

Abstract
The relationship between impaired calcium sensing, dysregulated parathyroid hormone (PTH) secretion, and parathyroid cell proliferation in parathyroid neoplasia is not understood. We previously reported that a GTPase activating protein, regulator of G-protein signaling 5 (RGS5) is overexpressed in a subset of parathyroid tumors associated with primary hyperparathyroidism (PHPT), and that RGS5 can inhibit signaling from the calcium-sensing receptor (CASR). In vivo, we found that RGS5-null mice have abnormally low PTH levels. To gain a better understanding of the potential role of RGS5 overexpression in parathyroid neoplasia and PHPT and to investigate whether inhibition of CASR signaling can lead to parathyroid neoplasia, we created and characterized a transgenic mouse strain over-expressing RGS5 specifically in the parathyroid gland. These mice develop hyperparathyroidism, bone changes reflective of elevated PTH, and parathyroid neoplasia. Further, expression of exogenous RGS5 in normal human parathyroid cells results in impaired signaling from CASR and negative feedback on PTH secretion. These results provide evidence that RGS5 can modulate signaling from CASR and support a role for RGS5 in the pathogenesis of PHPT through inhibition of CASR signaling.
Keywords: Primary hyperparathyroidism, calcium sensing receptor, RGS5, parathyroid
Introduction
Primary hyperparathyroidism (PHPT) is a disorder of bone and mineral metabolism resulting from inappropriate secretion of the parathyroid hormone (PTH) by parathyroid tumors. While inappropriate secretion of PTH is the hallmark of all hyperparathyroid states, the interplay between pathologic hypersecretion of hormone and abnormal parathyroid cell proliferation is not well understood. Specifically, there is uncertainty as to whether the abnormal Ca+2-PTH feedback observed in PHPT is a cause or effect of parathyroid neoplasia.
The most widely held view of parathyroid tumorigenesis in PHPT holds that somatic mutations in parathyroid tissue lead to a proliferative and/or survival advantage in transformed clones. Frequent somatic mutational events known to drive parathyroid tumorigenesis in PHPT include the PTH-cyclin D1 (PRAD1) gene rearrangement and loss-of-function point mutations in the multiple endocrine neoplasia tumor suppressor gene MEN1 (1–5). Parathyroid-specific transgenic mouse models harboring these mutations demonstrate biochemical and skeletal features of PHPT with parathyroid neoplasia in the PTH-cyclin D1 model (6) and elevated calcium with parathyroid neoplasia in the MEN1 model (7). Evidence that abnormal PTH secretion occurs in advance of parathyroid adenoma formation and that the allosteric CASR modulator, cinacalcet, may inhibit parathyroid proliferation in the PTH-cyclin D1 transgenic model indicates that deficient calcium sensing and disrupted feedback to PTH occurs early in the pathogenic process of parathyroid cell hyperplastic expansion (8).
An alternative model for the origin of parathyroid tumors proposes that attenuated calcium sensing in parathyroid cells initially drives elevated secretion of PTH, followed by secondary parathyroid cell proliferation in response to chronic demand for increased PTH (9–13). Evidence in support of this hypothesis is largely correlative, based on expression studies in parathyroid tumor cells showing reduced CASR expression (14) and altered levels of molecules known to modulate CASR expression and signaling including the vitamin D receptor (VDR), heterotrimeric G protein (Gα) subtypes, regulator of G-protein signaling 5 protein (RGS5), and G protein coupled receptor 64 (GPR64) (15–18). Mouse models evaluating abnormal calcium sensing in the parathyroid gland have shown that loss or inhibition of CASR signaling can lead to elevated PTH and parathyroid hyperplasia but not parathyroid tumors (17, 19, 20). Heterozygous CASR knockout mice manifest mild hypercalcemia and elevated PTH without parathyroid gland abnormalities while homozygous CASR knockout mice present with severe PHPT and parathyroid hyperplasia without adenomas (19). Parathyroid-specific double knockout of Gαq/Gα11 phenocopies CASR-null mice with hypercalcemia, elevated PTH, and parathyroid cell hyperplasia without tumors (20).
Despite insights from these established models, it remains unclear why parathyroid tumors are insensitive to calcium feedback to PTH secretion and whether impaired CASR signaling can actually lead to parathyroid tumors. We previously reported that RGS5, a GTPase activating protein (GAP) that targets Gαq, is expressed in parathyroid tissue, is selectively up-regulated in parathyroid tumors relative to normal glands, and can inhibit calcium signaling through CASR (16). We found that RGS5-null mice display significantly reduced levels of circulating plasma PTH with preserved responses to changes in calcium, consistent with a constitutively elevated baseline of CASR signaling activity. These data suggested that opposing CASR signaling through increased expression of RGS5 in the parathyroid could result in inappropriate PTH secretion and afford a unique opportunity to test whether RGS5 can contribute to parathyroid neoplasia. To determine whether RGS5 overexpression in the parathyroid gland can lead to PHPT, we generated a strain of mice overexpressing RGS5 specifically in the parathyroid gland (PTG-RGS5) using a Cre-lox strategy. Characterization of this strain revealed that overexpression of RGS5 in the murine parathyroid gland results in elevated PTH secretion and parathyroid neoplasia as well as a bone phenotype reflecting abnormally elevated production of PTH. Additionally, overexpression of RGS5 increased PTH secretion by normal human parathyroid cells and reduced calcium signaling in a HEK-CASR stable cell line and in human normal parathyroid cells, mechanistically supporting our in vivo observations. The mouse model described here provides direct experimental support for the involvement of RGS5 in CASR signaling, PTH secretion, and neoplasia in parathyroid tissue. Further analysis of this model could reveal a novel mechanistic basis for hyperparathyroidism caused by inhibition of CASR signaling in the parathyroid gland.
Materials and Methods
A brief description of methods is provided here. For detailed protocols and reagents see supplemental material.
Generation of transgenic mice
Transgenic mice expressing RGS5 specifically in the parathyroid gland were produced by generating a strain harboring a ROSA26-targeted RGS5 floxed-Stop expression cassette and then crossing this line to PTH-Cre transgenic mice (Jackson Laboratory strain 129;FVB-Tg(PTH-Cre)4167Slib/J, stock number 005989). All studies were performed following a protocol approved by the Institutional Animal Care and Use Committees at Duke University and The University of Maryland, Baltimore. Detailed approaches for generation and validation of transgenic mouse and histologic, immunohistochemical and biochemical examination are provided in supplemental material section.
Bone micro-computed tomography (μCT) measurements
Assessment of mouse bone morphology by μCT was performed and are reported according to guidelines published in (21). Femurs from 12 month old male and female mice were scanned at a 10 μm voxel size, using a Skyscan 1172 system (Bruker MicroCT, Kontich, Belgium), as described previously (22, 23). The following settings were applied: X-ray power and tube current were 60kV and 0.167 mA, respectively. Beam hardening (20%) was reduced with a 0.5mm aluminum filter, ring artifact reduction was set at 5, and exposure time was 5.9 seconds. Final images were generated from an average of 10 pictures taken at 0.9° angles. Bone microarchitectural patterns were assessed in the cortical and trabecular regions of femurs in all mice using Skyscan software packages (NRecon, CtAn, and Dataviewer), as described previously (22, 23).
In vitro assays in HEK-CASR cells
Generation and maintenance of HEK293 cells stably expressing FLAG-tagged human CASR was described previously (15). Detailed description of assays performed with HEK-CASR cells are provided in supplemental material section (15).
Human parathyroid cell isolation
Normal human parathyroid glands were collected according to an IRB-approved protocol from deceased eucalcemic organ donors at the University of Maryland (HP-00053228). Informed consent was obtained from next of kin to allow harvesting of parathyroid glands for research after collection of donor organs for transplantation. Representative portions of resected parathyroid glands were formalin fixed and stained with haematoxylin and eosin to confirm parathyroid tissue identity. Isolation of primary human parathyroid cells was performed as described previously (15, 24). Dispersed cells were allowed to recover overnight at 37°C in KSFM media (Thermo Fisher Scientific# 37010–022) supplemented with the manufacturer’s provided media supplements, antibiotics and 1.25 mM Ca2+.
Human PTH measurement
Normal human parathyroid sells were transduced with LacZ (control) or RGS5 expressing lentivirus for 24 hours in KSFM media (Thermo Fisher Scientific# 37010–022) supplemented with media supplements, antibiotics and 1.25 mM Ca2+. Cells were then centrifuged and transferred to KSFM media with various concentrations of Ca2+ for 1 hour at 37°C. Supernatants were collected by centrifugation at 10,000 rpm, 5 min at 4°C and were stored at −80° C for determination of secreted PTH. Human intact PTH (1–84) in cell supernatants was measured in an ELISA assay (#60–3000, Immutopics, San Diego, CA), following standard curve generation per manufacturer’s instructions (15, 24). IC50 of Ca2+ on CASR-mediated suppression of PTH secretion was derived from curves generated in Prism software by four-parameter fitting with variable slopes.
Statistics
All data were analyzed in GraphPad Prism software version 7.0 (La Jolla, Calif). Parametric t-test or non-parametric Mann-Whitney t-test were used for statistical comparisons and P values less than 0.05 were considered significant. Statistical outliers were determined using the Grubb’s test.
Results
Generation of parathyroid-specific RGS5 overexpressing mice
A transgenic mouse strain engineered for conditional expression of a bicistronic RGS5-myc/eGFP transcript following Cre-mediated activation was generated as depicted in Figure 1A (25). Successful integration of the targeting construct into the murine ROSA26 locus was confirmed by PCR to demonstrate the presence of the bicistronic transgene (S1/S2 amplicon, Fig. 1B) and correct insertional orientation (G1/G2 amplicon, Fig. 1B) in tail snip genomic DNA from heterozygous and homozygous RGS5-myc/eGFPfl/fl mice. The RGS5-myc/eGFP transgene and cloning junctions were sequenced in their entirety in the targeting construct and in genomic DNA from transgenic founder animals. Amplification with the G1/S2 primer pair produces a 2.2 kb amplicon from RGS5-myc/eGFPfl/fl cells only upon excision of the PGK-Neo-STOP cassette from the transgene locus by Cre recombinase (Figure 1C). This result confirms the RGS5-myc/eGFPfl/fl transgenic strain as an appropriate Cre-dependent conditional model for parathyroid tissue-specific expression following cross-breeding to PTH-Cre mice (7). To demonstrate Cre-dependent excision of the PGK-Neo-STOP sequence and expression of RGS5, lung fibroblasts derived from RGS5-myc/eGFPfl/fl or WT littermate mice were transduced with a Cre-expressing lentiviral construct. RGS5-myc protein was detected in RGS5-myc/eGFPfl/fl lung fibroblasts transduced with Cre (Figure 1D), but not in lung fibroblasts derived from WT or PTH-Cre mice. Parathyroid gland-specific expression of the RGS5 transgene was confirmed in parathyroid tissue sections by staining for GFP (Figure 1E, middle panel) and PTH (Figure 1 F). Control (RGS5-myc/eGFPfl/fl and PTH-Cre) mice showed no evidence of RGS5-myc expression (Figure 1D) or GFP staining (Figure 1E, middle panel). PTG-RGS5 (RGS5-myc/eGFPfl/fl-PTH-Cre) mice showed higher expression of RGS5 than the endogenous RGS5 expressed by control littermates (RGS5-myc/eGFPfl/fl) (Figure 1E, right panel). PTG-RGS5 and control mice showed no significant differences in appearance, growth, weight, or serum sodium and potassium values (Supplemental table 1).
Figure 1.
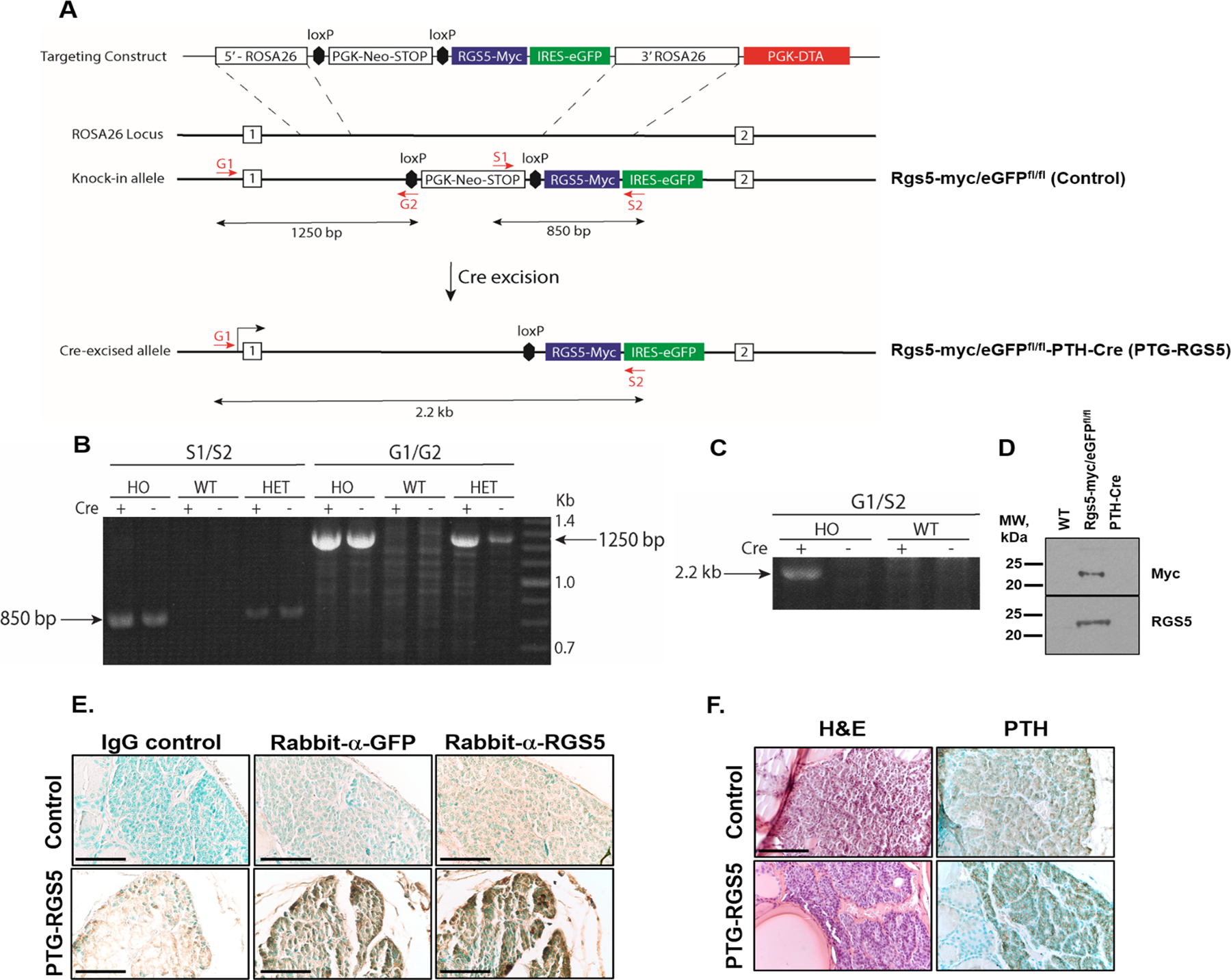
A. ROSA26 knock-in strategy for creating the parathyroid-specific RGS5 (PTG-RGS5) overexpressing transgenic mouse. B. PCR amplification of the RGS5 transgene from tail snips of homozygous (HO, RGS5-myc/eGFPfl/fl) and heterozygous (HET, RGS5-myc/eGFPfl/+) mice. The S1/S2 primers flank the RGS5 cDNA within the targeting vector and produce an 850 bp product specific to the transgenic mice. The G1/G2 primer combination demonstrates insertion into the ROSA26 locus. PCR from tail snip genomic DNA produces a 1250 bp product indicating correctly oriented transgene insertion in the HO and HET PTG-RGS5 strain. The presence or absence of Cre in mice does not affect the size of either product because the genomic DNA is extracted from tail. C. Cre-dependent recombination at the transgene in PTG-RGS5 mice. Lung fibroblasts from HO PTG-RGS5 or WT mice were transduced in culture with an NLS-Cre lentivirus and then genomic PCR was performed using G1 and S2 primers. In the presence of Cre, the PGK-Neo-STOP cassette is excised, bringing the G1 and S2 sites closer together and allowing amplification of the expected 2.2 kb product. In the absence of NLS-Cre, there is no detectable product. D. Western blot of murine lung fibroblasts from the indicated strains, after infection with the Cre lentivirus. In the RGS5-myc/eGFPfl/fl cells, a band of the correct size is recognized by both an anti-myc monoclonal antibody (9E10) and by an anti-RGS5 polyclonal antibody. E. Mouse tracheal blocks were sectioned and incubated with rabbit IgG control, rabbit anti-GFP or rabbit anti-RGS5 antibody followed by DAB staining (brown) and methylgreen nuclear counterstaining (green). Magnification is 400X and scale bar is 50 μm. F. Mouse tracheal blocks were sectioned and incubated with mouse anti-PTH antibody followed by DAB staining (brown) and methylgreen nuclear counterstaining (green) (right panels) or were stained with hematoxylin/eosin (left panels). Magnification is 400X. Scale bar: 50 μm.
PTG-RGS5 mice develop biochemical hyperparathyroidism and retain PTH responses to changes in serum calcium
We compared the plasma levels of calcium and PTH in PTG-RGS5 mice at 1 month of age. We found that transgenic mice show elevated calcium levels (PTG-RGS5, 0.981 ± 0.019 mmol/L versus control, 0.891 ± 0.0162 mmol/L, P<0.01). However, plasma PTH levels were not significantly different (PTG-RGS5, 33.83 ± 8.30 ng/L versus control, 41.29 ± 9.66 ng/L, P>0.05). Male and female PTG-RGS5 mice demonstrated features of HPT at 6 months of age (Figure 2A–D). Compared to control mice, male PTG-RGS5 mice demonstrated elevated plasma levels of PTH at both 6 (PTG-RGS5, 291.1 ± 26.93 ng/L versus control, 106.7 ± 12.73 ng/L, P<0.0001) and 12 months of age (PTG-RGS5, 336 ± 41.74 ng/L versus control, 165.3 ± 40.68 ng/L, P<0.01). Female PTG-RGS5 mice showed significantly elevated PTH at 12 months of age (PTG-RGS5, 188.7 ± 20.44 ng/L versus control, 86.89 ± 10.03 ng/L, P<0.0001), but not at 6 months of age. Plasma calcium levels in female and male mice were within the normal range at 6 months of age and did not vary significantly between PTG-RGS5 and control littermates. However, at 12 months of age, male but not female PTG-RGS5 mice demonstrated elevated plasma calcium (PTG-RGS5, 1.238 ± 0.012 mmol/L versus control, 1.194 ± 0.014 mmol/L, P<0.05). Intraperitoneal injection of calcium gluconate or the calcium chelator EGTA resulted in appropriately responsive modulation of plasma PTH in both PTG-RGS5 and control mice (Figures 2E and F). Although control and PTG-RGS5 mice had significantly different PTH levels at baseline (PTG-RGS5, 308.2 ± 62.34 ng/L versus control, 126.8 ± 13.45 ng/L, P<0.05), EGTA-induced hypocalcemia caused a similar, significant (6-fold) increase in plasma PTH (PTG-RGS5, 1756.0 ± 201.6 ng/L versus WT= 722.7 ± 146.1 ng/L, P<0.05). Calcium gluconate-induced hypercalcemia caused a similar, significant (3-fold) decrease in plasma PTH levels in both PTG-RGS5 and control mice (PTG-RGS5, 108 ± 32.41 ng/L versus control, 39.76 ± 20.58 ng/L, P>0.05). These data indicate that while mice with RGS5 overexpression in the parathyroid gland demonstrate altered baseline PTH levels under normocalcemic conditions, the PTH response to changes in ambient calcium is retained, reflecting an altered CASR setpoint.
Figure 2.
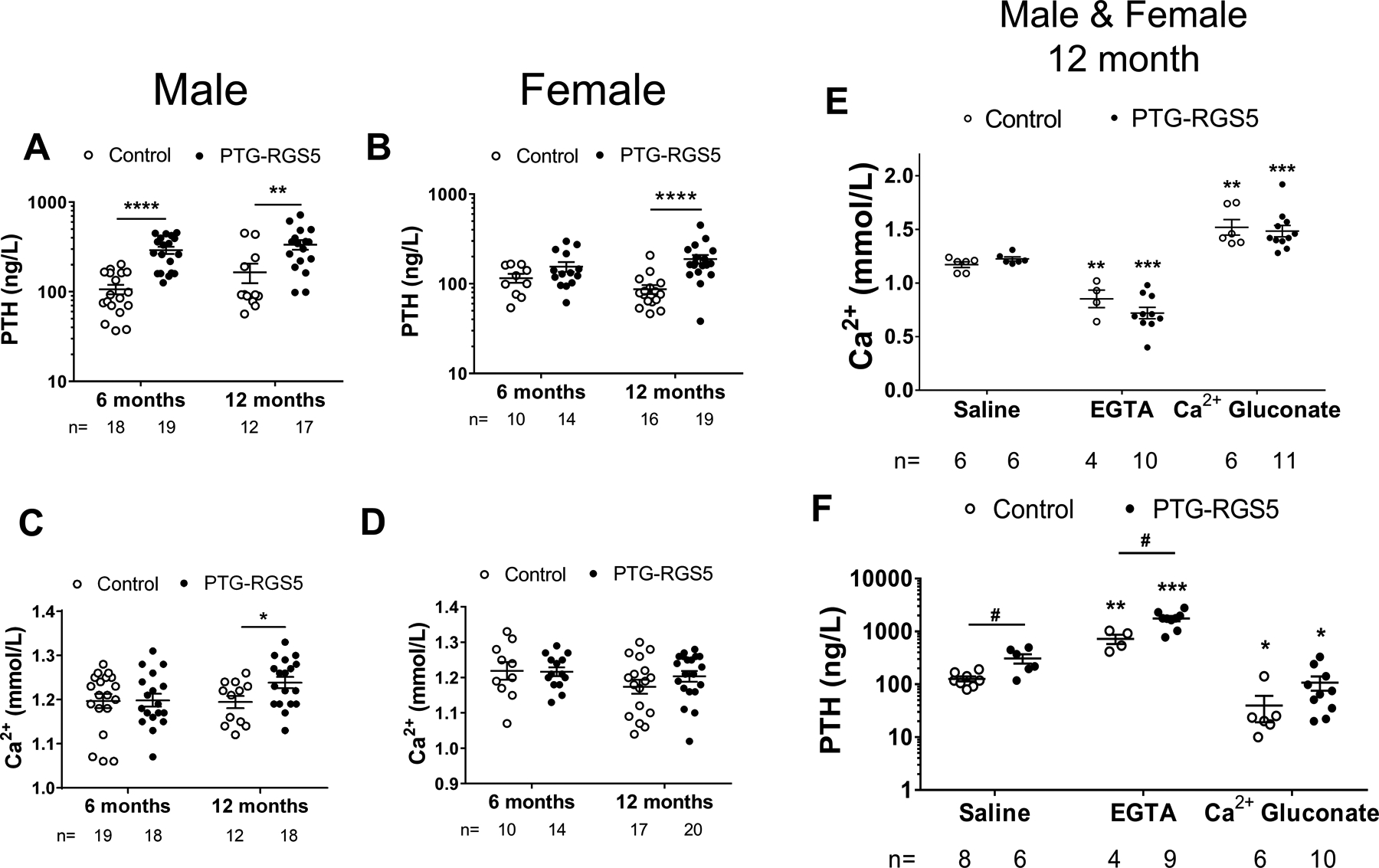
Biochemical assessment of PTG-RGS5 mice. Basal PTH (A-B) and calcium (C-D) levels in male and female control (open circles) and PTG-RGS5 (filled circles) mice at 6 and 12 months of age are shown. Data are mean ± SEM. Nonparametric Mann-Whitney t-test was used for analysis of A-D. *P < 0.05, **P < 0·01, ****P < 0.0001. (E-F) Dynamic calcium and PTH levels in response to hypercalcemic and hypocalcemic stimuli. Male and female 12-month-old mice were injected intraperitoneally with Saline, EGTA or Calcium gluconate, followed by (E) Ca2+ and (F) PTH measurement. Data are mean ± SEM. Nonparametric Mann-Whitney t-test was used for data analysis. Comparison to saline in each genotype is shown by *; Comparison between control and PTG-RGS5 mice is shown by #. *or # P < 0.05, **P < 0·01, ***P < 0.001.
PTG-RGS5 mice develop parathyroid neoplasia
A total of 90 glands from 45 6-month old PTG-RGS5 mice and 84 glands from 42 12-month PTG-RGS5 mice were examined in a blinded manner alongside sections from RGS5-myc/eGFPfl/fl (n=38 at 6 months, n=32 at 12 months), WT (n=16 at 6 months, n=21 at 12 months), and PTH-Cre (n=6 at 6 months, n=6 at 12 months) genotypes. At 6 months of age, 8 of 45 PTG-RGS5 mice demonstrated hypercellularity and increased gland size indicative of parathyroid gland hyperplasia with features suggestive of adenoma such as encapsulation and compression of adjacent hypercellular parathyroid tissue (Figure 3A). At 12 months, parathyroid adenomas were found in 9 of 42 12-month old PTG-RGS5 mice. Control mice demonstrated no abnormalities in the parathyroid glands at either age. The mean longest cross-sectional axis for the hyperplastic glands was 605.8 ± 30.57 microns compared to 313.5 ± 33.89 microns for glands from control mice (P = 0.0002). Assessment of the parathyroid gland volume revealed the relative enlargement of parathyroid glands in PTG-RGS5 mice compared to control littermates at 12 months of age (PTG-RGS5, 1.429×107 ± 1.406×106 μm3 versus control, 8.119×106 ± 1.001×106 μm3 , P<0.01) (Figure 3B). We evaluated CASR expression in mouse parathyroid glands qualitatively (Figure 3C) and quantitatively (Figure 3D) by both IHC-Fluorescence and IHC-DAB staining. Consistent with our earlier studies in human parathyroid adenomas (26), we did not observe reduced CASR expression in parathyroid tumors in PTG-RGS5 mice (Figure 3C&D).
Figure 3.
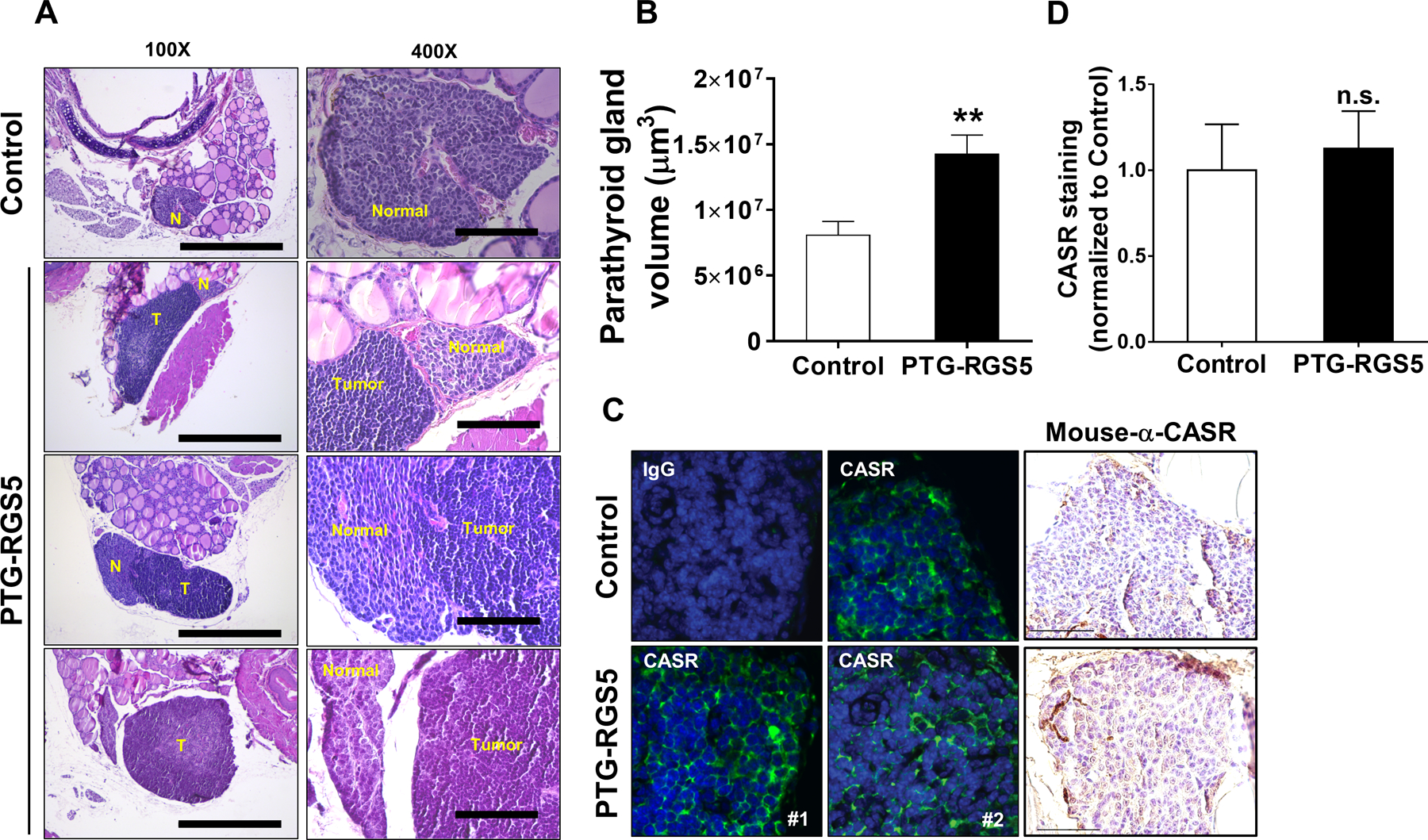
Histological assessment of parathyroid glands in PTG-RGS5 mice. A. (Left panel) Hyperplastic parathyroid glands from 3 different PTG-RGS5 mice are shown in comparison to a reference age-matched control mouse. Tracheal block 5-micron axial sections were stained with hematoxylin/eosin. Magnification is 100X. Scale bar = 500 μm. T = parathyroid tumor. N = normal parathyroid tissue. (Right panel) Higher magnification view (400X) of the same sections showing compressed or residual normal parathyroid tissue adjacent to each tumor. Scale bar = 100 μm. B. Tumor volume was measured as described in supplemental material section. Data are mean ± SEM. Nonparametric Mann-Whitney t-test was used for data analysis. **P < 0·01. C. CASR expression in normal and hyperplastic parathyroid glands. CASR was visualized by two methods: (Left panels) Immunofluorescent staining of control or tumors from two different PTG-RGS5 mice. The IgG control panel shows staining with an isotype-matched negative control primary and AlexaFluor488-conjugated secondary antibody. Blue = DAPI; Green = CASR. (Right panel) Anti-CASR antibody was used for IHC followed by DAB staining (brown) and hematoxylin nuclear counterstaining (blue). Magnification is 400X and scale bar is 50 μm. D. Quantitative measurement of DAB-stained pixels do not show a significant change in CASR expression between control and PTG-RGS5 mice. (n.s. not significant).
PTG-RGS5 mice demonstrate altered trabecular but not cortical bone
The bone in PHPT is characterized by high turnover, cortical bone loss, and preserved or increased trabecular bone (27, 28). To investigate skeletal changes in PTG-RGS5 mice with HPT, we assessed the microstructure of trabecular and cortical bone in femurs from 12-month old transgenic and control mice using μCT (Figure 4). Analysis of trabecular bone in PTG-RGS5 male and female mice showed significantly increased bone volume fraction (Male: PTG-RGS5, 10.6 ± 1.951 % versus control, 4.446 ± 0.5723 %, P<0.05; Female: PTG-RGS5, 6.755 ± 1.322 % versus control, 0.8462 ± 0.2862 %, P<0.01). In addition, there was a significant difference in trabecular separation (Male: PTG-RGS5, 0.2801 ± 0.01131 mm versus control, 0.3693 ± 0.0274 mm, P<0.01; Female: PTG-RGS5, 0.3492 ± 0.02459 mm versus control, 0.495 ± 0.02636 %, P<0.01) (Figure 4A–F and Supplemental table 2). In contrast, cortical bone parameters in PTG-RGS5 mice were similar to control mice of both genders (Figure 4G–L and Supplemental table 2). Change in trabecular bone volume, separation and number correlated with PTH levels in both male and female mice but not with plasma calcium (Supplemental table 3).
Figure 4.
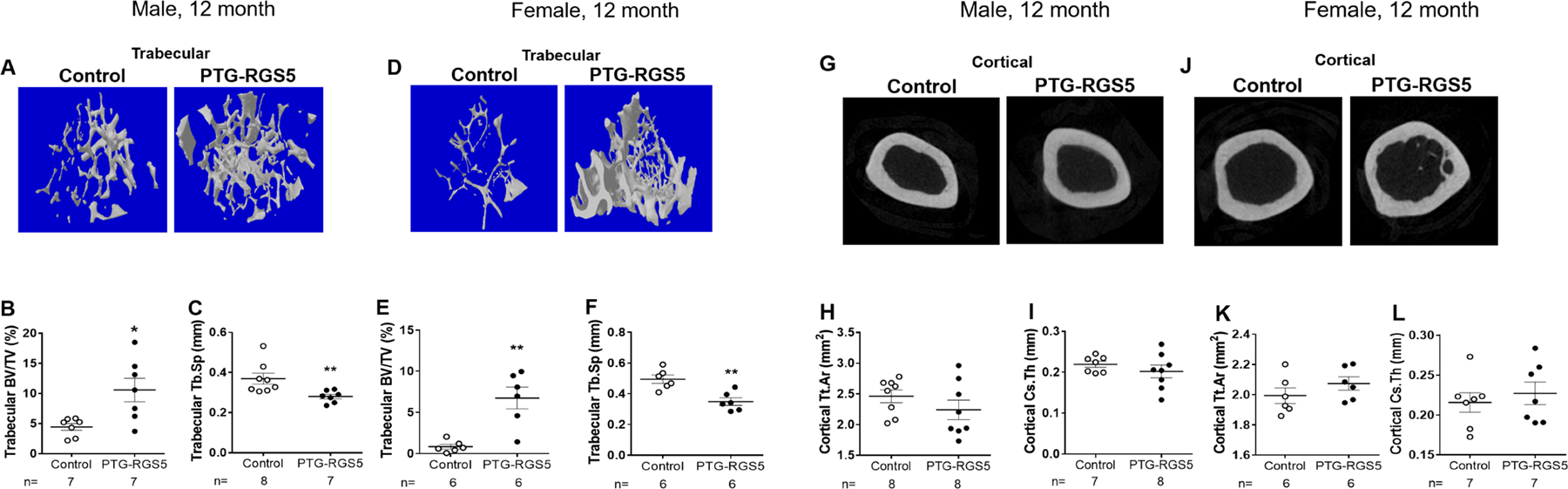
Bone histomorphometry in PTG-RGS5 transgenic mice. Femurs of male and female 12-month-old PTG-RGS5 and control mice were analyzed by μCT. (A&D) 3D images of trabecular bone, (B&E) trabecular bone volume (BV/TV%) and (C&F) trabeculae spaces (Tb.Sp) are shown. (G&J) Cross sectional images of cortical bones, (H&K) total cross-sectional area inside the periosteal envelope (Tt.Ar) and (I&L) average cortical thickness (Cs.Th) are shown. Data are mean ± SEM. Nonparametric Mann-Whitney t-test was used for data analysis. *P < 0.05, **P < 0.01.
Overexpression of RGS5 inhibits CASR signaling and PTH secretion from normal human parathyroid cells
We explored the mechanisms by which RGS5 modulates CASR intracellular signaling pathways in vitro. Using a HEK293 cell line that stably expresses CASR on its surface (15), we assessed the effect of RGS5 on CASR-mediated intracellular calcium release in response to calcium stimulation. RGS5 expression did not affect total (Figure 5A&B) or surface expression (Figure 5C) of CASR. However, RGS5 overexpression significantly inhibited CASR-mediated intracellular calcium flux (Figure 5D) and right-shifted the concentration-response curve of Ca2+ (potency (-LogEC50): RGS5, 2.90 ± 0.015 versus pcDNA3.1, 3.11 ± 0.017; P<0.0001) (Figure 5E). To assess the impact of RGS5 overexpression on PTH secretion in normal parathyroid cells, we transduced normal human parathyroid cells from deceased organ donors with lentiviruses expressing GFP reporter with either LacZ (control) or RGS5 (Figure 6A). Across a range of extracellular Ca2+ concentrations, PTH secretion by parathyroid cells overexpressing RGS5 was significantly greater than the control cells (Figure 6B). Such changes were more modest at higher Ca2+ concentrations and the potency of Ca2+ to suppress PTH secretion via CASR was unchanged ((-LogIC50): RGS5, 2.919 ± 0.045 versus control, 2.923 ± 0.065; P>0.05). The amplitude and duration of intracellular calcium signaling of CASR was reduced by RGS5 in these normal human parathyroid cells (Figure 6C). Although, at several extracellular Ca2+ concentrations control cells showed significantly higher calcium signaling, the potency of Ca2+ remained unchanged (potency (-LogEC50): RGS5, 2.324 ± 0.581 versus control, 2.584 ± 0.105; P>0.05) (Figure 6D). Overall, these data recapitulate the PTG-RGS5 mouse HPT phenotype, in which parathyroid-specific overexpression of RGS5 elevates basal plasma PTH levels.
Figure 5.
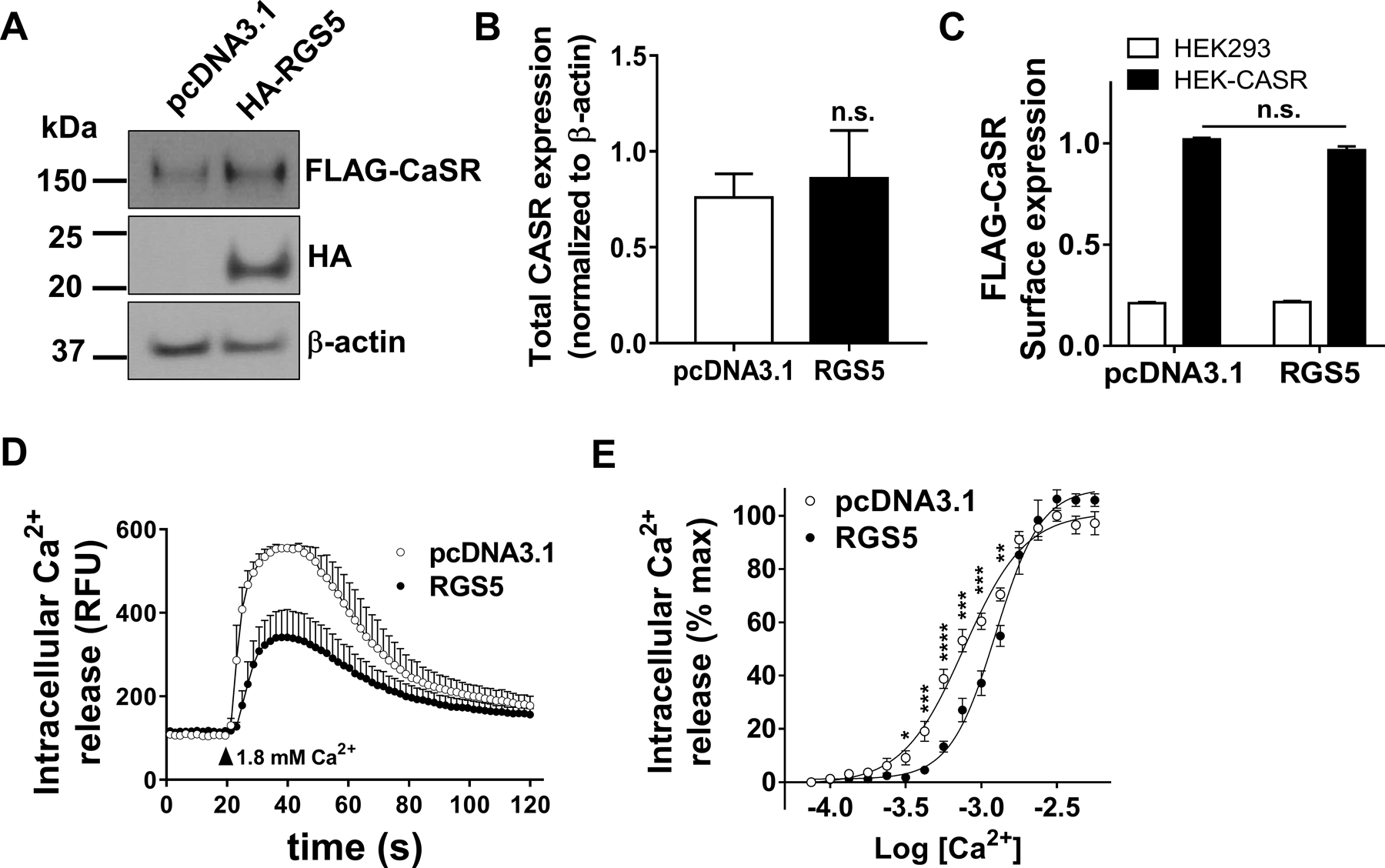
Effect of RGS5 on CASR signaling. HEK-CASR cells were transiently transfected with pcDNA3.1 control or RGS5 plasmid. Expression of RGS5 did not affect the total (A&B) or surface expression (C) of CASR as measured by WB and on-cell ELISA, respectively. D. Kinetic of CASR-mediated intracellular Ca2+ release was monitored 20 seconds before and 100 seconds after stimulation with 1.8 mM extracellular Ca2+. E. Concentration response curves of CASR-mediated intracellular calcium release were derived by GraphPad software as described in methods. Data in B-E are mean ± SEM from 5–6 independent experiments conducted in triplicate. T-test was used for data analysis. *P < 0.05, **P < 0·01, ***P < 0·001, ****P < 0·0001. (n.s. not significant).
Figure 6.
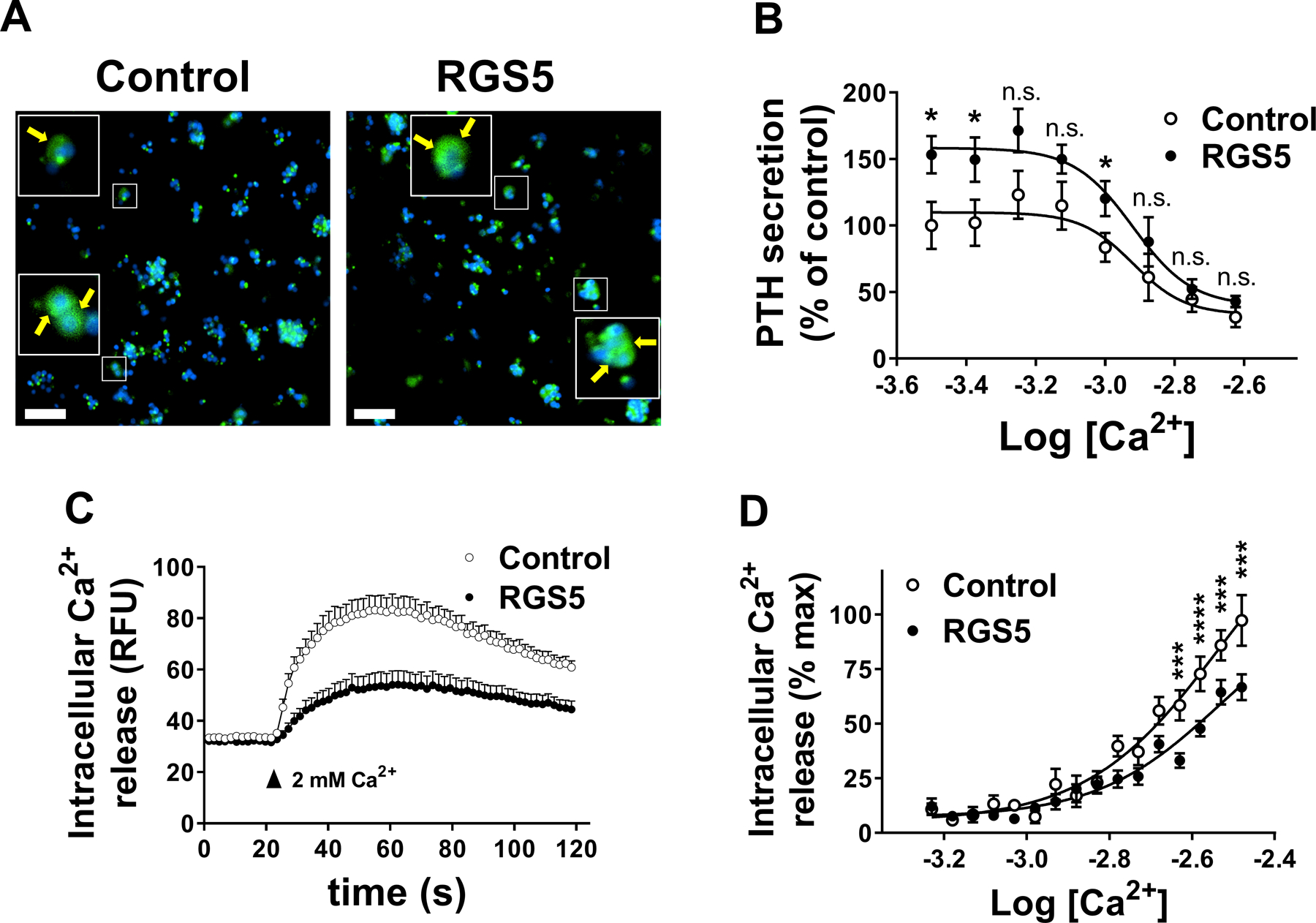
Effect of overexpression of RGS5 on PTH secretion by normal human parathyroid cells. Dispersed normal cadaveric parathyroid cells from eucalcemic donors were transduced with lentivirus particles expressing GFP reporter along with either LacZ (control) or RGS5 for 24 hrs. A. Cells were fixed and stained with DAPI nuclear dye (blue). GFP (green), co-expressed with transgenes by lentivirus is shown. Inset shows cytoplasmic staining of GFP. Scale bar: 50 μm. B. Cells were stimulated with increasing concentrations of extracellular Ca2+ and PTH secretion was measured in a colorimetric ELISA assay. Data are mean ± SEM from 3 independent experiments performed in triplicate. Nonparametric Mann-Whitney t-test was used for data analysis. *P < 0.05. (n.s. not significant). C. Kinetic of CASR-mediated intracellular Ca2+ release in human normal parathyroid cells was monitored 20 seconds before and 100 seconds after stimulation with 2 mM extracellular Ca2+. D. Concentration response curves of CASR-mediated intracellular calcium release were derived by GraphPad software as described in methods. Data are mean ± SEM from 3 independent experiments conducted in triplicate. Nonparametric Mann-Whitney t-test was used for data analysis. ***P < 0·001, ****P < 0·0001.
Discussion
Based on a gene expression study in human parathyroid tumors, we identified RGS5 as a regulator of CASR signaling in the parathyroid gland that when overexpressed leads to impaired signaling from CASR, and conversely, when deleted results in reduced baseline PTH levels in an RGS5 knockout mouse model (15, 16). We now extend these observations by showing that RGS5 overexpression specifically in the mouse parathyroid gland leads to elevated PTH, normal to mildly elevated calcium, skeletal changes, and parathyroid tumors in a unique and clinically relevant model of PHPT. In addition, we show that overexpression of RGS5 in normal human parathyroid tissue causes increased PTH secretion that is insensitive to calcium feedback. Collectively, these results demonstrate that elevated expression of RGS5 in the parathyroid gland can represent an alternative pathogenic mechanism in PHPT, providing experimental support to the notion that abnormal expression of molecules that regulate signaling through CASR may also contribute to parathyroid neoplasia.
PTG-RGS5 transgenic mice demonstrate elevated calcium levels without changes in baseline PTH levels as early as 1 month of age, consistent with attenuated CASR feedback to PTH (i.e. inappropriately normal PTH in the face of hypercalcemia). PTG-RGS5 transgenic mice demonstrate elevated baseline PTH secretion as early as 6 months of age, again consistent with attenuated CASR feedback to PTH. Despite elevated PTH in both sexes, only male PTG-RGS5 mice demonstrated elevated plasma calcium levels at 12 months of age. The reasons behind this age and gender related finding of normohormonal hypercalcemia followed by normocalcemic or secondary hyperparathyroidism evolving to classic hyperparathyroidism in male mice is unclear but will be of great interest for future study of the age and gender-specific natural history of HPT in this model. Importantly, this new mouse model may be well suited to the study of factors involved in the progression of secondary HPT and/or normocalcemic PHPT to hypercalcemic PHPT, a highly relevant scenario given the increasing prevalence of mild or asymptomatic PHPT in patients identified by routine serum calcium screening. Similar to our prior findings in mice lacking RGS5 (16), PTG-RGS5 transgenic mice retain dynamic responsiveness to experimentally induced hypo- and hyper-calcemia, indicating that constitutive expression of RGS5 leads to elevated basal PTH secretion but does not interfere with the ability to respond to changes in ambient calcium.
The increase in trabecular bone without change in cortical bone in the PTG-RGS5 mice is noteworthy since this phenotype mirrors the bone changes often found in normocalcemic or mild PHPT in humans (27, 29). The plasma level of PTH but not calcium correlated positively with trabecular bone changes, a finding consistent with effects of exogenous PTH administration on bone in other mouse models (30, 31). No gender difference in bone quality were observed, possibly reflecting the young age of the mice studied. The outcome of preserved cortical bone and increased trabecular bone in PTG-RGS5 mice is distinct from the bone loss phenotype reported in the cyclin D1-driven model of parathyroid neoplasia and PHPT (6). There are several possible explanations for this discrepancy. The mice assessed in the current study were younger (6–12 months) with normocalcemic or mildly hypercalcemic HPT, as opposed to the 22-month-old animals examined in the PTH-cyclin D1 model (6). It is possible that the bone changes observed in PTG-RGS5 mice are progressive and require a longer latency time beyond the 12-month endpoint used in our study. Future studies of this strain will examine age and gender effects on the natural history of bone disease in this model of HPT.
Mechanisms underlying the abnormal calcium-PTH negative feedback relationship in parathyroid tumors causative of PHPT have been most often attributed to decreased expression of the CASR (14, 32). More recently, signaling molecules downstream to CASR including Gα proteins and RhoGEFs have been implicated in the pathogenesis of abnormal calcium signaling in PHPT (16, 33). Our prior studies have shown that RGS5, a member of the R4 family of RGS proteins that accelerates GTP hydrolysis by Gα subunits, is overexpressed in parathyroid tumors in PHPT and can inhibit signaling from CASR without changing expression levels of the receptor itself. The demonstration that RGS5 can oppose CASR signaling and alter basal PTH secretion by normal parathyroid cells establishes an important alternative to the prevailing view that the altered calcium sensing observed in PHPT patients results only from reduced expression of CASR in parathyroid tumor cells. In our experiments, expression of CASR was unchanged by RGS5 overexpression. Although RGS5 inhibits signaling from CASR (i.e. signaling setpoint) in the HEK-CASR model and reduces CASR signaling at higher calcium concentrations in parathyroid cells, RGS5 only seems to affect basal secretion of PTH in human parathyroid glands without a setpoint shift. Thus our model currently falls short of providing direct proof of the setpoint mechanism of parathyroid tumorigenesis. Whether RGS5-induced elevated PTH production in the PTG-RGS5 mice is due to CASR inhibition or another mechanism is unclear. The fact that RGS5 has no other known effector function beyond GAP activity and that parathyroid tissue does not express appreciable levels of other GPCRs known to be modulated by RGS5, point to a CASR-mediated mechanism. Future studies will address this important question.
The fact that PTG-RGS5 transgenic mice develop parathyroid hyperplasia and parathyroid tumors directly supports a connection between abnormal PTH secretion and parathyroid neoplasia. Importantly, the evolution of parathyroid neoplasia observed in PTG-RGS5 mice supports a paradigm of disease progression from hyperplasia to tumor formation, possibly akin to the progression of secondary parathyroid hyperplasia to autonomous tertiary parathyroid tumor formation in patients with hyperparathyroidism due to renal failure (34). Recent data from our group showing that many tumors in PHPT are oligoclonal rather than uniformly monoclonal further supports the concept that underlying secondary parathyroid hyperplasia may evolve to parathyroid tumors in a subset of PHPT cases (24, 35). The PTG-RGS5 murine model described here provides a unique in vivo tool to define the molecular determinants of parathyroid tumor progression and evaluate potential management approaches to mitigate setpoint disruption as a driver of parathyroid tumor development.
Supplementary Material
Acknowledgements
We wish to thank Elizabeth A. Streeten, MD for critical review of the manuscript.
Grant Support: This work was supported in part by NIH grant 1R01DK088188–01 (JAO and JK) and PhRMA Foundation and American Cancer Society research grants (NB).
Footnotes
Disclosure Statement: The authors declare that no conflicts of interest exist.
References
- 1.Arnold A, Kim HG, Gaz RD, Eddy RL, Fukushima Y, Byers MG, Shows TB, and Kronenberg HM. Molecular cloning and chromosomal mapping of DNA rearranged with the parathyroid hormone gene in a parathyroid adenoma. J Clin Invest. 1989;83(6):2034–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Motokura T, Bloom T, Kim HG, Juppner H, Ruderman JV, Kronenberg HM, and Arnold A. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350(6318):512–5. [DOI] [PubMed] [Google Scholar]
- 3.Arnold A, Shattuck TM, Mallya SM, Krebs LJ, Costa J, Gallagher J, Wild Y, and Saucier K. Molecular pathogenesis of primary hyperparathyroidism. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2002;17 Suppl 2(N30–6. [PubMed] [Google Scholar]
- 4.Cromer MK, Starker LF, Choi M, Udelsman R, Nelson-Williams C, Lifton RP, and Carling T. Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J Clin Endocrinol Metab. 2012;97(9):E1774–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newey PJ, Nesbit MA, Rimmer AJ, Attar M, Head RT, Christie PT, Gorvin CM, Stechman M, Gregory L, Mihai R, et al. Whole-exome sequencing studies of nonhereditary (sporadic) parathyroid adenomas. J Clin Endocrinol Metab. 2012;97(10):E1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Imanishi Y, Hosokawa Y, Yoshimoto K, Schipani E, Mallya S, Papanikolaou A, Kifor O, Tokura T, Sablosky M, Ledgard F, et al. Primary hyperparathyroidism caused by parathyroid-targeted overexpression of cyclin D1 in transgenic mice. The Journal of clinical investigation. 2001;107(9):1093–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Libutti SK, Crabtree JS, Lorang D, Burns AL, Mazzanti C, Hewitt SM, O’Connor S, Ward JM, Emmert-Buck MR, Remaley A, et al. Parathyroid gland-specific deletion of the mouse Men1 gene results in parathyroid neoplasia and hypercalcemic hyperparathyroidism. Cancer Res. 2003;63(22):8022–8. [PubMed] [Google Scholar]
- 8.Imanishi Y, Kawata T, Kenko T, Wada M, Nagano N, Miki T, Arnold A, and Inaba M. Cinacalcet HCl suppresses Cyclin D1 oncogene-derived parathyroid cell proliferation in a murine model for primary hyperparathyroidism. Calcif Tissue Int. 2011;89(1):29–35. [DOI] [PubMed] [Google Scholar]
- 9.Parfitt AM, Braunstein GD, and Katz A. Radiation-associated hyperparathyroidism: comparison of adenoma growth rates, inferred from weight and duration of latency, with prevalence of mitosis. J Clin Endocrinol Metab. 1993;77(5):1318–22. [DOI] [PubMed] [Google Scholar]
- 10.Parfitt AM, and Fyhrie DP. Gompertzian growth curves in parathyroid tumours: further evidence for the set-point hypothesis. Cell Prolif. 1997;30(8–9):341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parfitt AM, Wang Q, and Palnitkar S. Rates of cell proliferation in adenomatous, suppressed, and normal parathyroid tissue: implications for pathogenesis. J Clin Endocrinol Metab. 1998;83(3):863–9. [DOI] [PubMed] [Google Scholar]
- 12.Mayer GP, and Hurst JG. Sigmoidal relationship between parathyroid hormone secretion rate and plasma calcium concentration in calves. Endocrinology. 1978;102(4):1036–42. [DOI] [PubMed] [Google Scholar]
- 13.Brown EM. Four-parameter model of the sigmoidal relationship between parathyroid hormone release and extracellular calcium concentration in normal and abnormal parathyroid tissue. The Journal of clinical endocrinology and metabolism. 1983;56(3):572–81. [DOI] [PubMed] [Google Scholar]
- 14.Kifor O, Moore FD Jr., Wang P, Goldstein M, Vassilev P, Kifor I, Hebert SC, and Brown EM. Reduced immunostaining for the extracellular Ca2+-sensing receptor in primary and uremic secondary hyperparathyroidism. The Journal of clinical endocrinology and metabolism. 1996;81(4):1598–606. [DOI] [PubMed] [Google Scholar]
- 15.Balenga N, Azimzadeh P, Hogue JA, Staats PN, Shi Y, Koh J, Dressman H, and Olson JA Jr. Orphan Adhesion GPCR GPR64/ADGRG2 Is Overexpressed in Parathyroid Tumors and Attenuates Calcium-Sensing Receptor-Mediated Signaling. J Bone Miner Res. 2017;32(3):654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koh J, Dar M, Untch BR, Dixit D, Shi Y, Yang Z, Adam MA, Dressman H, Wang X, Gesty-Palmer D, et al. Regulator of G protein signaling 5 is highly expressed in parathyroid tumors and inhibits signaling by the calcium-sensing receptor. Molecular endocrinology. 2011;25(5):867–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pi M, Chen L, Huang M, Luo Q, and Quarles LD. Parathyroid-specific interaction of the calcium-sensing receptor and G alpha q. Kidney Int. 2008;74(12):1548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varrault A, Pena MS, Goldsmith PK, Mithal A, Brown EM, and Spiegel AM. Expression of G protein alpha-subunits in bovine parathyroid. Endocrinology. 1995;136(10):4390–6. [DOI] [PubMed] [Google Scholar]
- 19.Ho C, Conner DA, Pollak MR, Ladd DJ, Kifor O, Warren HB, Brown EM, Seidman JG, and Seidman CE. A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat Genet. 1995;11(4):389–94. [DOI] [PubMed] [Google Scholar]
- 20.Wettschureck N, Lee E, Libutti SK, Offermanns S, Robey PG, and Spiegel AM. Parathyroid-specific double knockout of Gq and G11 alpha-subunits leads to a phenotype resembling germline knockout of the extracellular Ca2+ -sensing receptor. Molecular endocrinology. 2007;21(1):274–80. [DOI] [PubMed] [Google Scholar]
- 21.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, and Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2010;25(7):1468–86. [DOI] [PubMed] [Google Scholar]
- 22.Moorer MC, Hebert C, Tomlinson RE, Iyer SR, Chason M, and Stains JP. Defective signaling, osteoblastogenesis and bone remodeling in a mouse model of connexin 43 C-terminal truncation. J Cell Sci. 2017;130(3):531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buo AM, Tomlinson RE, Eidelman ER, Chason M, and Stains JP. Connexin43 and Runx2 Interact to Affect Cortical Bone Geometry, Skeletal Development, and Osteoblast and Osteoclast Function. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2017;32(8):1727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Y, Hogue J, Dixit D, Koh J, and Olson JA Jr. Functional and genetic studies of isolated cells from parathyroid tumors reveal the complex pathogenesis of parathyroid neoplasia. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(8):3092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nyabi O, Naessens M, Haigh K, Gembarska A, Goossens S, Maetens M, De Clercq S, Drogat B, Haenebalcke L, Bartunkova S, et al. Efficient mouse transgenesis using Gateway-compatible ROSA26 locus targeting vectors and F1 hybrid ES cells. Nucleic Acids Res. 2009;37(7):e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koh J, Dar M, Untch BR, Dixit D, Shi Y, Yang Z, Adam MA, Dressman H, Wang X, Gesty-Palmer D, et al. Regulator of G-protein signaling 5 is highly expressed in parathyroid tumors and inhibits signaling by the calcium-sensing receptor. Molecular Endo. 2011;25(5):867–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dempster DW, Silverberg SJ, Shane E, and Bilezikian JP. In: Bilezikian JP, Marcus R, Levine MA, Marcocci C, Silverberg SJ, and Potts JT Jr. eds. The Parathyroids. Amsterdam: Elsevier; 2015:429–45. [Google Scholar]
- 28.Silverberg SJ, Shane E, de la Cruz L, Dempster DW, Feldman F, Seldin D, Jacobs TP, Siris ES, Cafferty M, Parisien MV, et al. Skeletal disease in primary hyperparathyroidism. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1989;4(3):283–91. [DOI] [PubMed] [Google Scholar]
- 29.Dempster DW, Muller R, Zhou H, Kohler T, Shane E, Parisien M, Silverberg SJ, and Bilezikian JP. Preserved three-dimensional cancellous bone structure in mild primary hyperparathyroidism. Bone. 2007;41(1):19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gunness-Hey M, and Hock JM. Increased trabecular bone mass in rats treated with human synthetic parathyroid hormone. Metab Bone Dis Relat Res. 1984;5(4):177–81. [DOI] [PubMed] [Google Scholar]
- 31.Iida-Klein A, Zhou H, Lu SS, Levine LR, Ducayen-Knowles M, Dempster DW, Nieves J, and Lindsay R. Anabolic action of parathyroid hormone is skeletal site specific at the tissue and cellular levels in mice. J Bone Miner Res. 2002;17(5):808–16. [DOI] [PubMed] [Google Scholar]
- 32.Farnebo F, Hoog A, Sandelin K, Larsson C, and Farnebo LO. Decreased expression of calcium-sensing receptor messenger ribonucleic acids in parathyroid adenomas. Surgery. 1998;124(6):1094–8; discussion 8–9. [DOI] [PubMed] [Google Scholar]
- 33.Pi M, Spurney RF, Tu Q, Hinson T, and Quarles LD. Calcium-sensing receptor activation of rho involves filamin and rho-guanine nucleotide exchange factor. Endocrinology. 2002;143(10):3830–8. [DOI] [PubMed] [Google Scholar]
- 34.Fukuda N, Tanaka H, Tominaga Y, Fukagawa M, Kurokawa K, and Seino Y. Decreased 1,25-dihydroxyvitamin D3 receptor density is associated with a more severe form of parathyroid hyperplasia in chronic uremic patients. J Clin Invest. 1993;92(3):1436–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi Y, Azimzadeh P, Jamingal S, Wentworth S, Ferlitch J, Koh J, Balenga N, and Olson JA Jr. Polyclonal origin of parathyroid tumors is common and is associated with multiple gland disease in primary hyperparathyroidism. Surgery. 2018;163(1):9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.