

Abstract
Cardiac amyloidosis (CA) is a disease characterized by the deposition of misfolded protein deposits in the myocardial interstitium. While advanced CA confers significant morbidity and mortality, the magnitude of deposition and ensuing clinical manifestations vary greatly. Thus, an improved understanding of disease pathogenesis at both cellular and functional levels would afford critical insights that may improve outcomes. This review will summarize contemporary therapies for the two major types of CA, transthyretin (ATTR) and light-chain (AL) amyloidosis, and outline the capacity of imaging modalities to both diagnose CA, inform prognosis, and follow response to available therapies. We explore the current landscape of echocardiography, cardiac magnetic resonance, and bone scintigraphy in the assessment of functional and cellular parameters of dysfunction in CA throughout disease pathogenesis. Finally, we examine the impact of concurrent advances in both therapeutics and imaging on future research questions that improve our understanding of underlying disease mechanisms. Multi-modal imaging in CA affords an indispensable tool to offer individualized treatment plans and improve outcomes in patients with CA.
Introduction
The systemic amyloidoses are a group of diseases characterized by the deposition of insoluble, misfolded protein fibrils (amyloid) within the extracellular space of different organs thereby interfering with structure and function1. Cardiac amyloidosis (CA) occurs when amyloid fibrils accumulate within the extracellular space between cardiomyocytes, fundamentally altering cardiac structure and eliciting systolic and diastolic dysfunction manifesting clinically as heart failure. CA remains the major cause of mortality and morbidity in patients with amyloidosis, irrespective of the misfolded protein driving the pathogenesis of their disease2. The type of amyloidosis is determined by the protein that misfolds with nomenclature designating an “A” for amyloidosis followed by the affected protein. While over 30 different proteins have been implicated in systemic amyloidosis, the overwhelming majority of CA is caused by misfolded immunoglobulin light chain (AL) and transthyretin (ATTR) protein.
AL amyloidosis is a systemic disease with protean manifestations caused by deposition of misfolded immunoglobulin light chain produced by an abnormal clonal proliferation of bone marrow plasma cells3. AL amyloidosis is a rare disease with an estimated incidence of 3 – 12 per million person-years4, which translates to approximately 1 in 50,000 to 1 in 100,000. Given its multi-organ involvement, the clinical presentation is diverse. Cardiac AL amyloidosis (AL-CA) can span the spectrum from minimally symptomatic to severe circulatory collapse and overall is present in approximately 50–75% of cases2,5. Other organs commonly affected include the kidneys, liver, and nervous system. Amyloid deposition in soft tissues may present with pathognomonic but rare signs of macroglossia and periorbital bruising. AL amyloidosis with concomitant heart failure historically conferred a 6-month survival if untreated6, however the prognosis of AL amyloidosis has improved significantly in recent years with the advent of high dose melphalan (HDM) based chemotherapy followed by autologous stem cell transplantation (ASCT) and other highly active anti-plasma cell therapeutics. With contemporary treatments, median survival can exceed 10 years in patients with AL amyloidosis following successful SCT7.
ATTR amyloidosis is caused by misfolding of the liver-derived transthyretin (TTR) protein. The name TTR (also known as prealbumin) is derived from its function as a transport hormone for both thyroxine and the retinol (vitamin A)-retinol-binding protein complex. Pathogenicity occurs through either inherited mutation or other poorly defined mechanisms associated with aging that result in TTR tetramer dissociation into monomers and oligomers which misfold and assemble into amyloid fibrils3. Once deposited in the nervous system, ATTR amyloidosis causes a progressive, length-dependent, small fiber sensory peripheral polyneuropathy and autonomic neuropathy. Cardiac deposition results in progressive wall thickening, impaired systolic and diastolic function, and heart failure as in AL-CA. ATTR amyloidosis is further sub-defined by the genetic sequence of TTR as genetically wild-type (abbreviated wtATTR) or hereditary/variant (hATTR or ATTRv). The prevalence of ATTR amyloidosis is unclear but is thought to be significantly underdiagnosed, with autopsy data suggesting that TTR amyloid deposits can been identified in up to 25% of elderly individuals (over the age of 80–85 years) at post-mortem8. Wild-type ATTR (formally known as senile systemic amyloidosis) presents later in life (after age 65 years) and predominantly affects males with the heart often being the only visceral organ involved. Non-cardiac sequelae commonly observed in wtATTR include carpal tunnel syndrome, lumbar spinal stenosis, and tendinopathies. Conversely, hereditary ATTR (hATTR) is an autosomal dominant condition that presents at varying age with different manifestations that primarily depend upon the specific TTR variant. Clinical features comprise a combination of cardiomyopathy (ATTR-CA), autonomic, and peripheral neuropathies9. Over 120 mutations in the TTR gene have been identified as potentially pathogenic but only a handful are implicated in the majority of hATTR cases10. These variants are abbreviated by the wild-type amino acid followed by the residue number followed by the substitution (e.g Val122Ile). Prognosis in ATTR amyloidosis varies depending on subtype but compared to AL amyloidosis, is comparably better. Later in life onset and slower disease progression in wtATTR confer a median survival of 3–5 years following diagnosis without treatment. The different variants of hATTR give rise to variable prognoses, with only a 16% overall four-year survival observed with Val122Ile (valine to isoleucine at position 122) mutations, compared to 40% and 79% four-year survival seen with Thr60Ala (threonine to alanine at position 60) and Val30Met (valine to methionine at position 30) mutations respectively11.
CA presents as a heterogenous and complex pathophysiology, with perturbations in numerous parameters of cardiac, neurological, and renal function interacting variably in different individuals. An improved understanding of the disease pathogenesis at both cellular and functional levels is the key to improve outcomes. The proper application of available imaging techniques serves to elucidate the different mechanisms that contribute to disease progression in CA and how they produce the varying phenotypes recognized. Imaging allows us to comprehensively phenotype individuals to personalize their treatment strategies. This review will summarize contemporary therapies for CA and present the role of specific imaging modalities in the assessment of disease progression or response to treatment as tools for clinical care and research applications.
Overview of treatment of amyloidosis
Currently available treatments for AL amyloidosis
Treatments for AL amyloidosis are directed against the plasma cell and aim to reduce the concentration of free light chains to remove the pathogenic monoclonal protein from the circulation. Chemotherapeutics derived from agents approved for multiple myeloma form the mainstay of regimens. The principal therapeutic decision in AL amyloidosis involves the determination of eligibility for HDM/ASCT – a strategy with potential toxicities that is reserved for selected patients best administered in experienced centers. That said, successful HDM/ASCT can induce clinically meaningful hematological response rates of 40–60% and prolong survival to exceed 15 years for many patients12. Other agents commonly used alone without ASCT or as pre-transplantation/post-transplant adjuvant strategies include proteasome inhibitors (bortezomib), alkylating agents (melphalan, cyclophosphamide), immune-modulatory agents (IMiDs, lenolidamide), and monoclonal biologics specific to the plasma cell (daratumumab). Daratumumab recently received approved in January 2021 from the US Food and Drug Administration (FDA) for systemic amyloidosis representing the first therapeutic approved for this disease.
Currently available treatments for ATTR Amyloidosis
Historically, solid organ transplantation was the only available treatment option for those with ATTR amyloidosis. Cardiac transplantation for those with wtATTR was most often infeasible as patients presented at advanced age limiting eligibility. In hATTR, liver transplantation successfully removed the pathological TTR protein from the circulation, but the treatment was found to be limited by observations of progressive amyloid cardiomyopathy and polyneuropathy post-transplant13. This resulted in the development of specific pharmacological strategies designed to knock-down TTR protein synthesis or stabilize the protein to prevent misfolding. These investigations have yielded regulatory approval of different agents for ATTR amyloidosis, each of which have shown positive results in phase 3 clinical trials, as summarized in Table 1.
Table 1 –
Table of currently available therapies for ATTR amyloidosis
| Drug name (ROA) | Trial | Population | Patient demographics - Age (median), subtype, race | Inclusion criteria (cardiac) | Follow up | Primary (and secondary) endpoints | Adverse events |
|---|---|---|---|---|---|---|---|
| Patisiran (IV every 3 weeks) |
APOLLO | 148 drug; 77 placebo | 62 years, hATTR (100%) White (74%), Asian (23%), Black (2%) |
NYHA I-II ↑ LVWT |
18 months | ↑ PN score (↑ QOL) |
Peripheral oedema (30%) Infusion reaction (20%) |
| Inotersen (SC every week) |
NEURO-TTR | 112 drug, 60 placebo | 59 years, hATTR (100%) White (92%), Black (2%) |
NYHA I-II LVWT ≥13mm |
15 months | ↑ PN score ↑ QOL |
Glomerulonephritis (3%), Low platelets (2%) Excess deaths (5 vs 0) |
| Tafamidis (PO daily) |
ATTR-ACT | 264 drug, 177 placebo | 75 years, wtATTR (76%), hATTR (24%) White (80%), Black (14%) |
NYHA I-III History of HF NT-proBNP ≥600 6MWT >100m |
30 months | ↓ all-cause mortality ↓ CV-related hospitalisations |
None known |
| Diflunisal | Berk et al | 64 drug, 66 placebo | 60 years, hATTR (100%) White (79%), Asian (11%), Black (5%) |
NYHA I-III Anticoagulation |
24 months | ↑ PN score (↑ QOL and modified BMI) |
4 drug-related discontinuations (GI bleeding, HF, glaucoma, nausea) |
(ROA: Route of administration, IV: intravenous, SC: subcutaneous, PO: oral, hATTR: hereditary transthyretin amyloidosis, wtATTR: wild-type transthyretin amyloidosis, NYHA: New York Heart Association, LVWT: left ventricular wall thickness, NT-proBNP: n-terminal pro-brain natriuretic peptide, 6MWT: 6-minute walk test, HF: heart failure), PN score: peripheral neuropathy score as measured by the modified neuropathy impairment score plus 7 nerve tests (mNIS+7), QOL: quality of life, BMI: body mass index, GI: gastrointestinal)
Patisiran (Alnylam Pharmaceuticals) is an intravenously administered, encapsulated, small interfering RNA (siRNA) that specifically degrades TTR messenger RNA (mRNA) to lower TTR protein production. Benefitted by intravenous dosing once every 3 weeks, early studies found it to be well-tolerated and resulted in upto a 90% reduction in TTR protein production14. It gained U.S FDA approval for the treatment of hATTR with polyneuropathy symptoms in 2018 following results of the APOLLO trial, which demonstrated significant dose-dependent improvement in neurological symptoms and quality of life in those receiving patisiran compared to placebo at 18 months15. While APOLLO also demonstrated improvements in cardiac functional parameters including global longitudinal strain, wall thickness, and natriuretic peptides with patisiran in a subgroup of ATTR-CA patients, the study was not sufficiently powered to conclusively assess hard endpoints such as mortality or cardiovascular hospitalization. Further, patients with New York Heart Association (NYHA) class III and IV symptoms were excluded. For this reason, a specific trial of patisiran among patients with dominant ATTR cardiomyopathy (APOLLO-B, clinicaltrials.gov identifier NCT03997383) is presently underway.
Inotersen (Akcea/Ionis Therapeutics) is a subcutaneously administered anti-sense oligodeoxynucleotide that similarly degrades TTR mRNA (albeit by a different mechanism as patisiran) thereby reducing TTR production by the liver. The phase-3 NEURO-TTR trial demonstrated improvements in neurological symptoms for inotersen compared to placebo with adverse events related to glomerulonephritis and thrombocytopenia (3% and 1% respectively) prompting FDA approval in 2018 but with a mandated Risk Evaluation and Mitigation Strategy16. In a small study of 8 patients with ATTR-CA, inotersen demonstrated stabilization of left ventricular (LV) wall thickness, LV mass, and echocardiographic global systolic strain at 1 year17.
Tafamidis (Pfizer) is an orally administered small molecule that selectively binds to the thyroxine-binding sites of TTR to slow the dissociation of TTR tetramers into monomers. It was shown to slow progression of peripheral neurological impairment in TTR-related polyneuropathy18 and was approved for early-stage disease in the European Union in 2011, but not in the US. The phase 3 clinical trial ATTR-ACT demonstrated a lower risk of all-cause mortality (absolute risk reduction of 13%) and cardiovascular hospitalisations (relative risk reduction of 32%) with tafamidis compared to placebo for patients with ATTR-CA. Reductions in the decline of functional capacity and quality of life were also demonstrated19 prompting FDA approval in 2019 as the first and still only agent for ATTR-CA.
Diflunisal is a non-steroidal anti-inflammatory drug which also acts to stabilise TTR degradation through binding to its thyroxine binding sites. It has been shown to improve symptoms of hATTR-related neuropathy in a phase 3 clinical trial20 and may confer disease slowing and survival benefits comparable to that seen with tafamadis21. Administration is limited to selected patients with normal renal and platelet function.
Therapies for ATTR amyloidosis in development.
The therapeutic landscape for ATTR-CA is rich and varied. Other agents in development are likely also to show efficacy in ATTR-CA based on prior reported results. Acoramidis (formerly known as AG10) is a TTR stabiliser in phase 3 trial (ATTRIBUTE, NCT03860935, Eidos Therapeutics) with a structure derived from a protective TTR mutation interaction. The drug is well-tolerated with a very high degree of in vitro TTR stabilisation22. Vutrisiran (HELIOS-B, NCT04153149, Alylam) and AKCEA-TTR-LRx (CARDIO-TTRansform, NCT04136171, Akcea) are emerging therapies also in phase 3 trial for ATTR-CA targeting similar gene expression pathways to patisiran and inotersen, respectively23,24.
Clinical challenges of treatment and role of imaging
While contemporary therapies in AL-CA and ATTR-CA are highly effective in ameliorating clinical outcomes, several challenges in both treatment selection and monitoring efficacy remain. First, no robust and specific imaging markers of cardiac response in either type have been identified. Response to chemotherapy in AL-CA is assessed both hematologically (reduction in the affected light chain, normalization of the bone marrow) and through changes in end-organ function. For CA, measurement of the N-terminal fragment of brain natriuretic peptide (NT-proBNP) or intact BNP are useful with a 30% reduction from baseline defined as a “cardiac response”12. Changes in natriuretic peptides are influenced by renal impairment, a common co-morbidity, by fluid balance changes, and by specific chemotherapy agents. Thus, an imaging biomarker that reflects a cardiac response (or lack thereof) would be of great utility to provide evidence of structural or functional changes with treatment that are associated with circulating biomarker variation. In ATTR-CA, while change in prealbumin concentration is being explored as a circulating marker for treatment efficacy, similarly there are no specific imaging biomarkers that indicate efficacy of treatment. Second, while clinical symptoms and outcomes clearly improve with effective treatment, structural changes in the heart that may reflect amyloid fibril resorption have not been definitively shown. Here again, imaging may prove indispensable as a means to explore hypotheses regarding the mechanism of actions of the different therapeutic strategies at a myocardial structure level.
Overview of imaging in cardiac amyloidosis
Echocardiography with strain quantification.
Echocardiography is widely accessible, safe, relatively inexpensive, and a standard of care imaging test for all patients with heart failure. Characteristic findings in both AL-CA and ATTR-CA include biventricular wall thickening with non-dilated, non-compliant ventricles. Interventricular wall thickness (IVWT) of >12mm may raise suspicion of CA in the absence of aortic valve disease and significant hypertension25, though cardiac involvement can be present with a lesser degree of wall thickening. Symmetrical LV wall thickening is more common in AL-CA compared to more asymmetrical patterns that have been observed with ATTR-CA with the maximal hypertrophy seen within the septum demonstrating either a sigmoid shape (70% of cases) or reverse septal curvature (30% of cases)3,26. One retrospective study found that LV wall thickening and diastolic dysfunction develop over 3 years, with significant abnormalities in each seen 1-year preceding a diagnosis of CA27. Ejection fraction (EF) is widely used to quantify LV function but reflects global radial contractility which is often preserved until end-stage disease in CA. Stroke volume index and myocardial contraction fraction (ratio of stroke volume to myocardial wall volume) identify the presence of CA and confer prognosis28,29, but changes in longitudinal function, which occur earlier, can be measured more reliably through longitudinal strain imaging (LSI). LSI may distinguish CA from other cardiomyopathies by observation of a characteristic “bulls-eye” pattern in the strain polar map owing to preservation of apical LV function relative to basal function in CA2. Different LSI analysis platforms (from different vendors) generate regional and global LSI results that can vary slightly, but the apical to basal ratio corrects for this variation within an individual patient and has been shown to be sensitive (92%) and specific (82%) in discriminating CA from LV hypertrophy30. In respect to diastolic dysfunction, initial infiltration in the LV is characterized by impaired relaxation, which then progresses to restrictive physiology that is almost invariably present in advanced disease. Similar patterns of systolic and diastolic dysfunction with increase in free wall thickness are also observed in the right ventricle. The effects of systolic and diastolic ventricular dysfunction with reduced cardiac output along with direct amyloid infiltration of atrial walls can cause blood stasis and risk of thrombus formation31. Contemporary assessment of atrial strain may indicate markers of the risk for incident atrial fibrillation in AL-CA32.
Cardiac magnetic resonance.
Cardiac magnetic resonance (CMR) imaging is well-established in distinguishing between different cardiomyopathies through the use of “tissue characterisation” techniques in order to yield information on the composition of myocardial tissue. Differences between normal and pathological myocardium can be determined both with and without the use of contrast agents. “Intrinsic” changes in native magnetic myocardial properties (myocardial T1, T2 and T2*) can be observed in pathological processes including CA, with elevated native T1 having a high sensitivity (92%) and specificity (91%) to detect CA33. The late gadolinium enhancement (LGE) technique utilises the “extrinsic” administration of a gadolinium-based contrast agent. These contrast agents accumulate within the extracellular space of areas of myocardial injury, fibrosis, or amyloid fibril infiltration. In CA, the extracellular amyloid protein deposition increases the interstitial compartment between myocardial cells, giving rise to a highly characteristic appearance on LGE imaging, with initially subendocardial, followed by transmural LGE alongside abnormal gadolinium kinetics and concurrent (or inverted) nulling of both the blood pool and myocardium on Look Locker or inversion time (TI) scout sequences34. LGE can visualize the continuum of cardiac amyloid infiltration with high sensitivity (86%) and sensitivity (92%)35 and is the main tool currently used in CMR to assess the presence of cardiac amyloidosis. However, LGE has limitations: it is not readily quantitative, rendering it a difficult metric to track changes over time, and in patients with severe reduction in the kidney function (estimated glomerular filtration rate, eGFR, <30 ml/min - a relatively common finding in patients with systemic amyloidosis, especially of AL type) certain gadolinium-based contrast agents (GBCA groups 1 and 3) are contraindicated.
T1 mapping has the potential to overcome some of these limitations. T1 mapping directly measures a quantitative signal from the myocardium, the myocardial T1 recovery time, in a pixel-wise manner either pre- or post-contrast. In CA, native (pre-contrast) myocardial T1 tracks amyloid infiltration within the heart, with elevated native T1 shown to offer a high diagnostic accuracy for CA in single centre studies33. However, this does not fully differentiate the fundamental pathological processes (oedema, fibrosis or amyloid infiltration), owing to the concept that native T1 comprises a composite myocardial signal from both myocytes and interstitium. It is important to note that measured T1 values in milliseconds depend upon the magnetic field strength (longer with 3.0T vs 1.5T, for example) and by acquisition technique. Interpretation and generalization across studies must therefore account for these variables. The administration of GBCAs adds another dimension to CMR tissue characterization, enabling the measurement of the extracellular volume (ECV) from the ratio of signal change in blood and myocardium after contrast and the blood volume of distribution (defined as 1.0 minus the hematocrit)36. Cardiac amyloidosis is the exemplar of interstitial disease, and this is reflected by massive ECV elevation in the patients with cardiac amyloidosis (generally greater than 42%). It has been shown to be highly sensitive (92%) and specific (82%) for diagnosing ATTR-CA and correlates well with a variety of markers of disease activity37,38. ECV may also be useful to evaluate patients with a high pre-test probability of CA where standard clinical investigations or patterns of LGE suggest no cardiac involvement, highlighting a potential role to detect subtle changes as an early marker of disease39.
Nuclear imaging/bone scintigraphy.
Since the 1980s it has been recognised that the hearts of patients affected by cardiac amyloidosis can take up certain 99mTc-phosphate derivatives in a selective fashion. These initial observations were followed many years later, in 2005, by a seminal paper showing the diagnostic potential of 99mTc 2,3-dicarboxypropane-2, 1-diphosphonate (DPD) in identifying ATTR amyloidosis40. Subsequent studies have confirmed the high sensitivity to detect ATTR amyloidosis41 and showed that mild uptake of 99mTc-DPD (Grade 1) may be noted in patients with other subtypes of CA (i.e., AL, Amyloid A amyloidosis, and Apolipoprotein A1)42. Bone scintigraphy is positive in only 40% of patients with AL-CA and grade 2 can be found in up to 10% of advanced AL-CA patients42 thus concurrent testing for a plasma cell disorder (PCD) is required for accurate clinical interpretation. Recently, bone scintigraphy has dramatically changed the diagnostic approach to ATTR amyloidosis after the publication of a large, multicentre trial showing the ability of bone scintigraphy to enable the accurate diagnosis of ATTR-CA without the need for confirmatory biopsy in patients who do not have a monoclonal gammopathy42. If cardiac amyloidosis is suspected clinically or based on echocardiography/CMR, blood and urine must be analyzed for evidence of a PCD by free-light chain assay and immunofixation electrophoresis. In the absence of a PCD, 99mTc-PYP/DPD/HMDP cardiac scintigraphy should be considered. In the presence of a Grade 2 or 3 positive 99mTc-PYP/DPD/HMDP cardiac scan without evidence of a PCD in blood and urine, a diagnosis of ATTR-CA can be made without a biopsy (specificity and positive predictive value >98%). For this reason, scintigraphy has been positioned as a first diagnostic imaging test in suspected patients with ATTR-CA in recent clinical treatment algorithms43. It is important to note that scintigraphy cannot be interpreted without plasma cell marker data and that efficacy has only been established in referral populations of moderate to high pre-test likelihood of CA. Utility of these radiotracers as a means to screen for ATTR-CA has not yet been clearly defined and is the subject of ongoing investigation. Table 2 compares the advantages and disadvantages of these three imaging modalities.
Table 2 –
Comparisons of benefits and limitations between transthoracic echocardiography, cardiac magnetic resonance and bone scintigraphy.
| Transthoracic echocardiography | Cardiac Magnetic Resonance | Bone Scintigraphy | |
|---|---|---|---|
| Cost | $ | $$$$$$$$ | $$$$ |
| Benefits | Takes 20 minutes Portable, readily available in hospital and community centres Non-invasive, no exposure to ionising radiation Safe in pregnancy |
Image quality unaffected by body habitus No exposure to ionising radiation Provides information on cardiac function and tissue characterisation Can distinguish between different aetiologies of heart muscle disease |
High diagnostic sensitivity and specificity for cardiac involvement early in disease Can distinguish ATTR from AL amyloidosis Unaffected by body habitus Not operator-dependant |
| Limitations | Image quality affected by body habitus Image quality affected by experience of operator No information on tissue characterisation |
Takes 45–60 minutes Requires injection of contrast Only available in specialist centres Image quality affected by patient factors (poor breathing technique, arrhythmia) Patient may not tolerate if claustrophobic Rare risk of nephrogenic systemic fibrosis in patients with renal dysfunction receiving gadolinium-based contrasts Not safe in pregnancy |
Only available in centres with access to a gamma camera Requires injection of radioactive tracers Exposure to ionising radiation Time-sensitive – need to wait 2–4 hours after injection of tracer before scanning Not safe in pregnancy or breastfeeding |
Costs estimates are taken from NHS tariffs (2020/21) and may differ in different countries.
Cardiac positron emission tomography (PET) imaging
Cardiac positron emission tomography (PET) was identified as another nuclear imaging modality with diagnostic capabilities in CA following its utility in evaluating brain amyloidosis. PET can measure myocardial blood flow and metabolism, while directly imaging amyloid deposits following administration of tracers including 18F-florbetapir, 18F-florbetaben, 11-C-Pittsburgh compound B (11C-PIB), and 18F-flutemetamol. Small preliminary studies utilising these tracers have demonstrated increased standardised uptake values (SUV) in CA when compared to controls44–46, with some data highlighting potential benefits in differentiating AL-CA from ATTR-CA47,48. Research thus far has primarily focussed on retention index (RI) to quantify the degree of tracer uptake; a calculation of the fractional or percentage change in SUV between the early and late stages of PET imaging. Tracer uptake assessment has been shown to most strongly correlate with both CMR and echocardiographic parameters in diagnosing the AL subtype49 and highlights reductions in tracer uptake correlating with successful treatment in AL-CA50, which corroborates studies using 11C-PIB to demonstrate the degree of tracer uptake as an independent predictor of clinical outcomes51. The only known meta-analysis of cardiac PET in CA included almost 100 patients, calculating a combined sensitivity and specificity for disease detection of 95% and 98%, respectively52.
Cardiac Computed Tomography (CT) imaging
Evidence for the use of cardiac CT in diagnosing CA remains limited but primarily driven by post-iodine contrast imaging in combination with dual-energy equilibrium CT to measure the myocardial iodine concentration (MIC). Pilot studies of CA patients identified increased myocardial attenuation53 and higher ECV54 compared to controls; the latter also shown to correlate well with concurrent CMR ECV quantification55. MIC was more recently found to distinguish CA from other cardiomyopathies with a reported sensitivity and specificity as high as 100% and 92% respectively56. Although CT is not a first-line modality employed in the diagnosis of CA, it may provide an alternative to CMR when limited by factors including claustrophobia or implantable cardiac devices. CT-ECV determination obtained during imaging for transcutaneous aortic valve replacement (TAVR) for severe aortic stenosis has recently been reported as a means to identify CA in this specific population where the co-existence of CA and AS can approach 10–15%57.
Prognosis and tracking treatment response with imaging
Predicting prognosis
The stratification of prognosis is an essential initial step to determine an appropriate treatment plan for a patient with CA. Biomarker schemes to prognosticate survival for AL-CA and ATTR-CA utilizing a combination of plasma troponins, natriuretic peptides, and renal function are well established. While biomarkers serve to integrate the entire multi-system organ functional status at a given time point (i.e., cardiac and renal), they only indirectly capture the inherent functional or morphological impact of amyloid deposition on the heart. For this reason, specific imaging markers confer additive information to biomarkers and can refine risk prediction. Change in echocardiographic and CMR parameters of systolic and diastolic dysfunction, including strain parameters, have been shown to correlate with prognosis in both AL and ATTR amyloidosis in numerous studies58–60. The addition of global longitudinal strain (GLS) to biomarkers clearly refines the prediction of risk61. Mitral and tricuspid annular plane systolic excursion (MAPSE and TAPSE) and GLS are surrogates for longitudinal systolic function that can typically track disease progression from early to very advanced infiltration28,60,62. The typical apical sparing pattern seen with GLS has been shown to be particular prognostic, while the worst outcomes seen in patients with a loss of radial function, as demonstrated by reduced EF63. Quantification of myocardial infiltration through ECV measurement remains a strong predictor of outcomes in both AL-CA and ATTR-CA. Other tissue characterisation parameters including transmurality of LGE and markers of oedema (elevated native T1 and T2) offer prognostic benefit predominantly for the AL-CA subtype39,64–67. The degree of scintigraphy uptake may also confer risk68, though quantification is challenging owing to relative uptake of the heart vs. bone structures.
Tracking changes over time - echocardiography
Data regarding serial echocardiographic changes over time following treatments for both AL and ATTR amyloidosis are predominantly limited to retrospective analyses over different time periods with varying results. A summary of these studies can be found in Table 3.
Table 3 –
Comparison of studies that image cardiac amyloidosis over time.
| Study | Subtype | Imaging modality | Study design, patient numbers | Time interval between imaging | Results (of statistical significance) |
|---|---|---|---|---|---|
| Meier-Ewert et al, 2011 | AL, post-chemo | Echo | Retrospective, 55 patients | 1.3 years (non-responders), 3.1 years (responders) | 43% of responders & 24% of non-responders# had >1mm reduction in IVWT |
| Madan et al, 2012 | AL, post-chemo /SCT | Echo | Retrospective, 148 patients | 4.25 years | 41% had >2mm reduction in IVWT or >20% improvement to EF |
| Amano et al, 2017 | AL, post-chemo | Echo | Retrospective, 29 patients | 0.65 years | LV size, SVI and CI reduced and diastolic wall strain worsened in those that died compared to survivors |
| Salinaro et al, 2017 | AL, post-chemo | Echo | Retrospective, 61 patients | 1 year | Improvement in apical/basal strain ratio and relative apical sparing in responders compared to non-responders# |
| Tuzovic et al, 2017 | AL, post-chemo | Echo | Registry data, 41 patients | 0.25 years | No significant change in parameters |
| Pun et al, 2018 | AL, post-chemo/SCT | Echo | Retrospective, 34 patients | 1 year | Small reduction in EF. Otherwise no significant change in parameters |
| Hwang et al, 2020 | AL, post-chemo (26% also had SCT) | Echo | Retrospective: 38 patients; prospective: 34 patients | 0.25, 0.5, 1 and 2 years | Increase in mitral E/e’ ratio and decrease in GLS from 3–6 months in those who died/heart transplant |
| Solomon et al, 2018 | Hereditary ATTR, post-patisiran | Echo | APOLLO trial sub-analysis, 126 patients | 0.75 and 1.5 years | Reduction in mean LV wall thickness, reduced GLS and increased cardiac output in patisiran group compared to placebo |
| Zadok et al, 2020 | AL and ATTR pre-diagnosis | Echo | Retrospective, 42 AL and 33 ATTR | Upto 3 years before diagnosis | Increased wall thickening, diastolic dysfunction present >3 years, significant diastolic dysfunction and LV deterioration seen between 1–3 years before diagnosis |
| Martinez-Naharro et al, 2019 | AL, post-chemo | CMR | Retrospective, 31 patients | Variable | ECV regression seen in 13 patients (92% following responsive chemotherapy), ECV correlated with improved LV mass, LA area and diastolic function parameters |
| Fontana et al, 2020 | ATTR, post-patisiran (75% also diflunisal) | CMR | Retrospective, 16 patients | 0, 12 months | Significant reduction in ECV (adjusted mean difference −6.2%) |
AL: systemic amyloidosis, ATTR: transthyretin amyloidosis, SCT: stem cell transplantation,
non-responders and responders = “haematological response to chemotherapy”
IVWT: interventricular wall thickness, SV: stroke volume index, EF: ejection fraction, CI: cardiac index
AL-CA:
One short-term study (1 year follow-up interval) of patients with AL amyloidosis found improvements in apical/basal strain ratio and relative apical sparing among those who responded to chemotherapy as compared to non-responders61. Another short-term study found LV size and stroke volume decreased and diastolic wall strain worsened in those who did not respond to chemotherapy69. Similar retrospective studies of AL-CA failed to demonstrate significant changes in these cardiac parameters over a similar follow up period70,71. Studies of longer follow-up periods demonstrated 1–2mm improvement in septal wall thickness over approximately 3–4 years7,72, with a larger study of 148 patients also showing a 20% improvement in EF among responders to chemotherapy7. Only one study of AL offered both retrospective and prospective analysis. Over a 2-year period, 72 patients were studied with up to 4 serial echocardiograms. Progressive worsening of mitral E/e’ and GLS were seen at 3–6 months and beyond prior to death or heart transplantation73. Figure 1 demonstrates a clinical case example of improvement in AL-CA following successful chemotherapy as measured by improved LV strain parameters.
Figure 1 –
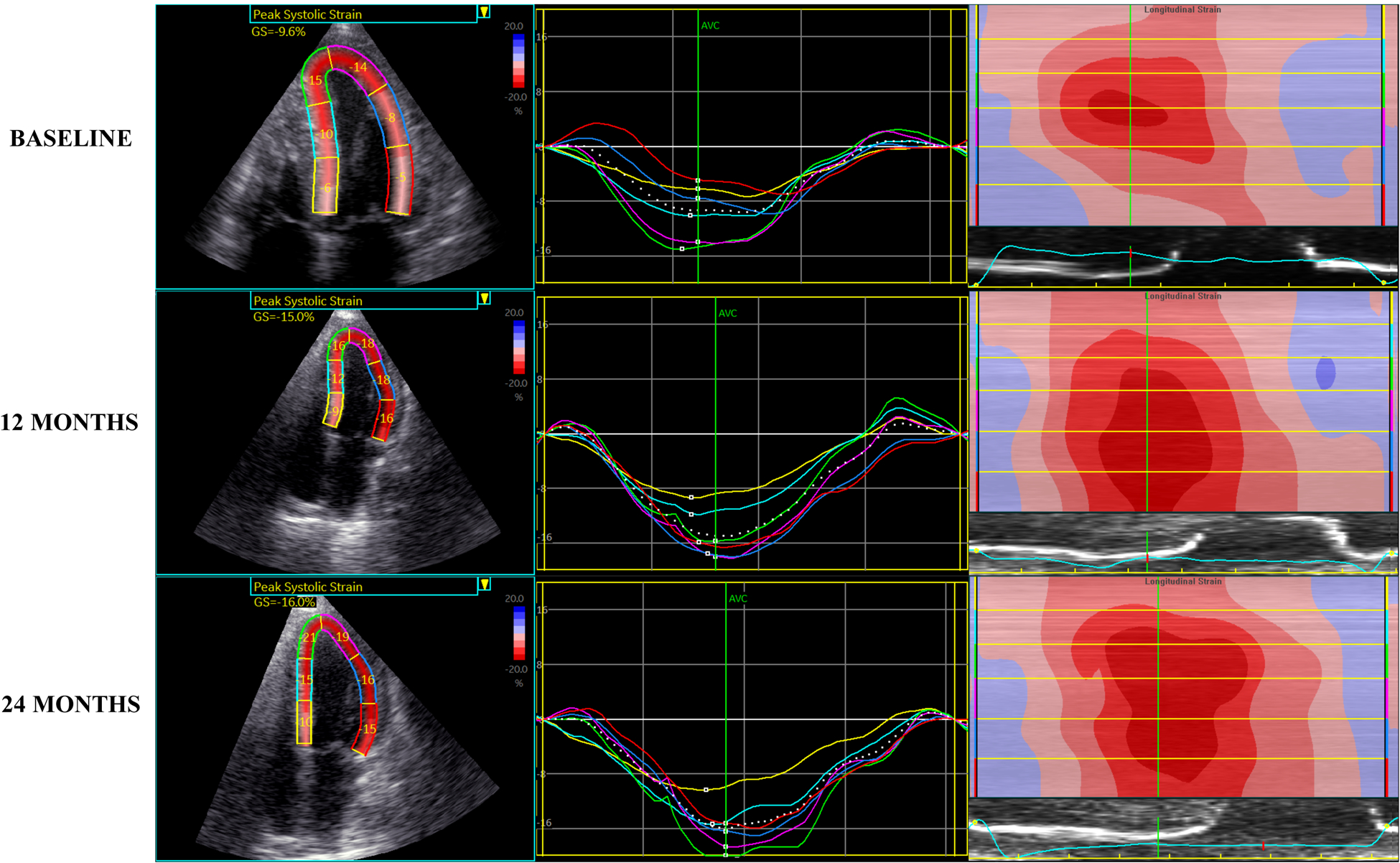
Regression of AL amyloidosis demonstrated by improved left ventricular strain parameters on echocardiography at baseline, 12 months and 24 months post-treatment with chemotherapy
ATTR-CA:
In ATTR-CA, echocardiographic changes in response to treatment vary depending upon treatment. A sub-analysis of the APOLLO trial of patisiran involving 90 patients found improvements in GLS and LV wall thickness after 18 months compared to placebo74 although no changes in these parameters were seen with inotersen16. That said, a small CMR-based unblinded analysis showed preservation of wall thickness and mass with inotersen albeit without a comparison cohort16. The ATTR-ACT trial demonstrated a reduction in the rate of decline of stroke volume after 30 months with tafamidis treatment compared to placebo19, while diflunisal appears to attenuate the decrement in GLS associated with wtATTR-CA75. One study reported echocardiographic changes prior to a diagnosis of both AL and ATTR amyloidosis, noting changes in IVWT and diastolic dysfunction present up to 3 years and significant diastolic impairment and EF deterioration progressing over 1–3 years prior to diagnosis27.
Tracking changes over time - CMR
Limited data are available on the potential role of CMR for tracking changes over time in AL-CA and ATTR-CA. Studies demonstrating changes in CMR parameters are limited to small patient cohorts and the two main studies are shown in Table 3. One study of AL-CA patients following chemotherapy was the first to show that regression of cardiac amyloid following substantial response to chemotherapy is a relatively common phenomenon, occurring in 42% of patients in the study cohort66. Reductions in LV mass, left atrial area and diastolic parameters were also seen with ECV regression. Importantly, worsening biventricular function and wall thickness were seen with ECV progression in this study66. Figure 2 demonstrates a case example of disease regression following chemotherapy in AL-CA with reduction in native T1, ECV and regression in LGE on serial CMR studies in one individual. Similar findings were shown in a study of patients with ATTR-CA, where treatment with patisiran was associated with a significant fall in ECV after 12 months of initiating therapy76. Interestingly, there was also a 19.6% reduction in cardiac tracer uptake in those receiving patisiran on DPD scintigraphy76, although the authors urged caution in attributing this result solely to CA regression, without confirmatory evidence of CMR or biochemical cardiac improvement. At present, there is no evidence that reduction in cardiac tracer uptake on DPD scintigraphy is a reflection of cardiac amyloid regression as it may represent redistribution of tracer uptake between different compartments.
Figure 2 –
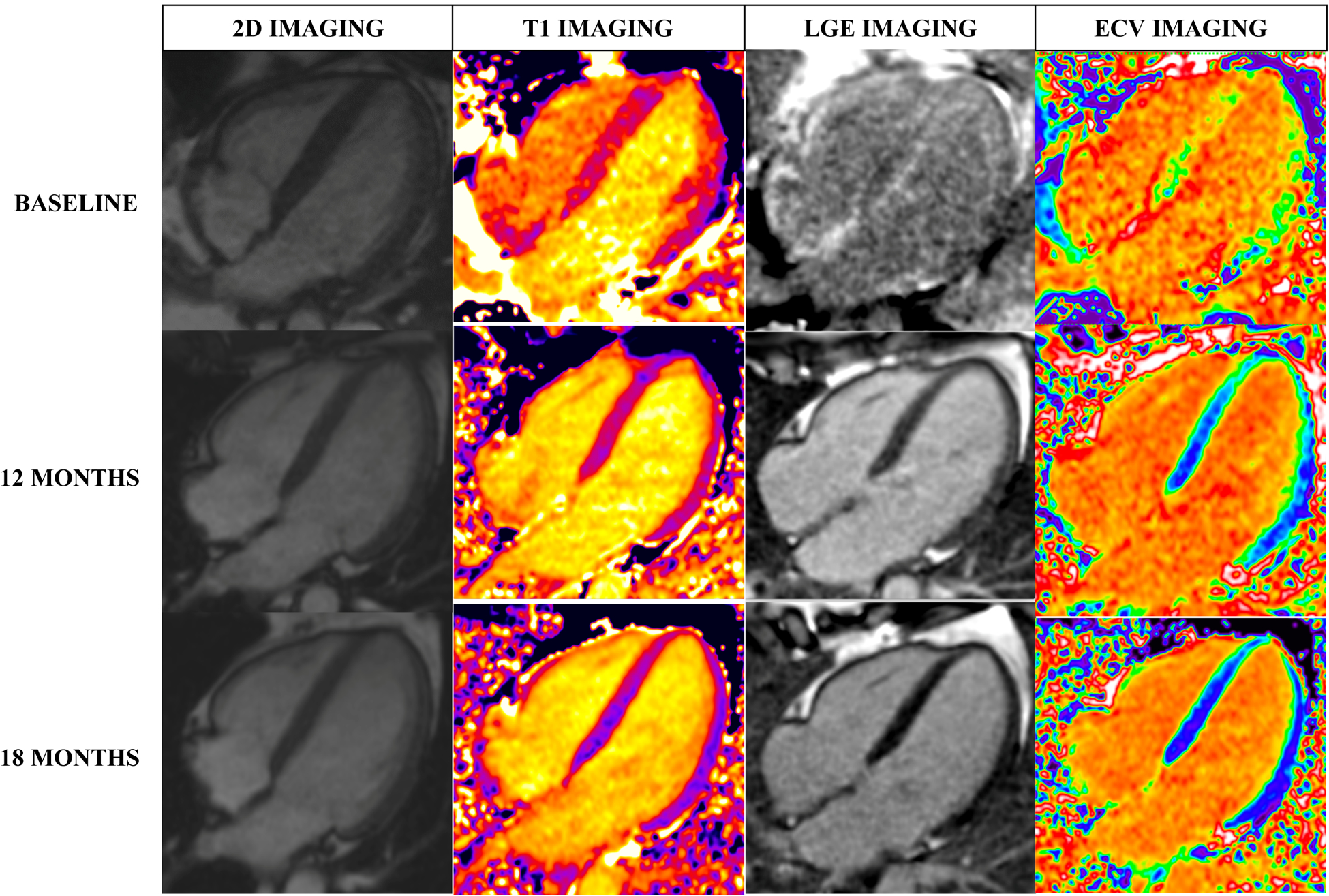
Regression of amyloidosis demonstrated on CMR in a patient with AL amyloidosis with serial scans at baseline, 12 months and 18 months post-treatment with chemotherapy. Reductions in left ventricular wall thickness (on 2D imaging) and native T1, LGE and ECV within the myocardium are seen. (T1: T1 tissue characterisation, LGE: late-gadolinium enhancement, ECV: extra-cellular volume)
Tracking changes over time – scintigraphy/PET
There are a paucity of available data describing the utility of nuclear techniques to follow disease activity; rather, these modalities are utilized primarily as a means to establish the presence of CA. Figure 3 highlights a rare example where ATTR-CA disease progression can be appreciated on serial DPD scintigraphy, although the general concept is that once definitive uptake has been identified, visualizable changes are unlikely to occur77. Indeed, in the aforementioned report that demonstrated changes in CMR ECV with patisiran therapy, no changes in DPD were identified serially among individual patients. That said, isolated case reports have noted PYP change with treatment78.
Figure 3 –
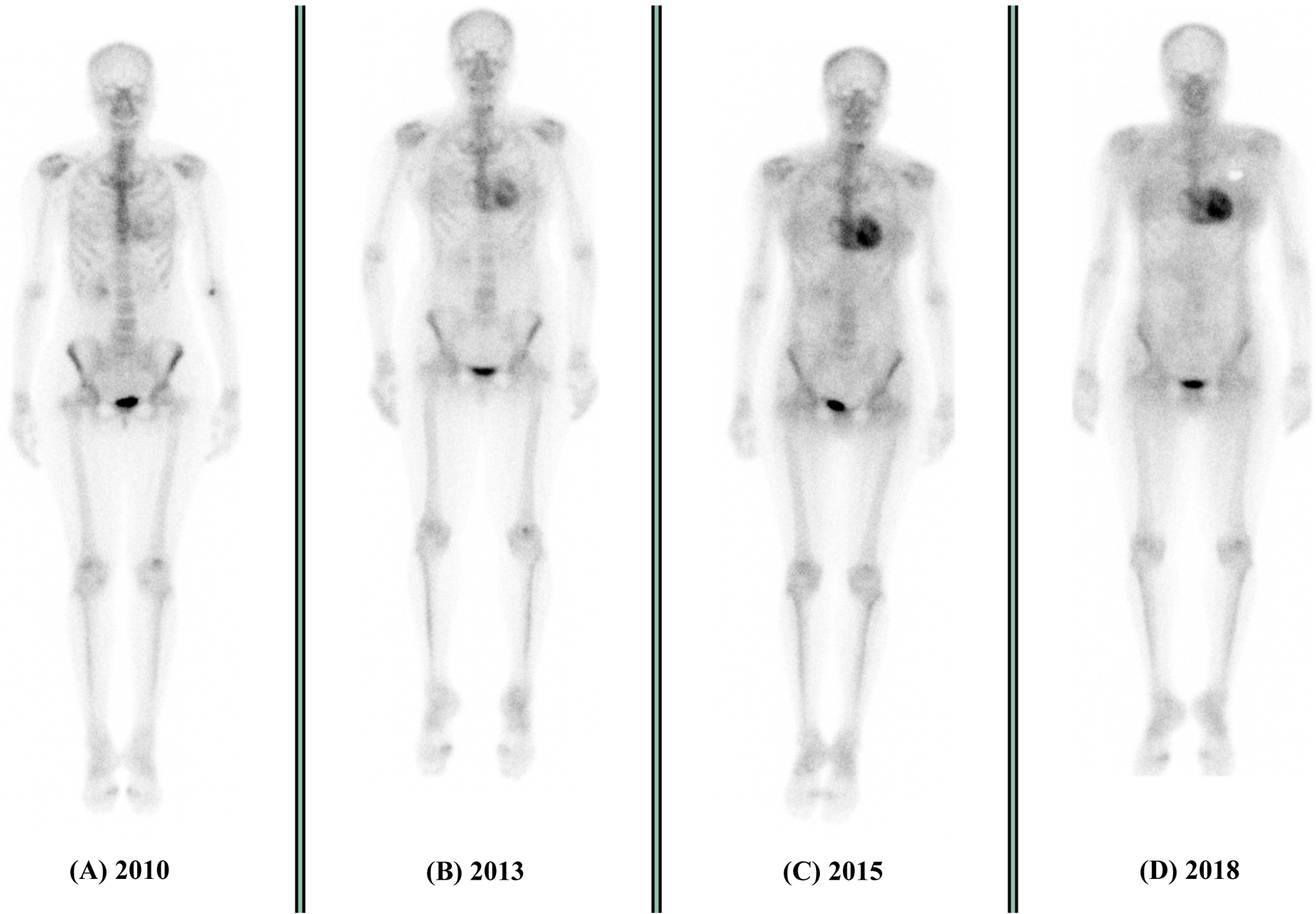
Progression of ATTR amyloidosis on DPD scintigraphy over a 9-year period. At baseline (A), there is minimal tracer uptake within the heart with high uptake in extra-cardiac tissues (demonstrated by a well-defined skeletal system). Over the next 5 years, progressive tracer uptake is seen within the heart, with a loss of definition to the extra-cardiac tissues (described as Perugini grade 1 (B) and Perugini grade 2 (C)). At 9 years there is the highest level of cardiac uptake with near complete loss of extra-cardiac tissue uptake (D)
Cardiac PET imaging similarly has been principally utilized as a means to identify CA among those with suspected disease44,79 or cardiac involvement among pathological TTR allele carriers80. Data also demonstrate the capacity of 18F-florbetapir to identify extra-cardiac amyloid deposits, particularly pulmonary, and vital organ involvement81. While the RI is a continuous variable that should lend itself to serial imaging and a capacity to differentiate smaller increments of change, data have not yet been reported.
Summary Recommendations - How to select and apply imaging in CA
1). Diagnosis of cardiac amyloidosis
Given that a significant proportion of patients with amyloidosis present with clinical heart failure, echocardiography should remain the standard of care imaging test to identify any suspicious features of CA such as biventricular wall thickening, non-compliant ventricles or IVWT of >12mm in the absence of abnormal loading conditions25. An apical sparing longitudinal strain pattern should significantly raise suspicion of unrecognized CA. If subsequent suspicions are raised on echocardiography but clinical features indicate a more widespread differential diagnosis, CMR should form the next most appropriate investigation. CMR is particularly applicable in younger (i.e. age < 60 years) patients, without a known TTR variant, in whom the diagnosis of ATTRwt is rare. Tissue characterisation and contrast imaging can discriminate the different morphological and functional features of different cardiomyopathies (e.g., hypertrophic cardiomyopathy, hypertensive remodelling, inflammatory myopathies, storage diseases, and/or iron overload diseases) which would otherwise be indistinguishable by echocardiography. Alternatively, if the clinical history is specifically suggestive of ATTR-CA (particularly in older patients), then bone scintigraphy should be undertaken following echocardiography, given its high sensitivity for ATTR-CA. However, it is important to note that a negative bone scintigraphy scan does not exclude CA; in such cases, patients should then undergo further testing with CMR or direct tissue sampling by biopsy if the clinical suspicion of either AL-CA or ATTR-CA remains present, given that bone scintigraphy can be positive in some cases of AL-CA and negative in rare variants of ATTR-CA. Irrespective of the imaging selection of CMR or nuclear imaging, concurrent testing for a plasma cell disorder is required.
2). Prognosis: Echo and CMR
Echocardiographic and CMR measures of impaired longitudinal function such as apical sparing in GLS, myocardial contraction fraction, MAPSE, and TAPSE represent a poorer prognosis in all subtypes of CA, but the poorest outcomes are noted in patients with reduced EF, which represents impaired radial function, reduced stroke volume index and end-stage disease. CMR evidence of significantly elevated ECV, transmurality of LGE and elevated markers of oedema (elevated native T1 and T2) are markers of a poor prognosis in AL-CA. In ATTR-CA, higher levels of ECV correlate with severity of disease and subsequent poorest outcomes. Table 4 summarises these parameters along with the cut-off values reported to confer a poorer prognosis in CA.
Table 4 –
Imaging parameters associated with poorer prognosis in cardiac amyloidosis seen on both echocardiography and cardiac magnetic resonance (CMR).
| Parameters – Echocardiography | Thresholds associated with worse prognosis |
|---|---|
| Global longitudinal strain (GLS) | −12% or greater (less negative) * |
| Apical/basal strain ratio | 2.1 or more |
| Mitral annular plane systolic excursion (MAPSE) | 8.8mm or less |
| Tricuspid annular plane systolic excursion (TAPSE) | 14mm or less |
| Myocardial contraction fraction | 0.25 or less |
| Parameters – CMR | |
| Pre-contrast myocardial T1 (1.5 T field strength; using shMOLLI) |
> 1044ms |
| Extra-cellular volume (ECV) | > 0.45 |
| Late-gadolinium enhancement (LGE) | Transmural pattern |
Thresholds for each parameter based on available data is shown (shMOLLI: Shortened Modified Look Lockers Inversion).
cut-offs used for prognosis in current studies range between −10% to 14%
3). Response to therapy: Echo and CMR
Cautious interpretation of current echocardiographic studies (summarised in Table 3) may offer utility to track responses to treatment in AL-CA. Treatment non-responders may be identifiable early, with rapid deterioration in LV dimensions, stroke volume, cardiac index, mitral E/e’ or GLS demonstrated within 6 months following treatment initiation. Conversely an efficacious response to chemotherapy may be observed with slow improvements in GLS and apical/basal strain ratio by 12 months and improved IVWT and EF over 3–4 years. GLS may offer an accurate and reproducible measure of LV function to track changes over a reasonable follow-up period. CMR studies have demonstrated both ECV regression alongside improvements in LV functional parameters following therapeutic chemotherapy in AL-CA, representing a direct relationship between amyloid burden and its impact on LV function66. The underlying mechanisms of this relationship remain unclear and is clearly an area that requires further investigation.
Data in ATTR-CA over time are limited but given the aforementioned study of patients on patisiran, improvements in GLS and IVWT on echocardiography and reductions in both ECV and DPD uptake may allow treatment efficacy to be monitored over time76. Importantly, ECV regression following patisiran in combination with diflunisal was seen alongside an 86% knockdown in serum TTR which, if reproducible on serial studies, may represent a direct measure of amyloid regression within the heart. The presence of concurrent change in an imaging and circulating biomarker further substantiates treatment efficacy and mechanism of action. Table 5 and Figure 4 highlight the major parameters that have been shown to worsen or improve in treatment non-responders and responders.
Table 5 –
Imaging parameters used to monitor disease progression or regression following response to therapy in cardiac amyloidosis seen on both echocardiography and cardiac magnetic resonance (CMR).
| Parameters – Echocardiography | Changes in non-responders to treatment | Changes in responders to treatment |
|---|---|---|
| Interventricular wall thickness (IVWT) | No change/worsening | 1–2mm reduction over 3–4 years |
| Left ventricular ejection fraction (LVEF) | No change/reduction | >20% improvement over 4 years |
| Global longitudinal strain (GLS) | Worsening | Improvement |
| Apical/basal strain ratio | No change | Overall reduction over 1 year |
| Stroke volume indexed (SVi) | Reduction of 10 ml/m2 | No change/improvement |
| E/e’ | Increase to > 20 | No change/reduction |
| Parameters – CMR | ||
| Extra-cellular volume | > 5% increase | > 5% reduction |
| Left ventricular mass | Overall increase | Overall reduction |
We would suggest similar cut-offs for both AL and ATTR cardiac amyloidosis but it is worth noting that the cut-off values suggested within the table have not been well validated at present.
Figure 4 –
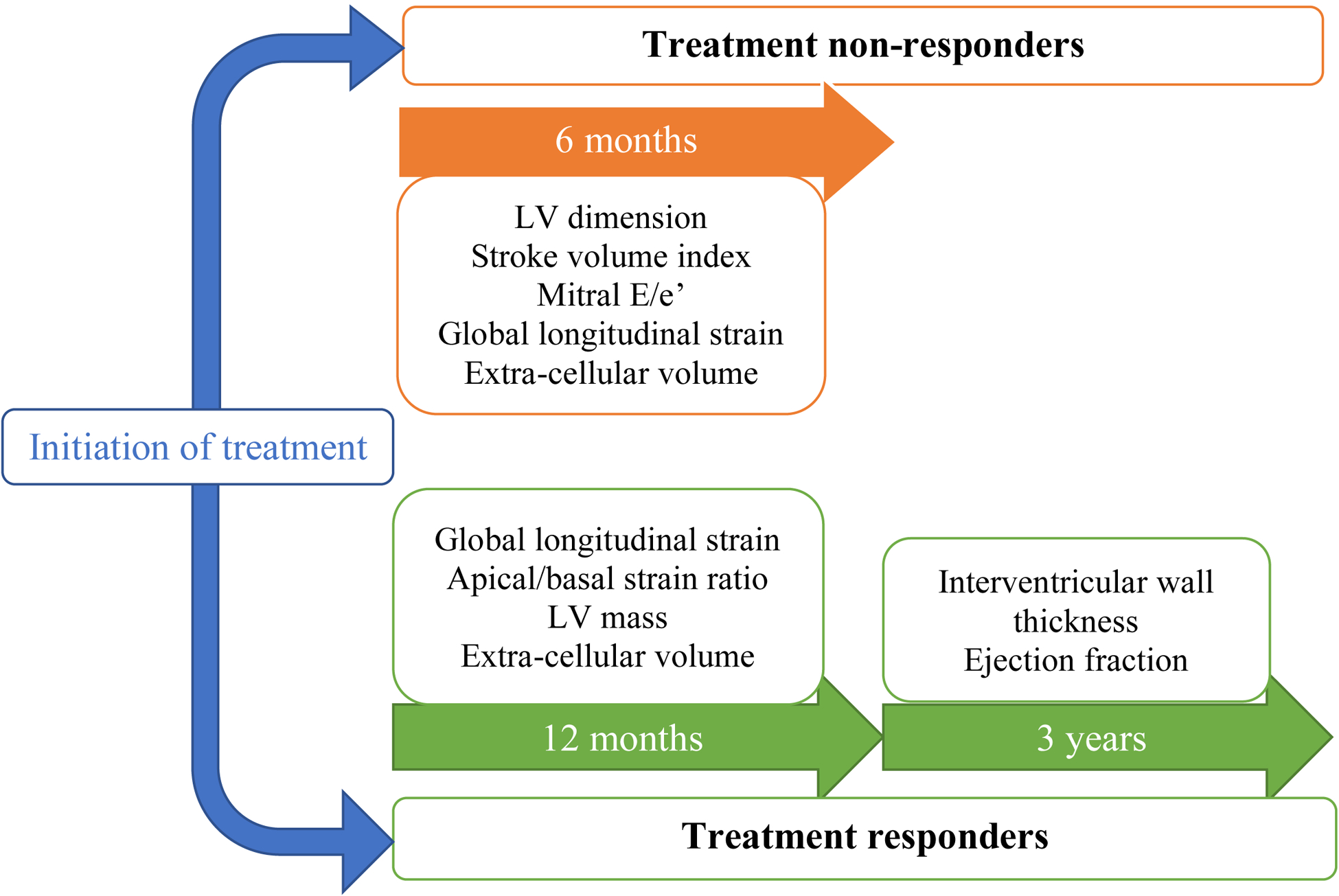
Imaging parameters measured on both echocardiography and cardiac magnetic resonance which have been shown to worsen over 6 months in treatment non-responders (orange boxes; top) and improve over 1–3 years in treatment responders (green boxes; bottom). (LV: left ventricle)
Implications for future clinical trials
The selection of appropriate primary and secondary endpoints for clinical trials, particularly those designed to qualify an agent for registration, is a painstaking process. Traditionally, regulatory agencies have favoured hard clinical outcomes (mortality, hospitalizations, clinical severity scores) over surrogates such as biomarkers or imaging metrics. That stated, imaging endpoints are incredibly important because they can afford insight into the mechanisms by which a novel intervention elicits a desired clinical outcome. In CA, certain imaging markers bear mention in this regard. GLS (by echocardiography or CMR) appears to confer a readout on ventricular systolic function above that of circulating biomarkers or measurements of cardiac ultrastructure. An agent that evidences both concordant and proportional changes in GLS with clinical and patient centred outcomes is more convincingly effective. Similarly, CMR ECV affords a means to measure microstructural changes in the interstitium which (if not related to resolution of fibrosis) likely relate to resorption of amyloid fibrils. We submit that a standardised approach to acquiring and measuring imaging markers such as these, would validate their use as secondary endpoints to enhance the rigor of a clinical trial and provide further substantiation of drug mechanisms.
Conclusions
Given the heterogeneous nature of CA, comprised of different types each resulting from a different pathophysiological process, a spectrum of clinical disease presents in each individual. A comprehensive phenotype of the systemic impact of amyloid deposition is afforded by imaging conferring critical information to calibrate treatment regimens in a personalized manner. Imaging biomarkers enable and support future research into the efficacy of specific drug therapies for CA. Imaging offers the capacity to disentangle the cardiac contribution from other systemic pathologies (renal, autonomic nervous, pulmonary) commonly encountered in amyloidosis. By leveraging the right cardiac imaging test, in the right patient, at the right time, we can better delineate the underlying cardiac structural and functional derangements that impact clinical outcomes.
Disclosures
Dr. Ruberg acknowledges research grant support from NIH/NHLBI (R01 HL139671), Alnylam Pharmaceuticals, Akcea Therapeutics, Eidos Therapeutics, and Pfizer, and consulting relationship with Alexion Pharmaceuticals.
Dr. Fontana acknowledges consulting relationships with Alnylam Pharmaceuticals, Akcea Therapeutics, Eidos Therapeutics, Intellia, Pfizer, and Alexion and research grant support from the British heart foundation fellowship FS/18/21/33447, Pfizer, and Eidos.
Dr. Patel has nothing to disclose.
Abbreviations and acronyms
- 11C-PIB
11-C-Pittsburgh compound B
- AL
light chain amyloidosis
- AS
aortic stenosis
- ASCT
autologous stem cell transplantation
- ATTR
transthyretin amyloidosis
- hATTR/ATTRv
hereditary transthyretin amyloidosis
- wtATTR
wild-type transthyretin amyloidosis
- BNP
brain natriuretic peptide
- CA
cardiac amyloidosis
- CMR
cardiac magnetic resonance
- CT
computed tomography
- DPD
2,3-dicarboxypropane-2, 1-diphosphonate
- EF
ejection fraction
- ECV
extra-cellular volume
- eGFR
estimated glomerular filtration rate
- FDA
Food and Drug Administration
- GBCA
gadolinium-based contrast agents
- GLS
global longitudinal strain
- HDM
high dose melphalan
- HMDP
hydroxymethylene diphosphonate
- IMiD
immunomodulatory agents
- IVWT
interventricular wall thickness
- LGE
late gadolinium enhancement
- LSI
longitudinal strain imaging
- LV
left ventricle
- MAPSE
mitral annular plane systolic excursion
- MIC
myocardial iodine concentration
- NT-proBNP
N-terminal fragment of brain natriuretic peptide
- NYHA
New York Heart Association
- PCD
plasma cell disorder
- PET
positron emission tomography
- PYP
pyrophosphate
- RI
retention index
- RNA
ribonucleic acid
- siRNA
small interfering ribonucleic acid
- mRNA
messenger ribonucleic acid
- SCT
stem-cell transplantation
- SUV
standardised uptake values
- TAPSE
tricuspid annular plane systolic excursion
- TAVR
transcutaneous aortic valve replacement
- TTR
transthyretin
REFERENCES
- 1.Lachmann HJ, Hawkins PN. Systemic amyloidosis. Curr Opin Pharmacol 2006;6:214–220. doi: 10.1016/j.coph.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Martinez-Naharro A, Hawkins PN, Fontana M. Cardiac amyloidosis. Clin Med Lond Engl 2018;18:s30–s35. doi: 10.7861/clinmedicine.18-2-s30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siddiqi OK, Ruberg FL. Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med 2018;28:10–21. doi: 10.1016/j.tcm.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv 2018;2:1046–1053. doi: 10.1182/bloodadvances.2018016402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chacko L, Martone R, Cappelli F, Fontana M. Cardiac Amyloidosis: Updates in Imaging. Curr Cardiol Rep 2019;21:108. doi: 10.1007/s11886-019-1180-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, Kurland LT. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992;79:1817–1822. [PubMed] [Google Scholar]
- 7.Madan S, Kumar SK, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Dingli D, Rajkumar SV, Hogan WJ, Leung N, et al. High-dose melphalan and peripheral blood stem cell transplantation for light-chain amyloidosis with cardiac involvement. Blood 2012;119:1117–1122. doi: 10.1182/blood-2011-07-370031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cornwell GG, Murdoch WL, Kyle RA, Westermark P, Pitkänen P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med 1983;75:618–623. doi: 10.1016/0002-9343(83)90443-6. [DOI] [PubMed] [Google Scholar]
- 9.Sattianayagam PT, Hahn AF, Whelan CJ, Gibbs SDJ, Pinney JH, Stangou AJ, Rowczenio D, Pflugfelder PW, Fox Z, Lachmann HJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J 2012;33:1120–1127. doi: 10.1093/eurheartj/ehr383. [DOI] [PubMed] [Google Scholar]
- 10.Merlini G, Westermark P. The systemic amyloidoses: clearer understanding of the molecular mechanisms offers hope for more effective therapies. J Intern Med 2004;255:159–178. doi: 10.1046/j.1365-2796.2003.01262.x. [DOI] [PubMed] [Google Scholar]
- 11.Arruda-Olson AM, Zeldenrust SR, Dispenzieri A, Gertz MA, Miller FA, Bielinski SJ, Klarich KW, Scott CG, Grogan M. Genotype, echocardiography, and survival in familial transthyretin amyloidosis. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis 2013;20:263–268. doi: 10.3109/13506129.2013.845745. [DOI] [PubMed] [Google Scholar]
- 12.Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood 2020;136:2620–2627. doi: 10.1182/blood.2020006913. [DOI] [PubMed] [Google Scholar]
- 13.Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology 2010;75:324–327. doi: 10.1212/WNL.0b013e3181ea15d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suhr OB, Coelho T, Buades J, Pouget J, Conceicao I, Berk J, Schmidt H, Waddington-Cruz M, Campistol JM, Bettencourt BR, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis 2015;10:109. doi: 10.1186/s13023-015-0326-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang C-C, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 16.Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Planté-Bordeneuve V, Barroso FA, Merlini G, Obici L, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379:22–31. doi: 10.1056/NEJMoa1716793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benson MD, Dasgupta NR, Rissing SM, Smith J, Feigenbaum H. Safety and efficacy of a TTR specific antisense oligonucleotide in patients with transthyretin amyloid cardiomyopathy. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis 2017;24:219–225. doi: 10.1080/13506129.2017.1374946. [DOI] [PubMed] [Google Scholar]
- 18.Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Planté-Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceição IM, Schmidt HH-J, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012;79:785–792. doi: 10.1212/WNL.0b013e3182661eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018;379:1007–1016. doi: 10.1056/NEJMoa1805689. [DOI] [PubMed] [Google Scholar]
- 20.Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, Heneghan MA, Gorevic PD, Litchy WJ, Wiesman JF, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013;310:2658–2667. doi: 10.1001/jama.2013.283815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenblum H, Castano A, Alvarez J, Goldsmith J, Helmke S, Maurer MS. TTR (Transthyretin) Stabilizers Are Associated With Improved Survival in Patients With TTR Cardiac Amyloidosis. Circ Heart Fail 2018;11:e004769. doi: 10.1161/CIRCHEARTFAILURE.117.004769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Judge DP, Heitner SB, Falk RH, Maurer MS, Shah SJ, Witteles RM, Grogan M, Selby VN, Jacoby D, Hanna M, et al. Transthyretin Stabilization by AG10 in Symptomatic Transthyretin Amyloid Cardiomyopathy. J Am Coll Cardiol 2019;74:285–295. doi: 10.1016/j.jacc.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 23.Habtemariam BA, Karsten V, Attarwala H, Goel V, Melch M, Clausen VA, Garg P, Vaishnaw AK, Sweetser MT, Robbie GJ, et al. Single-Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting N-acetylgalactosamine-Small Interfering Ribonucleic Acid Conjugate, Vutrisiran, in Healthy Subjects. Clin Pharmacol Ther 2021;109:372–382. doi: 10.1002/cpt.1974. [DOI] [PubMed] [Google Scholar]
- 24.Viney NJ, Tai L-J, Jung S, Yu RZ, Guthrie S, Baker BF, Geary RS, Schneider E, Guo S, Monia BP. Phase 1 Investigation of a Ligand-Conjugated Antisense Oligonucleotide with Increased Potency for the Treatment of Transthyretin Amyloidosis. J Card Fail 2019;25:S80–S81. doi: 10.1016/j.cardfail.2019.07.228. [DOI] [Google Scholar]
- 25.Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, Berk JL, Plante-Bordeneuve V, Schmidt HHJ, Merlini G. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J Am Coll Cardiol 2015;66:2451–2466. doi: 10.1016/j.jacc.2015.09.075. [DOI] [PubMed] [Google Scholar]
- 26.González-López E, Gagliardi C, Dominguez F, Quarta CC, de Haro-Del Moral FJ, Milandri A, Salas C, Cinelli M, Cobo-Marcos M, Lorenzini M, et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J 2017;38:1895–1904. doi: 10.1093/eurheartj/ehx043. [DOI] [PubMed] [Google Scholar]
- 27.Itzhaki Ben Zadok O, Eisen A, Shapira Y, Monakier D, Iakobishvili Z, Schwartzenberg S, Abelow A, Ofek H, Kazum S, Ben-Avraham B, et al. Natural History and Disease Progression of Early Cardiac Amyloidosis Evaluated by Echocardiography. Am J Cardiol 2020;133:126–133. doi: 10.1016/j.amjcard.2020.07.050. [DOI] [PubMed] [Google Scholar]
- 28.Chacko L, Martone R, Bandera F, Lane T, Martinez-Naharro A, Boldrini M, Rezk T, Whelan C, Quarta C, Rowczenio D, et al. Echocardiographic phenotype and prognosis in transthyretin cardiac amyloidosis. Eur Heart J 2020;41:1439–1447. doi: 10.1093/eurheartj/ehz905. [DOI] [PubMed] [Google Scholar]
- 29.Rubin J, Steidley DE, Carlsson M, Ong M-L, Maurer MS. Myocardial Contraction Fraction by M-Mode Echocardiography Is Superior to Ejection Fraction in Predicting Mortality in Transthyretin Amyloidosis. J Card Fail 2018;24:504–511. doi: 10.1016/j.cardfail.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phelan D, Collier P, Thavendiranathan P, Popović ZB, Hanna M, Plana JC, Marwick TH, Thomas JD. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart Br Card Soc 2012;98:1442–1448. doi: 10.1136/heartjnl-2012-302353. [DOI] [PubMed] [Google Scholar]
- 31.Feng D, Edwards WD, Oh JK, Chandrasekaran K, Grogan M, Martinez MW, Syed IS, Syed II, Hughes DA, Lust JA, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation 2007;116:2420–2426. doi: 10.1161/CIRCULATIONAHA.107.697763. [DOI] [PubMed] [Google Scholar]
- 32.Lohrmann Graham, Patel Monica Arun, Brauneis Dina, Sanchorawala Vaishali, Sarosiek Shayna, Vellanki Nirupama, Siddiqi Omar K., Ruberg Frederick L., Gopal Deepa M. Left Atrial Mechanics Associates With Paroxysmal Atrial Fibrillation in Light-Chain Amyloidosis Following Stem Cell Transplantation. JACC CardioOncology 2020;2:721–731. doi: 10.1016/j.jaccao.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karamitsos TD, Piechnik SK, Banypersad SM, Fontana M, Ntusi NB, Ferreira VM, Whelan CJ, Myerson SG, Robson MD, Hawkins PN, et al. Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 2013;6:488–497. doi: 10.1016/j.jcmg.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 34.Maceira Alicia Maria, Joshi Jayshree, Prasad Sanjay Kumar, Moon James Charles, Enrica Perugini, Idris Harding, Sheppard Mary Noelle, Poole-Wilson Philip Alexander, Hawkins Philip Nigel, Pennell Dudley John. Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation 2005;111:186–193. doi: 10.1161/01.CIR.0000152819.97857.9D. [DOI] [PubMed] [Google Scholar]
- 35.Zhao L, Tian Z, Fang Q. Diagnostic accuracy of cardiovascular magnetic resonance for patients with suspected cardiac amyloidosis: a systematic review and meta-analysis. BMC Cardiovasc Disord 2016;16:129. doi: 10.1186/s12872-016-0311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banypersad Sanjay M, Sado Daniel M, Flett Andrew S, Gibbs Simon DJ, Pinney Jennifer H, Maestrini Viviasna, Cox Andrew T, Fontana Marianna, Whelan Carol J, Wechalekar Ashutosh D., et al. Quantification of Myocardial Extracellular Volume Fraction in Systemic AL Amyloidosis. Circ Cardiovasc Imaging 2013;6:34–39. doi: 10.1161/CIRCIMAGING.112.978627. [DOI] [PubMed] [Google Scholar]
- 37.Martinez-Naharro A, Kotecha T, Norrington K, Boldrini M, Rezk T, Quarta C, Treibel TA, Whelan CJ, Knight DS, Kellman P, et al. Native T1 and Extracellular Volume in Transthyretin Amyloidosis. JACC Cardiovasc Imaging 2019;12:810–819. doi: 10.1016/j.jcmg.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 38.Knight DS, Zumbo G, Barcella W, Steeden JA, Muthurangu V, Martinez-Naharro A, Treibel TA, Abdel-Gadir A, Bulluck H, Kotecha T, et al. Cardiac Structural and Functional Consequences of Amyloid Deposition by Cardiac Magnetic Resonance and Echocardiography and Their Prognostic Roles. JACC Cardiovasc Imaging 2019;12:823–833. doi: 10.1016/j.jcmg.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 39.Martinez-Naharro A, Treibel TA, Abdel-Gadir A, Bulluck H, Zumbo G, Knight DS, Kotecha T, Francis R, Hutt DF, Rezk T, et al. Magnetic Resonance in Transthyretin Cardiac Amyloidosis. J Am Coll Cardiol 2017;70:466–477. doi: 10.1016/j.jacc.2017.05.053. [DOI] [PubMed] [Google Scholar]
- 40.Perugini E, Guidalotti PL, Salvi F, Cooke RMT, Pettinato C, Riva L, Leone O, Farsad M, Ciliberti P, Bacchi-Reggiani L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 2005;46:1076–1084. doi: 10.1016/j.jacc.2005.05.073. [DOI] [PubMed] [Google Scholar]
- 41.Hutt DF, Fontana M, Burniston M, Quigley A-M, Petrie A, Ross JC, Page J, Martinez-Naharro A, Wechalekar AD, Lachmann HJ, et al. Prognostic utility of the Perugini grading of 99mTc-DPD scintigraphy in transthyretin (ATTR) amyloidosis and its relationship with skeletal muscle and soft tissue amyloid. Eur Heart J Cardiovasc Imaging 2017;18:1344–1350. doi: 10.1093/ehjci/jew325. [DOI] [PubMed] [Google Scholar]
- 42.Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016;133:2404–2412. doi: 10.1161/CIRCULATIONAHA.116.021612. [DOI] [PubMed] [Google Scholar]
- 43.Kittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, Nair AP, Nativi-Nicolau J, Ruberg FL, American Heart Association Heart Failure and Transplantation Committee of the Council on Clinical Cardiology. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation 2020;142:e7–e22. doi: 10.1161/CIR.0000000000000792. [DOI] [PubMed] [Google Scholar]
- 44.Antoni G, Lubberink M, Estrada S, Axelsson J, Carlson K, Lindsjö L, Kero T, Långström B, Granstam S-O, Rosengren S, et al. In vivo visualization of amyloid deposits in the heart with 11C-PIB and PET. J Nucl Med Off Publ Soc Nucl Med 2013;54:213–220. doi: 10.2967/jnumed.111.102053. [DOI] [PubMed] [Google Scholar]
- 45.Dorbala S, Vangala D, Semer J, Strader C, Bruyere JR, Di Carli MF, Moore SC, Falk RH. Imaging cardiac amyloidosis: a pilot study using 18F-florbetapir positron emission tomography. Eur J Nucl Med Mol Imaging 2014;41:1652–1662. doi: 10.1007/s00259-014-2787-6. [DOI] [PubMed] [Google Scholar]
- 46.Dietemann S, Nkoulou R. Amyloid PET imaging in cardiac amyloidosis: a pilot study using 18F-flutemetamol positron emission tomography. Ann Nucl Med 2019;33:624–628. doi: 10.1007/s12149-019-01372-7. [DOI] [PubMed] [Google Scholar]
- 47.Rosengren S, Skibsted Clemmensen T, Tolbod L, Granstam S-O, Eiskjær H, Wikström G, Vedin O, Kero T, Lubberink M, Harms HJ, et al. Diagnostic Accuracy of [11C]PIB Positron Emission Tomography for Detection of Cardiac Amyloidosis. JACC Cardiovasc Imaging 2020;13:1337–1347. doi: 10.1016/j.jcmg.2020.02.023. [DOI] [PubMed] [Google Scholar]
- 48.Genovesi D, Vergaro G, Giorgetti A, Marzullo P, Scipioni M, Santarelli MF, Pucci A, Buda G, Volpi E, Emdin M. [18F]-Florbetaben PET/CT for Differential Diagnosis Among Cardiac Immunoglobulin Light Chain, Transthyretin Amyloidosis, and Mimicking Conditions. JACC Cardiovasc Imaging 2021;14:246–255. doi: 10.1016/j.jcmg.2020.05.031. [DOI] [PubMed] [Google Scholar]
- 49.Cuddy SAM, Bravo PE, Falk RH, El-Sady S, Kijewski MF, Park M-A, Ruberg FL, Sanchorawala V, Landau H, Yee AJ, et al. Improved Quantification of Cardiac Amyloid Burden in Systemic Light Chain Amyloidosis: Redefining Early Disease? JACC Cardiovasc Imaging 2020;13:1325–1336. doi: 10.1016/j.jcmg.2020.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kircher M, Ihne S, Brumberg J, Morbach C, Knop S, Kortüm KM, Störk S, Buck AK, Reiter T, Bauer WR, et al. Detection of cardiac amyloidosis with 18F-Florbetaben-PET/CT in comparison to echocardiography, cardiac MRI and DPD-scintigraphy. Eur J Nucl Med Mol Imaging 2019;46:1407–1416. doi: 10.1007/s00259-019-04290-y. [DOI] [PubMed] [Google Scholar]
- 51.Lee S-P, Suh H-Y, Park S, Oh S, Kwak S-G, Kim H-M, Koh Y, Park J-B, Kim H-K, Cho H-J, et al. Pittsburgh B Compound Positron Emission Tomography in Patients With AL Cardiac Amyloidosis. J Am Coll Cardiol 2020;75:380–390. doi: 10.1016/j.jacc.2019.11.037. [DOI] [PubMed] [Google Scholar]
- 52.Kim YJ, Ha S, Kim Y-I. Cardiac amyloidosis imaging with amyloid positron emission tomography: A systematic review and meta-analysis. J Nucl Cardiol Off Publ Am Soc Nucl Cardiol 2020;27:123–132. doi: 10.1007/s12350-018-1365-x. [DOI] [PubMed] [Google Scholar]
- 53.Deux J-F, Mihalache C-I, Legou F, Damy T, Mayer J, Rappeneau S, Planté-Bordeneuve V, Luciani A, Kobeiter H, Rahmouni A. Noninvasive detection of cardiac amyloidosis using delayed enhanced MDCT: a pilot study. Eur Radiol 2015;25:2291–2297. doi: 10.1007/s00330-015-3642-2. [DOI] [PubMed] [Google Scholar]
- 54.Treibel TA, Bandula S, Fontana M, White SK, Gilbertson JA, Herrey AS, Gillmore JD, Punwani S, Hawkins PN, Taylor SA, et al. Extracellular volume quantification by dynamic equilibrium cardiac computed tomography in cardiac amyloidosis. J Cardiovasc Comput Tomogr 2015;9:585–592. doi: 10.1016/j.jcct.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee H-J, Im DJ, Youn J-C, Chang S, Suh YJ, Hong YJ, Kim YJ, Hur J, Choi BW. Myocardial Extracellular Volume Fraction with Dual-Energy Equilibrium Contrast-enhanced Cardiac CT in Nonischemic Cardiomyopathy: A Prospective Comparison with Cardiac MR Imaging. Radiology 2016;280:49–57. doi: 10.1148/radiol.2016151289. [DOI] [PubMed] [Google Scholar]
- 56.Chevance V, Damy T, Tacher V, Legou F, Ridouani F, Luciani A, Kobeiter H, Rahmouni A, Deux J-F. Myocardial iodine concentration measurement using dual-energy computed tomography for the diagnosis of cardiac amyloidosis: a pilot study. Eur Radiol 2018;28:816–823. doi: 10.1007/s00330-017-4984-8. [DOI] [PubMed] [Google Scholar]
- 57.Scully PR, Patel KP, Saberwal B, Klotz E, Augusto JB, Thornton GD, Hughes RK, Manisty C, Lloyd G, Newton JD, et al. Identifying Cardiac Amyloid in Aortic Stenosis: ECV Quantification by CT in TAVR Patients. JACC Cardiovasc Imaging 2020;13:2177–2189. doi: 10.1016/j.jcmg.2020.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koyama J, Falk RH. Prognostic significance of strain Doppler imaging in light-chain amyloidosis. JACC Cardiovasc Imaging 2010;3:333–342. doi: 10.1016/j.jcmg.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 59.Lee Chuy K, Drill E, Yang JC, Landau H, Hassoun H, Nahhas O, Chen CL, Yu AF, Steingart RM, Liu JE. Incremental Value of Global Longitudinal Strain for Predicting Survival in Patients With Advanced AL Amyloidosis. JACC CardioOncology 2020;2:223–231. doi: 10.1016/j.jaccao.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Siepen FA dem, Bauer R, Voss A, Hein S, Aurich M, Riffel J, Mereles D, Röcken C, Buss SJ, Katus HA, et al. Predictors of survival stratification in patients with wild-type cardiac amyloidosis. Clin Res Cardiol Off J Ger Card Soc 2018;107:158–169. doi: 10.1007/s00392-017-1167-1. [DOI] [PubMed] [Google Scholar]
- 61.Salinaro F, Meier-Ewert HK, Miller EJ, Pandey S, Sanchorawala V, Berk JL, Seldin DC, Ruberg FL. Longitudinal systolic strain, cardiac function improvement, and survival following treatment of light-chain (AL) cardiac amyloidosis. Eur Heart J Cardiovasc Imaging 2017;18:1057–1064. doi: 10.1093/ehjci/jew298. [DOI] [PubMed] [Google Scholar]
- 62.Bodez D, Ternacle J, Guellich A, Galat A, Lim P, Radu C, Guendouz S, Bergoend E, Couetil J-P, Hittinger L, et al. Prognostic value of right ventricular systolic function in cardiac amyloidosis. Amyloid 2016;23:158–167. doi: 10.1080/13506129.2016.1194264. [DOI] [PubMed] [Google Scholar]
- 63.Senapati A, Sperry BW, Grodin JL, Kusunose K, Thavendiranathan P, Jaber W, Collier P, Hanna M, Popovic ZB, Phelan D. Prognostic implication of relative regional strain ratio in cardiac amyloidosis. Heart Br Card Soc 2016;102:748–754. doi: 10.1136/heartjnl-2015-308657. [DOI] [PubMed] [Google Scholar]
- 64.Fontana M, Pica S, Reant P, Abdel-Gadir A, Treibel TA, Banypersad SM, Maestrini V, Barcella W, Rosmini S, Bulluck H, et al. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation 2015;132:1570–1579. doi: 10.1161/CIRCULATIONAHA.115.016567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Banypersad SM, Fontana M, Maestrini V, Sado DM, Captur G, Petrie A, Piechnik SK, Whelan CJ, Herrey AS, Gillmore JD, et al. T1 mapping and survival in systemic light-chain amyloidosis. Eur Heart J 2015;36:244–251. doi: 10.1093/eurheartj/ehu444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martinez-Naharro A, Abdel-Gadir A, Treibel TA, Zumbo G, Knight DS, Rosmini S, Lane T, Mahmood S, Sachchithanantham S, Whelan CJ, et al. CMR-Verified Regression of Cardiac AL Amyloid After Chemotherapy. JACC Cardiovasc Imaging 2018;11:152–154. doi: 10.1016/j.jcmg.2017.02.012. [DOI] [PubMed] [Google Scholar]
- 67.Kotecha T, Martinez-Naharro A, Treibel TA, Francis R, Nordin S, Abdel-Gadir A, Knight DS, Zumbo G, Rosmini S, Maestrini V, et al. Myocardial Edema and Prognosis in Amyloidosis. J Am Coll Cardiol 2018;71:2919–2931. doi: 10.1016/j.jacc.2018.03.536. [DOI] [PubMed] [Google Scholar]
- 68.Castaño A, Bokhari S, Maurer MS. Prognosis Using Planar Imaging in Cardiac Amyloidosis-Reply. JAMA Cardiol 2017;2:704–705. doi: 10.1001/jamacardio.2016.5888. [DOI] [PubMed] [Google Scholar]
- 69.Amano M, Izumi C, Nishimura S, Kuroda M, Sakamoto J, Tamaki Y, Enomoto S, Miyake M, Tamura T, Kondo H, et al. Predictors of Prognosis in Light-Chain Amyloidosis and Chronological Changes in Cardiac Morphology and Function. Am J Cardiol 2017;120:2041–2048. doi: 10.1016/j.amjcard.2017.08.024. [DOI] [PubMed] [Google Scholar]
- 70.Tuzovic M, Kobayashi Y, Wheeler M, Barrett C, Liedtke M, Lafayette R, Schrier S, Haddad F, Witteles R. Functional Cardiac Recovery and Hematologic Response to Chemotherapy in Patients With Light-Chain Amyloidosis (from the Stanford University Amyloidosis Registry). Am J Cardiol 2017;120:1381–1386. doi: 10.1016/j.amjcard.2017.07.025. [DOI] [PubMed] [Google Scholar]
- 71.Pun SC, Landau HJ, Riedel ER, Jordan J, Yu AF, Hassoun H, Chen CL, Steingart RM, Liu JE. Prognostic and Added Value of Two-Dimensional Global Longitudinal Strain for Prediction of Survival in Patients with Light Chain Amyloidosis Undergoing Autologous Hematopoietic Cell Transplantation. J Am Soc Echocardiogr Off Publ Am Soc Echocardiogr 2018;31:64–70. doi: 10.1016/j.echo.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meier-Ewert HK, Sanchorawala V, Berk J, Finn KT, Skinner M, Seldin DC, Ruberg FL. Regression of cardiac wall thickness following chemotherapy and stem cell transplantation for light chain (AL) amyloidosis. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis 2011;18 Suppl 1:130–131. doi: 10.3109/13506129.2011.574354048. [DOI] [PubMed] [Google Scholar]
- 73.Hwang I-C, Koh Y, Park J-B, Yoon YE, Kim H-L, Kim H-K, Kim Y-J, Cho G-Y, Sohn D-W, Lee S-P. Time trajectory of cardiac function and its relation with survival in patients with light-chain cardiac amyloidosis. Eur Heart J Cardiovasc Imaging 2020. doi: 10.1093/ehjci/jeaa146. [DOI] [PubMed] [Google Scholar]
- 74.Solomon SD, Adams D, Kristen A, Grogan M, González-Duarte A, Maurer MS, Merlini G, Damy T, Slama MS, Brannagan TH, et al. Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients With Hereditary Transthyretin-Mediated Amyloidosis. Circulation 2019;139:431–443. doi: 10.1161/CIRCULATIONAHA.118.035831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lohrmann G, Pipilas A, Mussinelli R, Gopal DM, Berk JL, Connors LH, Vellanki N, Hellawell J, Siddiqi OK, Fox J, et al. Stabilization of Cardiac Function With Diflunisal in Transthyretin (ATTR) Cardiac Amyloidosis. J Card Fail 2020;26:753–759. doi: 10.1016/j.cardfail.2019.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fontana M, Martinez-Naharro A, Chacko L, Rowczenio D, Gilbertson JA, Whelan CJ, Strehina S, Lane T, Moon J, Hutt DF, et al. Reduction in CMR Derived Extracellular Volume With Patisiran Indicates Cardiac Amyloid Regression. JACC Cardiovasc Imaging 2021;14:189–199. doi: 10.1016/j.jcmg.2020.07.043. [DOI] [PubMed] [Google Scholar]
- 77.Castaño A, DeLuca A, Weinberg R, Pozniakoff T, Blaner WS, Pirmohamed A, Bettencourt B, Gollob J, Karsten V, Vest JA, et al. Serial scanning with technetium pyrophosphate (99mTc-PYP) in advanced ATTR cardiac amyloidosis. J Nucl Cardiol Off Publ Am Soc Nucl Cardiol 2016;23:1355–1363. doi: 10.1007/s12350-015-0261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Groothof D, Nienhuis HLA, Bijzet J, Houwerzijl EJ, van den Berg MP, Glaudemans AWJM, Slart RHJA, Hazenberg BPC. Regression of Bone-Tracer Uptake in Cardiac Transthyretin Amyloidosis. Mayo Clin Proc 2020;95:417–418. doi: 10.1016/j.mayocp.2019.10.036. [DOI] [PubMed] [Google Scholar]
- 79.Lee S-P, Lee ES, Choi H, Im H-J, Koh Y, Lee M-H, Kwon J-H, Paeng JC, Kim H-K, Cheon GJ, et al. 11C-Pittsburgh B PET Imaging in Cardiac Amyloidosis. JACC Cardiovasc Imaging 2015;8:50–59. doi: 10.1016/j.jcmg.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 80.Pilebro B, Arvidsson S, Lindqvist P, Sundström T, Westermark P, Antoni G, Suhr O, Sörensen J. Positron emission tomography (PET) utilizing Pittsburgh compound B (PIB) for detection of amyloid heart deposits in hereditary transthyretin amyloidosis (ATTR). J Nucl Cardiol Off Publ Am Soc Nucl Cardiol 2018;25:240–248. doi: 10.1007/s12350-016-0638-5. [DOI] [PubMed] [Google Scholar]
- 81.Khor YM, Cuddy S, Harms HJ, Kijewski MF, Park M-A, Robertson M, Hyun H, Di Carli MF, Bianchi G, Landau H, et al. Quantitative [18F]florbetapir PET/CT may identify lung involvement in patients with systemic AL amyloidosis. Eur J Nucl Med Mol Imaging 2020;47:1998–2009. doi: 10.1007/s00259-019-04627-7. [DOI] [PMC free article] [PubMed] [Google Scholar]