

Abstract
Introduction:
The safety and efficacy of bintrafusp alfa, a first-in-class bifunctional fusion protein composed of the extracellular domain of the transforming growth factor β (TGF-β) receptor II (a TGF-β “trap”) fused to a human immunoglobulin G1 antibody blocking programmed death-ligand 1 (PD-L1), was evaluated in patients with advanced NSCLC.
Methods:
This expansion cohort of NCT02517398, an ongoing, phase 1, open-label trial, includes 80 patients with advanced NSCLC that progressed after platinum doublet therapy or after platinum-based adjuvant or neoadjuvant treatment and those who also have not received previous immunotherapy. Patients were randomized at a one-to-one ratio to receive either bintrafusp alfa 500 mg or the recommended phase 2 dosage of 1200 mg every 2 weeks. The primary end point was the best overall response (by Response Evaluation Criteria in Solid Tumors 1.1 as adjudicated by independent review committee) and was assessed by the objective response rate (ORR).
Results:
A total of 80 patients were randomized to receive bintrafusp alfa 500 or 1200 mg (n = 40 each). Median follow-up was 51.9 weeks (IQR, 19.6–74.0). The ORR in all patients was 21.3% (17 of 80). The ORR was 17.5% (seven of 40) and 25.0% (10 of 40) for the 500 mg dose and the 1200 mg dose (recommended phase 2 dose), respectively. At the 1200 mg dose, patients with PD-L1–positive and PD-L1–high (≥80% expression on tumor cells) had ORRs of 36.0% (10 of 27) and 85.7% (six of seven), respectively. Treatment-related adverse events occurred in 55 of the 80 patients (69%) and were graded as greater than or equal to 3 in 23 of the 80 patients (29%). Of the 80 patients, eight (10%) had a treatment-related adverse event that led to treatment discontinuation; no treatment-related deaths occurred.
Conclusions:
Bintrafusp alfa had encouraging efficacy and manageable tolerability in patients with NSCLC previously treated with platinum.
Keywords: Bintrafusp alfa, M7824, TGF-β, PD-L1, NSCLC
Introduction
Antibodies targeting programmed death protein 1 (PD-1) or programmed death-ligand 1 (PD-L1) as first- or second-line treatments have achieved regulatory approvals after positive outcomes from phase 3 studies in patients with advanced or metastatic NSCLC.1 These include two anti–PD-1 antibodies (nivolumab and pembrolizumab) and one anti–PD-L1 antibody (atezolizumab), which revealed improved overall survival (OS) and safety compared with docetaxel. In these studies of patients who were not selected for PD-L1 expression, objective response rates (ORRs) with these agents ranged from 14% to 20%, median progression-free survival (PFS) ranged from 2.3 to 4.0 months, and median OS ranged from 9.2 to 13.8 months. Groups of patients with high PD-L1 expression (defined as ≥50% with the 22C3 assay) had ORRs from 30% to 31%, median PFS that ranged from 4.2 to 5.6 months and median OS that ranged from 17.1 to 20.6 months.2-4
Transforming growth factor β (TGF-β) is a cytokine involved in promoting tumor immune evasion and tumor progression through effects on both the innate and adaptive immune systems. TGF-β–mediated signaling in the tumor microenvironment (TME) has been reported to promote invasiveness, migration, and metastasis through multiple mechanisms, including epithelial-mesenchymal transition, an integral mechanism for disease progression and metastasis.5 TGF-β may also impact extrinsic processes in tumor cells that alter the local TME, including fibrosis, epithelial-mesenchymal transition, and tumor angiogenesis.5 Increased TGF-β plasma levels have been observed in patients with NSCLC compared with individuals without lung cancer, and these high levels are an indicator of poor survival.6,7 The immune-related actions of the TGF-β pathway are nonredundant with PD-L1 signaling, which works to inhibit cytotoxic T cells that are already active and present in the TME. TGF-β activity may also attenuate the efficacy of, or even lead to, resistance to anti–PD-L1 therapies.8-10 Preclinical studies have found that the combination of a TGF-β inhibitor and anti–PD-L1 reduced TGF-β signaling in stromal cells facilitated T-cell penetration into the center of the tumor, and elicited robust antitumor immunity and tumor regression.11-13 The TGF-β pathway, therefore, presents an attractive target for inhibition, especially when combined with anti–PD-L1.
Bintrafusp alfa (M7824) is a first-in-class bifunctional fusion protein composed of the extracellular domain of the human TGF-β receptor II (which functions as a TGF-β “trap”) fused through a flexible linker to the C-terminus of each heavy chain of an immunoglobulin G1 antibody blocking PD-L1. Preclinical studies have shown that bintrafusp alfa reduces TGF-β signaling within the TME.14 In MC38 syngeneic mouse models, bintrafusp alfa treatment resulted in greater reductions in tumor volume compared with either TGF-β trap or anti–PD-L1 antibody treatment alone.13 Other preclinical studies revealed that bintrafusp alfa treatment increased the density of CD8+ tumor-infiltrating lymphocytes in tumor-bearing mice and reversed epithelial-mesenchymal transition in human lung cancer cells.13,15 In a phase 1 dose-escalation study (NCT02517398), bintrafusp alfa had a manageable safety profile and revealed encouraging antitumor activity, including one ongoing confirmed complete response in a group of patients (n = 19) with heavily pretreated advanced solid tumors.16,17
To obtain further exposure-response data after the dose-escalation part of this phase I study, patients in this cohort were randomized to receive 500 mg or 1200 mg doses, the latter being the recommended phase 2 dose.17 On the basis of complete PD-L1 target occupancy and TGF-β trapping,16 both doses are expected to provide full pharmacologic effects.17 A manuscript that describes in detail the data associated with 1200 mg every 2 weeks as the recommended phase 2 dosage is currently under preparation.17,18
Materials and Methods
Study Design and Participants
This expansion cohort of a larger ongoing, phase 1, open-label trial (NCT02517398) will evaluate whether bintrafusp alfa is tolerable and has clinical activity in patients with metastatic or recurrent NSCLC that has progressed on previous therapies. In this dose-expansion cohort, patients included were aged 18 years or older and had histologically confirmed stage IIIb or IV or recurrent NSCLC. Patients were required to have disease progression during or after a minimum of two cycles of one course of platinum-based combination therapy administered for the treatment of metastatic disease or have disease progression within 6 months of completion of platinum-based adjuvant, neoadjuvant, or definitive chemotherapy or concomitant chemoradiation regimen for locally advanced disease. Patients with EGFR, anaplastic lymphoma kinase, or ROS1 mutations (determined by a local laboratory or a central laboratory if local testing is not available) were eligible if they had received at least one line of tyrosine kinase inhibitor therapy but did not require previous systemic chemotherapy. Patients also must not have received previous checkpoint inhibitor therapy, and archival tumor tissue or fresh tumor biopsy was required for participation. Patients with active or previously treated central nervous system metastases were not included unless they had fully recovered from treatment, revealed no progression for at least 2 months, and did not require continued steroid therapy. Full eligibility criteria are provided in the Supplementary Methods. Patients were enrolled per a protocol approved by the principal investigator and coordinating investigator of the trial and relevant regulatory authorities. International standards of Good Clinical Practice and the Declaration of Helsinki were followed. Written informed consent was provided by patients or their representatives. Ethics committees at all participating institutions approved the protocol.
Randomization
Patients provided informed consent and were enrolled by study investigators according to eligibility criteria. They were then randomized in a one-to-one ratio to receive bintrafusp alfa 500 mg or 1200 mg through permuted block randomization without stratification, which assigned unique treatment vial numbers to patients. The vial numbers were also linked to corresponding doses through a Good Manufacturing Practice–qualified system.
Procedures
Patients received bintrafusp alfa 500 or 1200 mg intravenously over 1 hour every 2 weeks until confirmed progressive disease, unacceptable toxicity, or any criteria for trial withdrawal occurred (Supplementary Methods). Dose reduction was not permitted; however, interruption or discontinuation of bintrafusp alfa was allowed if grade 2 or higher treatment-related adverse events (TRAEs), infusion-related reactions (IRRs), or severe or life-threatening adverse events (AEs) occurred.
Clinical activity was assessed by radiographic imaging 6 weeks after treatment initiation and every 6 weeks thereafter for the first year and then every 12 weeks. Tumor responses were assessed according to the Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 and adjudicated by an independent end point review committee (IRC). Responses were confirmed by imaging at or more than 4 weeks from the first documentation of response, and progressive disease was confirmed by imaging between 4 and 6 weeks after progression had been diagnosed.
Safety was assessed in all patients at each trial visit, and then at 28 days after the last study dose. Assessments included the following: (1) occurrence of AEs, (2) occurrence of AEs of special interest, (3) concurrent medications, (4) physical examination, and (5) clinical laboratory tests (hematology and serum chemistry). AEs were coded according to the Medical Dictionary for Regulatory Activities terms version 21.0. AEs and laboratory tests were classified by grade according to the National Cancer Institute Common Terminology Criteria for AEs version 4.03. Any AEs that were potentially immune-related or IRRs were considered AEs of special interest. Owing to the mechanism of action of bintrafusp alfa, hyperkeratosis, keratoacanthoma (KA), and squamous cell carcinoma of the skin were also considered AEs of special interest. Premedication with an antihistamine and paracetamol (acetaminophen) approximately 30 to 60 minutes before each dose of bintrafusp alfa was mandatory for the first two infusions and was administered at the investigator’s discretion thereafter. If a grade 2 or higher IRR occurred during the first two infusions, premedication was mandatory.
Archival formalin-fixed paraffin-embedded tumor samples were collected at baseline in this study. Biomarker analysis was performed for all available tumor samples from this cohort. PD-L1 expression was assessed retrospectively by immunohistochemistry using antibody clone 73-10 (PD-L1 immunohistochemistry 73-10 pharmDx; Dako, Carpinteria, CA); expression was categorized as PD-L1–negative (<1% PD-L1+ tumor cells), PD-L1–positive (≥1% PD-L1+ tumor cells), PD-L1–low (1%–<80% PD-L1+ tumor cells), or PD-L1–high (≥80% PD-L1+ tumor cells). PD-L1 expression categories were determined post hoc. Previous studies have shown that the PD-L1–high expression cutoff for the 73-10 assay (≥80% PD-L1+ tumor cells) is comparable to the greater than or equal to 50% cutoff for the 22C3 PD-L1 assay (manuscript in preparation) because both assays select a similar patient population at their respective cutoffs.19 Further information regarding methodologies for biomarker assessments, including plasma TGF-β1 concentrations, tumor immune phenotypes, and expression of genes and gene signatures associated with bintrafusp alfa’s mechanism of action in pretreated tumor samples, are provided in the Supplementary Methods.
Outcomes
The primary end point for the dose-expansion cohorts was the best overall response (BOR) per RECIST 1.1, as adjudicated by the IRC, and was evaluated by confirmed ORR in the overall population. Secondary end points included safety, BOR according to RECIST 1.1 per investigator assessment, and pharmacokinetic (PK) parameters. Exploratory end points included the following: (1) duration of response; (2) PFS; (3) time to progression according to RECIST 1.1 and per IRC; (4) immune-related efficacy (BOR, PFS, and time to progression) according to immune-related RECIST 1.1 per IRC; (5) OS; (6) biomarkers; (7) pharmacodynamic markers; and (8) changes in patient-reported outcomes assessed by Patient Global Impression of Severity and NSCLC-Symptom Assessment Questionnaire. All radiographic scans for patients enrolled in the study were sent from trial sites to the central reading facility for evaluation per RECIST 1.1.
Statistical Analysis
Enrollment of 80 patients was planned and randomized in a one-to-one ratio to bintrafusp alfa 500 or 1200 mg. This trial was not designed to test the efficacy differences between the two doses; these two doses were included for PK exposure-response evaluation.17 After 30 patients were enrolled in the 1200 mg group, the study had 82% power to rule out a less than or equal to 20% ORR when the true ORR was 40% with one-sided 0.1 alpha. Forty patients in each treatment arm were included for PK and statistical considerations for this analysis. Efficacy and safety were analyzed in all randomized patients who received at least one dose of the treatment. ORR, defined as the proportion of patients with a confirmed complete or partial response, was calculated using a two-sided 95% Clopper-Pearson confidence interval (CI). Disease control rate (DCR) was defined as the proportion of patients with a BOR of complete response, partial response, non–complete response or non–progressive disease, or stable disease. Time-to-event end points were estimated using the Kaplan-Meier method. All statistical analyses were performed using SAS version 9.1.3 or higher or R version 2.10.1 or higher. This trial is registered with ClinicalTrials.gov, number NCT02517398.
Results
Between October 18, 2016, and April 12, 2017, 80 eligible patients were enrolled at 25 sites and were randomly assigned to receive 500 or 1200 mg doses of bintrafusp alfa (n = 40 each) (Fig. 1) (Supplementary Methods) The data cutoff date was July 23, 2018. All 80 patients had received more than one dose of treatment and were included in all efficacy and safety analyses. The median age in the overall population was 64 years (range, 38–85 y). Most patients were male (n = 57 [71.3%]), had an Eastern Cooperative Oncology Group performance status of 1 (n = 60 [75.0%]), and had a history of smoking (n = 62 [77.5%]). Most patients in the 500 mg and 1200 mg groups were from Europe (57.5%) and the Asia Pacific region (62.5%), respectively. Owing to the design of randomization by treatment arm, baseline characteristics were generally similar, although slight numerical differences were observed. Histologic findings were squamous and nonsquamous in 16 (20.0%) and 64 patients (80.0%), respectively. A total of 10 patients (15.9%) had tumors with EGFR mutations and one patient (1.3%) had a ROS1 rearrangement. PD-L1 expression, assessed retrospectively using the 73-10 assay, was evaluable in 75 patients (93.8%). Among the randomized patients, 58 (72.5%) had a PD-L1–positive tumor, including 13 (16.3%) with PD-L1–high expression, and the median duration of first-line, platinum-based chemotherapy was 13.1 weeks (range, 6.0–120.4 wk). Additional patient and disease characteristics and previous anticancer regimens are summarized in Table 1.
Figure 1.
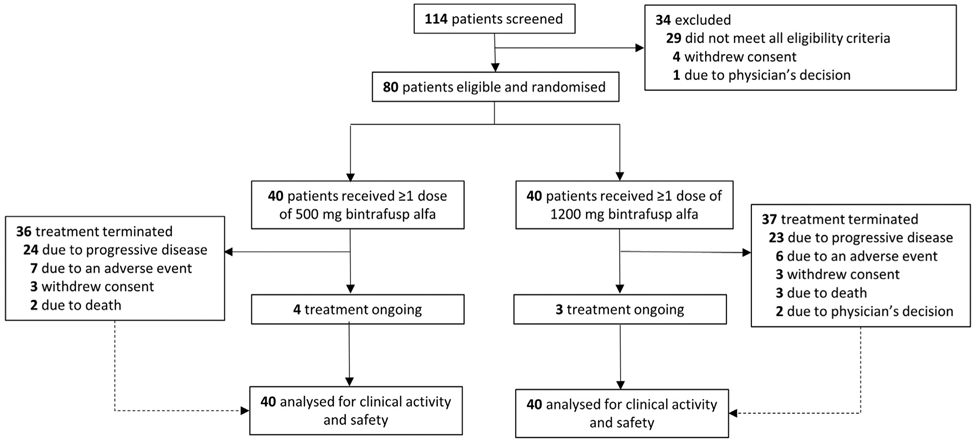
Trial profile. * Programmed death-ligand 1 expression data are not available for two and three patients in the 500 mg and 1200 mg treatment arms, respectively, and were not included in efficacy subgroup analyses.
Table 1.
Patient Baseline Characteristics
| Characteristic | 500 mg (n = 40) | 1200 mg (n = 40) | Overall (N = 80) |
|---|---|---|---|
| Age, median (range), y | 66 (43-85) | 61 (38-79) | 64 (38-85) |
| Sex, n (%) | |||
| Male | 24 (60.0) | 33 (82.5) | 57 (71.3) |
| Female | 16 (40.0) | 7 (17.5) | 23 (28.8) |
| Geographic region, n (%) | |||
| Asia and Pacific | 15 (37.5) | 25 (62.5) | 40 (50.0) |
| Europe | 23 (57.5) | 13 (32.5) | 36 (45.0) |
| North America | 2 (5.0) | 2 (5.0) | 4 (5.0) |
| ECOG PS, n (%) | |||
| 0 | 13 (32.5) | 7 (17.5) | 20 (25.0) |
| 1 | 27 (67.5) | 33 (82.5) | 60 (75.0) |
| Histology, n (%) | |||
| Squamous | 7 (17.5) | 9 (22.5) | 16 (20.0) |
| Nonsquamousa | 33 (82.5) | 31 (77.5) | 64 (80.0) |
| Number of previous anticancer regimens, n (%)b | |||
| 1 | 28 (70.0) | 35 (87.5) | 63 (78.8) |
| 2 | 8 (20.0) | 4 (10.0) | 12 (15.0) |
| ≥3 | 4 (10.0) | 1 (2.5) | 5 (6.3) |
| Type of previous anticancer therapies for metastatic or locally advanced disease | |||
| Cytotoxic therapy | 39 (97.5) | 35 (87.5) | 74 (92.5) |
| Monoclonal antibodies therapy | 4 (10.0) | 1 (2.5) | 5 (6.3) |
| Small molecules | 6 (15.0) | 1 (2.5) | 7 (8.8) |
| Intent of therapy | |||
| Neoadjuvant | 1 (2.5) | 2 (5.0) | 3 (3.8) |
| Adjuvant | 5 (12.5) | 7 (17.5) | 12 (15.0) |
| Metastatic | 40 (100.0) | 36 (90.0) | 76 (95.0) |
| PD-L1 tumor expression, n (%)c | |||
| Negative (<1%) | 7 (17.5) | 10 (25.0) | 17 (21.3) |
| Positive (≥1%) | 31 (77.5) | 27 (67.5) | 58 (72.5) |
| Low (1% to <80%) | 25 (62.5) | 20 (50.0) | 45 (56.3) |
| High (≥80%) | 6 (15.0) | 7 (17.5) | 13 (16.3) |
| Not evaluable | 2 (5.0) | 3 (7.5) | 5 (6.3) |
| EGFR mutation, n of N (%)d | 6 of 33 (18.2) | 4 of 31 (12.9) | 10 of 64 (15.6) |
| ALK translocation, n of N (%)e | 0 of 34 (0) | 0 of 32 (0) | 0 of 66 (0) |
| ROS1 rearrangement, n of N (%)f | 1 of 28 (3.6) | 0 of 24 (0) | 1 of 52 (1.9) |
| Smoking status, n (%) | |||
| Never smoker | 13 (32.5) | 5 (12.5) | 18 (22.5) |
| Current or former smoker | 27 (67.5) | 35 (87.5) | 62 (77.5) |
Nonsquamous histology comprised adenocarcinoma (500 mg group, n = 31; 1200 mg group, n = 28), mixed adenocarcinoma and sarcomatoid (1200 mg group, one patient), poorly differentiated adenosquamous carcinoma (500 mg group, one patient), poorly differentiated carcinoma (500 mg group, one patient), poorly differentiated NSCLC (1200 mg group, one patient), and not specified (1200 mg group, one patient).
Includes previous platinum-based chemotherapy and chemoradiation therapy.
PD-L1 positivity was defined by PD-L1 expression of any intensity greater than or equal to 1% of tumor cells detected by IHC using a proprietary assay (PD-L1 IHC 73-10 pharmDx; Dako, Carpinteria, CA).
Percentage was calculated on the basis of the number of patients with nonsquamous or other histology minus one patient with poorly differentiated adenosquamous carcinoma classified as other (n = 64).
Percentage calculated on the basis of the number of patients tested for ALK translocation.
Percentage calculated on the basis of the number of patients tested for ROS1 rearrangement.
ALK, anaplastic lymphoma kinase; ECOG PS, Eastern Cooperative Oncology Group performance status; IHC, immunohistochemistry; PD-L1, programmed death-ligand 1.
In the overall population, patients received a median of five doses of bintrafusp alfa (interquartile range [IQR], 3.0–14.0 doses) for a median duration of 11.9 weeks (IQR, 5.6–31.9 weeks). At the data cutoff, the median duration of follow-up was 51.9 weeks (IQR, 19.6–74.0 weeks), and seven patients (8.8%) remained on treatment (n = 3 [7.5%] at 1200 mg and n = 4 [10.0%] at 500 mg). Most patients received chemotherapy after the discontinuation of bintrafusp alfa treatment (Supplementary Table 1).
In the overall population, 17 had a confirmed partial response according to RECIST 1.1 adjudicated by the IRC, resulting in an ORR of 21.3%. For those who received 500 mg and 1200 mg doses, the ORRs were 17.5% (seven of 40) and 25.0% (10 of 40), respectively (Table 2). The DCR was 40.0% (32 of 80) overall and was 32.5% (n = 13) and 47.5% (n = 19) for patients who received 500 and 1200 mg, respectively. At data cutoff, responses were ongoing in 10 of 80 patients (12.5%) in the overall population, including three of 40 (7.5%) and seven of 40 (17.5%) in the 500 and 1200 mg groups, respectively (Fig. 2A and B). Most (nine of 10) ongoing responses were observed in patients who had been receiving bintrafusp alfa for more than 12 months. The median duration of response was 14.1 months (95% CI: 7.0–not reached [NR]) in the overall population. For patients receiving 500 and 1200 mg doses, the median duration of response was 14.1 months (95% CI: 7.0–NR) and NR (95% CI: 4.2–NR), respectively (Table 2). Responses on the basis of investigator assessment were comparable to IRC-assessed responses (Supplementary Tables 2 and 3). Responses measured by immune-related RECIST 1.1 were not discussed owing to the lack of pseudoprogression in this study and the ineligibility of certain patients’ responses to be measured by these criteria.
Table 2.
IRC-Adjudicated Efficacy According to RECIST 1.1
| Parameter | 500 mg (n = 40) | 1200 mg (n = 40) | Overall (N = 80) |
|---|---|---|---|
| BOR, n (%) | |||
| Complete response | 0 | 0 | 0 |
| Partial response | 7 (17.5) | 10 (25.0) | 17 (21.3) |
| Stable disease | 5 (12.5) | 8 (20.0) | 13 (16.3) |
| Non-complete response or non-progressive diseasea | 1 (2.5) | 1 (2.5) | 2 (2.5) |
| Progressive disease | 22 (55.0) | 17 (42.5) | 39 (48.8) |
| Not evaluable | 5 (12.5) | 4 (10.0) | 9 (11.3) |
| ORR, n (%) | 7 (17.5) | 10 (25.0) | 17 (21.3) |
| DCR, n (%)b | 13 (32.5) | 19 (47.5) | 32 (40) |
| ORR in PD-L1-evaluable patients, n of N (%) | |||
| All | 7 of 38 (18.4) | 10 of 37 (27.0) | 17 of 75 (22.7) |
| PD-L1 negativec | 1 of 7 (14.3) | 0 of 10 (0) | 1 of 17 (5.9) |
| PD-L1 positived | 6 of 31 (19.4) | 10 of 27 (37.0) | 16 of 58 (27.6) |
| Lowe | 4 of 25 (16.0) | 4 of 20 (20.0) | 8 of 45 (17.8) |
| Highf | 2 of 6 (33.3) | 6 of 7 (85.7) | 8 of 13 (61.5) |
| Median PFS (95% CI), mo | |||
| All | 1.4 (1.3-4.2) | 4.0 (1.3-9.5) | 2.6 (1.3-4.2) |
| PD-L1 negativec | 1.4 (0.3-10.9) | 1.3 (0.9-4.0) | 1.3 (1.3-4.0) |
| PD-L1 positived | 1.4 (1.2-4.2) | 9.5 (2.6-15.2) | 2.7 (1.4-9.6) |
| Lowe | 1.4 (1.2-4.2) | 5.5 (1.3-11.0) | 2.6 (1.3-9.5) |
| Highf | 1.3 (0.2-NR) | 15.2 (1.3-NR) | 15.2 (1.0-NR) |
| Median DOR (95% CI), mo | 14.1 (7.0-NR) | NR (4.2-NR) | 14.1 (7.0-NR) |
Persistence of one or more nontarget lesions or maintenance of tumor marker level above the normal limits.
DCR included those patients who achieved a complete response, partial response, or stable disease (including non-complete response or non-progressive disease).
PD-L1 expression on less than 1% of tumor cells.
PD-L1 expression on greater than or equal to 1% of tumor cells.
PD-L1 expression on 1% to less than 80% of tumor cells.
PD-L1 expression on greater than or equal to 80% of tumor cells.
BOR, best overall response; CI, confidence interval; DCR, disease control rate; DOR, duration of response; IRC, independent review committee; NR, not reached; ORR, objective response rate; PD-L1, programmed death-ligand 1; PFS, progression-free survival; RECIST, Response Evaluation Criteria in Solid Tumors.
Figure 2.

Change from baseline in the sum of target lesion diameters over time according to RECIST 1.1 and as adjudicated by the IRC in patients treated with (A) 500 mg and (B) 1200 mg of bintrafusp alfa. IRC, independent review committee. NE, not evaluable; PD, progressive disease; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease. *Patient had a greater than 200% increase in target lesion diameter from baseline.
Overall, 74 of 80 patients (92.5%) had an AE (any grade); 55 of 80 (68.8%) had a TRAE (500 mg, 27 of 40 [67.5%]; 1200 mg, 28 of 40 [70.0%]), of which the most common (occurring in ≥10% patients) were pruritus (21.3%), maculopapular rash (18.8%; reported by the investigator as MedDRA version 21.0 preferred term [PT] of “rash maculopapular”), decreased appetite (12.5%), asthenia (11.3%), and rash (10.0%; reported by the investigator as MedDRA version 21.0 PT of “rash”) (Fig. 3) (Supplementary Table 4). One of 80 patients (1.3%; 500 mg) had an IRR, which was grade 1. Immune-related AEs occurred in 14 of 80 patients (17.5%). TRAEs occurred in 23 of 80 patients (28.8%; 500 mg, 13 of 40 [32.5%]; 1200 mg, 10 of 40 [25.0%]), of which two were grade 4 (2.5%; one of 40 [2.5%] each per dose). The most common grade 3 TRAEs were skin and subcutaneous tissue disorders (nine of 80 [11.3%]), mostly maculopapular rash (six of 80 [7.5%]), and increased lipase (four of 80 [5.0%]). Grade 4 TRAEs occurred in two patients and were asymptomatic laboratory abnormalities and did not lead to treatment discontinuation; they included hypokalemia and decreased blood magnesium level in one patient (1.25%; 500 mg) and increased amylase and lipase levels without symptoms of clinical pancreatitis in the other patient (1.25%; 1200 mg) (Supplementary Table 4). Skin lesion events (seven of 80 [8.8%]), which were potentially TGF-β–mediated, included hyperkeratosis (two of 80 [2.5%]), KA (four of 80 [5.0%]), and squamous cell carcinoma of the skin (two of 80 [2.5%]), most of which were grade 1 or 2. One patient had both a KA and squamous cell carcinoma of the skin. These skin lesions occurred irrespective of bintrafusp alfa dose and did not impact response to treatment (four of seven affected patients had a partial response). Patients who developed these lesions were adequately managed with simple excision when indicated and did not require discontinuation from the treatment or trial. TRAEs led to permanent treatment discontinuation in eight of 80 patients (10.0%). Reasons for permanent treatment discontinuation include adrenal insufficiency (one patient), colitis (one patient; reported by the investigator as MedDRA v21.0 PT of “colitis” on the basis of specific biopsy findings), autoimmune colitis (one patient; reported by the investigator as MedDRA v21.0 PT of “autoimmune colitis” on the basis of specific biopsy findings), cholestasis (one patient), increased alanine aminotransferase (one patient), increased aspartate aminotransferase (one patient), increased gamma-glutamyl transferase (one patient), blood alkaline phosphatase increased (one patient), and maculopapular rash (three patients). No treatment-related deaths occurred.
Figure 3.

Treatment-related adverse events of any grade occurring in 3% or more patients or of grade 3 or worse in any patients.
Across all patients, the median PFS was 1.6 months (95% CI: 1.3–4.2 mo), the 12-month PFS rate was 20.1% (95% CI: 11.2%−30.8%), and the median OS was 13.6 months (95% CI: 10.9–NR). Median PFS in the 500 and 1200 mg groups was 1.4 months (95% CI: 1.3–4.2 mo) and 2.7 months (95% CI: 1.3–8.1 mo), respectively; and the 12-month PFS rates were 16.3% (95% CI: 5.6%−31.9%) and 23.6% (95% CI: 10.9%−39.0%) (Fig. 4A) (Supplementary Fig. 1). Median OS in the 500 mg and 1200 mg groups was 10.9 months (95% CI: 4.5–NR) and 15.6 months (95% CI: 12.0–NR), respectively; 12-month OS rates were 44.6% (95% CI: 28.5%–59.5%) and 65.5% (95% CI: 47.0%–78.9%) (Fig. 4B) (Supplementary Fig. 1).
Figure 4.

(A) Progression-free survival (IRC adjudicated) and (B) overall survival in PD-L1-evaluable patients who received bintrafusp alfa 1200 mg. IRC, independent review committee; NR, not reached; PD-L1, programmed death-ligand 1; CI, confidence interval.
Efficacy by PD-L1 expression was also evaluated. For patients receiving the recommended phase 2 dose of 1200 mg, the ORR was 37.0% in patients with PD-L1–positive tumors (response in 10 of 27 patients) and 85.7% in patients with PD-L1–high tumor expression (response in six of seven patients) (Table 2). Median PFS in the 1200 mg group was 9.5 months (95% CI: 2.6–15.2 mo) in patients with PD-L1–positive tumors, and 15.2 months (95% CI: 1.3-NR) in patients with PD-L1–high tumor expression (Fig. 4A); 12-month PFS rates were 35.4% (95% CI: 16.6%–54.9%) and 68.6% (95% CI: 21.3%–91.2%), respectively. OS was also improved with increasing PD-L1 expression (Fig. 4B). These trends of increased ORR, PFS, and OS at higher PD-L1 expression levels were also observed for the 500 mg group (Supplementary Figs. 1 and 2). Responses occurred in patients with squamous or nonsquamous tumor histologic types, and similar to the 1200 mg group, clinical activity was improved with increasing PD-L1 expression for both histologic types (Supplementary Fig. 3). Efficacy by EGFR mutation status was assessed in patients with nonsquamous tumor histology (n = 64). Of 10 patients with tumors that were positive for EGFR mutations, one patient had a response to bintrafusp alfa treatment; this patient had received the 1200 mg dose and had PD-L1–high expression on tumor cells. A total of 12 of 54 patients without EGFR mutations had a response in this study, six patients received the 500 mg dose and six patients received the 1200 mg dose. In the single patient who had a ROS1 rearrangement (500 mg subgroup; PD-L1–low expressing tumor), BOR was stable disease.
The tumor immune phenotype was evaluable at baseline in archival tumor samples from 69 patients. Of these, 17 (25.0%) were immune-inflamed, six (8.7%) were immune-desert, 40 (58.0%) were immune-excluded, two (2.9%) were indeterminate, and four (5.8%) were not annotated. Responses were found in tumors with immune-inflamed (six [35.3%]) and immune-excluded phenotypes (eight [20.0%]; Supplementary Fig. 4). In addition, a best response of stable disease occurred in two patients (11.8%) with the immune-inflamed phenotype and 10 patients (25.0%) with the immune-excluded phenotype. Analyses of gene expression (Supplementary Figs. 5 and 6) revealed that no genes or gene signatures related to the immune system or TGF-β pathway were associated with response in these patients regardless of PD-L1 expression level.
Discussion
Data from this phase 1 expansion cohort revealed that bintrafusp alfa monotherapy has encouraging efficacy and manageable tolerability in patients with advanced platinum-experienced NSCLC, particularly in patients with PD-L1–high tumors. In previous analyses, the recommended phase 2 dose of 1200 mg was selected on the basis of data from two clinical trials, supported by population PK and exposure-response modeling and simulation (manuscript in preparation).17 In this cohort, efficacy, as measured by ORR, DCR, PFS, and OS, was numerically higher in patients who received the 1200 mg dose compared with the 500 mg dose; however, the cohort was not powered to compare the efficacy between dose subgroups. The confirmed ORR data, as assessed by an independent review of 21.3% in the overall patient population, is encouraging. In previous studies of avelumab (anti–PD-L1 antibody) and pembrolizumab (anti–PD-1 antibody), in patients with previously treated metastatic NSCLC unselected for PD-L1 expression, ORRs were 12.0% and 18.0%, respectively.20,21 At the recommended phase 2 dose, bintrafusp alfa had an ORR per IRC of 25.0% in the overall population and 85.7% in patients with PD-L1–high tumors.
Clinical activity was observed across PD-L1 expression levels and tumor histologic types (squamous and nonsquamous). In this study, PD-L1 expression on tumor cells was analyzed using the 73-10 antibody clone assay, which has been used in studies of avelumab.20,22,23 A recent comparison of the 73-10 assay and the 22C3 assay, used in pembrolizumab trials, revealed that the 73-10 assay has greater sensitivity than the 22C3 assay, identifying a slightly higher proportion of patients with PD-L1–positive tumors from NSCLC samples.19,24 This suggests that the greater than or equal to 80% with the 73-10 assay identifies a comparable subgroup to the greater than or equal to 50% cutoff with the 22C3 assay. In a previous study, the proportion of advanced NSCLC samples identified as having PD-L1–high expression using the greater than or equal to 80% cutoff of 73-10 was 23.6%, which is comparable to the 20.3% of samples identified using the greater than or equal to 50% cutoff of 22C3.19 In separate phase 3 trials in patients with platinum-treated NSCLC (JAVELIN Lung 200 and KEYNOTE-010), the greater than or equal to 80% cutoff for 73-10 and greater than or equal to 50% cutoff for 22C3 identified similar proportions of patients (29% versus 28%, respectively).4,25 In this cohort, relatively few patients had PD-L1–high tumors (16.3%) compared with previous experience; enrollment of patients with PD-L1–high tumors might have been impacted by the use of pembrolizumab as first-line treatment for patients with PD-L1–high tumors26 and by the fact that patients in this cohort were required to be immunotherapy naive.
Historical responses in patients with PD-L1–positive tumors (≥1% cutoff, 22C3 assay) have ranged from 18.0% to 27.6%; with PD-L1–high tumors (≥50% cutoff, 22C3 assay), they have ranged from 29.1% to 43.9%.4,21 In this study, patients receiving the recommended phase 2 dose with PD-L1–positive tumors (n = 27) had an ORR of 37.0% and patients with PD-L1–high expression (seven patients) had an encouraging ORR of 85.7%; these ORRs seem favorable compared with previous studies, with the limitation of small sample size.
Analyses of PFS were also encouraging. The median PFS reported for patients with PD-L1–positive NSCLC treated with anti–PD-1 or anti–PD-L1 antibodies has ranged from 2.8 to 5.2 months.3,4,25,27 The median PFS reported here in patients with PD-L1–positive tumors who received the recommended phase 2 dose was 9.5 months; the median PFS in those who had PD-L1–high tumors was 15.2 months. This trend was also seen in OS analyses (Fig. 4).
Preclinical data suggest that improved clinical efficacy may be observed by targeting both TGF-β and PD-L1 pathways compared with either pathway alone. Furthermore, as a bifunctional fusion protein, bintrafusp alfa may have improved efficacy compared with simply treating patients separately with both PD-L1 and TGF-β–targeting agents.13 This is hypothesized to occur with bintrafusp alfa binding to PD-L1 in the TME, which may facilitate local TGF-β trapping more effectively than administering these treatments separately.
In the immune-profiling analyses, responses were observed in patients with immune-excluded tumors and immune-inflamed tumors. Of note, there were few tumor samples categorized as immune-excluded, and larger sample sets will be needed to replicate the current findings. Although previous studies would suggest that patients with immune-inflamed tumors were more responsive to PD-L1 targeting,11 these immunophenotyping analyses further suggest a novel mechanism of action within the local TME compared with PD-L1 targeting alone. In this study, although tumor immune phenotypes were assessed using specific criteria on the basis of published work,11 it is recognized that there are no validated or broadly accepted methods to determine immune phenotypes, which may limit these analyses.
The overall safety profile of bintrafusp alfa is similar to that of established checkpoint inhibitors, except for skin lesion TRAEs (KAs, squamous cell carcinoma of the skin, and hyperkeratosis), which were manageable and did not lead to treatment discontinuation. KAs are rarely reported with established checkpoint inhibitors. However, KAs and squamous cell carcinoma of the skin are associated with BRAF inhibition and inhibited TGF-β signaling.28,29 In addition, similar skin lesions have been described in individuals with certain inherited mutations or truncations of TGF-β, such as Ferguson-Smith disease.30 Therefore, the occurrence of KAs in this study is presumably related to the function of the TGF-β trap portion of bintrafusp alfa. Similarly, in previous phase 1 studies of fresolimumab, an anti–TGF-β monoclonal antibody, rates of KAs, hyperkeratosis, and squamous cell carcinoma of the skin were similar, all of which resolved after treatment completion, demonstrating that they were not autonomous malignancies.31,32 Although no cases of pneumonitis were reported for this cohort, incidences have been reported with bintrafusp alfa in other cohorts.33 Therefore, we think it is too early to hypothesize on the impact of TGF-β inhibitions on individual events typically associated with checkpoint inhibitors.34
In conclusion, the clinical activity of bintrafusp alfa observed in this expansion cohort of patients with advanced or metastatic NSCLC that had progressed after standard first-line treatment is encouraging, particularly in patients with tumors that had higher PD-L1 expression. These data formed the basis for further trials, evaluating bintrafusp alfa in various NSCLC treatment settings, including a recently initiated phase 2 study of bintrafusp alfa versus pembrolizumab as first-line treatment for patients with PD-L1–high advanced NSCLC (ClinicalTrials.gov number NCT03631706).
Supplementary Material
Acknowledgments
LP-A was funded by ISCIII (PI14/01964, PIE15/00076, PI17/00778, and DTS17/00089) and CIBERONC (CD16/12/00442) and cofunded by Fonds européen de développement régional from Regional Development European Funds (European Union). Medical writing support was provided by Abhijith Thippeswamy, of ClinicalThinking, which was also funded by Merck KGaA and GlaxoSmithKline in accordance with Good Publication Practice guidelines (http://www.ismpp.org/gpp3). Merck KGaA, Darmstadt, Germany, provided the study drug and worked with investigators on the trial design and plan, collection and analysis of data, and interpretation of results. The authors thank Christian Ihling, of Merck KGaA, for his substantial contribution to the immune phenotype analysis.
Footnotes
Disclosure: Dr. Paz-Ares is an external member on the board of Genomica; has received honoraria from Adacap, Amgen, AstraZeneca, Bayer, Blueprint, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Incyte, Ipsen, Lilly, Merck KGaA (Darmstadt, Germany), Merck Sharp & Dohme, Novartis, Pfizer, PharmaMar, Roche, Sanofi, Servier, and Sysmex; is cofounder and board member of Altum Sequencing; and has served in nonfinancial senior leadership roles in other medical societies, research groups, foundations, and political pressure groups. Dr. Kim has received research funding from AstraZeneca-KHIDI. Dr. Felip has served as a speaker for AbbVie, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Eli Lilly, Medscape, Merck KGaA, Merck Sharp & Dohme, Novartis, Pfizer, Prime Oncology, Roche, and Takeda, and has served in advisory roles for all of the aforementioned companies in addition to BerGenBio, Blueprint Medicines, Guardant Health, Janssen, Samsung, and Touch Time. Dr. D.H. Lee has received honoraria from AbbVie, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, ChongKeunDang, CJ Healthcare, Eli Lilly, Genexine, Janssen, Merck KGaA, Merck Sharp & Dohme, Mundipharma, Novartis, Ono, Pfizer, Roche, Samyang Bio-Pharm, ST Cube, and Takeda. Dr. K.H. Lee has served in advisory roles for AstraZeneca, Bristol-Myers Squibb, and Merck Sharp & Dohme. Dr. Lin has received honoraria from BeiGene, Boehringer Ingelheim, Daiichi-Sankyo, Novartis, Roche, and Takeda. Alvarez has received honoraria from Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Novartis, PharmaMar, and Roche/Genentech. Drs. Dussault and Ojalvo are employees of EMD Serono Research and Development Institute, Inc. (a business of Merck KGaA). Mr. Helwig is an employee of Merck KGaA, Darmstadt, Germany. Dr. Gulley reports that his institute has a Cooperative Research and Development Agreement with EMD Serono Research and Development Institute, Inc.; is a coprimary investigator on the Cooperative Research and Development Agreement but with no personal financial interests; and is an unpaid member of the expert oncology faculty that advises EMD Serono Research and Development Institute, Inc. Dr. Cho has received research funding from AstraZeneca, Bayer, Champions Oncology, Dizal Pharma, Dong-A ST, Janssen, Merck Sharp & Dohme, MOGAM Institute, Novartis, Ono, and Yuhan; has provided consultancy to AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Janssen, Merck Sharp & Dohme, Novartis, Ono, Pfizer, Roche, Takeda, and Yuhan; has stock in TheraCanVac Inc., Gencurix Inc., and Bridgebio therapeutics; and receives royalties from Champions Oncology. The remaining authors declare no conflict of interest.
Data availability statement
For all new products or new indications approved in both the European Union and the US after January 1, 2014, Merck KGaA, Darmstadt, Germany, will share patient-level and study-level data after deidentification and redacted study protocols and clinical study reports from clinical trials in patients. These data will be shared with qualified scientific and medical researchers, on the researcher’s request, as necessary, for conducting legitimate research. Such requests must be submitted in writing to the company’s data sharing portal. More information can be found at https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. Where Merck KGaA has a coresearch, codevelopment, comarketing, or copromotion agreement or where the product has been out-licensed, it is recognized that the responsibility for disclosure may be dependent on the agreement between parties. Under these circumstances, Merck KGaA will endeavor to gain agreement to share data in response to requests.
References
- 1.Ettinger DS, Woods DE, Aisner DL, et al. Non-small cell lung cancer, version 5.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2017;15:504–535. [DOI] [PubMed] [Google Scholar]
- 2.Horn L, Spigel DR, Vokes EE, et al. Nivolumab versus docetaxel in previously treated patients with advanced non-small-cell lung cancer: two-year outcomes from two randomized, open-label, phase III trials (CheckMate 017 and CheckMate 057). J Clin Oncol. 2017;35:3924–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–1550. [DOI] [PubMed] [Google Scholar]
- 5.Akhurst RJ. Targeting TGF-β signaling for therapeutic gain. Cold Spring Harb Perspect Biol. 2017;9:a022301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barthelemy-Brichant N, David JL, Bosquée L, et al. Increased TGFbeta1 plasma level in patients with lung cancer: potential mechanisms. Eur J Clin Investig. 2002;32:193–198. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Shen C, Wang X, et al. Prognostic value of TGF-β in lung cancer: systematic review and meta-analysis. BMC Cancer. 2019;19:691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferris RL, Lenz HJ, Trotta AM, et al. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat Rev. 2018;63:48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hugo W, Zaretsky JM, Sun L, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravi R, Noonan KA, Pham V, et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat Commun. 2018;9:741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–543. [DOI] [PubMed] [Google Scholar]
- 13.Lan Y, Zhang D, Xu C, et al. Enhanced preclinical anti-tumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci Transl Med. 2018;10:eaan5488. [DOI] [PubMed] [Google Scholar]
- 14.Knudson KM, Hicks KC, Luo X, Chen J-Q, Schlom J, Gameiro SR. M7824, a novel bifunctional anti-PD-L1/TGFβ TRAP fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology. 2018;7:e1426519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.David JM, Dominguez C, McCampbell KK, Gulley JL, Schlom J, Palena C. A novel bifunctional anti-PD-L1/TGF-β TRAP fusion protein. Oncoimmunology. 2017;6:e1349589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strauss J, Heery CR, Schlom J, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFβ, in advanced solid tumors. Clin Cancer Res. 2018;24:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vugmeyster Y, Wilkins J, Harrison-Moench E, et al. Selection of the recommended phase 2 dose (RP2D) for M7824 (MSB0011359C). J Clin Oncol. 2018;36(suppl 16):2566.29945529 [Google Scholar]
- 18.Paz-Ares LG, Kim TM, Vicente Baz D, et al. Results from a second-line (2L) NSCLC cohort treated with M7824 (MSB0011359C), a bifunctional fusion protein targeting TGF-β and PD-L1. J Clin Oncol. 2018;36(suppl 15):9017. [Google Scholar]
- 19.Feng Z, Schlichting M, Helwig C, et al. Comparative study of two PD-L1 expression assays in patients with non-small cell lung cancer (NSCLC). J Clin Oncol. 2017;35(suppl 15):e20581. [Google Scholar]
- 20.Gulley JL, Rajan A, Spigel DR, et al. Avelumab for patients with previously treated metastatic or recurrent non-small-cell lung cancer (JAVELIN Solid Tumor): dose-expansion cohort of a multicentre, open-label, phase 1b trial. Lancet Oncol. 2017;18:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. [DOI] [PubMed] [Google Scholar]
- 22.Apolo AB, Infante JR, Balmanoukian A, et al. Avelumab, an anti-programmed death-ligand 1 antibody, in patients with refractory metastatic urothelial carcinoma: results from a multicenter, phase lb study. J Clin Oncol. 2017;35:2117–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17:1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsao M, Kerr K, Yatabe Y, Hirsch FR. PL 03.03 Blueprint 2: PD-L1 immunohistochemistry comparability study in real-life, clinical samples. J Thorac Oncol. 2017;12(suppl 2):S1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barlesi F, Vansteenkiste J, Spigel D, et al. Avelumab versus docetaxel in patients with platinum-treated advanced non-small-cell lung cancer (JAVELIN Lung 200): an open-label, randomised, phase 3 study. Lancet Oncol. 2018;19:1468–1479. [DOI] [PubMed] [Google Scholar]
- 26.Pai-Scherf L, Blumenthal GM, Li H, et al. FDA approval summary: pembrolizumab for treatment of metastatic non-small cell lung cancer: first-line therapy and beyond. Oncologist. 2017;22:1392–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837–1846. [DOI] [PubMed] [Google Scholar]
- 28.Rose AM, Sansom OJ, Inman GJ. Loss of TGF-β signaling drives cSCC from skin stem cells—more evidence. Cell Cycle. 2017;16:386–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibney GT, Messina JL, Fedorenko IV, Sondak VK, Smalley KS. Paradoxical oncogenesis and the long term consequences of BRAF inhibition in melanoma. Nat Rev Clin Oncol. 2013;10:390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goudie DR, D’Alessandro M, Merriman B, et al. Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat Genet. 2011;43:365–369. [DOI] [PubMed] [Google Scholar]
- 31.Lacouture ME, Morris JC, Lawrence DP, et al. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol Immunother. 2015;64:437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris JC, Tan AR, Olencki TE, et al. Phase I studyof GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFb) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9:e90353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoo C, Oh DY, Choi H, et al. M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGF-β, in Asian patients with pretreated biliary tract cancer: preliminary results from a phase 1 trial. Ann Oncol. 2018;29(suppl 8):viii258–viii259. [Google Scholar]
- 34.Nishino M, Giobbie-Hurder A, Hatabu H, Ramaiya NH, Hodi FS. Incidence of programmed cell death 1 inhibitor-related pneumonitis in patients with advanced cancer: a systematic review and meta-analysis. JAMA Oncol. 2016;2:1607–1616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For all new products or new indications approved in both the European Union and the US after January 1, 2014, Merck KGaA, Darmstadt, Germany, will share patient-level and study-level data after deidentification and redacted study protocols and clinical study reports from clinical trials in patients. These data will be shared with qualified scientific and medical researchers, on the researcher’s request, as necessary, for conducting legitimate research. Such requests must be submitted in writing to the company’s data sharing portal. More information can be found at https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. Where Merck KGaA has a coresearch, codevelopment, comarketing, or copromotion agreement or where the product has been out-licensed, it is recognized that the responsibility for disclosure may be dependent on the agreement between parties. Under these circumstances, Merck KGaA will endeavor to gain agreement to share data in response to requests.