

Abstract
Objectives
HIV-associated lipoatrophy has been associated with mitochondrial dysfunction induced by nucleoside reverse transcriptase inhibitor therapy. We hypothesize that lipid profiles and markers of mitochondrial function will improve in HIV-lipoatrophic patients switched to the nucleotide analogue tenofovir.
Methods
Ten patients receiving stavudine, lamivudine and lopinavir/ritonavir (Kaletra®) for over 6 years were switched from stavudine to tenofovir for 48 weeks. Subcutaneous fat tissue biopsies, fasting metabolic tests, HIV RNA, CD4 cell count and whole body dual energy X-ray absorptiometry (DEXA) scans were obtained at study entry and week 48. Mitochondrial DNA (mtDNA) copies/cell and mitochondrial morphology were assessed in adipose tissue biopsies, mtDNA 8-oxo-deoxyguanine in peripheral blood mononuclear cells, and glutathione (GSH) and F2-isoprostane in plasma.
Results
There was no change in limb fat mass by DEXA; however, trunk fat mass increased by 18.9% (P = 0.01). Fasting total cholesterol decreased by 33 mg/dL (P = 0.005) and serum glucose decreased by 4 mg/dL (P = 0.039). mtDNA copies/cell increased from 386 to 1537 (P < 0.001). Transmission electron microscopy showed that mitochondrial cristae were lacking or poorly defined at study entry, whereas mitochondrial inner structures were more well defined and outer membranes were intact at 48 weeks. Oxidative damage decreased in 8/10 patients, GSH increased and F2-isoprostane decreased.
Conclusions
The results from this study demonstrate that systemic and peripheral fat mitochondria improve in patients switched to tenofovir following long-term exposure to stavudine, while continuing protease inhibitor therapy.
Keywords: adipose, HIV, lipodystrophy, metabolic, mitochondria
Introduction
One of the main long-term adverse effects of HIV treatment is lipodystrophy, a metabolic syndrome that manifests phenotypically as areas of fat depletion (lipoatrophy) or accumulation. Lipodystrophy has been associated with decreased quality of life, depression, disincentive to maintain antiretroviral therapy and metabolic abnormalities such as hyperlipidaemia and insulin resistance.1
Initially, protease inhibitors (PIs) were believed to be the main cause of lipodystrophy,2,3 but lipodystrophy was later found to occur in PI-naive patients as well.4,5 Several studies since then have linked lipoatrophy with the use of nucleoside reverse transcriptase inhibitors (NRTIs).6,7 Drugs in the NRTI class inhibit mitochondrial DNA (mtDNA) polymerase-γ, which can lead to mtDNA depletion and/or impairment in the production of mitochondrial enzymes and proteins involved in oxidative phosphorylation. This latter effect can further lead to increased oxidative stress.1,8 Substantial decreases in mtDNA content have been observed in the subcutaneous fat of NRTI-treated lipoatrophic patients in comparison with controls.9–11 Morphological correlates include dense mitochondrial proliferation and abnormal cristae architecture, particularly in those treated with stavudine.10 The severity of mtDNA depletion and the relative risk of developing subcutaneous fat wasting appear to be greatest in those treated with stavudine.12
Interestingly, depleted subcutaneous fat and mtDNA can be replenished in some patients who switch to less toxic NRTIs.13–15 Overall, tenofovir (an NRTI), abacavir and lamivudine are weak inhibitors of mtDNA polymerase-γ, and thus they exhibit less mitochondrial toxicity than stavudine, didanosine and zidovudine, which are more potent inhibitors.16–19 Despite the well-known toxicities associated with stavudine, it remains widely used in resource-limited settings.
This study was designed to assess changes in peripheral fat and metabolic parameters among virologically controlled patients who switched from stavudine to tenofovir, while continuing lamivudine and lopinavir/ritonavir. An additional component of the study was to investigate the effect of the NRTI substitution on mtDNA copies/cell and mitochondrial morphology in subcutaneous adipose tissue, levels of 8-oxo-deoxyguanine mitochondrial oxidative damage in peripheral blood mononuclear cells (PBMCs) and markers of oxidative stress in plasma.
Materials and methods
Study population
HIV-infected subjects were recruited from a cohort enrolled in the Abbott-sponsored M97-720 clinical trial at Northwestern University who had been on the stavudine, lamivudine and lopinavir/ritonavir regimens for at least 300 weeks. All subjects in the M97-720 study were advised to switch from stavudine to tenofovir in response to the emerging literature, which associated stavudine with metabolic toxicities. The subjects who agreed to switch from stavudine to tenofovir and were willing to undergo fat biopsies were eligible for this study. Subjects provided written informed consent following approval of the study by the Institutional Review Board at Northwestern University and at the University of Hawaii where laboratory assays were performed. Twelve subjects were enrolled in this study: two of whom withdrew following relocation.
Assessments of body composition
Subjects were evaluated for clinical lipodystrophy and mitochondrial function prior to switching from stavudine to tenofovir (study entry) and again 48 weeks post-tenofovir switch. The assessments were performed in a fasting state (no food or beverage for at least 8 h). Clinical assessments included a physical examination for lipoatrophy in the arms, legs and face, and for fat accumulation in the abdomen, neck and breasts. Body mass index (BMI) was calculated {weight (kg)/[height (m)]2}.20
Body composition was assessed by whole body dual energy X-ray absorptiometry (DEXA) measurements with a total body scanner (QDR-2000 Hologic, Waltham, MA, USA). DEXA measurements included arm, leg, limb (both arms plus both legs) and trunk fat mass (g) and fat percentage in each body area.
Subjects completed a questionnaire about their dietary habits, use of lipid-lowering agents and body mass supplements, frequency of exercise, smoking, alcohol use and a self-assessment of bodily changes at study entry and 48 weeks post-drug switch.
Laboratory assessments
Fasting blood specimens were collected for measurements of total cholesterol, high-density lipoprotein (HDL) cholesterol, calculated low-density lipoprotein (LDL) cholesterol, triglycerides, serum glucose and insulin. Samples for venous lactate measurement were drawn without a tourniquet into chilled heparin tubes and processed within 30 min. Plasma HIV-1 RNA quantification (Ultrasensitive Roche Amplicor Monitor version 1.0 assay) and CD4 cell counts were performed/determined.
Fat biopsy procedures
Subcutaneous adipose (fat) tissue was collected from the proximal lateral thigh by the same dermatologist using a 4.0 mm Sklar Tru-Punch disposable biopsy punch instrument. A scalpel was used to cut off the skin and the remaining fat was placed into RNAlater (Ambion, Austin, TX, USA) at 4°C for isolating RNA and stored at −70°C. The instrument was reinserted deeper into the same biopsy site and another piece of fat was removed and cut in half. Half of the specimen was placed in RNAlater and frozen for DNA isolation and the other half was stored in 100% glutaraldehyde (4°C) for electron microscopy. Fat tissue was stored at the site and shipped to the University of Hawaii Laboratory for assaying within 4 weeks.
PBMC isolation
Blood was collected in EDTA vacutainer tubes and PBMCs were isolated over a Ficoll-Paque gradient and washed three times with PBS. Plasma was stored at −70°C for assessment of mitochondrial markers involved in oxidative stress. Cells were viably cryopreserved.
mtDNA quantification
Analysis of mtDNA copies/cell was conducted by absolute quantitative real-time PCR. DNA was extracted from frozen adipose tissue previously stored at −70°C using a Blood and Tissue DNA Kit (Qiagen, Inc., USA). Standardization of real-time PCR was performed using LightCycler FastStart DNA Master SYBR Green I with the Roche LightCycler instrument (Roche, Indianapolis, IN, USA), as described previously.21 Each sample and standard were run in duplicate (20 µL reaction volume) and contained: SYBR Green Master Mix, 10 pM of mitochondrial or genomic primers and ∼10 ng DNA of sample. PCR cycling conditions were: denaturation at 95°C for 10 min, amplification for 40 cycles at 95°C for 10 s, 58°C for 5 s and 72°C for 5 s, melt curve analysis beginning at 65°C and increasing temperature half a degree every 30 s for 60 cycles. The results were then analysed with Version 4.0 LightCycler software (Roche).
Mitochondrial 8-oxo-7-hydrodeoxyguanosine (8-oxo-dG)
Mitochondrial 8-oxo-dG damage was assessed using a Gene-Specific Repair Assay.22,23 Ten micrograms of PBMC DNA was isolated with a DNeasy Blood and Tissue Kit (Qiagen, Inc.). DNA was then digested with PvuII (New England BioLabs, Inc., Ipswich, MA, USA) overnight to linearize mtDNA. Digested DNA was halved: 5 µg of DNA was treated with human 8-oxoguanine DNA glycosylase (hOGG1) for 1 h at 37°C in a reaction volume of 15 µL and then for 1 h at 65°C for enzyme deactivation, and the remaining 5 µg of DNA was left untreated and stored at 4°C. For analysis, 4 µL of 1× Alkaline Agarose Loading Dye (Boston Bioproducts, MA, USA) was added, and cleaved and non-cleaved products were resolved on a 0.75% alkaline agarose gel. DNA was transferred to nylon (+) membranes using standard Southern blot methodology. Human mitochondrial probes specific for cytochrome b were labelled with digoxigenin-dUTP (Roche) by linear PCR amplification. Primer sequences were: DigFor, GCT ACC TTC ACG CCA A (14 976–15 001); and DigRev, CCG TTT CGT GCA AGA AT (15 357–15 341). Blots were hybridized overnight at 45°C and processed for chemiluminescent detection following Roche protocols. Finally, membranes were developed on a chemilumi imager (Roche) using LumiAnalyst software (Roche). Mitochondrial 8-oxo-dG damage was quantified by calculating break frequencies (BFs) based on the Poisson distribution of DNA treated with the hOGG1 repair enzyme and DNA untreated.
Glutathione (GSH) and F2-isoprostane
GSH was processed from human plasma using a Glutathione Assay Kit (Cayman Chemical, Ann Arbor, MI, USA) and measured at 405 nm on a microplate reader (Molecular Devices). F2-isoprostane was purified from human plasma using an 8-Isoprostane Affinity Purification Kit (Cayman Chemical) and assessed by an 8-Isoprostane Competitive Enzyme Immunoassay Kit (Cayman Chemical), then measured at 412 nm. Samples were plated in triplicate and analysed with SoftMax Pro software. Final quantities of GSH and F2-isoprostane were calculated according to kit instructions.
Transmission electron microscopy (TEM)
Subcutaneous adipose tissue was fixed at 4°C in 3% paraformaldehyde and 3% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 (Electron Microscopy Sciences, Hatfield, PA, USA), then post-fixed in 1% osmium tetroxide. Tissues were embedded in LX-112 epoxy resin (Ladd Research, Williston, VT, USA), sectioned (80 nm), mounted on slot or mesh grids and double stained with uranyl acetate and lead citrate. Images were viewed at ×16 000–31 500 on a Zeiss LEO 912 energy-filtering TEM. Toluidine Blue-stained adipocytes and cell area were evaluated at ×10 magnification on a Zeiss Axiovert 200 and analysed using AxioVision Rel. 4.4 software.
Statistical analysis
A Wilcoxon signed-rank test for non-parametric data was conducted to analyse changes in fat content and metabolic parameters in patients from study entry to week 48. Changes in peripheral limb fat and mtDNA quantities were correlated using Spearman's correlation coefficient. A Student's t-test assessed significant differences at study entry and week 48 for mtDNA, 8-oxo-dG, GSH, F2-isoprostane values and adipocyte cell area. Additionally, a Pearson product moment correlation was calculated to determine whether a significant relationship exists across various oxidative damage and mtDNA parameters.
Results
The results of the 10 subjects who completed the study are presented. At study entry, the subjects had been on a regimen of stavudine, lamivudine and lopinavir/ritonavir for a median of 358 weeks (∼6.9 years). Subjects were male with a mean age of 39 years. Two subjects had lipoatrophy, two had fat accumulation and six subjects had both lipoatrophy and fat accumulation. All subjects had HIV-1 RNA levels <50 copies/mL and the median study entry CD4 count was 781 cells/mm3. Viral suppression below 50 copies/mL was maintained in all subjects, and there were no significant changes in CD4 cell counts during the study. Clinical assessments (BMI, lipoatrophy and fat accumulation), fasting cholesterol and triglycerides, glucose, lactate, insulin, fat mass (limb, trunk and total) and percentage leg fat by DEXA at study entry and 48 weeks after stavudine-to-tenofovir switch are presented in Table 1. Median fasting total cholesterol significantly decreased from 204 to 173 mg/dL (mean change −33 mg/dL, P = 0.005) and serum glucose decreased from 93.5 to 91.5 mg/dL (mean change −4 mg/dL, P = 0.039). Median trunk fat mass increased by 18.9% (P = 0.01) from 9568 to 11 273 g. However, there was no change in limb or total fat mass or percentage of leg fat. Three subjects reported taking lipid-lowering agents at study entry and continued this therapy throughout the study. No changes in diet, exercise or use of body mass building supplements were reported.
Table 1.
Clinical, metabolic and DEXA analysis parameters at study entry and week 48
| Week 0 [median (range)] | Week 48 [median (range)] | Mean change (%) | P value* | |
|---|---|---|---|---|
| Body mass index (kg/m2) | 27.2 (21.8–41) | 27.2 (21.9–41.3) | ||
| Clinical lipoatrophy (%) | 80 | 80 | ||
| Clinical fat accumulation (%) | 80 | 70 | ||
| Total cholesterol (mg/dL) | 204 (170–299) | 173 (145–237) | −33 | 0.005* |
| LDL cholesterol (mg/dL) | 116 (85–210) | 105 (70–400) | 2.8 | |
| HDL cholesterol (mg/dL) | 46 (30–87) | 42 (26–64) | −4.5 | |
| Triglycerides (mg/dL) | 220 (88–408) | 178 (91–426) | −25 | |
| Glucose (mg/dL) | 93.5 (84–104) | 91.5 (81–104) | −4 | 0.039* |
| Insulin (µU/mL) | 9.5 (6–38) | 16 (0.9–61) | 9.4 | |
| Lactate (mmol/L) | 0.8 (0.4–1.7) | 0.9 (0.4–1) | 0.6 | |
| DEXA analysis | ||||
| leg fat (%) | 12.7 (7–36.8) | 12.9 (7.2–39.2) | 0.7 | |
| limb fat mass (g) | 4925 (1908–24 410) | 4930 (2140–22 408) | −392# (−2%) | |
| trunk fat mass (g) | 9568 (2714–20 266) | 11 273 (3410–24 777) | 1589 (18.9%) | 0.01* |
| total fat mass (g) | 18 885 (6721–45 152) | 200 047 (7090–48 473) | 1161 |
*Statistically significant with P < 0.05. Wilcoxon signed-rank test for non-parametric comparisons.
#No correlation between change in limb fat mass and change in fat mtDNA (Spearman's coefficient = 0.85).
mtDNA quantities in fat tissue significantly increased (P < 0.001) from a mean of 386 ± 167 copies/cell at study entry to a mean of 1537 ± 342 copies/cell at 48 weeks post stavudine-to-tenofovir switch (Table 2). No correlation was found between changes in mtDNA levels and those in limb fat mass (Spearman's correlation coefficient = 0.85).
Table 2.
Mitochondrial and oxidative stress parameters at study entry (week 0) and week 48 post-drug switch
| Patient | Adipose tissue mtDNA (copies/cell) |
8-oxo-dG (BF)a
|
Glutathione (µM) |
F2-isoprostane (pg/mL) |
||||
|---|---|---|---|---|---|---|---|---|
| week 0 | week 48 | week 0 | week 48 | week 0 | week 48 | week 0 | week 48 | |
| 1 | 514 | 1564 | 0.18 | 0.27 | 0.58 | UDb | 157.65 | 119.67 |
| 2 | 460 | 1255 | 0.76 | 0.42 | UD | 0.99 | 190.64 | 182.65 |
| 3 | 351 | 2016 | 0.33 | 0.23 | UD | 0.07 | 184.53 | 9.03 |
| 4 | 663 | 1379 | 0.67 | 0.38 | 0.87 | 1.10 | 163.90 | 45.94 |
| 5 | 206 | 1650 | 0.52 | 0.2 | 0.94 | 2.19 | 1.33 | 95.69 |
| 6 | 163 | 797 | 0.59 | 0.01 | 1.34 | 1.28 | 189.31 | 0.80 |
| 7 | 599 | 1688 | 0.68 | 0.57 | NAc | NA | NA | NA |
| 8 | 306 | 1904 | 0.07 | 0.05 | UD | UD | 2.40 | 145.74 |
| 9 | 255 | 1570 | 1.76 | 0.93 | 1.34 | 2.44 | 139.01 | 4.94 |
| 10 | 339 | 1550 | 0.02 | 0.03 | 1.41 | 0.05 | 15.05 | 23.63 |
| Mean ± SD | 386 ± 167* | 1537 ± 342* | 0.56 ± 0.50 | 0.31 ± 0.28 | 1.08 ± 0.33 | 1.16 ± 0.93 | 115.98 ± 84.00 | 69.78 ± 68.03 |
aBF, break frequency calculation.
bUD, undetectable; concentration <50 nM to be detected.
cNA, sample not available for quantification.
*P < 0.001.
Although differences in oxidatively damaged mtDNA did not reach statistical significance, a reduction was observed in 8 of 10 patients, improving by ∼40% from a mean BF of 0.56 ± 0.50 at study entry to 0.31 ± 0.28 at week 48. Average GSH was 1.08 ± 0.33 µM at study entry and improved to an average of 1.16 ± 0.93 µM at 48 weeks. Mean F2-isoprostane decreased from 115.98 ± 84.00 pg/mL at study entry to 69.78 ± 68.03pg/mL at 48 weeks, but this did not reach statistical significance. Table 2 shows calculated BFs, GSH and F2-isoprostane values for all patients at study entry and week 48. No significant correlation was found between 8-oxo-dG, and/or GSH, and/or F2-isoprostane quantities.
Morphological studies of adipose tissue by TEM collected at study entry (Figure 1a and b) showed mitochondria at ×16 000 magnification that appeared small and with poorly defined inner membrane structures as well as damaged cristae. Furthermore, the majority of mitochondria possessed disrupted outer membranes (Figure 1b). However, TEM of adipose tissues following stavudine switch to tenofovir showed mitochondria that appeared larger and with well-defined inner membrane cristae (Figure 1c and d). At ×31 500 magnification, defined intact outer double membranes were visible (Figure 1d). Mitochondrial diameters ranged from 200 to 400 nm with one to three mitochondria (25% disrupted) at study entry and two to four mitochondria (none disrupted) at week 48 in each field at ×16 000 magnification (four fields/patient were evaluated). Five adipocytes/patient from random fields at ×10 magnification at study entry and week 48 were selected for analysis. Mean adipocyte area ± SD significantly increased (P < 0.001) from 7008.4 ± 4635 µm2 at study entry to 10 310.9 ± 3959 µm2 at week 48.
Figure 1.
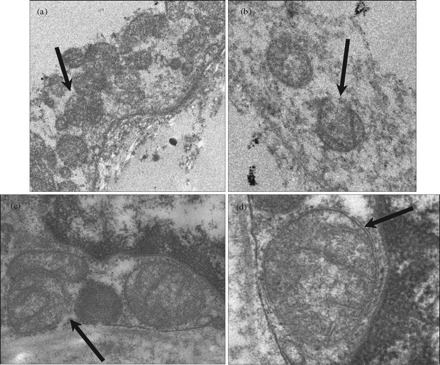
Morphological assessment of mitochondria in subcutaneous adipose tissue at study entry and week 48. Transmission electron micrographs depicting mitochondria with poorly defined inner membrane structures and broken outer membranes, see arrows, at ×16 000 magnification (a and b) in gluteal subcutaneous adipose tissue from patients at study entry. Mitochondrial morphology dramatically improves demonstrating membrane structural integrity at ×16 000 (c) and ×31 500 magnification (c) 48 weeks following tenofovir substitution.
Discussion
In this study, we show that mtDNA levels and morphology improved in the adipose tissue of patients switched from stavudine to tenofovir, while remaining on lamivudine and lopinavir/ritonavir. There was a reduction in 8-oxo-dG oxidative damage found in fat mtDNA, an increase in plasma GSH and a decrease in F2-isoprostane. Accordingly, there was a significant increase in mtDNA quantities observed. Metabolically, total cholesterol and serum glucose decreased, whereas trunk fat increased. Virological suppression was maintained in all subjects and CD4+ cell counts remained unchanged.
Subcutaneous adipose tissue mtDNA levels significantly increased from study entry to week 48, suggesting that switching from stavudine to tenofovir decreases mtDNA inhibition. These findings are similar to those of other studies of stavudine drug switches to zidovudine or abacavir demonstrating improved mtDNA levels.24 In turn, transcription of oxidative phosphorylation proteins and enzymes should increase and overall mitochondrial function should improve. Importantly, the morphological improvement of mitochondria in adipose tissue parallels the improvements seen in mtDNA levels. At study entry, when patients were on the stavudine-containing regimen, mitochondria appeared abnormal, with poorly defined inner membrane structures and often lysed outer membranes; however, 48 weeks after substituting with tenofovir, mitochondria appeared healthier, having well-defined cristae architecture and intact double outer membrane layers. Our results are consistent with previous studies that have found abnormal mitochondria architecture in the subcutaneous adipose tissue of patients treated with stavudine.10 Ultrastructural changes in mitochondria have been shown to depend on the concentration of stavudine,25 which may explain the severe loss of mitochondrial structural integrity in this group of patients since they have been exposed to stavudine for >6 years.
Oxidative stress is defined as an imbalance between the production of reactive oxygen species (ROS) produced during oxidative phosphorylation and cellular antioxidant defences.26 Perhaps of particular concern is ROS causing damage to mtDNA,27 since it is located in close proximity to the electron transport chain, and is thus more susceptible to ROS. Therefore, measuring oxidatively damaged mtDNA (8-oxo-dG), quantities of ROS and quantities of antioxidant defences that combat cellular damage by ROS can characterize the present condition of oxidative stress occurring in vivo. In this study, 8-oxo-dG, GSH and F2-isoprostane parameters were evaluated in patients at study entry and week 48.
One of the most common oxidative and mutagenic lesions to mtDNA is 8-oxo-dG. Elevated levels of 8-oxo-dG have been detected in muscle tissue of HIV-infected patients,28 mouse liver29 and monkey fetal tissues exposed to zidovudine.30 In addition, photodegradation products of zidovudine have been shown to cause 8-oxo-dG formation in vitro.31 In this study, we demonstrated a decrease in 8-oxo-dG mtDNA oxidative damage in the adipose tissue of HIV-infected patients switched to a different antiretroviral regimen. Eight of the ten patients who were evaluated exhibited an average 40% decrease in oxidative damage at week 48 compared with study entry, suggesting that tenofovir either reduces the production of oxygen radicals via oxidative phosphorylation, or reverses damage through stimulating or increasing transcription of components involved in base-excision repair, such as hOGG1. Although this reduction of systemic oxidative damage at week 48 did not reach statistical significance, the observed improvement may explain the significant increase in mtDNA content or vice versa. This decrease in mitochondrion-specific oxidative DNA damage led us to evaluate GSH and F2-isoprostane levels.
GSH is a tripeptide antioxidant that serves as an electron donor to GSH peroxidases in the reduction of hydroperoxides (H2O2) to water (H2O). Alterations in GSH concentration and redox status [ratio of oxidized GSH (GSSH)/GSH] have been associated with elevated levels of oxidative stress in mitochondria.32 Reduced levels of GSH, determined by increased levels of GSSH, have been shown to directly correlate with increased oxidative damage to mtDNA.33 Our results support these previous studies, showing that mean GSH increased after tenofovir switch, while mtDNA 8-oxo-dG oxidative damage decreased. Although it may be unclear whether GSH is depleted in the cytosol and/or in mitochondria, systemic GSH concentration improved by 7.4% in patients 48 weeks following tenofovir substitution.
8-Isoprostane is a class of F2-isoprostane prostaglandin-like compounds with biological activity as a potent pulmonary and renal vasoconstrictor.34 Plasma from normal healthy adults contains modest amounts of 8-isoprostane, ∼40–100 pg/mL, which increases with age.35 HIV-lipoatrophic patients have presented increased 8-isoprostane levels.36 In our study, average F2-isoprostane levels were considerably elevated compared with normal adults and then improved to normal levels at week 48. Patients experienced about a 40% improvement in 8-isoprostane levels in plasma 48 weeks after switching to the tenofovir regimen, suggesting improvements in systemic oxidative stress.
In addition to the mitochondrial improvements noted after the stavudine-to-tenofovir switch, there were modest improvements in metabolic parameters. Median fasting cholesterol and median glucose decreased; however, no changes were found in peripheral limb fat, triglycerides and cholesterol fractions. Some of these findings are contrary to other studies in which significant improvements in limb fat, total and LDL cholesterol and triglycerides were demonstrated in patients who switched from an antiretroviral regimen containing a thymidine nucleoside analogue (zidovudine or stavudine) to either tenofovir or abacavir.37–40 Perhaps, the disparate findings in our study are linked to the longer exposure of our patients to stavudine: a median of almost 7 years versus as short as 4–6 months in the other studies. It is possible that the longer stavudine exposure in this study led to such severe adipocyte and pre-adipocyte depletion that the 48 week period of follow-up was too short to demonstrate an improvement in limb fat. Further, our study lacks a control arm comprising patients who would continue to receive stavudine treatment and could then be compared with patients switched to tenofovir. Such a comparison might have allowed us to demonstrate that loss of limb fat ceased with discontinuation of stavudine. There was an unexpected increase in trunk fat mass in this study. The reasons for this observation are unclear and deserve further investigation. Nevertheless, it is reassuring that it was not associated with adverse changes in fasting glucose or lipids. A larger sample size would have yielded more generalizable results.
Despite these limitations, this small study does demonstrate improvements in mtDNA content and morphology, and improved oxidative markers in 10 patients switched from stavudine to tenofovir. Furthermore, these results suggest that switching to tenofovir treatment may improve mitochondrial structure and DNA integrity. Although no significant changes were found in limb fat, an unexpected significant increase in trunk fat was observed, along with modest improvements in total cholesterol and glucose levels.
In conclusion, the results from this study demonstrate that systemic and peripheral fat mitochondria improve in patients switched from stavudine to tenofovir, while maintaining PI treatment. Continued use of stavudine is still very common in resource-limited settings and is likely to be associated with significant mitochondrial dysfunction at some point during therapy and alternatives should be considered for long-term therapy.
Funding
This study was funded by Abbott Laboratories. The funding source had no role in the design, analysis and interpretation of the study results.
Transparency declarations
M. G. is a consultant for Abbott and Oncolys Biopharma. R. L. M. has received grant support from Abbott and Bristol-Myers Squibb and is a consultant to Gilead and Bristol-Myers Squibb. B. D. is an Abbott employee and owns Abbott stock. G. A. M. serves as a consultant and on the Speaker Bureaus for GlaxoSmithKline, Bristol-Myers Squibb, Gilead and Abbott. The remaining authors have none to declare.
Acknowledgements
Preliminary findings of this research were presented at the Eighth International Workshop on Adverse Drug Reactions in Lipodystrophy in HIV, San Francisco, CA, USA, 2006 (abstract 49 in Antivir Ther 2006; 11 Suppl 3: L32).
References
- 1.Gerschenson M. Brinkman K. Mitochondrial dysfunction in AIDS and its treatment. Mitochondrion. 2004;4:763–77. doi: 10.1016/j.mito.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 2.Carr A. Cooper DA. Images in clinical medicine. Lipodystrophy associated with an HIV-protease inhibitor. N Engl J Med. 1998;339:1296. doi: 10.1056/NEJM199810293391806. [DOI] [PubMed] [Google Scholar]
- 3.Carr A. Samaras K. Burton S. et al. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;12:F51–8. doi: 10.1097/00002030-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Madge S. Kinloch-de-Loes S. Mercey D. et al. Lipodystrophy in patients naive to HIV protease inhibitors. AIDS. 1999;13:735–7. doi: 10.1097/00002030-199904160-00020. [DOI] [PubMed] [Google Scholar]
- 5.Carr A. Miller J. Law M. et al. A syndrome of lipoatrophy, lactic acidaemia and liver dysfunction associated with HIV nucleoside analogue therapy: contribution to protease inhibitor-related lipodystrophy syndrome. AIDS. 2000;14:F25–32. doi: 10.1097/00002030-200002180-00001. [DOI] [PubMed] [Google Scholar]
- 6.Haubrich RH. Riddler S. DiRienzo G. et al. Metabolic outcomes of ACTG 5142: a prospective, randomized, Phase III trial of NRTI-, PI-, and NNRTI-sparing regimens for initial treatment of HIV-1 infection. Abstracts of the Fourteenth Conference on Retroviruses and Opportunistic Infections; 2007; Los Angeles, CA, USA. Alexandria, VA, USA: Foundation for Retrovirology and Human Health; p. 78. Abstract 38. [Google Scholar]
- 7.Cameron DW. da Silva BA. Arribas JR. et al. A 96-week comparison of lopinavir-ritonavir combination therapy followed by lopinavir-ritonavir monotherapy versus efavirenz combination therapy. J Infect Dis. 2008;198:234–40. doi: 10.1086/589622. [DOI] [PubMed] [Google Scholar]
- 8.Kohler JJ. Lewis W. A brief overview of mechanisms of mitochondrial toxicity from NRTIs. Environ Mol Mutagen. 2007;48:166–72. doi: 10.1002/em.20223. [DOI] [PubMed] [Google Scholar]
- 9.Shikuma CM. Hu N. Milne C. et al. Mitochondrial DNA decrease in subcutaneous adipose tissue of HIV-infected individuals with peripheral lipoatrophy. AIDS. 2001;15:1801–9. doi: 10.1097/00002030-200109280-00009. [DOI] [PubMed] [Google Scholar]
- 10.Nolan D. Hammond E. Martin A. et al. Mitochondrial DNA depletion and morphologic changes in adipocytes associated with nucleoside reverse transcriptase inhibitor therapy. AIDS. 2003;17:1329–38. doi: 10.1097/00002030-200306130-00007. [DOI] [PubMed] [Google Scholar]
- 11.Gerschenson M. Shiramizu B. LiButti DE. et al. Mitochondrial DNA levels of peripheral blood mononuclear cells and subcutaneous adipose tissue from thigh, fat and abdomen of HIV-1 seropositive and negative individuals. Antivir Ther. 2005;10(Suppl 2):M83–9. [PubMed] [Google Scholar]
- 12.Saint-Marc T. Partisani M. Poizot-Martin I. et al. A syndrome of peripheral fat wasting (lipodystrophy) in patients receiving long-term nucleoside analogue therapy. AIDS. 1999;13:1659–67. doi: 10.1097/00002030-199909100-00009. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Benayas T. Blanco F. de la Cruz JJ. et al. Replacing stavudine by abacavir reduces lactate levels and may improve lipoatrophy. AIDS. 2003;17:921–4. doi: 10.1097/00002030-200304110-00021. [DOI] [PubMed] [Google Scholar]
- 14.John M. McKinnon EJ. James IR. et al. Randomized, controlled, 48-week study of switching stavudine and/or protease inhibitors to combivir/abacavir to prevent or reverse lipoatrophy in HIV-infected patients. J Acquir Immune Defic Syndr. 2003;33:29–33. doi: 10.1097/00126334-200305010-00005. [DOI] [PubMed] [Google Scholar]
- 15.McComsey GA. Ward DJ. Hessenthaler SM. et al. Improvement in lipoatrophy associated with highly active antiretroviral therapy in human immunodeficiency virus-infected patients switched from stavudine to abacavir or zidovudine: the results of the TARHEEL study. Clin Infect Dis. 2004;38:263–70. doi: 10.1086/380790. [DOI] [PubMed] [Google Scholar]
- 16.Birkus G. Hitchcock MJ. Cihlar T. Assessment of mitochondrial toxicity in human cells treated with tenofovir: comparison with other nucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother. 2002;46:716–23. doi: 10.1128/AAC.46.3.716-723.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallant JE. Staszewski S. Pozniak AL. et al. Efficacy and safety of tenofovir DF vs stavudine in combination therapy in antiretroviral-naive patients: a 3-year randomized trial. JAMA. 2004;292:191–201. doi: 10.1001/jama.292.2.191. [DOI] [PubMed] [Google Scholar]
- 18.Gallant JE. Staszewski S. Pozniak AL. et al. Efficacy and safety of tenofovir DF vs. stavudine in combination therapy in antiretroviral-naive patients: a 3-year randomized trial. JAMA. 2004;292:191–201. doi: 10.1001/jama.292.2.191. [DOI] [PubMed] [Google Scholar]
- 19.Claas GJ. Julg B. Goebel FD. et al. Metabolic and anthropometric changes one year after switching from didanosine/stavudine to tenofovir in HIV-infected patients. Eur J Med Res. 2007;12:54–60. [PubMed] [Google Scholar]
- 20. AIDS Clinical Trials Group. Diagnostic Criteria for Abnormalities of Fat Distribution. http://www.actgnetwork.org. (10 March 2009, date last accessed) [Google Scholar]
- 21.Shiramizu B. Shikuma KM. Kamemoto L. et al. Placenta and cord blood mitochondrial DNA toxicity in HIV-infected women receiving nucleoside reverse transcriptase inhibitors during pregnancy. J Acquir Immune Defic Syndr. 2003;32:370–4. doi: 10.1097/00126334-200304010-00004. [DOI] [PubMed] [Google Scholar]
- 22.Anson RM. Bohr VA. Gene-specific and mitochondrial repair of oxidative DNA damage. Methods Mol Biol. 1999;113:257–79. doi: 10.1385/1-59259-675-4:257. [DOI] [PubMed] [Google Scholar]
- 23.Anson RM. Mason PA. Bohr VA. Gene-specific and mitochondrial repair of oxidative DNA damage. Methods Mol Biol. 2006;314:155–81. doi: 10.1385/1-59259-973-7:155. [DOI] [PubMed] [Google Scholar]
- 24.McComsey GA. Paulsen DM. Lonergan JT. et al. Improvements in lipoatrophy, mitochondrial DNA levels and fat apoptosis after replacing stavudine with abacavir or zidovudine. AIDS. 2005;19:15–23. doi: 10.1097/00002030-200501030-00002. [DOI] [PubMed] [Google Scholar]
- 25.Medina DJ. Tsai CH. Hsiung GD. et al. Comparison of mitochondrial morphology, mitochondrial DNA content, and cell viability in cultured cells treated with three anti-human immunodeficiency virus dideoxynucleosides. Antimicrob Agents Chemother. 1994;38:1824–8. doi: 10.1128/aac.38.8.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Betteridge DJ. What is oxidative stress? Metabolism. 2000;49:3–8. doi: 10.1016/s0026-0495(00)80077-3. [DOI] [PubMed] [Google Scholar]
- 27.Hashiguchi K. Bohr VA. de Souza-Pinto NC. Oxidative stress and mitochondrial DNA repair: implications for NRTIs induced DNA damage. Mitochondrion. 2004;4:215–22. doi: 10.1016/j.mito.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 28.de la Asuncion JG. del Olmo ML. Sastre J. et al. AZT treatment induces molecular and ultrastructural oxidative damage to muscle mitochondria. Prevention by antioxidant vitamins. J Clin Invest. 1998;102:4–9. doi: 10.1172/JCI1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de la Asuncion JG. del Olmo ML. Sastre J. et al. Zidovudine (AZT) causes an oxidation of mitochondrial DNA in mouse liver. Hepatology. 1999;29:985–7. doi: 10.1002/hep.510290353. [DOI] [PubMed] [Google Scholar]
- 30.Bialkowska A. Bialkowski K. Gerschenson M. et al. Oxidative DNA damage in fetal tissues after transplacental exposure to 3′-azido-3′-deoxythymidine (AZT) Carcinogenesis. 2000;21:1059–62. doi: 10.1093/carcin/21.5.1059. [DOI] [PubMed] [Google Scholar]
- 31.Iwamoto T. Hiraku Y. Oikawa S. et al. Oxidative DNA damage induced by photodegradation products of 3(′)-azido-3(′)-deoxythymidine. Arch Biochem Biophys. 2003;416:155–63. doi: 10.1016/s0003-9861(03)00316-3. [DOI] [PubMed] [Google Scholar]
- 32.Jocelyn PC. Dickson J. Glutathione and the mitochondrial reduction of hydroperoxides. Biochim Biophys Acta. 1980;590:1–12. doi: 10.1016/0005-2728(80)90141-3. [DOI] [PubMed] [Google Scholar]
- 33.Esteve JM. Mompo J. Garcia de la Asuncion J. et al. Oxidative damage to mitochondrial DNA and glutathione oxidation in apoptosis: studies in vivo and in vitro. FASEB J. 1999;13:1055–64. doi: 10.1096/fasebj.13.9.1055. [DOI] [PubMed] [Google Scholar]
- 34.Morrow JD. Roberts LJ., II The isoprostanes. Current knowledge and directions for future research. Biochem Pharmacol. 1996;51:1–9. doi: 10.1016/0006-2952(95)02072-1. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z. Ciabattoni G. Creminon C. et al. Immunological characterization of urinary 8-epi-prostaglandin F2 alpha excretion in man. J Pharmacol Exp Ther. 1995;275:94–100. [PubMed] [Google Scholar]
- 36.McComsey GA. Morrow JD. Lipid oxidative markers are significantly increased in lipoatrophy but not in sustained asymptomatic hyperlactatemia. J Acquir Immune Defic Syndr. 2003;34:45–9. doi: 10.1097/00126334-200309010-00006. [DOI] [PubMed] [Google Scholar]
- 37.Moyle GJ. Sabin CA. Cartledge J. et al. A randomized comparative trial of tenofovir DF or abacavir as replacement for a thymidine analogue in persons with lipoatrophy. AIDS. 2006;20:2043–50. doi: 10.1097/01.aids.0000247574.33998.03. [DOI] [PubMed] [Google Scholar]
- 38.Milinkovic A. Martinez E. Lopez S. et al. The impact of reducing stavudine dose versus switching to tenofovir on plasma lipids, body composition and mitochondrial function in HIV-infected patients. Antivir Ther. 2007;12:407–15. [PubMed] [Google Scholar]
- 39.Carr A. Workman C. Smith DE. et al. Abacavir substitution for nucleoside analogs in patients with HIV lipoatrophy: a randomized trial. JAMA. 2002;288:207–15. doi: 10.1001/jama.288.2.207. [DOI] [PubMed] [Google Scholar]
- 40.Cherry CL. Lal L. Thompson KA. et al. Increased adipocyte apoptosis in lipoatrophy improves within 48 weeks of switching patient therapy from stavudine to abacavir or zidovudine. J Acquir Immune Defic Syndr. 2005;38:263–7. [PubMed] [Google Scholar]