

CONSPECTUS:
The total synthesis of structurally complex natural products has challenged and inspired generations of chemists and remains an exciting area of active research. Despite their history as privileged bioactivity-rich scaffolds, the use of natural products in drug-discovery has waned. This shift is driven by their relatively low abundance hindering isolation from natural sources and the challenges presented by their synthesis. Recent developments in biocatalysis have resulted in the application of enzymes for the construction of complex molecules. From the inception of the Narayan lab in 2015, we have focused on harnessing the exquisite selectivity of enzymes alongside contemporary small molecule-based approaches to enable concise chemoenzymatic routes to natural products.
We have focused on enzymes from various families that perform selective oxidation reactions. For example, we have targeted xyloketal natural products through a strategy that relies on a chemo- and site-selective biocatalytic hydroxylation. Members of the xyloketal family are characterized by polycyclic ketal cores and demonstrate potent neurological activity. We envisioned assembling a representative xyloketal natural product (xyloketal D) involving a biocatalytically generated ortho-quinone methide intermediate. The non-heme iron (NHI) dependent monooxygenase ClaD was used to perform the benzylic hydroxylation of a resorcinol precursor, the product of which can undergo spontaneous loss of water to form an ortho-quinone methide under mild conditions. This intermediate was trapped using a chiral dienophile to complete the total synthesis of xyloketal D.
A second class of biocatalytic oxidation that we have employed in synthesis is the hydroxylative dearomatization of resorcinol compounds using flavin-dependent monooxygenases (FDMOs). We anticipated that the catalyst-controlled site- and stereoselectivity of FDMOs would enable the total synthesis of azaphilone natural products. Azaphilones are bioactive compounds characterized by a pyranoquinone bicyclic core and a fully substituted chiral carbon atom. We leveraged the stereodivergent reactivity of FDMOs AzaH and AfoD to achieve the enantioselective synthesis of trichoflectin enantiomers, deflectin 1a, and lunatoic acid. We also leveraged FDMOs to construct tropolone and sorbicillinoid natural products. Tropolones are a structurally diverse class of bioactive molecules characterized by an aromatic cycloheptatriene core bearing an α-hydroxyketone moiety. We developed a two-step, biocatalytic cascade to the tropolone natural product stipitatic aldehyde starting using the FDMO TropB and a NHI monooxygenase TropC. The FDMO SorbC obtained from the sorbicillin biosynthetic pathway was used in the concise total synthesis of a urea sorbicillinoid natural product.
Our long-standing interest in using enzymes to carry out C–H hydroxylation reactions has also been channeled for the late-stage diversification of complex scaffolds. For example, we have used Rieske oxygenases to hydroxylate the tricyclic core common to paralytic shellfish toxins. The systemic toxicity of these compounds can be reduced by adding hydroxyl and sulfate groups, which improves their properties and potential as therapeutic agents. The enzymes SxtT, GxtA, SxtN, and SxtSUL, were used to carry out selective C–H hydroxylation and O-sulfation in saxitoxin and related structures. We conclude this account with a discussion of existing challenges in biocatalysis and ways we can currently address them.
Graphical Abstract
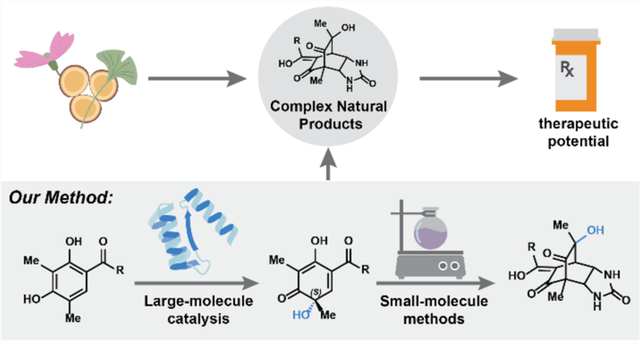
1. Introduction
Extracts of plants and microbes are a rich source of complex organic compounds used in traditional medicine to remedy a host of human diseases.5 Small molecules are often responsible for these curative effects and can provide starting points in developing pharmaceutical agents.5 Natural products and their derivatives are the basis for more than half of all the FDA approved anticancer drugs and antibiotics.5 However, the challenges associated with the synthesis of natural products have resulted in small-molecule based drug discovery efforts to focus on synthetic molecule libraries.5 The synthetic difficulties posed by the complex architecture of natural products also hinder efforts to synthesize and investigate natural-product analogs for drug-discovery efforts.5
Recent developments in biocatalysis have resulted in our potential to streamline the challenges associated with synthesizing complex natural products (Figure 1a).6,7 Advances in DNA sequencing and bioinformatic technologies have enabled the identification of secondary metabolite gene clusters that provide a blueprint for Nature’s synthetic approach.8 Parallel advances in protein engineering and evolution strategies have allowed for manipulating these enzymes in heterologous expression systems to affect specific chemical reactions.9 Most importantly, enzymes enable access to chemical scaffolds previously intractable using traditional small-molecule based catalytic methods.10
Figure 1.
a. Natural products form a rich source of complex organic compounds with high therapeutic potential. b. Biocatalysis workflow.
Biocatalytic methods offer several key advantages over conventional synthetic methods in mediating chemical transformations.7 These benefits include increased safety, sustainability, and procedural simplicity, in addition to high selectivity profiles in a given chemical reaction.11 Consequently, biocatalytic methods are becoming blended in the mainstream synthetic organic chemistry repertoire.12,13 The advantages of enzymes in synthesis are exemplified by the recent work published by Merck and Codexis in their development of a five-step biocatalytic cascade of the HIV drug islatravir.14
Our research integrates the catalyst-controlled selectivity of enzymes along with the best tools of contemporary synthetic organic chemistry to enable rapid access to complex chemical architectures.3,15–23 We are particularly interested in the application of chemoenzymatic strategies for the synthesis of bioactive natural products and unnatural analogs of these compounds that remain underexplored due to the synthetic challenges presented by their complex structures to enable access to these fruitful sources of bioactive compounds.1,2,4
This account presents examples of chemoenzymatic synthesis of complex natural products developed in our lab since 2015.1–4,22 Our team is particularly motivated by enzymes that carry out selective oxidation reactions. The enzymes employed in our total synthesis endeavors belong to the following classes of enzymes: α-ketoglutarate (α-KG) non-heme iron (NHI)-dependent oxygenases,1 flavin-adenine dinucleotide dependent oxygenases (FDMOs),2,4 and Rieske oxygenases.3,22 The key oxygenation chemistry in each synthesis is mediated by NHI-dependent enzymes or FDMOs. In particular, C–H hydroxylation and oxidative dearomatization are used in concert with traditional synthetic methods to enable access to natural products and their analogs.1,2,4
2. Biocatalysis in organic synthesis
We are motivated by enzymes that have the potential to solve reactivity and selectivity challenges in synthesis. Starting by identifying reactions that are complexity-generating and challenging to achieve, we next question if Nature has developed a strategy for mastering the transformation in question and identify enzymes reported within secondary metabolite gene clusters with a putative or demonstrated ability to carry out the target reaction.24–28 As a starting point, we obtain a plasmid that encodes for the biocatalysts of interest through gene synthesis, from the source organism or, from others that have worked with the target biocatalyst.21 Common host organisms used for gene overexpression include E. coli and P. pastoris. These organisms are transformed with a given plasmid and used to produce the enzyme of interest (Figure 1b).
Within the academic community, several biocatalysis research groups, including ours, have repositories of plasmids that encode for the production of biocatalysts and glycerol stocks of heterologous hosts ready to produce enzymes on demand.1–4 Such glycerol stocks can be used to inoculate cultures grown for enzyme production and are easily distributed upon request.1–4 In this framework, the catalyst cost is minimal. Some equipment common to biochemistry laboratories is required for chemists to easily produce biocatalysts on their own, including a temperature-controlled shaking incubator, flasks for cultures, and reagents for making media and buffers used for cultures and cell lysis and/or enzyme purification.29 The ability to produce the enzyme in house allows for enhanced ease of use and provides flexibility in optimizing the platform for a specific reaction, such as choosing to run reactions in either whole cells or with crude cell lysate.21
Following the production of enzymes, biocatalytic reactions can be carried out in different workflows on either analytical or preparative scales.21 Similar to traditional chemical methods, biocatalytic reactions can be run in a variety of formats. For example, reactions can be performed with the biocatalyst in multiple forms, including (1) whole-cells expressing enzymes in live cultures (referred to as fermentation or biotransformation), (2) cells containing enzyme collected from cultures and resuspended in a reaction buffer, (3) harvesting and lysing the cells to form crude cell lysate, or (4) purified enzyme. Using the purified enzyme of interest for in-vitro reactions allows for more precise control over biocatalyst stoichiometry but adds additional steps towards obtaining the biocatalyst. Post biocatalytic transformation, the products can be extracted using liquid-liquid extraction with common organic solvents and purified and characterized using standard methods employed by synthetic chemistry labs.12
3. Chemoenzymatic total synthesis of xyloketal natural products
3.1. Introduction to xyloketal natural products
The xyloketals are a class of structurally complex natural products isolated from a mangrove fungus of the Xylaria species.30 Each member contains at least one 5,6-bicyclic ketal attached to an aromatic core (Figure 2a).30 Isolated xyloketals are chiral with absolute stereochemistry conserved across the family.31 Xyloketals possess a bicyclic ketal cis-fused with a methyl group on the five-membered ring syn to the hydrogen and methyl substituents at the 5,6-ring fusion, presenting a unique synthetic challenge (Figure 2a).31 Although this class of molecules is relatively small, its members possess a wide range of structural diversity and biological activities (see xyloketals A, B, and D, Figure 2a). Notably, xyloketals A-D have demonstrated acetylcholine esterase (AchE) inhibition.32 AchE is an essential enzyme in the human nervous system which transforms the active neurotransmitter acetylcholine into inactive choline. Patients identified with Alzheimer’s disease or Parkinson’s disease show decreased acetylcholine levels in nervous system tissue.32 One strategy proven to be highly useful in diagnosing and treating these diseases is AchE inhibition.32 Suppression of AchE activity decreases acetylcholine’s metabolism, thereby increasing the neurotransmitter levels in the brain. In addition to AchE inhibition, select xyloketals have demonstrated L-calcium channel inhibition, radical-scavenging activity, antioxidant activity, and neuroprotective effects.31,32
Figure 2.
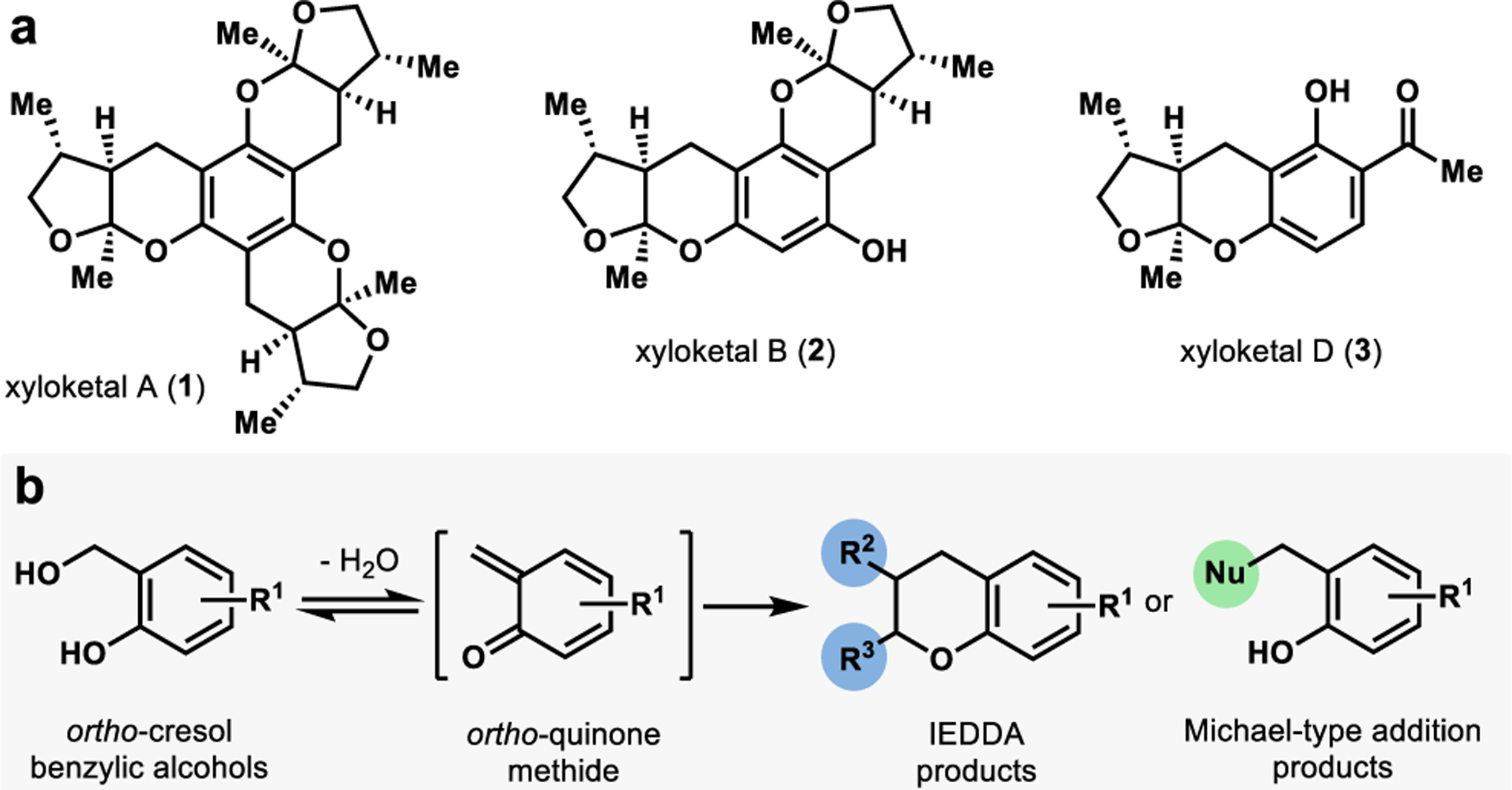
a. Representative xyloketal natural products. b.ortho-Quinone methide formation and diversification from o-cresol benzylic alcohols.
Many of the strategies developed towards the synthesis of this class of molecules have centered around constructing the bicyclic ketal moiety. One member, (−)-xyloketal D, has been previously synthesized through gold-catalyzed cycloisomerization (Sarkar and Panda),33 Michael-addition ketalization sequence (Florke),34 and through a Diels-Alder cycloaddition (Wilson).35 The Wilson strategy utilizes an ortho-quinone methide (o-QM) derived from a morpholine-based precursor.35 Under thermal conditions, the o-QM is generated and captured by a chiral dienophile, leading to a 37% combined yield of 4 products.35 These products are in a 2:1 ratio of ketal to spiroketal isomers, which stem from the dienophile’s isomerization under the thermal reaction conditions.35 We hypothesized that this o-QM strategy could be improved by generating a more active precursor through biocatalytic oxidation.
3.2. Biocatalytic o-quinone methide formation
There are several reported examples in Nature in which o-QMs are implicated in the biosynthesis of natural products.36,37 These reactive intermediates can be captured with nucleophiles or dienophiles through Michael-type additions or inverse-electron demand Diels-Alder (IEDDA) cycloadditions, respectively (Figure 2b). Interception of o-QMs allows for rapid building of molecular complexity and has been employed in the total synthesis of several natural products. Conventional methods for generating o-QMs often require harsh conditions such as strongly acidic38 or basic39 conditions or photolysis40 using low wavelength (< 300 nm) UV light, microwave,41 heat,42 or transition metal catalysts.43 In Nature, one approach to access o-QMs is benzylic hydroxylation of ortho-cresol compounds, followed by loss of water under mild physiological conditions.25 We were inspired by this method and sought to develop a biocatalytic platform for benzylic hydroxylation of ortho-cresol compounds.
One class of enzymes that can affect this oxidation is non-heme iron (NHI) alpha-ketoglutarate dependent oxygenases.44 These enzymes are capable of activating molecular oxygen at iron contained in the enzyme active site.45 Upon substrate binding, an iron(IV)-oxo intermediate capable of hydrogen abstraction is generated (Figure 3).45 The resulting substrate radical can recombine with the iron(III)-hydroxy intermediate in a process referred to as the rebound hydroxylation.45 A similar process occurs in heme-containing enzymes, such as cytochrome P450 hydroxylases; however, these enzymes often require an external reductase to supply electrons for the reduction of O2 and can present challenges in scalability. Conversely, α-KG is oxidized to reduce O2 in reactions with NHI enzymes, which have been carried out on preparative scale.1,7,46 These advantages allow for a synthetically useful platform for benzylic hydroxylation and subsequent diversification of products.1
Figure 3.
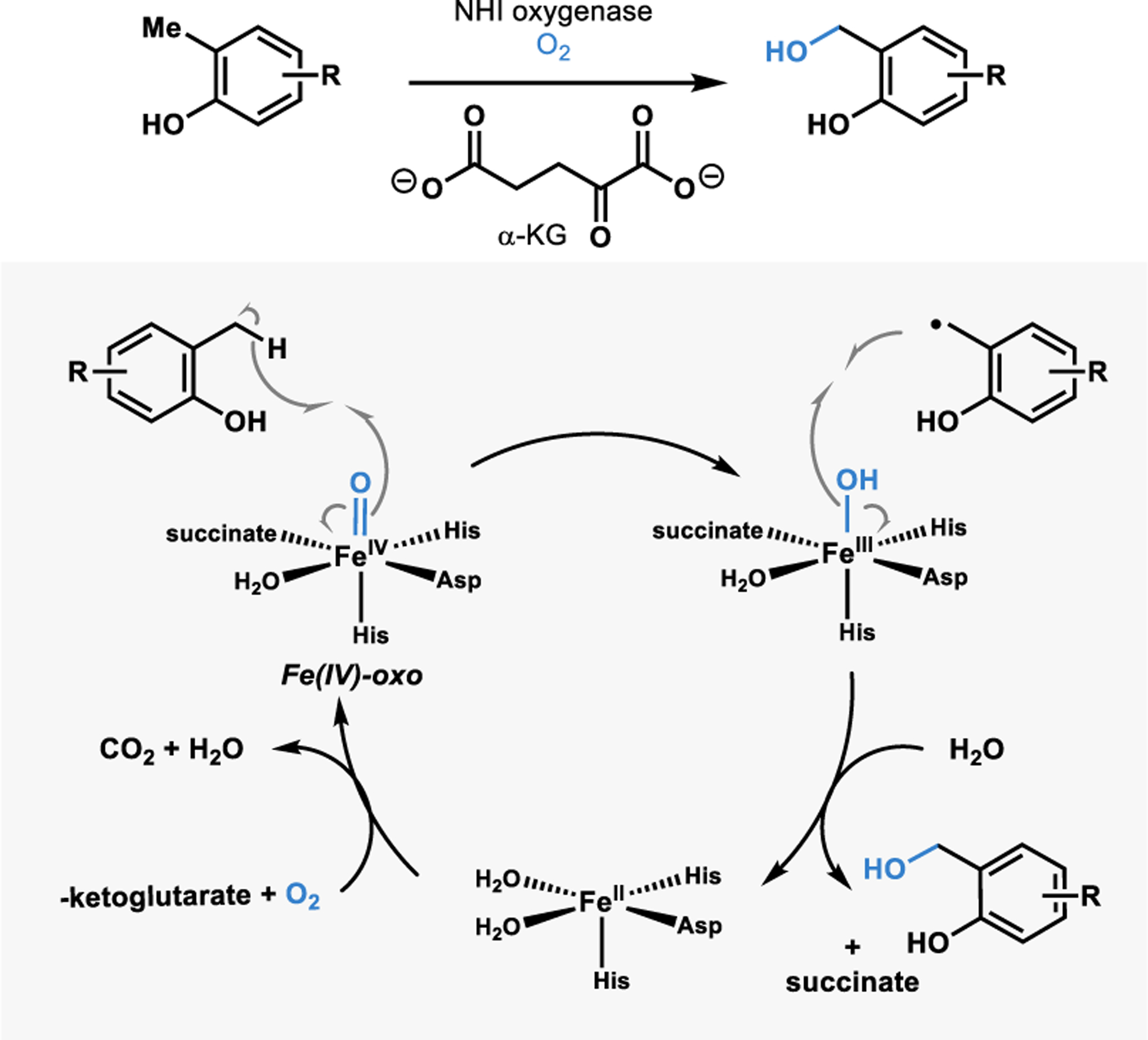
Non-heme iron a-KG dependent benzylic hydroxylation.
Benzylic hydroxylation of ortho-cresol compounds provides a direct route to access o-QMs through the loss of water (Figure 2b).25 Further elaboration of o-QMs can often be performed in one-pot chemoenzymatic cascades due to mild conditions under which enzymatic production of benzylic alcohols with NHI enzymes occurs. In this strategy, nucleophile or dienophile is added directly to the enzymatic reaction mixture to combine with the in situ generated o-QM, eliminating the need to isolate the benzylic alcohol. This one-pot strategy allows for the rapid building of molecular complexity, and we have employed this strategy in the total synthesis of xyloketal natural products (vide infra).
3.3. Chemoenzymatic synthesis of (−)-xyloketal D
Spearheaded by Doyon and Perkins, our lab used a NHI monooxygenase-centered strategy to achieve the total synthesis of (−)-xyloketal D (Figure 4).1 This was carried out using either CitB or ClaD (54% sequence identity), which were previously reported to perform benzylic hydroxylation in fungal metabolite biosynthesis.24,25 Cox and coworkers implicated CitB in citrinin biosynthesis in Monascus ruber,24 and ClaD was reported by Li and coworkers to hydroxylate the penultimate step of peniphenone and penilactone biosynthesis in Penicillium crustosum.25 Heterologous expression in E. coli provided access to these enzymes that have demonstrated promiscuity in the benzylic hydroxylation of more than thirty resorcinol substrates.1 Resorcinol 4, the precursor to (−)-xyloketal D, was converted to the corresponding benzylic alcohol 5 in excellent yield by ClaD. Following enzymatic oxidation, dienophile 735 was added to the reaction mixture along with benzene, and the mixture was refluxed at 80 °C to affect the desired cycloaddition. This chemoenzymatic cascade generated (−)-xyloketal D (3) in 64% yield in a 2:1 diastereomeric ratio. Products resulting from the C6 o-QM (e.g. 8, Figure 4) were not observed, likely due to strong hydrogen bonding interactions between the hydrogen of the C6 hydroxyl and the C5 carbonyl’s oxygen.1 Additionally, we do not observe the formation of spiroketal products (e.g. 10, Figure 4) as reported by Wilson and coworkers in their synthesis of xyloketal D.35 This is attributed to the ease of conversion of benzylic alcohols to o-QMs in our systems compared to the morpholine derived precursor used by Wilson and coworkers.35 This allows lower reaction temperatures to be employed and thus avoids isomerization of the dienophile.
Figure 4.
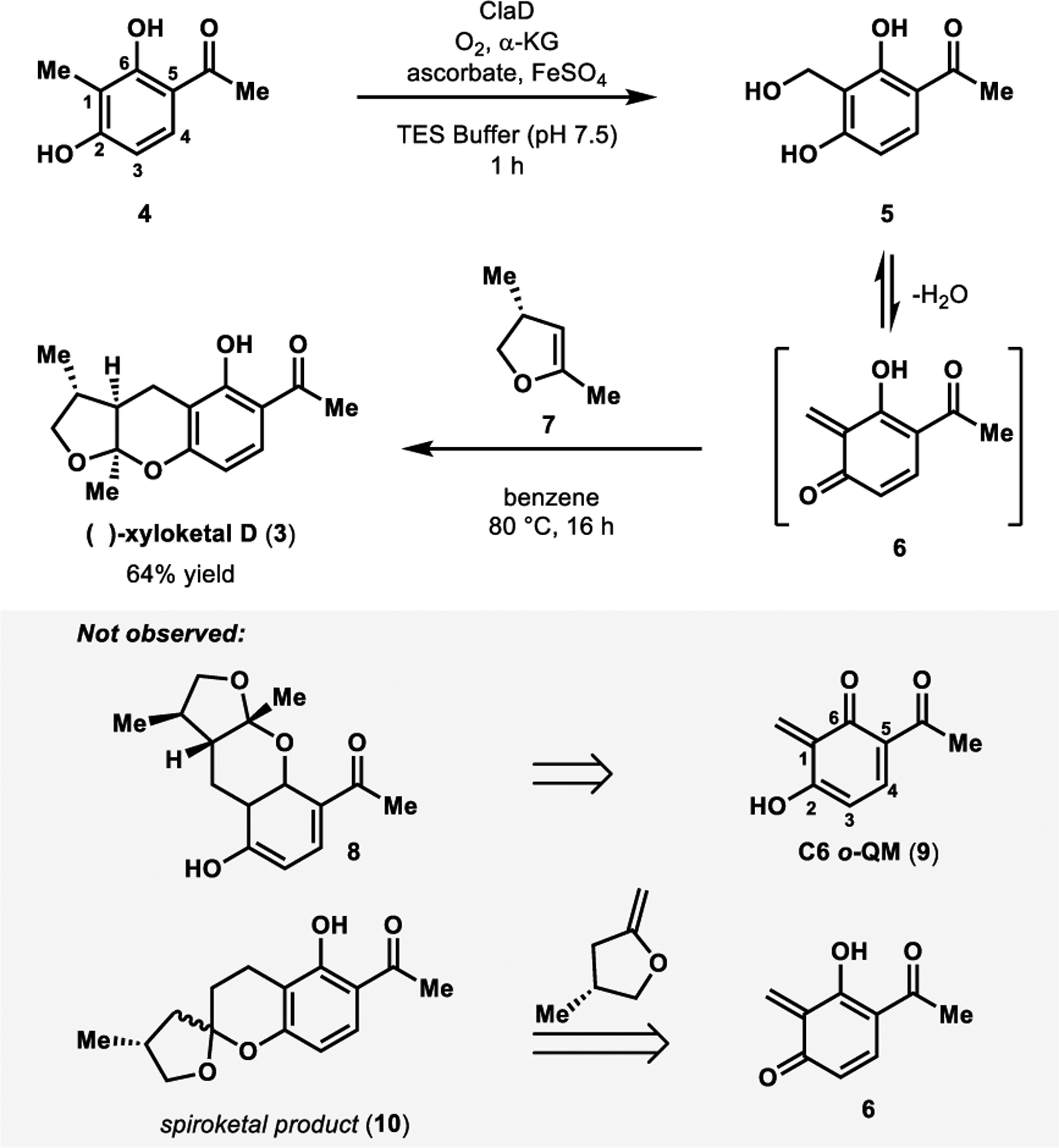
One-pot chemoenzymatic total synthesis of (−)-xyloketal D.
Our chemoenzymatic synthesis of (−)-xyloketal D is performed in a one-pot manner, providing rapid access to the enantioenriched product.21 The modularity of this method also enables facile analog generation. This important synthetic attribute is useful for the evaluation of xyloketal D as a potential therapeutic agent. Furthermore, xyloketal D’s chemoenzymatic synthesis is expected to set the stage for the future total synthesis of additional xyloketal family members using an analogous sequence.
The developed method for biocatalytic access to ortho-quinone methides has the potential to enable the synthesis of numerous other natural products. Retrosynthetic analysis of natural products that could arise from a cycloaddition or nucleophilic addition to an o-QM will identify substrates for enzymatic benzylic hydroxylation. We anticipate that bioinformatic and experimental profiling across NHI enzyme families will pinpoint enzymes that can carry out efficient and selective hydroxylation on desired substrates in the path to other complex molecules.
4. Chemoenzymatic total synthesis involving biocatalytic dearomatization
4.1. Introduction to oxidative dearomatization
The ortho or para-quinol motif (12–14, Figure 5a) obtained via oxidative dearomatization of phenolic compounds forms several biologically important natural products’ core structure.47–49 Chemical oxidants including IIII, IV, PbIV, and CuI reagents have been developed to carry out oxidative dearomatization reactions.47,49 These oxidants have been used in a variety of complex molecule synthesis.47,49 However, small-molecule reagents that mediate oxidative dearomatization often suffer from several disadvantages. For example, these oxidants are typically required in stoichiometric quantities and generate stoichiometric by-products.47 Additionally, small-molecule based oxidants can provide stereocontrol; however, achieving reagent-controlled site-selectivity is a tremendous challenge. Furthermore, side reactions such as dimerization, rearomatization, and rearrangement50 are often observed under the reaction conditions (Figure 5a). Enantioselective versions of oxidative dearomatization reactions have been developed using chiral hypervalent iodine reagents and superstoichiometric amounts of chiral metal complexes.47 However, a catalytic platform for controlling site- and stereoselective oxidative dearomatization remains elusive to small molecule-based approaches.
Figure 5.
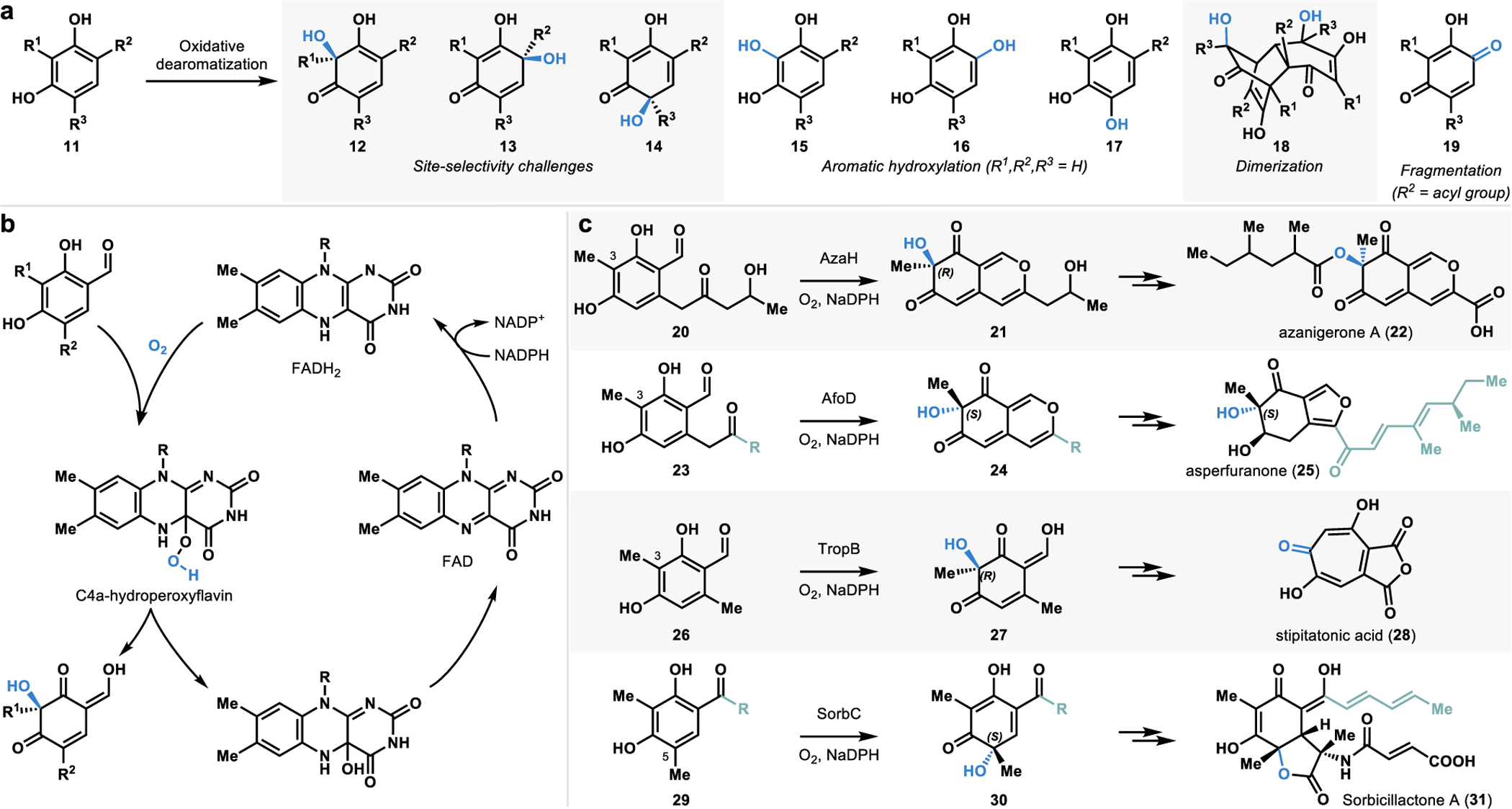
a. Challenges associated with chemical methods of oxidative dearomatization. b. Catalytic mechanism of FDMOs. c. Catalytic activity of FDMOs AzaH, AfoD, TropB, and SorbC.
To solve the limitations posed by the traditional methods, we chose to develop a biocatalytic platform to carry out oxidative dearomatization reactions.2,4 Flavin-dependent monooxygenases (FDMOs) have been implicated in the stereodetermining steps of several biosynthetic pathways.19 Such monooxygenases bind flavin and require molecular oxygen and a nicotinamide cofactor for catalysis (Figure 5b).19 FDMOs present an ideal platform for oxidative dearomatization reactions as these operate under mild conditions with high levels of the site- and stereoselectivity.19,21 Our research in this area was guided by the work of Cox, Tang, Wang, and Watanabe groups.2,26–28,51,52 Tang and coworkers characterized the FDMO AzaH from a silent A. niger gene cluster and demonstrated its role in the C3-hydroxylative dearomatization of resorcinol 20 to form the dearomatized azaphilone intermediate 21 in the biosynthesis of azanigerone A (22, Figure 5c).26 The initial stereoselectivity of AzaH was not reported; however, studies from our lab demonstrated that the newly formed stereocenter possesses the “R” configuration.2 Wang and coworkers identified another FDMO, AfoD, which was implicated in the biosynthesis of asperfuranone (25, Figure 5c).27 This enzyme was initially proposed to catalyze enantioselective oxidative dearomatization of a substrate (23) similar to the native substrate of AzaH (20).27,53 Research from our lab demonstrated that AfoD results in oxidative dearomatization with the same site-selectivity yet complementary stereoselectivity to AzaH.2 Cox and coworkers characterized the function of the FDMO TropB included in the gene cluster encoding for the tropolone natural product stipitatonic acid (28, Figure 5c).51,54 TropB catalyzes the hydroxylative dearomatization of resorcinol 26 to afford quinol product 27.4 The Cox group also identified the FDMO SorbC from the sorbicillactone A (31, Figure 5c) biosynthetic pathway and demonstrated its efficacy in mediating a C5 hydroxylative dearomatization of its native substrate 29 to form the chiral quinol product 30.28 Although they perform similar hydroxylative dearomatization reactions, SorbC offers different site-selectivity compared to AzaH, AfoD, and TropB.4
We began our studies in oxidative dearomatization by heterologously expressing these FDMOs in E. coli and subsequently conducting analytical scale biocatalytic reactions to profile the substrate scope of each enzyme.2 We then developed preparative-scale biocatalytic reactions towards the chemoenzymatic total synthesis of azaphilone natural products, a urea sorbicillinoid natural product, and a tropolone natural product stipitatic aldehyde, respectively (vide infra).2,4
4.2. Chemoenzymatic total synthesis of azaphilone natural products
Azaphilones are biologically active fungal natural products structurally characterized by a pyranoquinone bicyclic core and a fully-substituted carbon atom.55 These natural products bear diverse structural elements that are responsible for a range of interesting biological actions, including anticancer,56 anti-inflammatories,57 and antiviral58 activities (select examples in Figure. 6a). Despite their high utility, the synthesis of azaphilones has proven challenging due to their nonaromatic cyclic structures and the presence of a congested stereocenter.55 Previously reported syntheses of azaphilones typically relied on the oxidative dearomatization of prefunctionalized resorcinol intermediates using hypervalent iodine reagents59 or Pb(OAc)4,60 enabling access to racemic azaphilones. To date, however, there is a single reported method of the enantioselective synthesis of azaphilone core using a small-molecule copper-oxo (−)-sparteine mediated dearomatization strategy reported by Porco and coworkers.47 This state-of-the-art methodology requires superstoichiometric quantities of both the oxidant and the chiral alkaloid ligand.47,61
Figure 6.
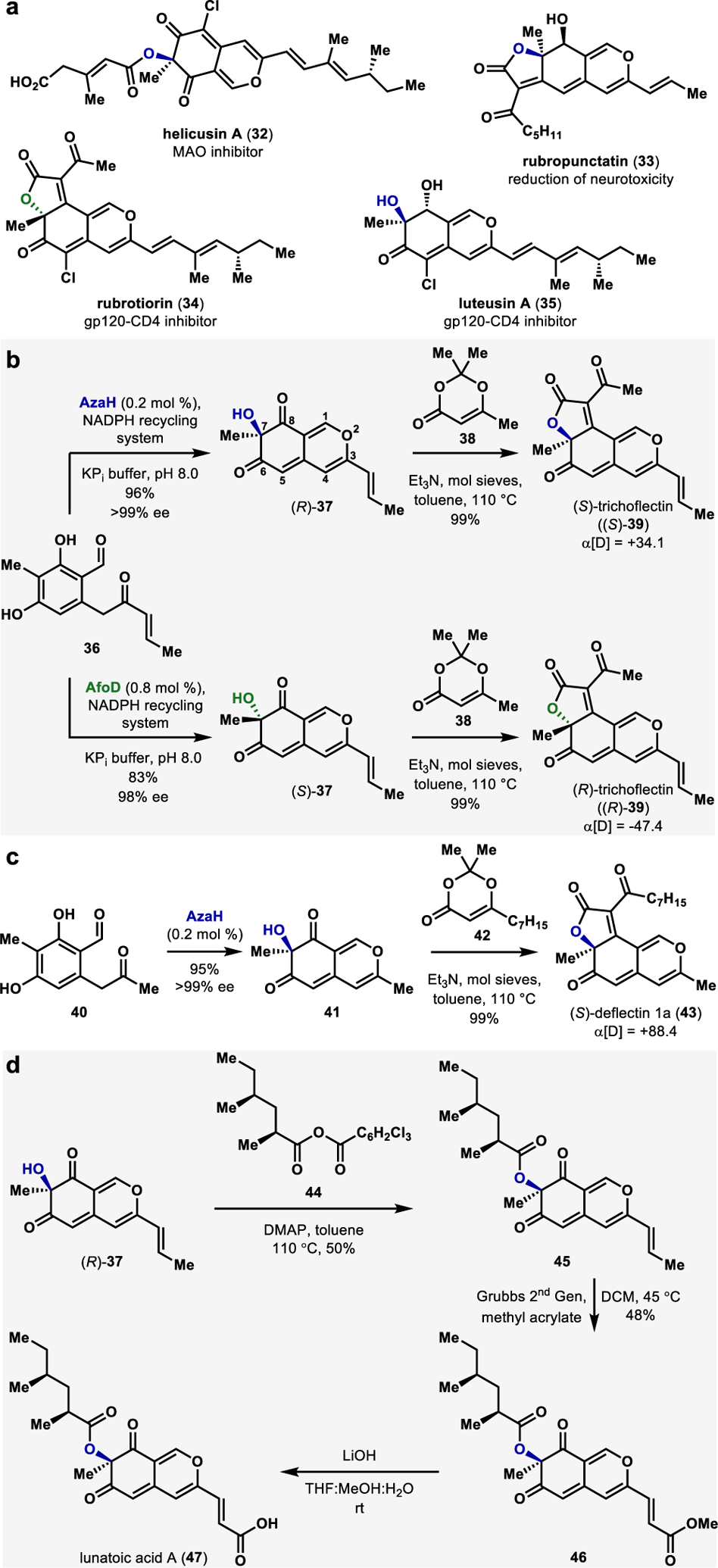
a. Representative examples of bioactive azapilones. b.Chemoenzymatic total synthesis of trichoflectin. c. Chemoenzymatic total synthesis of deflectin. d. Chemoenzymatic total synthesis of lunatoic acid.
The pivotal discovery of complimentary stereoselectivity provided by FDMOs AzaH and AfoD enabled us to pursue a stereodivergent chemoenzymatic total synthesis of the angular azaphilone natural product trichoflectin (39, Figure 6b).2 Trichoflectin was first isolated by Sterner in 1998 and exhibits moderate antimicrobial activity and inhibition of 1,8-dihydroxynaphthalene (DHN)-melanin biosynthesis in certain fungi.62 We envisioned setting the C7-stereocenter of trichoflectin using stereoselective hydroxylative dearomatization of enone 36 mediated by a FDMO.2 Following dearomatization, we envisioned constructing the butanolide ring through acylation of the C7-hydroxyl group, followed by a spontaneous intramolecular Knoevenagel condensation (Figure 6b).63
Hydroxylative dearomatization of enone 36 with AzaH afforded the azaphilone intermediate (R)-37 in 96% yield and in >99% ee (Figure 5b).2 Acylation of (R)-37 using the acylketene generated in-situ from precursor compound 38 provided (S)-trichoflectin, (S)-39, in 99% yield.2 Notably, we were able to exclusively access the angular tricycle and found no traces of the linear regioisomeric product.2 We attribute this to a difference in the electronic properties of the bicyclic core and have observed experimentally that the C8 carbonyl is more electrophilic than C6 (Figure 6b).2 An analogous sequence beginning with hydroxylative dearomatization of enone 36 with AfoD resulted in (R)-trichoflectin, (R)-39, in 98% ee. Access to each enantiomer of this compound provided evidence for the structural revision of the natural product from (S)- to (R)- configuration at C7 based on optical rotation, X-ray crystal data, and comparison of calculated and measured CD spectra.2
We next turned our attention to a chemoenzymatic total synthesis of (S)-deflectin 1a (43, Figure 6c).64 Deflectins are reported to possess inhibitory activity of bacteria and erythrocytes in addition to bearing cytotoxic activity.64 This class also shares a very similar structural scaffold to trichoflectins.64 We began our chemoenzymatic synthesis with hydroxylative dearomatization of methyl ketone 40 to produce the bicyclic intermediate 41 in 95% yield and >99% ee.2 Acylation and subsequent Knoevenagel condensation with acylketene derived from 42 delivered (S)-deflectin-1a (43) in 87% yield.2 Further analysis of this synthetically prepared natural product also provided evidence for its structural revision from (R)- to (S)-configuration at C7.
The final azaphilone natural product constructed using this methodology is lunatoic acid A (47), which exhibits antibacterial and antifungal properties (Figure 6d).65 We envisioned building lunatoic acid A from the intermediate 37, which was obtained through hydroxylative dearomatization with AzaH en-route to (S)-trichoflectin, (S)-39 (Figure 6b).2 Starting with 37, we planned to access the carboxylic acid moiety in lunatoic acid through a cross-metathesis reaction and envisioned appending the chiral aliphatic ester via acylation of the C7-hydroxyl group.2,61 The first challenge that we encountered was the acylation of the tertiary alcohol.2 Acyl chlorides or symmetric anhydrides consistently failed to provide the desired ester.2 Most conditions afforded only the starting material, and forcing conditions resulted in the decomposition of 37. We were finally successful in carrying out the desired transformation using Yamaguchi’s conditions.66 However, after reaction optimization, a maximum of 50% yield of intermediate 45 was obtained, starting with the mixed anhydride 44.67 We then carried out a cross-olefin metathesis with methyl acrylate and Grubbs second-generation catalyst to access methyl ester 46 in a 48% yield.68 We discovered that performing the metathesis before esterification resulted in starting material decomposition, indicating that the protection of the C7-hydroxyl group increases the compound’s stability to the metathesis conditions.2 The mass of Lunatoic acid A was observed by LCMS following the saponification of 46 using LiOH.2 However, sufficient quantities of the pure compound for NMR characterization could not be obtained. This observed instability parallels the reported nature of the acid stability in the isolation literature; methylation of cell-culture extracts was carried out to obtain lunatoic acid A methyl ester, which was characterized in place of the free acid.2,65 All spectroscopic analysis of our synthetic methyl ester match reported values, confirming the correct original assignment of the C7 stereocenter.2,65
4.3. Chemoenzymatic total synthesis of stipitatic aldehyde and urea sorbicillinoid
Tropolones are a structurally diverse class of bioactive fungal natural products characterized by an aromatic cycloheptatriene core bearing an α-hydroxyketone functionality (e.g., 49, Figure 7a).54,69 Synthetic approaches towards tropolones often rely on ring expansion through two-electron rearrangements,70 or via radical based approaches from ortho-dearomatized catechols.71 Although highly successful, the existing synthetic methods to access tropolones suffer from significant limitations in their generality, making it challenging to access all tropolone based natural products.70 In contrast, Nature has devised efficient methods to access tropolones in its biosynthetic machinery.4,72 We developed a two-step, biocatalytic cascade to the tropolone natural product stipitatic aldehyde starting with the resorcinol-derived quinol 27.4,72 Hydroxylative dearomatization of 26 using TropB affords quinol intermediate 27 (Figure 5c). The quinol intermediate 27 undergoes oxidation by an α-KG dependent NHI enzyme TropC to form the radical intermediate 48.4,72 We propose that this radical intermediate undergoes rearrangement resulting in a net ring expansion to form stipitatic aldehyde (49).72
Figure 7.
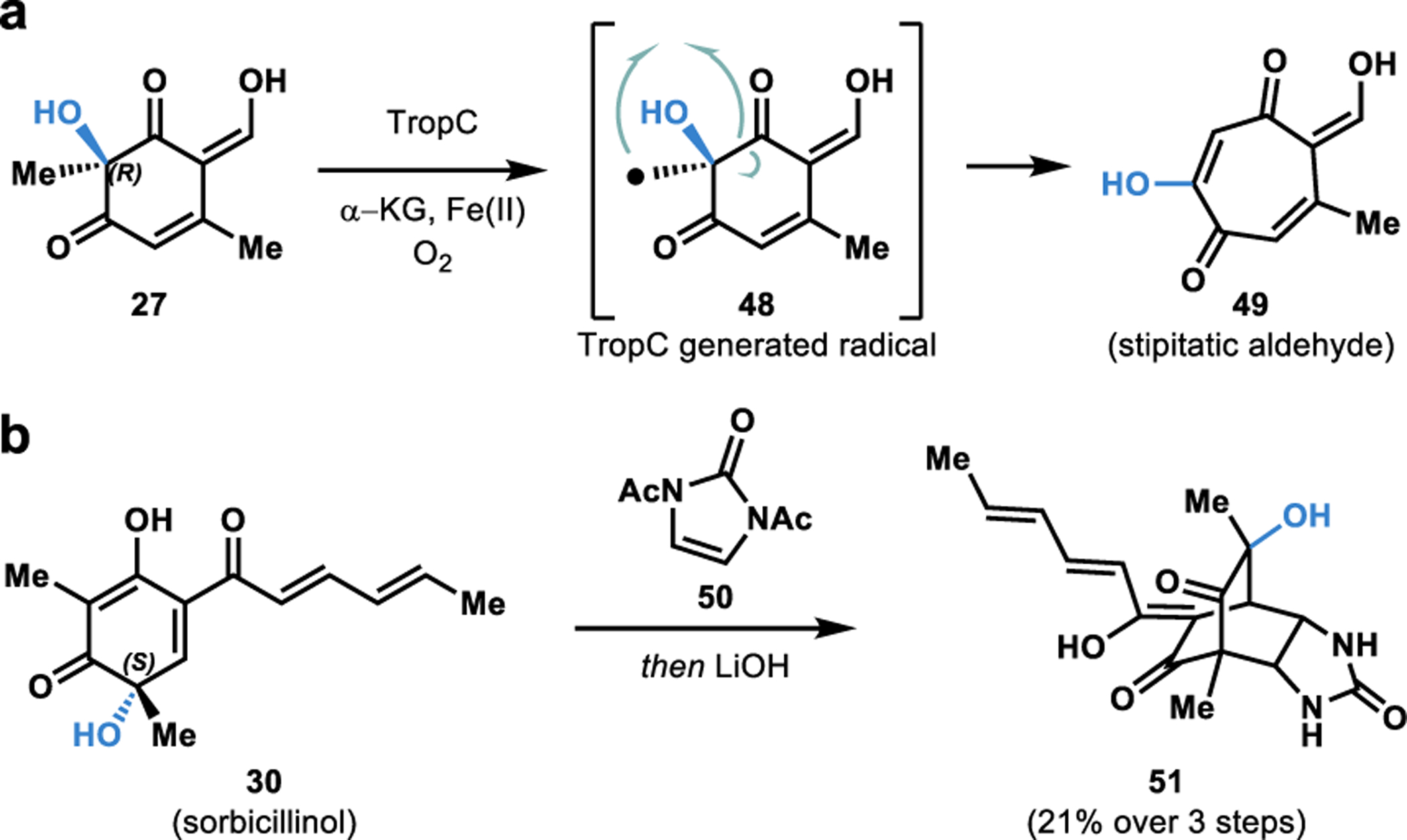
a. Chemoenzymatic total synthesis of stipitatic aldehyde. b. Chemoenzymatic total synthesis of urea sorbicillinoid.
Sorbicillinol (30) forms a core structural component of the vertinoid class of fungal hexaketide-derived bioactive metabolites.73 Sorbicillinoid urea natural product 51 demonstrated antibiotic activity when it was first isolated by Cabrera and coworkers from the fungus P. marquandii.73 We accessed sorbicillinol (30) via SorbC mediated dearomatization of resorcinol substrate 29 (Figure 5c). Sorbicillinol (30) was exposed to bisacylated urea 50, leading to a facile [4+2] cycloaddition reaction.4 Saponification of the acetate groups was carried out on the crude reaction mixture to afford urea sorbicillinoid 51 in 21% yield over three steps.4 This chemoenzymatic total synthesis is an excellent example of rapidly building molecular complexity using biocatalysis. The FDMO SorbC was also utilized by Gulder and coworkers in the stereoselective total synthesis of bissorbicillinoid natural products.74
5. Selective functionalization of paralytic shellfish toxins
Late-stage C–H functionalization of complex bioactive organic molecules offers a streamlined route to access analogs for drug-discovery efforts.13 Enzymes often offer outstanding levels of chemo, site- and stereoselectivity for C–H functionalization as compared to small molecule based methods.13 Saxitoxin (STX, 53, Figure 8) is a paralytic shellfish toxin naturally produced by freshwater cyanobacteria and marine dinoflagellates.75 The picomolar toxicity exhibited by paralytic shellfish toxins stem from their high affinity for voltage-gated ion channels. Structurally related saxitoxin analogs bearing a higher level of oxygenation or at least one sulfate group, such as gonyautoxin 5 (GTX 5, 56, Figure 8), have reduced systemic toxicity and thus possess greater potential as safe pharmaceutical agents.75,76
Figure 8.
Enzymatic access to saxitoxin analogs.
Several analogs of paralytic shellfish toxins possessing varying levels of toxicity are known that are differently functionalized at the C11, C12, C13, and the N1 positions in the complex tricyclic bisguanidinium ion-containing core (Figure 8).75 Accessing such analogs through traditional synthetic methods can present a challenge that requires de-novo synthesis of each saxitoxin analog with a different oxidation pattern.75 In contrast, the use of enzymes to carry out the divergent synthesis of analogs from a common advanced intermediate provides a streamlined route, significantly reducing the time and effort for a given campaign.3,13,17,22
From tricyclic intermediates, we have developed enzymatic sequences to access hydroxylated and sulfated saxitoxin analogs (Figure 8).3,22 We have characterized the Reiske oxygenases SxtT and GxtA that each perform a C–H hydroxylation in a site- and stereoselective manner.3,22 For example, the enzyme SxtT performs the stereoselective hydroxylation of β-saxitoxinol (52, Figure 8), directly generating saxitoxin (53).22 The enzyme GxtA mediates the stereoselective conversion of saxitoxin (53) to 11-β-hydroxy saxitoxin (54).3,22 In addition to selective C–H hydroxylation catalysts, the paralytic shellfish toxin biosynthetic pathway also provides biocatalysts for selective sulfation.3 We characterized the enzyme SxtN from Aphanizomenon sp. NH-5,77,78 which mediates the exclusive sulfation of the carbamate in saxitoxin (53), leading to the formation of GTX 5 (56).3 The enzyme GxtA also carries out an analogous hydroxylation of GTX5 (56) to afford the natural product toxin M1β (57).3 We have also characterized the chemistry and substrate promiscuity of the O-sulfotransferase SxtSUL from Microseira wollei, which effectively carries out the sulfation of 11-β-hydroxy saxitoxin (54) to GTX 3 (55) and mediates a similar reaction to transform M1β (57) to toxin C2 (58).3,22 Characterization of the enzymes SxtT, GxtA, StxN, and SxtSUL constitutes a powerful new approach for the direct functionalization of saxitoxin core.3,17,22 Due to the high degree of systemic toxicity exhibited by saxitoxin and related paralytic shellfish toxins, we carried out these reactions in micromolar scales and analyzed product formation by comparison with authentic product standards via LCMS and MS/MS analysis.3,22 We anticipate that this work will pave the way for the development of novel saxitoxin analogs with enhanced therapeutic potential.
6. Conclusions
The powerful chemistry offered by biocatalytic reactions has resulted in increased adoption of biocatalytic methods by the synthetic community and presents new opportunities for blending small and large molecule catalysts to achieve the most efficient route toward target molecules.79 However, physical enzyme availability as well as limited data on an enzyme’s function and substrate scope can often be a significant hindrance to purely synthetic chemists that are willing to embrace enzymatic catalysis.13 To tap into the vast array of catalysts for which sequences are known, it is essential to accelerate the characterization of the chemistry of this treasure trove of biocatalysts to provide a roadmap for how the synthetic community can apply these tools in synthetic routes.80 We anticipate this gap will be filled by solutions developed by both academic and industrial scientists. We predict that expanding access to characterized biocatalytic enzymes will increase their adoption in chemistry.
This account summarizes the chemoenzymatic total synthesis of complex natural products developed by our group within the past five years.1–4 As demonstrated in this account, the use of biocatalysis in conjunction with contemporary synthetic methods can enable access to concise syntheses of complex natural products that are not easily accessible using traditional chemical methods alone. Biocatalytic synthetic methods have significantly expanded the repertoire of transformations in organic chemists’ toolbox, allowing greater access to chemical space than previously possible.6 Furthermore, the procedural simplicity of such reactions has enabled their gradual adoption by the mainstream synthetic community.6 Consequently, chemoenzymatic methods are becoming increasingly commonplace in academic and industrial settings,11 and a variety of complex natural products are now accessible by combining the best of small- and large- molecule catalysts.79 This synthetic renaissance stands to change the way we think about making molecules.
ACKNOWLEDGMENTS
We are grateful to our present and former colleagues and collaborators who have been involved in the presented research over the past five years. This research was supported by generous funds from the University of Michigan Life Sciences Institute, University of Michigan Department of Chemistry, the National Institutes of Health R35 GM124880, Sloan Foundation, Research Corporation Cottrell Scholars Program, and the Dreyfus Foundation. E.O.R. and J.A.Y. acknowledge support from NSF graduate research fellowships (DGE 1841052). J.B.P. acknowledges support from Ruth L. Kirschstein National Research Service Award (1F31GM139387-01).
Biographies
Suman Chakrabarty graduated with a B.Sc. degree in 2011 from St. Joseph’s College and an M.Sc. degree in Chemistry in 2013 from Christ University, both based in Bangalore, India. He began his doctoral research in 2013 at the University of Nebraska-Lincoln (UNL) under the mentorship of Prof. James M. Takacs. At UNL, Suman developed rhodium-catalyzed asymmetric hydroboration reactions to form chiral tertiary boronic esters. After obtaining his Ph.D. in 2019, he joined Prof. Alison Narayan’s lab as a postdoctoral fellow at the University of Michigan, where he is currently developing axially stereoselective biocatalytic C–C bond-forming reactions.
Evan O. Romero attended Calvin University and earned a B.S. in Chemistry in 2018. While there, he conducted research with Prof. Carolyn Anderson developing gold-catalyzed migrations and rearrangements of propargyloxypyridines. Evan also carried out undergraduate research as a Chemical and Synthetic Development Intern at Bristol-Myers Squibb under the supervision of Dr. Steven Wisniewski. Evan began his graduate studies in Prof. Alison Narayan’s lab at the University of Michigan in 2018, where he is currently developing chemoenzymatic total synthesis of natural products using non-heme iron-dependent enzymes.
Joshua B. Pyser graduated in 2016 with a B.S. degree in chemistry and biomolecular science from Clarkson University in Potsdam, NY. In the same year, he began his doctoral studies with Prof. Alison Narayan of the University of Michigan, where he is currently a Ph.D. candidate preparing to defend his thesis in 2021. His current project involves rapid compound library generation through the use of biocatalysts and chemoenzymatic cascades.
Jessica A. Yazarians graduated with a B.S. in biochemistry from Florida Gulf Coast University in 2016, with a research focus on natural product synthesis under the mentorship of Professor Gregory R. Boyce. She started her doctoral studies in 2016 at the University of Michigan under the direction of Professor Alison Narayan developing a general platform for biocatalytic phenolic cross-coupling reactions.
Alison R. H. Narayan is a native Michigander with a B.S. in chemistry from the University of Michigan. She holds a Ph.D. in organic chemistry from the University of California, Berkeley, where she worked with Prof. Richmond Sarpong. She then worked with Prof. David Sherman as a Life Sciences Research Foundation Postdoctoral Fellow. Alison joined the Department of Chemistry and the Life Sciences Institute at the University of Michigan as an assistant professor in 2015. Her research team develops biocatalytic tools for complex molecule synthesis.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Doyon TJ; Perkins JC; Baker Dockrey SA; Romero EO; Skinner KC; Zimmerman PM; Narayan ARH Chemoenzymatic o-Quinone Methide Formation. J. Am. Chem. Soc 2019, 141, 20269–20277. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights the use of o-quinone methides generated through biocatalytic oxidation in the chemoenzymatic total synthesis of xyloketal natural products.
- (2).Pyser JB; Baker Dockrey SA; Rodríguez Benítez A; Joyce LA; Wiscons RA; Smith JL; Narayan ARH Stereodivergent, Chemoenzymatic Synthesis of Azaphilone Natural Products. J. Am. Chem. Soc 2019, 141, 18551–18559 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights the use of sequence similarity network (SSN) analysis to identify stereodivergent biocatalysts and their applications in the chemoenzymatic total synthesis of azaphilone natural products.
- (3).Lukowski AL; Denomme N; Hinze ME; Hall S; Isom LL; Narayan ARH Biocatalytic Detoxification of Paralytic Shellfish Toxins. ACS Chem. Biol 2019, 14, 941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights the use of enzymes used for carrying out selective C-H hydroxylation and sulfation of paralytic shellfish toxins.
- (4).Baker Dockrey SA; Lukowski AL; Becker MR; Narayan ARH Biocatalytic site- and enantioselective oxidative dearomatization of phenols. Nat. Chem 2018, 10, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights the use of enzymes derived from natural product biosynthetic pathways in the chemoenzymatic total synthesis of azaphilone, tropolone, and sorbicillinoid natural products.
- (5).Harvey AL; Edrada-Ebel R; Quinn RJ The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov 2015, 14, 111–129. [DOI] [PubMed] [Google Scholar]
- (6).Devine PN; Howard RM; Kumar R; Thompson MP; Truppo MD; Turner NJ Extending the application of biocatalysis to meet the challenges of drug development. Nat. Rev. Chem 2018, 2, 409–421. [Google Scholar]
- (7).Zhang X; King-Smith E; Dong L-B; Yang L-C; Rudolf JD; Shen B; Renata H Divergent synthesis of complex diterpenes through a hybrid oxidative approach. Science 2020, 369, 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rodríguez Benítez A; Narayan ARH Frontiers in Biocatalysis: Profiling Function across Sequence Space. ACS Cent. Sci 2019, 5, 1747–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chen K; Arnold FH Engineering new catalytic activities in enzymes. Nat. Catal 2020, 3, 203–213. [Google Scholar]
- (10).Chen K; Huang X; Kan SBJ; Zhang RK; Arnold FH Enzymatic construction of highly strained carbocycles. Science 2018, 360, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Abdelraheem EMM; Busch H; Hanefeld U; Tonin F Biocatalysis explained: from pharmaceutical to bulk chemical production. React. Chem. Eng 2019, 4, 1878–1894. [Google Scholar]
- (12).Clouthier CM; Pelletier JN Expanding the organic toolbox: a guide to integrating biocatalysis in synthesis. Chem. Soc. Rev 2012, 41, 1585–1605. [DOI] [PubMed] [Google Scholar]
- (13).Chakrabarty S; Wang Y; Perkins JC; Narayan ARH Scalable biocatalytic C–H oxyfunctionalization reactions. Chem. Soc. Rev 2020, 49, 8137–8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Huffman MA; Fryszkowska A; Alvizo O; Borra-Garske M; Campos KR; Canada KA; Devine PN; Duan D; Forstater JH; Grosser ST; Halsey HM; Hughes GJ; Jo J; Joyce LA; Kolev JN; Liang J; Maloney KM; Mann BF; Marshall NM; McLaughlin M; Moore JC; Murphy GS; Nawrat CC; Nazor J; Novick S; Patel NR; Rodriguez-Granillo A; Robaire SA; Sherer EC; Truppo MD; Whittaker AM; Verma D; Xiao L; Xu Y; Yang H Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 2019, 366, 1255–1259. [DOI] [PubMed] [Google Scholar]
- (15).Chun SW; Narayan ARH Biocatalytic, Stereoselective Deuteration of α-Amino Acids and Methyl Esters. ACS Catal 2020, 10, 7413–7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lukowski AL; Liu J; Bridwell-Rabb J; Narayan ARH Structural basis for divergent C–H hydroxylation selectivity in two Rieske oxygenases. Nat. Commun 2020, 11, 2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lukowski AL; Mallik L; Hinze ME; Carlson BM; Ellinwood DC; Pyser JB; Koutmos M; Narayan ARH Substrate Promiscuity of a Paralytic Shellfish Toxin Amidinotransferase. ACS Chem. Biol 2020, 15, 626–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dockrey SAB; Suh CE; Rodríguez Benítez A; Wymore T; Brooks III CL; Narayan ARH Positioning-Group-Enabled Biocatalytic Oxidative Dearomatization. ACS Cent. Sci 2019, 5, 1010–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Baker Dockrey SA; Narayan ARH Flavin-dependent biocatalysts in synthesis. Tetrahedron 2019, 75, 1115–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Rodríguez Benítez A; Tweedy SE; Baker Dockrey SA; Lukowski AL; Wymore T; Khare D; Brooks CL; Palfey BA; Smith JL; Narayan ARH Structural Basis for Selectivity in Flavin-Dependent Monooxygenase-Catalyzed Oxidative Dearomatization. ACS Catal 2019, 9, 3633–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Baker Dockrey SA; Doyon TJ; Perkins JC; Narayan ARH Whole-cell biocatalysis platform for gram-scale oxidative dearomatization of phenols. Chem. Biol. Drug. Des 2019, 93, 1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lukowski AL; Ellinwood DC; Hinze ME; DeLuca RJ; Du Bois J; Hall S; Narayan ARH C–H Hydroxylation in Paralytic Shellfish Toxin Biosynthesis. J. Am. Chem. Soc 2018, 140, 11863–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chun SW; Hinze ME; Skiba MA; Narayan ARH Chemistry of a Unique Polyketide-like Synthase. J. Am. Chem. Soc 2018, 140, 2430–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).He Y; Cox RJ The molecular steps of citrinin biosynthesis in fungi. Chem. Sci 2016, 7, 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Fan J; Liao G; Kindinger F; Ludwig-Radtke L; Yin W-B; Li S-M Peniphenone and Penilactone Formation in Penicillium crustosum via 1,4-Michael Additions of ortho-Quinone Methide from Hydroxyclavatol to γ-Butyrolactones from Crustosic Acid. J. Am. Chem. Soc 2019, 141, 4225–4229. [DOI] [PubMed] [Google Scholar]
- (26).Zabala AO; Xu W; Chooi YH; Tang Y Characterization of a silent azaphilone gene cluster from Aspergillus niger ATCC 1015 reveals a hydroxylation-mediated pyran-ring formation. Cell Chem. Biol 2012, 19, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chiang Y-M; Szewczyk E; Davidson AD; Keller N; Oakley BR; Wang CCC A Gene Cluster Containing Two Fungal Polyketide Synthases Encodes the Biosynthetic Pathway for a Polyketide, Asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc 2009, 131, 2965–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fahad A. a.; Abood A; Fisch KM; Osipow A; Davison J; Avramović M; Butts CP; Piel J; Simpson TJ; Cox RJ Oxidative dearomatisation: the key step of sorbicillinoid biosynthesis. Chem. Sci 2014, 5, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).On advances and challenges in biocatalysis. Nat. Catal 2018, 1, 635–636. [Google Scholar]
- (30).Whalley AJS The xylariaceous way of life. Mycol. Res 1996, 100, 897–922. [Google Scholar]
- (31).Roy B; Rout N; Kuila P; Sarkar D Synthesis and structural anomaly of xyloketals-unique benzoxacycles: A review. J Heterocyclic Chem 2020; 1–20. [Google Scholar]
- (32).Su J; Liu H; Guo K; Chen L; Yang M; Chen Q Research Advances and Detection Methodologies for Microbe-Derived Acetylcholinesterase Inhibitors: A Systemic Review. Molecules 2017, 22, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Panda B; Sarkar TK Gold Catalysis: Regio- and Stereoselective Total Synthesis of Xyloketals D and G and the Related Natural Product Alboatrin. J. Org. Chem 2013, 78, 2413–2421. [DOI] [PubMed] [Google Scholar]
- (34).Krohn K; Riaz M; Flörke U Synthesis of Xyloketals, Natural Products from the Mangrove Fungus Xylaria sp. Eur. J. Org. Chem 2004, 2004, 1261–1270. [Google Scholar]
- (35).Pettigrew JD; Freeman RP; Wilson PD Total synthesis of (−)-xyloketal D and its enantiomer - Confirmation of absolute stereochemistry. Can. J. Chem 2004, 82, 1640–1648. [Google Scholar]
- (36).Bai W-J; David JG; Feng Z-G; Weaver MG; Wu K-L; Pettus TRR The Domestication of ortho-Quinone Methides. Acc. Chem. Res 2014, 47, 3655–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Singh MS; Nagaraju A; Anand N; Chowdhury S ortho-Quinone methide (o-QM): a highly reactive, ephemeral and versatile intermediate in organic synthesis. RSC Adv 2014, 4, 55924–55959. [Google Scholar]
- (38).Batsomboon P; Phakhodee W; Ruchirawat S; Ploypradith P Generation of ortho-Quinone Methides by p-TsOH on Silica and Their Hetero-Diels–Alder Reactions with Styrenes. J. Org. Chem 2009, 74, 4009–4012. [DOI] [PubMed] [Google Scholar]
- (39).Chen M-W; Cao L-L; Ye Z-S; Jiang G-F; Zhou Y-G A mild method for generation of o-quinone methides under basic conditions. The facile synthesis of trans-2,3-dihydrobenzofurans. Chem. Commun 2013, 49, 1660–1662. [DOI] [PubMed] [Google Scholar]
- (40).Chiang Y; Kresge AJ; Zhu Y Flash Photolytic Generation of ortho-Quinone Methide in Aqueous Solution and Study of Its Chemistry in that Medium. J. Am. Chem. Soc 2001, 123, 8089–8094. [DOI] [PubMed] [Google Scholar]
- (41).González-Pelayo S; López LA Microwave-Assisted Generation and Capture by Azoles of ortho-Quinone Methide Intermediates under Aqueous Conditions. Eur. J. Org. Chem 2017, 2017, 6003–6007. [Google Scholar]
- (42).Sugimoto H; Nakamura S; Ohwada T Generation and Application of o-Quinone Methides Bearing Various Substituents on the Benzene Ring. Adv. Synth. Catal 2007, 349, 669–679. [Google Scholar]
- (43).Stokes SM; Ding F; Smith PL; Keane JM; Kopach ME; Jervis R; Sabat M; Harman WD Formation of o-Quinone Methides from η2-Coordinated Phenols and Their Controlled Release from a Transition Metal To Generate Chromans. Organometallics 2003, 22, 4170–4171. [Google Scholar]
- (44).Herr CQ; Hausinger RP Amazing Diversity in Biochemical Roles of Fe(II)/2-Oxoglutarate Oxygenases. Trends Biochem. Sci 2018, 43, 517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Martinez S; Hausinger RP Catalytic Mechanisms of Fe(II)-and 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zwick CR III; Renata H Harnessing the biocatalytic potential of iron- and α-ketoglutarate-dependent dioxygenases in natural product total synthesis. Nat. Prod. Rep 2020, 37, 1065–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Zhu J; Grigoriadis NP; Lee JP; Porco JA Synthesis of the Azaphilones Using Copper-Mediated Enantioselective Oxidative Dearomatization. J. Am. Chem. Soc 2005, 127, 9342–9343. [DOI] [PubMed] [Google Scholar]
- (48).Wu W-T; Zhang L; You S-L Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Chem. Soc. Rev 2016, 45, 1570–1580. [DOI] [PubMed] [Google Scholar]
- (49).Roche SP; Porco JA Jr. Dearomatization Strategies in the Synthesis of Complex Natural Products. Angew. Chem. Int. Ed 2011, 50, 4068–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Dong S; Zhu J; Porco JA Enantioselective Synthesis of Bicyclo[2.2.2]octenones Using a Copper-Mediated Oxidative Dearomatization/[4 + 2] Dimerization Cascade. J. Am. Chem. Soc 2008, 130, 2738–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Abood A; Al-Fahad A; Scott A; Hosny AE-DMS; Hashem AM; Fattah AMA; Race PR; Simpson TJ; Cox RJ Kinetic characterisation of the FAD dependent monooxygenase TropB and investigation of its biotransformation potential. RSC Adv 2015, 5, 49987–49995. [Google Scholar]
- (52).Sato M; Winter JM; Kishimoto S; Noguchi H; Tang Y; Watanabe K Combinatorial Generation of Chemical Diversity by Redox Enzymes in Chaetoviridin Biosynthesis. Org. Lett 2016, 18, 1446–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Somoza AD; Lee K-H; Chiang Y-M; Oakley BR; Wang CCC Reengineering an Azaphilone Biosynthesis Pathway in Aspergillus nidulans To Create Lipoxygenase Inhibitors. Org. Lett 2012, 14, 972–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Davison J; al Fahad A; Cai M; Song Z; Yehia SY; Lazarus CM; Bailey AM; Simpson TJ; Cox RJ Genetic, molecular, and biochemical basis of fungal tropolone biosynthesis. PNAS 2012, 109, 7642–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Gao J-M; Yang S-X; Qin J-C Azaphilones: Chemistry and Biology. Chem. Rev 2013, 113, 4755–4811. [DOI] [PubMed] [Google Scholar]
- (56).Wang W; Liao Y; Chen R; Hou Y; Ke W; Zhang B; Gao M; Shao Z; Chen J; Li F Chlorinated Azaphilone Pigments with Antimicrobial and Cytotoxic Activities Isolated from the Deep Sea Derived Fungus Chaetomium sp NA-S01-R1. Mar. Drugs 2018, 16, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Yasukawa K; Takahashi M; Natori S; Kawai K; Yamazaki M; Takeuchi M; Takido M Azaphilones inhibit tumor promotion by 12-O-tetradecanoylphorbol-13-acetate in two-stage carcinogenesis in mice. Oncology 1994, 51, 108–1122. [DOI] [PubMed] [Google Scholar]
- (58).Matsuzaki K; Tahara H; Inokoshi J; Tanaka H; Masuma R; Omura S New brominated and halogen-less derivatives and structure-activity relationship of azaphilones inhibiting gp120-CD4 binding. J Antibiot (Tokyo) 1998, 51, 1004–1011. [DOI] [PubMed] [Google Scholar]
- (59).Zhu JL; Germain AR; Porco JA Synthesis of azaphilones and related molecules by employing cycloisomerization of o-alkynylbenzaldehydes. Angew. Chem. Int. Ed 2004, 43, 1239–1243. [DOI] [PubMed] [Google Scholar]
- (60).Chong R; Gray RW; King RR; Whalley WB The synthesis of (±) mitorubrin. J. Chem. Soc. D 1970, 101a–101a. [DOI] [PubMed] [Google Scholar]
- (61).Zhu J; Porco JA Asymmetric Syntheses of (−)-Mitorubrin and Related Azaphilone Natural Products. Org. Lett 2006, 8, 5169–5171. [DOI] [PubMed] [Google Scholar]
- (62).Thines E; Anke H; Sterner O Trichoflectin, a Bioactive Azaphilone from the Ascomycete Trichopezizella nidulus. J. Nat. Prod 1998, 61, 306–308. [DOI] [PubMed] [Google Scholar]
- (63).Peixoto PA; Boulangé A; Ball M; Naudin B; Alle T; Cosette P; Karuso P; Franck X Design and Synthesis of Epicocconone Analogues with Improved Fluorescence Properties. J. Am. Chem. Soc 2014, 136, 15248–15256. [DOI] [PubMed] [Google Scholar]
- (64).Anke H; Kemmer T; Höfle G Deflectins, new antimicrobial azaphilones from Aspergillus deflectus. J Antibiot (Tokyo) 1981, 34, 923–928. [DOI] [PubMed] [Google Scholar]
- (65).Nukina M; Marumo S Lunatoic acid A and B, aversion factor and its related metabolite of cochliobolus lunata. Tetrahedron Lett 1977, 2603–2606. [Google Scholar]
- (66).Kawanami Y; Ito Y; Kitagawa T; Taniguchi Y; Katsuki T; Yamaguchi M Asymmetric alkylation of carboxyamides by using trans-2,5-disubstituted pyrrolidines as chiral auxiliaries. Tetrahedron Lett 1984, 25, 857–860. [Google Scholar]
- (67).Myers AG; Yang BH; Chen H; McKinstry L; Kopecky DJ; Gleason JL Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of Highly Enantiomerically Enriched Carboxylic Acids, Alcohols, Aldehydes, and Ketones. J. Am. Chem. Soc 1997, 119, 6496–6511. [Google Scholar]
- (68).Blackwell HE; O’Leary DJ; Chatterjee AK; Washenfelder RA; Bussmann DA; Grubbs RH New Approaches to Olefin Cross-Metathesis. J. Am. Chem. Soc 2000, 122, 58–71. [Google Scholar]
- (69).Cox R Oxidative rearrangements during fungal biosynthesis. Nat. Prod. Rep 2014, 31, 1405–1424. [DOI] [PubMed] [Google Scholar]
- (70).Liu N; Song W; Schienebeck CM; Zhang M; Tang W Synthesis of Naturally Occurring Tropones and Tropolones. Tetrahedron 2014, 70, 9281–9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Barbier M; Barton DHR; Devys M; Topgi RS A simple synthesis of the tropone nucleus. J. Chem. Soc., Chem. Commun 1984, 743–744. [Google Scholar]
- (72).Doyon JD; Skinner KC; Yang D; Mallik L; Wymore T; Koutmos M; Zimmerman PM; Narayan ARH Radical Tropolone Biosynthesis, 2020. ChemRxiv Preprint. 10.26434/chemrxiv.12780044.v1. [DOI]
- (73).Cabrera GM; Butler M; Rodriguez MA; Godeas A; Haddad R; Eberlin MN A sorbicillinoid urea from an intertidal Paecilomyces marquandii. J. Nat. Prod 2006, 69, 1806–1808. [DOI] [PubMed] [Google Scholar]
- (74).Sib A; Gulder TAM Stereoselective Total Synthesis of Bisorbicillinoid Natural Products by Enzymatic Oxidative Dearomatization/Dimerization. Angew. Chem. Int. Ed 2017, 56, 1288812891. [DOI] [PubMed] [Google Scholar]
- (75).Thottumkara AP; Parsons WH; Du Bois J Saxitoxin. Angew. Chem. Int. Ed 2014, 53, 5760–5784. [DOI] [PubMed] [Google Scholar]
- (76).Walker JR; Novick PA; Parsons WH; McGregor M; Zablocki J; Pande VS; Du Bois J Marked difference in saxitoxin and tetrodotoxin affinity for the human nociceptive voltage-gated sodium channel (Nav1.7). PNAS 2012, 109, 18102–18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Mihali TK; Kellmann R; Neilan BA Characterisation of the paralytic shellfish toxin biosynthesis gene clusters in Anabaena circinalis AWQC131C and Aphanizomenon sp. NH-5. BMC Biochem 2009, 10, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Cullen A; D’Agostino PM; Mazmouz R; Pickford R; Wood S; Neilan BA Insertions within the Saxitoxin Biosynthetic Gene Cluster Result in Differential Toxin Profiles. ACS Chem. Biol 2018, 13, 3107–3114. [DOI] [PubMed] [Google Scholar]
- (79).Hauer B Embracing Nature’s Catalysts: A Viewpoint on the Future of Biocatalysis. ACS Catal 2020, 10, 8418–8427. [Google Scholar]
- (80).Baltz RH Natural product drug discovery in the genomic era: realities, conjectures, misconceptions, and opportunities. J. Ind. Microbiol. Biotechnol 2019, 46, 281–299. [DOI] [PubMed] [Google Scholar]