532. 10.1021/bi0477283. [DOI] [PubMed] [Google Scholar]
- Grewer C. Investigation of the α1-Glycine Receptor Channel-Opening Kinetics in the Submillisecond Time Domain. Biophys. J. 1999, 77, 727–738. 10.1016/s0006-3495(99)76927-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder M.; Zieglgänsberger W.; Dodt H.-U. Neocortical Long-Term Potentiation and Long-Term Depression: Site of Expression Investigated by Infrared-Guided Laser Stimulation. J. Neurosci. 2002, 22, 7558–7568. 10.1523/JNEUROSCI.22-17-07558.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit D. L.; Augustine G. J. Distribution of Functional Glutamate and GABA Receptors on Hippocampal Pyramidal Cells and Interneurons. J. Neurophysiol. 2000, 84, 28–38. 10.1152/jn.2000.84.1.28. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M.; Ellis-Davies G. C. R.; Nemoto T.; Miyashita Y.; Iino M.; Kasai H. Dendritic Spine Geometry Is Critical for AMPA Receptor Expression in Hippocampal CA1 Pyramidal Neurons. Nat. Neurosci. 2001, 4, 1086–1092. 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J. V.; Zhang F.; Ford P. C. Multi-Photon Excitation in Uncaging the Small Molecule Bioregulator Nitric Oxide. Philos. Trans. R. Soc., A 2013, 371, 20120129. 10.1098/rsta.2012.0129. [DOI] [PubMed] [Google Scholar]
- Ciuciu A. I.; Korzycka K. A.; Lewis W. J. M.; Bennett P. M.; Anderson H. L.; Flamigni L. Model Dyads for 2PA Uncaging of a Protecting Group via Photoinduced Electron Transfer. Phys. Chem. Chem. Phys. 2015, 17, 6554–6564. 10.1039/C4CP05812G. [DOI] [PubMed] [Google Scholar]
- Zhao M.; McGarry L. M.; Ma H.; Harris S.; Berwick J.; Yuste R.; Schwartz T. H. Optical Triggered Seizures Using a Caged 4-Aminopyridine. Front. Neurosci. 2015, 9, 25. 10.3389/fnins.2015.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A.; Makara J. K.; Magee J. C. Compartmentalized Dendritic Plasticity and Input Feature Storage in Neurons. Nature 2008, 452, 436–441. 10.1038/nature06725. [DOI] [PubMed] [Google Scholar]
- Katona G.; Kaszás A.; Turi G. F.; Hájos N.; Tamás G.; Vizi E. S.; Rózsa B. Roller Coaster Scanning Reveals Spontaneous Triggering of Dendritic Spikes in CA1 Interneurons. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 2148–2153. 10.1073/pnas.1009270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiovini B.; Turi G. F.; Katona G.; Kaszás A.; Pálfi D.; Maák P.; Szalay G.; Szabó M. F.; Szabó G.; Szadai Z.; Káli S.; Rózsa B. Dendritic Spikes Induce Ripples in Parvalbumin Interneurons during Hippocampal Sharp Waves. Neuron 2014, 82, 908–924. 10.1016/j.neuron.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Granger A. J.; Nicoll R. A. Expression Mechanisms Underlying Long-Term Potentiation: A Postsynaptic View, 10 Years On. Philos. Trans. R. Soc., B 2014, 369, 20130136. 10.1098/rstb.2013.0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya R. Input Transformation by Dendritic Spines of Pyramidal Neurons. Front. Neuroanat. 2014, 8, 1–18. 10.3389/fnana.2014.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt H.-U.; Schierloh A.; Eder M.; Zieglgänsberger W. Circuitry of Rat Barrel Cortex Investigated by Infrared-Guided Laser Stimulation. Neuroreport 2003, 14, 623–627. 10.1097/00001756-200303240-00020. [DOI] [PubMed] [Google Scholar]
- Brivanlou I. H.; Dantzker J. L. M.; Stevens C. F.; Callaway E. M. Topographic Specificity of Functional Connections from Hippocampal CA3 to CA1. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 2560–2565. 10.1073/pnas.0308577100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A.; Magee J. C. Integrative Properties of Radial Oblique Dendrites in Hippocampal CA1 Pyramidal Neurons. Neuron 2006, 50, 291–307. 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Araya R.; Eisenthal K. B.; Yuste R. Dendritic Spines Linearize the Summation of Excitatory Potentials. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 18799–18804. 10.1073/pnas.0609225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bort G.; Gallavardin T.; Ogden D.; Dalko P. I. From One-Photon to Two-Photon Probes: “Caged” Compounds, Actuators, and Photoswitches. Angew. Chem., Int. Ed. 2013, 52, 4526–4537. 10.1002/anie.201204203. [DOI] [PubMed] [Google Scholar]
- Molnár P.; Nadler J. V. γ-Aminobutyrate, α-Carboxy-2-Nitrobenzyl Ester Selectively Blocks Inhibitory Synaptic Transmission in Rat Dentate Gyrus. Eur. J. Pharmacol. 2000, 391, 255–262. 10.1016/s0014-2999(00)00106-0. [DOI] [PubMed] [Google Scholar]
- Donato L.; Mourot A.; Davenport C. M.; Herbivo C.; Warther D.; Léonard J.; Bolze F.; Nicoud J.-F.; Kramer R. H.; Goeldner M.; Specht A. Water-Soluble, Donor–Acceptor Biphenyl Derivatives in the 2-(o-Nitrophenyl)Propyl Series: Highly Efficient Two-Photon Uncaging of the Neurotransmitter γ-Aminobutyric Acid at λ=800 Nm. Angew. Chem., Int. Ed. 2012, 51, 1840–1843. 10.1002/anie.201106559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menge C.; Heckel A. Coumarin-Caged DG for Improved Wavelength-Selective Uncaging of DNA. Org. Lett. 2011, 13, 4620–4623. 10.1021/ol201842x. [DOI] [PubMed] [Google Scholar]
- Amatrudo J. M.; Olson J. P.; Lur G.; Chiu C. Q.; Higley M. J.; Ellis-Davies G. C. R. Wavelength-Selective One- and Two-Photon Uncaging of GABA. ACS Chem. Neurosci. 2014, 5, 64–70. 10.1021/cn400185r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantevari S.; Matsuzaki M.; Kanemoto Y.; Kasai H.; Ellis-Davies G. C. R. Two-Color, Two-Photon Uncaging of Glutamate and GABA. Nat. Methods 2010, 7, 123–125. 10.1038/nmeth.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigo F. F.; Papageorgiou G.; Corrie J. E. T.; Ogden D. Laser Photolysis of DPNI-GABA, a Tool for Investigating the Properties and Distribution of GABA Receptors and for Silencing Neurons in Situ. J. Neurosci. Methods 2009, 181, 159–169. 10.1016/j.jneumeth.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Sikder A.; Banerjee M.; Singha T.; Mondal S.; Datta P. K.; Anoop A.; Singh N. D. P. A Natural Alkaloid, β-Carboline, as a One- and Two-Photon Responsive Fluorescent Photoremovable Protecting Group: Sequential Release of the Same or Different Carboxylic Acids. Org. Lett. 2020, 22, 6998–7002. 10.1021/acs.orglett.0c02549. [DOI] [PubMed] [Google Scholar]
- Papageorgiou G.; Corrie J. E. T. Effects of Aromatic Substituents on the Photocleavage of 1-Acyl-7-Nitroindolines. Tetrahedron 2000, 56, 8197–8205. 10.1016/S0040-4020(00)00745-6. [DOI] [Google Scholar]
- Canepari M.; Nelson L.; Papageorgiou G.; Corrie J. E. T.; Ogden D. Photochemical and Pharmacological Evaluation of 7-Nitroindolinyl-and 4-Methoxy-7-Nitroindolinyl-Amino Acids as Novel, Fast Caged Neurotransmitters. J. Neurosci. Methods 2001, 112, 29–42. 10.1016/S0165-0270(01)00451-4. [DOI] [PubMed] [Google Scholar]
- Papageorgiou G.; Ogden D.; Kelly G.; Corrie J. E. T. Synthetic and Photochemical Studies of Substituted 1-Acyl-7-Nitroindolines. Photochem. Photobiol. Sci. 2005, 4, 887–896. 10.1039/B508756B. [DOI] [PubMed] [Google Scholar]
- Warther D.; Gug S.; Specht A.; Bolze F.; Nicoud J.-F.; Mourot A.; Goeldner M. Two-Photon Uncaging: New Prospects in Neuroscience and Cellular Biology. Bioorg. Med. Chem. 2010, 18, 7753–7758. 10.1016/j.bmc.2010.04.084. [DOI] [PubMed] [Google Scholar]
- Papageorgiou G.; Ogden D. C.; Barth A.; Corrie J. E. T. Photorelease of Carboxylic Acids from 1-Acyl-7-Nitroindolines in Aqueous Solution: Rapid and Efficient Photorelease of L-Glutamate [4]. J. Am. Chem. Soc. 1999, 121, 6503–6504. 10.1021/ja990931e. [DOI] [Google Scholar]
- Ellis-Davies G. C. R. Useful Caged Compounds for Cell Physiology. Acc. Chem. Res. 2020, 53, 1593–1604. 10.1021/acs.accounts.0c00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoryak O. D.; Sul J.-Y.; Haydon P. G.; Ellis-Davies G. C. R. Synthesis of a Caged Glutamate for Efficient One- and Two-Photon Photorelease on Living Cells. Chem. Commun. 2005, 3664–3666. 10.1039/B504922A. [DOI] [PubMed] [Google Scholar]
- Armstrong C.; Krook-Magnuson E.; Oijala M.; Soltesz I. Closed-Loop Optogenetic Intervention in Mice. Nat. Protoc. 2013, 8, 1475–1493. 10.1038/nprot.2013.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. K.; Gschwind T.; Nguyen T. M.; Bui A. D.; Felong S.; Ampig K.; Suh D.; Ciernia A. V.; Wood M. A.; Soltesz I. Optogenetic Intervention of Seizures Improves Spatial Memory in a Mouse Model of Chronic Temporal Lobe Epilepsy. Epilepsia 2020, 61, 561–571. 10.1111/epi.16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csizmadia I. G.; Mucsi Z.; Szalay G.; Kaszas A.; Lukacsne Haveland C.; Majercsik O.; Attila P.; Katona G.; Rozsa J. B.; Gundisch D.; Chiovini B.; Palfi D.. Use of Photocleavable Compounds. U.S. Patent 20,140,234,883 A1, 2014.
- Morgante P.; Guruge C.; Ouedraogo Y. P.; Nesnas N.; Peverati R. Competition Between Cyclization and Unusual Norrish Type I and Type II Nitro-Acyl Migration Pathways in the Photouncaging of 1-Acyl-7-Nitroindoline Revealed by Computations. ChemRxiv 2020, 10.26434/chemrxiv.11991651.v3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papageorgiou G.; Corrie J. E. T. Synthesis of an Anionically Substituted Nitroindoline-Caged GABA Reagent That Has Reduced Affinity for GABA Receptors. Tetrahedron 2007, 63, 9668–9676. 10.1016/j.tet.2007.07.030. [DOI] [Google Scholar]
- Aidas K.; Angeli C.; Bak K. L.; Bakken V.; Bast R.; Boman L.; Christiansen O.; Cimiraglia R.; Coriani S.; Dahle P.; Dalskov E. K.; Ekström U.; Enevoldsen T.; Eriksen J. J.; Ettenhuber P.; Fernández B.; Ferrighi L.; Fliegl H.; Frediani L.; Hald K.; Halkier A.; Hättig C.; Heiberg H.; Helgaker T.; Hennum A. C.; Hettema H.; Hjertenaes E.; Høst S.; Høyvik I.-M.; Iozzi M. F.; Jansík B.; Jensen H. J. A.; Jonsson D.; Jørgensen P.; Kauczor J.; Kirpekar S.; Kjaergaard T.; Klopper W.; Knecht S.; Kobayashi R.; Koch H.; Kongsted J.; Krapp A.; Kristensen K.; Ligabue A.; Lutnaes O. B.; Melo J. I.; Mikkelsen K. V.; Myhre R. H.; Neiss C.; Nielsen C. B.; Norman P.; Olsen J.; Olsen J. M. H.; Osted A.; Packer M. J.; Pawlowski F.; Pedersen T. B.; Provasi P. F.; Reine S.; Rinkevicius Z.; Ruden T. A.; Ruud K.; Rybkin V. V.; Sałek P.; Samson C. C. M.; de Merás A. S.; Saue T.; Sauer S. P. A.; Schimmelpfennig B.; Sneskov K.; Steindal A. H.; Sylvester-Hvid K. O.; Taylor P. R.; Teale A. M.; Tellgren E. I.; Tew D. P.; Thorvaldsen A. J.; Thøgersen L.; Vahtras O.; Watson M. A.; Wilson D. J. D.; Ziolkowski M.; Ågren H. The Dalton Quantum Chemistry Program System. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 269–284. 10.1002/wcms.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford CT, 2016.
- Monson P. R.; McClain W. M. Polarization Dependence of the Two-Photon Absorption of Tumbling Molecules with Application to Liquid 1-Chloronaphthalene and Benzene. J. Chem. Phys. 1970, 53, 29–37. 10.1063/1.1673778. [DOI] [Google Scholar]
- Mcclain W. M. Excited State Symmetry Assignment through Polarized Two-Photon Absorption Studies of Fluids. J. Chem. Phys. 1971, 55, 2789–2796. 10.1063/1.1676494. [DOI] [Google Scholar]
- Sałek P.; Vahtras O.; Helgaker T.; Ågren H. Density-Functional Theory of Linear and Nonlinear Time-Dependent Molecular Properties. J. Chem. Phys. 2002, 117, 9630–9645. 10.1063/1.1516805. [DOI] [Google Scholar]
- Alam M. M.; Chattopadhyaya M.; Chakrabarti S. A Critical Theoretical Study on the Two-Photon Absorption Properties of Some Selective Triaryl Borane-1-Naphthylphenyl Amine Based Charge Transfer Molecules. Phys. Chem. Chem. Phys. 2011, 13, 9285–9292. 10.1039/c0cp02958k. [DOI] [PubMed] [Google Scholar]
- Molnár-Perl I.; Vasanits A. Stability and Characteristics of the O-Phthaldialdehyde/3-Mercaptopropionic Acid and o-Phthaldialdehyde/N-Acetyl-L-Cysteine Reagents and Their Amino Acid Derivatives Measured by High-Performance Liquid Chromatography. J. Chromatogr. A 1999, 835, 73–91. 10.1016/S0021-9673(98)01088-7. [DOI] [Google Scholar]
- Behrens C. J.; Van Den Boom L. P.; Heinemann U. Effects of the GABAA Receptor Antagonists Bicuculline and Gabazine on Stimulus-Induced Sharp Wave-Ripple Complexes in Adult Rat Hippocampus in Vitro. Eur. J. Neurosci. 2007, 25, 2170–2181. 10.1111/j.1460-9568.2007.05462.x. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Zhao Y.; Yang M.; Zeng S.; Shu Y. Regulation of Action Potential Waveforms by Axonal GABAA Receptors in Cortical Pyramidal Neurons. PLoS One 2014, 9, e100968 10.1371/journal.pone.0100968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H.; Chen T.-W.; Hu A.; Shields B. C.; Guo C.; Looger L. L.; Kim D. S.; Svoboda K. Thy1-GCaMP6 Transgenic Mice for Neuronal Population Imaging In Vivo. PLoS One 2014, 9, e108697 10.1371/journal.pone.0108697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G.; Mellor R. H.; Bernstein M.; Keller-Peck C.; Nguyen Q. T.; Wallace M.; Nerbonne J. M.; Lichtman J. W.; Sanes J. R. Imaging Neuronal Subsets in Transgenic Mice Expressing Multiple Spectral Variants of GFP. Neuron 2000, 28, 41–51. 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Chen T.-W.; Wardill T. J.; Sun Y.; Pulver S. R.; Renninger S. L.; Baohan A.; Schreiter E. R.; Kerr R. A.; Orger M. B.; Jayaraman V.; Looger L. L.; Svoboda K.; Kim D. S. Ultrasensitive Fluorescent Proteins for Imaging Neuronal Activity. Nature 2013, 499, 295–300. 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozsa B.; Zelles T.; Vizi E. S.; Lendvai B. Distance-Dependent Scaling of Calcium Transients Evoked by Backpropagating Spikes and Synaptic Activity in Dendrites of Hippocampal Interneurons. J. Neurosci. 2004, 24, 661–670. 10.1523/JNEUROSCI.3906-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Barone V.; Cossi M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. 10.1021/jp9716997. [DOI] [Google Scholar]
- Cossi M.; Rega N.; Scalmani G.; Barone V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- Frank É.; Mucsi Z.; Zupkó I.; Réthy B.; Falkay G.; Schneider G.; Wölfling J. Efficient Approach to Androstene-Fused Arylpyrazolines as Potent Antiproliferative Agents. Experimental and Theoretical Studies of Substituent Effects on BF3-Catalyzed Intramolecular [3 + 2] Cycloadditions of Olefinic Phenylhydrazones. J. Am. Chem. Soc. 2009, 131, 3894–3904. 10.1021/ja808636e. [DOI] [PubMed] [Google Scholar]
- Mucsi Z.; Szabó A.; Hermecz I.; Kucsman Á.; Csizmadia I. G. Modeling Rate-Controlling Solvent Effects. The Pericyclic Meisenheimer Rearrangement of N-Propargylmorpholine N-Oxide. J. Am. Chem. Soc. 2005, 127, 7615–7631. 10.1021/ja042227q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.