

ABSTRACT
Clostridioides difficile (C. difficile) infection is the most common cause of healthcare–associated infection and an important cause of morbidity and mortality among hospitalized patients. A comprehensive understanding of C. difficile infection (CDI) pathogenesis is crucial for disease diagnosis, treatment, and prevention. Here, we characterized gut microbial compositions and a broad panel of innate and adaptive immunological markers in 243 well-characterized human subjects (including 187 subjects with both microbiota and immune marker data), who were divided into four phenotype groups: CDI, Asymptomatic Carriage, Non-CDI Diarrhea, and Control. We found that the interactions between gut microbiota and host immune markers are very sensitive to the status of C. difficile colonization and infection. We demonstrated that incorporating both gut microbiome and host immune marker data into classification models can better distinguish CDI from other groups than can either type of data alone. Our classification models display robust diagnostic performance to differentiate CDI from Asymptomatic carriage (AUC~0.916), Non-CDI Diarrhea (AUC~0.917), or Non-CDI that combines all other three groups (AUC~0.929). Finally, we performed symbolic classification using selected features to derive simple mathematic formulas that explicitly quantify the interactions between the gut microbiome and host immune markers. These findings support the potential roles of gut microbiota and host immune markers in the pathogenesis of CDI. Our study provides new insights for a microbiome-immune marker-derived signature to diagnose CDI and design therapeutic strategies for CDI.
KEYWORDS: C. difficile infection, gut microbiome, host immune markers, machine learning
Introduction
Clostridioides difficile infection (CDI) is the most common cause of healthcare–associated infection and an important cause of morbidity and mortality among hospitalized patients1–3. Exposure to toxinogenic C. difficile can lead to a range of clinical outcomes ranging from asymptomatic colonization to mild diarrhea and more severe disease syndromes such as pseudomembranous colitis, toxic megacolon, bowel perforation, sepsis, and death4,5. Asymptomatic C. difficile carriage is mainly characterized by C. difficile colonization in the absence of symptoms of infection. The diagnosis of CDI is based on clinical signs and symptoms in combination with laboratory testing, including enzyme immunoassays (EIA) for TcdA and TcdB, nucleic acid amplification tests (NAAT), selective toxinogenic culture, cell cytotoxicity neutralization assay, and glutamate dehydrogenase EIA6–8. However, currently available approaches do not accurately differentiate CDI from diarrhea with another cause in a patient colonized with toxinogenic C. difficile.
Current treatment strategies for CDI, including vancomycin and fidaxomicin, have inconsistent cure rates and treatment failure or CDI recurrence may occur in approximately one-third of cases9,10. Antibiotic exposure is considered the most important factor predisposing patients to CDI11,12. In fact, treatments with antibiotics have a tremendous impact on the composition and functionality of the gut microbiota, and accordingly are associated with reduced colonization resistance against pathogens such as C. difficile13–15. It has been reported that several gut commensal bacteria may contribute to the prevention of C. difficile colonization and infection16,17. Once colonized, C. difficile can produce toxins that mediate a robust inflammatory response. Toxin A (TcdA) and toxin B (TcdB) are the primary virulence factors of C. difficile18 and act on intestinal epithelial cells first, inducing pro-inflammatory cytokines, loss of tight junctions, cell detachment and an impaired mucosal barrier19–21 leading to further exposure of immune cells to toxins. The innate and adaptive immune responses to CDI play crucial roles in disease onset, expression, severity, progression, and overall prognosis22,23. The innate immune defense mechanisms against C. difficile and its toxins include the commensal intestinal organisms, mucosal barrier, intestinal epithelial cells, and mucosal immune system24,25. TcdA and TcdB have multiple effects on the innate immune system, including inducing expression of numerous pro-inflammatory mediators (e.g., cytokines, chemokines and neuroimmune peptides) and the recruitment and activation of a variety of innate immune cells26,27. Adaptive immunity is also sufficient to provide some protection from CDI, likely via antibody-mediated neutralization of TcdA and TcdB28–31. The role of the immune response combined with the knowledge that a balanced microbiota can prevent colonization and infection demonstrates the importance of combining both gut microbiota and host immune markers in understanding the pathogenesis of CDI.
Machine learning has a great impact in many areas of medical research, as it offers a principled approach for developing sophisticated, automatic, and objective algorithms for analysis of complex data. Indeed, previous studies indicate that supervised learning can be successfully employed for clinical disease assessment for diverse disorders32–35. In our previous work, we found that specific immune markers, particularly G-CSF, can be used to distinguish adults with CDI from other groups including asymptomatic carriers and NAAT-negative patients with and without diarrhea36. Here, we leverage machine learning tools to integrate the host immune marker data and newly obtained gut microbiome data from subjects of the same cohort to identify collections of bacteria and immune markers that can be associated with CDI. Our aim is to quantify the role of intricate interactions between gut microbiota and immune response in CDI pathogenesis, which can inform the design of microbiome-immune marker-based diagnostic test and therapeutic strategies.
Results
Baseline demographic and clinical characteristics of participants
Our clinical cohort consists of 243 well-characterized recruited participants, who were divided into four groups (see Methods)36: (1) Control (n = 47); (2) Non-CDI Diarrhea (n = 44); (3) Asymptomatic Carriage (n = 40); (4) CDI (n = 112). The first three groups can be combined as the Non-CDI group. The entire clinical cohort had a mean ± SD age of 63.66 ± 14.85 year and was 48.15% female. Demographic data of the cohort are summarized in Table 1. In total, 187 participants (76.95%) had both gut microbiome and immune marker data available (see Supplementary Table 1).
Table 1.
Demographic characteristics of the enrolled subjects
| |
NAAT negative |
NAAT positive |
||
|---|---|---|---|---|
| Characteristics | Control (n = 47) | Non-CDI Diarrhea (n = 44) | Asymptomatic Carriage (n = 40) | CDI (n = 112) |
| Sex | ||||
| Female | 14 (29.79%) | 22 (50.00%) | 20 (50.00%) | 61 (54.46%) |
| Male | 33 (70.21%) | 22 (50.00%) | 20 (50.00%) | 51 (45.54%) |
| Age, Avg ± SD | 62.40 ± 12.33 | 63.07 ± 13.15 | 62.15 ± 17.25 | 64.99 ± 15.62 |
| Ethnicity | ||||
| Hispanic | 1 (2.13%) | 3 (6.82%) | 1 (2.50%) | 6 (5.36%) |
| Non-Hispanic | 38 (80.85%) | 37 (84.09%) | 31 (77.50%) | 96 (85.71%) |
| Unknown | 8 (17.02%) | 4 (9.09%) | 8 (20.00%) | 10 (8.93%) |
| Race | ||||
| White | 33 (70.21%) | 28 (63.64%) | 28 (70.00%) | 89 (79.46%) |
| Other | 4 (8.51%) | 10 (22.73%) | 3 (7.50%) | 23 (20.54%) |
| Unknown | 10 (21.28%) | 6 (13.64%) | 9 (22.50%) | 0 (0.00%) |
Microbial community structure
To compare the overall microbial community structure of the four groups, we first calculated the alpha diversity (i.e., the within-sample taxonomic diversity) of each sample at the genus level using four different measures: taxa richness (the observed number of different taxa present in the sample), Chao1 (abundance-based estimator of taxa richness), Evenness (the uniformity of the population size of each taxa present in the sample), and Shannon diversity index (estimator of taxa richness and evenness: more weight on richness). As shown in Figure 1(a-d), we found that taxa richness and Chao1 did not differ significantly among these groups. The gut microbiota of Non-CDI Diarrhea subjects showed lower evenness than that of the Control group. Shannon diversity was significantly lower in the Non-CDI Diarrhea and CDI groups than in the Control group.
Figure 1.
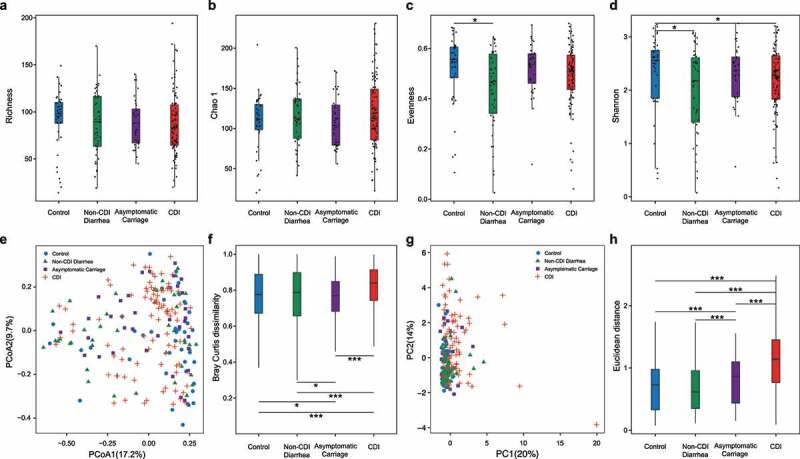
Comparing the diversity of the gut microbiota (and host immune markers) of subjects with different C.difficile infection/colonization statuses (Control, Non-CDI Diarrhea, Asymptomatic Carriage, and CDI). (a) Taxa richness. (b) Chao1. (c) Evenness. (d) Shannon index. (e) Principal Coordinates Analysis (PCoA) plot based on Bray–Curtis dissimilarities of microbial compositions. (f) Boxplot of the gut microbiome Bray–Curtis dissimilarity between subjects within each group. (g) Principal component analysis (PCA) plot of host immune marker concentrations. (h) Boxplot of the Euclidean distance for the host immune markers of subjects within each group. Statistical significance was determined by Mann–Whitney test, *P < .05, *P < .05, **P < .01, ***P < .001
To determine whether the gut microbial compositions of participants are affected by C. difficile infection/colonization status, we performed Principal Coordinates Analysis (PCoA) at the genus level using Bray–Curtis dissimilarity (which is a beta diversity measure to quantify the between-sample compositional dissimilarity). We found no distinct clusters corresponding to the four different phenotype groups (Figure 1e). Interestingly, by directly comparing the beta diversity of each group, we did find that the CDI group displays higher beta diversity than other groups (Figure 1f), indicating that the microbial compositions of participants within the CDI group vary more prominently than other groups. PERMANOVA (permutational multivariate analysis of variance) showed that the overall bacterial composition differed significantly among different groups based on the CDI status (P < .001; Supplementary Table 2), whereas other host factors such as age, sex, race and ethnicity had no significant effect on the microbiome composition.
To identify microbiome markers (i.e., certain taxa with very high discriminatory ability) to differentiate those different phenotype groups, we performed differential abundance analysis. In particular, we used ANCOM37 (analysis of composition of microbiomes) with a Benjamini-Hochberg correction, and adjusted for age and sex. We found that the abundances of 15 genera were significantly different between CDI and Asymptomatic Carriage groups (Figure 2a and Supplementary Table 3). Among the 15 genera, 4 of them (Veillonella, Enterobacter, Granulicatella, and Dialister) of these genera were enriched in the CDI group, while the other 11 genera (including Lactococcus, Dorea, Moryella, Stenotrophomonas, and Agathobacter) were enriched in the Asymptomatic Carriage group. We also found 16 differentially abundant genera between the Non-CDI Diarrhea group and the CDI group (Figure 2b and Supplementary Table 4). Of these, 10 genera (including Clostridioides, Enterobacter, Dialister, and Veillonella) were enriched in the CDI group, and the other 6 genera ([Eubacterium]_hallii_group, Collinsella, Agathobacter, Dorea, Stenotrophomonas, and Streptococcus) were enriched in the Non-CDI Diarrhea group. ANCOM analysis also enabled us to identify 40 genera (including Clostridioides and Veillonella) that have significant differential abundances between the CDI group and the whole Non-CDI group (Figure 2c and Supplementary Table 5). Note that a total of 6 differentially abundant genera were identified from all the three comparisons. Among them, Veillonella, Enterobacter and Dialister were enriched in the CDI group, while Dorea, Stenotrophomonas and Agathobacter were depleted in the CDI group.
Figure 2.
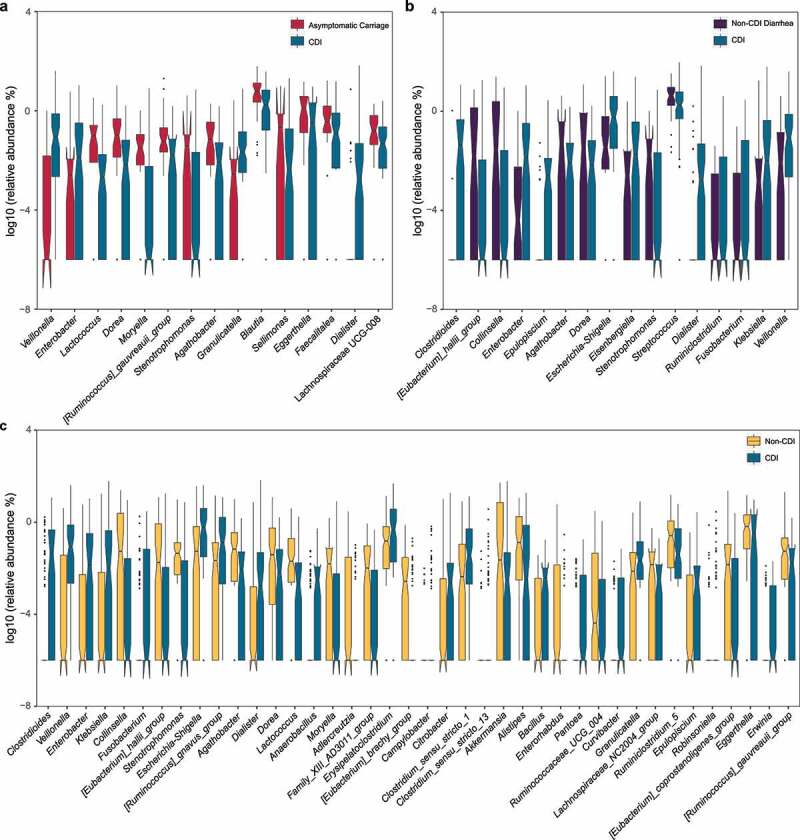
Relative abundances of differentially abundant genera identified by ANCOM in comparing different groups. (a) CDI vs. Asymptomatic Carriage. (b) CDI vs. Non-CDI Diarrhea. (c) CDI vs. Non-CDI. The top differentially abundant taxa were ranked based on their W statistics (a high “w score” generated by this test indicates the greater likelihood that the null hypothesis can be rejected, indicating the number of times a parameter is significantly different between groups) (from left to right). The relative abundance (%) are plotted on log10 scale. The notches in the boxplots show the 95% confidence interval around the median
Microbial correlation networks
As the gut microbiota is complex in both structure and function, this complexity can be well represented and modeled as networks. Network methods can be applied to microbiome studies to model the co-occurrence of microorganisms, find microbial relationships essential for community assembly or stability, and deduce the influence of various associations on the host health. To compare the microbial communities of the four groups at the network-level, we constructed the genus-level microbial correlation network for each group using SparCC38 (sparse correlations for compositional data). We found that the microbial correlation network of the CDI group has quite different structure compared to other groups (Figure 3). More precisely, it has fewer nodes and edges, lower average degree, but higher modularity (Supplementary Table 6). These indicate that the overall microbial correlations in the CDI group are much weaker than those in other groups.
Figure 3.
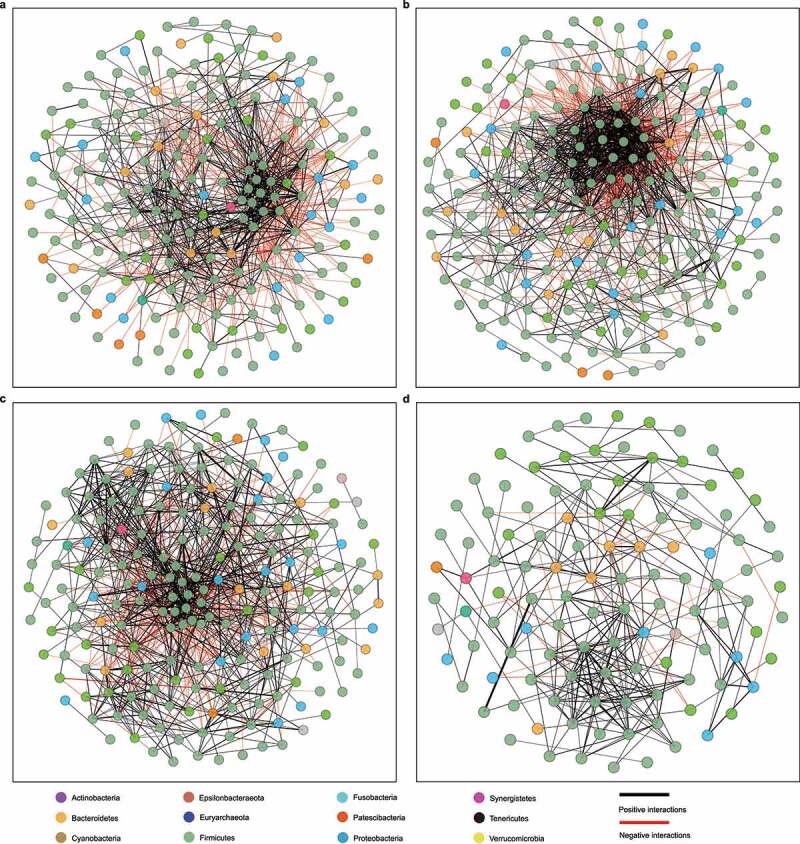
Microbial correlation networks of different groups. (a) Control. (b) Non-CDI Diarrhea. (c) Asymptomatic Carriage. (d) CDI. Nodes represent genera and are colored based on their phylum. Edges represent microbial correlations: green/red means positive/negative correlations, respectively. Edge thickness indicates correlation strength, and only the high-confidence interactions (p-value < 0.05) with high absolute correlation coefficients (> 0.3) were presented. For each group, we further identified the top-three most connected genera/nodes. They are Ruminococcus_1, Roseburia and Lachnospiraceae_UCG_008 for the Control group, [Ruminococcus]_torques_group, [Eubacterium]_hallii_group and Blautia for the Non-CDI Diarrhea group, Ruminiclostridium_5, Enterococcus and Lachnospiraceae_UCG_008 for the Asymptomatic Carriage group, and Alistipes, Ruminiclostridium_5 and Lachnoclostridium for the CDI group
To analyze these patterns in more detail, we used NetShift39 to identify potentially important “driver” taxa responsible for the change of microbial correlations. In the NetShift pipeline, the common taxa present in both ‘control’ and ‘case’ sample sets with a minimum abundance threshold are extracted to construct the microbial correlation networks. Then the ‘driver taxa’ are identified based on their Neighbor Shift (NESH) scores and the Betweenness Centrality (BC) measures. In a nutshell, the NESH score of a taxon/node is minimum when the associated partners of this node are the same in both ‘case’ and ‘control’ networks, intermediate when there is only a subset of the associated partners present in the ‘case’ network, and maximum when a completely new set of associated partners appear in the ‘case’ network. The Betweenness Centrality of a taxon/node quantifies its involvement in connecting other nodes in the network. A taxon with an altered set of edges (identified by a high NESH score), while still being increasingly important (i.e., with higher scaled BC in the ‘case’ network than in the ‘control’ network), necessarily holds a key significance in microbial interplay and is identified as a ‘driver’ taxon. This analysis revealed 24 potential driver taxa linked with the change of microbial correlations between CDI and Asymptomatic Carriage groups (Supplementary Figure 1). The top driver taxa were Alistipes, Clostridioides, Desulfovibrio, Eggerthella, Erysipelatoclostridium, Klebsiella, Odoribacter, Proteus, [Ruminococcus]_torques_group, Streptococcus, Vagococcus and Veillonella. We then identified 24 genera as potential driver taxa underlying the change of microbial correlations between CDI and Non-CDI Diarrhea groups (Supplementary figure 2). The top driver taxa were Alistipes, Buttiauxella, Citrobacter, Clostridium_sensu_stricto_13, Desulfovibrio, Klebsiella, Oscillibacter, Phascolarctobacterium, Streptococcus and Veillonella. Finally, Netshift analysis revealed 38 potential driver taxa underlying the change of microbial correlations between CDI and Non-CDI groups. The top driver taxa were Bifidobacterium, Clostridioides, Klebsiella, Oscillibacter, Streptococcus and Veillonella (Supplementary Figure 3). Together, these results suggested that certain bacterial taxa (e.g., Clostridioides, Klebsiella, Streptococcus and Veillonella) could play an important role in driving the changes of microbial correlations in subjects with different C. difficile infection/colonization status.
Host immune markers and CDI
To determine the systemic levels of proinflammatory cytokines in CDI, we measured the circulating levels of granulocyte-colony stimulating factor (G-CSF), interleukin-1β (IL-1β), IL-2, IL-4, IL-6, IL-8, IL-10, IL-13, IL-15, monocyte chemoattractant protein-1 (MCP-1), vascular endothelial growth factor-A (VEGF-A), tumor necrosis factor-alpha (TNF-α), and serum concentrations of immunoglobulin A (IgA), IgG, and IgM antibodies against C. difficile toxin A and toxin B as previously reported36. We previously demonstrated specific markers of these innate and adaptive immunity that can distinguish CDI from each of the other three groups36. In the current study, we are particularly interested in comparing the CDI group and the combined Non-CDI group. Based on the Mann-Whitney U test, we identified in total 11 immune markers that displayed significantly different concentrations in these two groups, including G-CSF, IL-4, IL-6, IL-8, IL-10, IL-15, TNF-α, MCP1, IgA anti-toxin A and B, and IgG anti-toxin A in blood (Supplementary Table 7). All of these immune markers had higher concentrations in the CDI group than in the Non-CDI group. Host immune marker variations between samples were evaluated using the Principal Component Analysis (PCA) (Figure 1g). PCA plot showed no clear clustering of those subjects based on immune marker concentrations. However, boxplot of Euclidean distance of immune marker profiles from CDI patients showed higher within-group variation than that in all the other three groups (Figure 1h). PERMANOVA analysis indicated that the immune homeostasis was significantly different among different groups based on the CDI status (P = .016; Supplementary Table 2).
Interactions between gut microbiome and host immune markers
To reveal the interactions between the gut microbiome and the host immune system, we calculated the correlations between microbial compositions and the circulating levels of host immune markers for each of the four groups. The results are shown in Figure 4 and Supplementary Figure 4. For the Control group, the most significant correlations were identified as Chiristensenellaceae R-7 group negatively correlated with TNFα, Bifidobacterium positively correlated with VEGFA and IL-13, Rothia positively correlated with IL-15, and Veillonella positively correlated with IL-4 (Figure 4a and Supplementary Figure 4). For the Non-CDI Diarrhea group, Ruminococcaceae UCG-011 was negatively correlated with IL-8 and IL-6, Defluviitaleaceae UCG-011 was positively correlated with IL-1β, and Blautia was negatively correlated with MCP1 levels (Figure 4b). For the Asymptomatic Carriage group, we found that Lactobacillus was negatively correlated with VEGFA, Akkermansia was positively correlated with IL-6, and Enterococcus was positively correlated with TNFα (Figure 4c). For the CDI group, negative correlations involved Akkermansia and IL-10, Lactococcus and G-CSF, while positive correlations involved Lactobacillus and IgG and IgA anti-toxin B (Figure 4d). Interestingly, none of these most significant correlations was universally present across different groups. This indicated that the interactions between gut microbiota and host immunological markers can be very sensitive to the status of C. difficile colonization and infection. Although the rudimentary correlation analysis cannot reveal any nonlinear interactions between gut microbiota and host immune markers, the result implies that the integration of gut microbiota and host immune markers might be quite useful for highly accurate classification of CDI.
Figure 4.
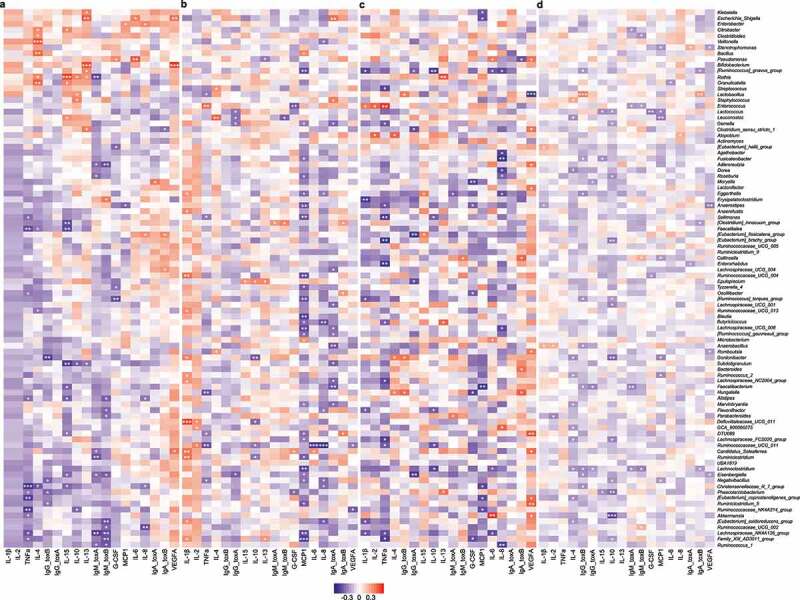
Correlations between gut microbial abundances and host immune markers in different groups, quantified by Spearman correlation with Benjamini-Hochberg correction. (a) Control. (b) Non-CDI Diarrhea. (c) Asymptomatic Carriage. (d) CDI. Rows represent genera; columns represent immune markers. The layout of the heatmap is followed the hierarchical clustering results of Control cohort (see Supplementary Figure 4). Red/blue represents positive/negative correlation, respectively. The intensity of the colors denotes the strength of the correlation. *α < 0.05, **α < 0.01, ***α < 0.001
Classification of CDI using host immune markers and gut microbiota
To determine whether host immune markers or gut microbiota could serve as biomarkers to classify subjects into different groups, we constructed a multi-class classifier based on random forests (RF). One of the most popular performance metrics of a classifier is the Area Under the receiver operating characteristic Curve (AUC). The performance of a multi-class classifier is measured by both micro-average and macro-average AUCs. We considered three different feature types: (1) host immune maker concentrations alone; (2) gut microbial compositions alone; and (3) the integration of (1) and (2) in our classification analysis. To eliminate confounding effects, we excluded the genus Clostridioides from our classification analysis. The immune marker-based classifier achieved macro-average AUC ~ 0.827 and micro-average AUC ~ 0.828 (Supplementary Figure 5a), which are quite comparable to the performance of microbiota-based classifier (Supplementary Figure 5b). Interestingly, integrating immune marker with gut microbiota showed much better classification performance (macro-average AUC ~ 0.926 and micro-average AUC ~ 0.869) (Supplementary Figure 5c).
Figure 5.
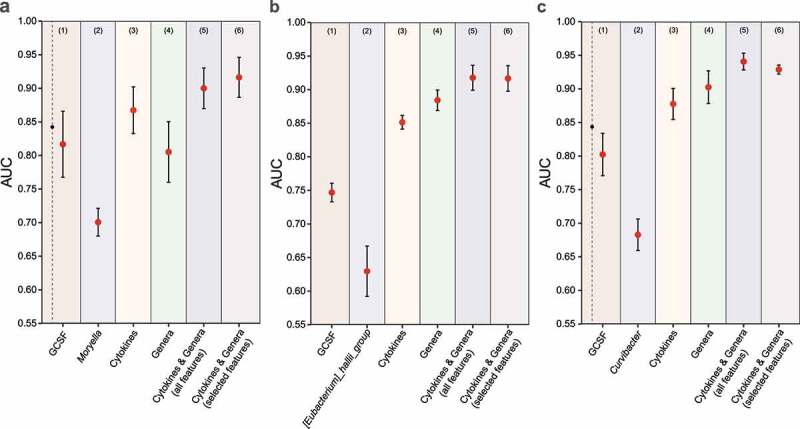
The performance of RF-based classification models based on various types of features in differentiating CDI from other groups. (a) CDI vs. Asymptomatic Carriage. (b) CDI vs. Non-CDI Diarrhea. (c) CDI vs. Non-CDI. For each classification task, we used different types of features: (1) the top-1 immune marker feature (based on mean decrease accuracy); (2) the top-1 genus feature; (3) all immune markers; (4) all genera; (5) integration of all immune markers and genera; (6) selected features from the set of all immune markers and genera. Error bars represent the standard errors of the means (SEM)
We further performed binary classifications to distinguish CDI subjects from Asymptomatic Carriage, Non-CDI Diarrhea, and Non-CDI subjects, using different feature types (Figure 5). The goal of this analysis was to assess whether any single taxon or immune marker could reliably differentiate CDI status. In the classification of CDI vs. Asymptomatic Carriage, we found that G-CSF and Moryella were the most important immune and microbial features based on mean decrease accuracy (MDA, the decrease in model accuracy from permuting the values in each feature), respectively (Supplementary Figure 6a-b). But the classification based on G-CSF (or Moryella) alone did not yield very high performance: mean AUC ~ 0.817 (or 0.701), respectively (Figure 5(a1, a2)). When we used all the immune markers (or all the genera) as features, we achieved mean AUC ~ 0.867 (or 0.805), respectively (Figure 5(a3, a4)). Interestingly, when we integrated all the host immune markers and gut microbial composition data together, we achieved a much higher performance with mean AUC ~ 0.900 (Figure 5(a5)). In order to select a subset of features that is as discriminatory as the whole set of features, we followed the “1-SE” rule (i.e., one chooses the model with fewest features such that its classification performance is less than one standard error away from that of the model with all the features), and selected the following 4 features: 2 bacterial genera (Moryella and Veillonella) and 2 immune markers (G-CSF and IL-6) in classifying CDI and Asymptomatic Carriage groups (Supplementary Figure 6:g-j). The RF classifier with those selected features displayed an outstanding classification performance, with mean AUC ~ 0.916 (Figure 5(a6)). Note that a significant negative correlation between Moryella and G-CSF was found in the Asymptomatic Carriage group (Figure 4c), which might contribute to the outstanding performance of the RF classifier with Moryella and G-CSF as selected features.
In the classification of CDI vs. Non-CDI Diarrhea groups, we found that G-CSF and [Eubacterium]_hallii_group are the top immune and microbial features, respectively (Supplementary Figure 6:c-d). But the classification based on G-CSF (or [Eubacterium]_hallii_group) alone did not perform very well: mean AUC ~ 0.747 (or ~ 0.630), respectively (Figure 5(b1, b2)). When we used all the immune marker (or all the microbial genera) as features, we achieved mean AUC ~ 0.851 (or ~ 0.884), respectively (Figure 5(b3, b4)). By integrating all features from both host immune marker and gut microbial genera, we further improved the classification performance to mean AUC ~ 0.918 (Figure 5(b5)). Following the “1-SE” rule, we selected the following 5 features: 3 genera: Enterococcus, Epulopiscium and [Eubacterium]_hallii_group; and 2 immune markers: G-CSF and IgA anti-toxin A (Supplementary Figure 6:h-k). The RF classifier with those selected features achieved mean AUC ~ 0.917 (Figure 5(b6)), which is quite comparable to that of using all the features. Note that Enterococcus was found to be significantly associated with G-CSF in the Non-CDI Diarrhea group (Figure 4b). This might partially explain the outstanding performance of the RF classifier with Enterococcus and G-CSF as selected features.
In the classification of CDI vs. Non-CDI groups, we found that G-CSF and Curvibacter are the top immune and microbial features, respectively (Supplementary Figure 6:e-f). Classification based on G-CSF (or Curvibacter) alone achieved mean AUC ~ 0.802 (or ~ 0.683), respectively (Figure 5(c1, c2)). When we used all the immune marker (or all the microbial genera) as features, we achieved mean AUC ~ 0.878 (or ~ 0.903), respectively (Figure 5(c3, c4)). Integrating all features from both host immune marker and gut microbial genera, we further improved the classification performance to mean AUC ~ 0.941 (Figure 5(c5)). Following the “1-SE” rule, we selected the following 10 features: 6 genera: Stenotrophomonas, Curvibacter, Enterobacter, Anaerobacillus, Fusobacterium and Veillonella; and 4 immune markers: G-CSF, IL-6, TNF-α and IgA anti-toxin B (Supplementary Figure 6:i-l). Classification with those well selected features achieved mean AUC ~ 0.929 (Figure 5(c6)).
Derive nonlinear interactions between gut microbiota and host immune markers using symbolic classification
As mentioned earlier, traditional correlation analysis cannot reveal any nonlinear interactions between gut microbiota and host immune markers. This fact and the outstanding classification results based on well-selected features prompt us to derive simple mathematical models to quantify the intricate interactions between gut microbiota and host immune markers. To achieve that, we leveraged symbolic classification (SC)40,41, a genetic programming technique that automatically searches the space of mathematical expressions to find the model that best fits a given dataset. The fitness function in SC is a maximization function, and the number of generations is chosen based on the saturation of the fitness score (Supplementary SFigure 7). Using the same set of selected features and trained with the entire dataset, the SC model outperformed logistic regression (LR) in differentiating CDI (see Table 2).
Table 2.
Diagnostic scores derived from symbolic classification (SC) and logistic regression (LR). For each subject , we calculate his/her diagnostic score (or ) based on one of the following formulas derived from SC (or LR), respectively. For SC, the class of subject is CDI if ; or Asymptomatic Carriage (or Non-CDI Diarrhea, Non-CDI) if . For LR, the class of subject is CDI if ; or Asymptomatic Carriage (or Non-CDI Diarrhea, Non-CDI) if . Here, both and were learned from the entire dataset. Features used here include: GCSF; IgA_toxA; IgA_toxB; IL6; TNFα; Anaerobacillus; Curvibacter; Enterobacter; Enterococcus; Epulopiscium; [Eubacterium]_haillii_group; Fusobacterium; Moryella; Stenotrophomonas; Veillonella. In particular, for each classification task (regardless of using SC or LR), the following selected features were: (1) CDI vs. Asymptomatic Carriage: ,, and ; (2) CDI vs. Non-CDI Diarrhea: , , , , and ; (3) CDI vs. Non-CDI: , , , , , , , , and . Note that in the calculation of precision, recall and F1-score, we can treat either CDI (or Asymptomatic Carriage, Non-CDI Diarrhea, Non-CDI) as the true positive. Results shown in the parenthesis represent the latter case
| Model | Diagnostic | Formula | Accuracy | Precision | Recall | F1-score |
|---|---|---|---|---|---|---|
| SC | CDI vs. Asymptomatic Carriage | 0.896 | 0.914 (0.840) | 0.949 (0.75) | 0.931(0.792) | |
| CDI vs. Non-CDI Diarrhea | 0.900 | 0.946 (0.826) | 0.897(0.905) | 0.921(0.864) | ||
| CDI vs. Non-CDI | 0.882 | 0.889 (0.878) | 0.821 (0.927) | 0.853 (0.902) | ||
| LR | CDI vs. Asymptomatic Carriage | 0.830 | 0.895 (0.667) | 0.872 (0.714) | 0.883 (0.690) | |
| CDI vs. Non-CDI Diarrhea | 0.800 | 0.814 (0.765) | 0.897 (0.619) | 0.854 (0.684) | ||
| CDI vs. Non-CDI | 0.813 | 0.841 (0.798) | 0.679 (0.908) | 0.752 (0.850) |
Indeed, as shown in Table 2, we derived a simple SC model with selected features, reaching a very high accuracy (0.896) in distinguishing CDI subjects from Asymptomatic Carriage. Basically, for each subject , we calculate the diagnostic score that will be used for CDI diagnosis: the class of subject is CDI if ; Asymptomatic Carriage, if . Similarly, we derived a SC model with accuracy of 0.900 (or 0.882) in distinguishing CDI from Non-CDI Diarrhea (or Non-CDI) with the corresponding diagnostic score shown in Table 2. To ensure the SC models learned from the entire dataset are not overfitting, we performed cross-validation. With different training sets, SC will derive different mathematical formulas (i.e., diagnostic scores). However, those SC models learned from different training datasets demonstrated quite robust performance in terms of Accuracy (ratio of the total number of correct predictions and the total number of predictions), Precision (the number of positive class predictions that actually belong to the positive class), Recall (the number of positive class predictions made out of all positive examples in the dataset) and F1-score (a weighted average of the precision and recall) (see Supplementary Table 8). More importantly, even trained with less data, the SC models still outperformed LR models learned from the entire dataset.
As shown in Table 2, in the formulas of the diagnostic score, we colored the gut microbiota (or host immune marker) features in red (or blue), respectively. It is clearly seen that any potential interactions between gut microbiota and host immune markers are completely ignored in the formulas derived from LR. But for the formulas derived from SC, nonlinear interactions between gut microbiota and host immune markers can be clearly seen. We emphasize that those nonlinear interactions are not always pairwise. Those explicit interaction terms could inform further mechanistic studies to further reveal the role of intricate interactions between gut microbiota and host immune markers in CDI pathogenesis.
Discussion
Our studies suggest that the interactions between gut microbiota and host immune markers are very sensitive to the status of C. difficile colonization and infection. We demonstrated that incorporating both gut microbiome and host immune marker data into classification models can better distinguish CDI from other patient groups. Our classification models display robust diagnostic performance to differentiate CDI from Asymptomatic carriage. Using selected features to derive simple mathematic formulas we can explicitly quantify the interactions between gut microbiome and host immune markers, the two key components of CDI pathogenesis.
Consistent with previous studies42–45, we found that the gut microbiota of CDI patients was characterized by lower Shannon diversity than that of the Control group. Interestingly, we observed an increased variation of both immune markers and gut microbial compositions in the CDI group with respect to other studied groups. This suggests that CDI is characterized by a significantly less stable microbiome and immune homeostasis. Our findings are in line with the Anna Karenina principle, which suggests that CDI linked changes in the microbiome and immune homeostasis are likely stochastic, leading to community instability46–48.
We identified several candidate driver taxa (e.g., Desulfovibrio, Klebsiella, Streptococcus and Veillonella) that played a key role in driving the changes of microbial correlation networks between CDI and Asymptomatic Carriage (or Non-CDI Diarrhea, Non-CDI) groups. Among those driver taxa, Streptococcus has previously been shown to produce lactate thus impacting C. difficile TcdA expression to alleviate CDI49. Previous study indicated that Desulfovibrio has a pathogenic role in ulcerative colitis due to its ability to generate sulfides50. Klebsiella bacteria have been increasingly shown to develop antimicrobial resistance, most recently to the class of antibiotics known as carbapenems51,52. It is thus possible that the CDI pathogenesis is further enforced by the enrichment of antagonistic bacteria present in the gut microbiome of CDI subjects.
We developed classification models aimed at differentiating CDI status based on host immune markers and gut microbiome data. We were able to identify specific immune and microbial features that could accurately distinguish CDI subjects. In addition, most of the selected features identified by feature selection were also differentially abundant genera and differentially expressed immune markers. From the classification of CDI and Asymptomatic Carriage, we were able to select a few features with outstanding discriminability, including Veillonella and Moryella. Interestingly, a positive relationship between Veillonella and CDI has been identified in recent studies53–56. An important role for Veillonella in CDI is supported by the fact that Veillonella species were associated with low coprostanol levels that correlated strongly with CDI53. A similar negative relationship between Moryella species and CDI has previously been observed57. Enterococcus, a feature selected from the classification of CDI vs. Non-CDI Diarrhea, has been reported to be associated with CDI due to vancomycin resistance58. Consistent with the findings from previous reports59,60, Epulopiscium was significantly enriched in the CDI group and played an important role in differentiating this comparison. Among those features selected from the classification of CDI and Non-CDI groups, Enterobacter and Fusobacterium have been considered as opportunistic pathogens involved in multiple diseases61,62.
Machine learning method has the potential to identify biomarkers and aid in the diagnosis of many diseases. However, the learnt relationships between predictors and outcome are typically nontransparent, especially non-linear methods (i.e., decision tree learning)63. Classical logistic regression is one of the most common machine learning models in medicine. Yet, it fails to solve non-linear problems where there are multiple or non-linear decision boundaries64. Furthermore, the log odds scale in LR is hard to interpret65. Symbolic classification based on genetic programming is an automated technique to derive formulas from features66. Using the selected features from the random forests model, we demonstrated that the mathematical formulas automatically derived from symbolic classification have robust diagnostic accuracy to differentiate CDI patients from Asymptomatic Carriage (or Non-CDI Diarrhea, and Non-CDI groups). Specifically, symbolic classification provides explicit mathematic formulas as its output, which significantly improves the transparency of the learned relationship between predictors and outcomes. We previously demonstrated the potential clinical utility of a specific immunological biomarker (i.e., G-CSF) for diagnosis of CDI36. This study leverages the same unique and well-characterized study cohort, allowing us to study integrated host immune marker and microbial signatures associated with CDI. The fundamental differences between this study and the previous one are the clinical utilization of comprehensive immunological and microbial markers to explore the pathogenesis of CDI, and to generate clinical diagnostic models to detect CDI.
We acknowledge the following limitations of this study. First, the 16S rRNA gene sequencing may not have captured additional insights associated with CDI at the species or strain level. Second, observed associations do not prove causal relationship, and further studies are needed to validate the mechanism underlying the observed associations between these biomarkers and CDI. Our findings support the potential role of gut microbiome and immune markers in CDI and may serve as a starting point for future mechanistic studies. Third, diarrhea itself can affect the gut microbiome. When we compare the gut microbial compositions of groups with and without diarrhea, it is hard to disentangle the impact of diarrhea from that of disease on the gut microbiota, especially when the diarrhea is caused by the disease (e.g., in the CDI group). Completely resolving this limitation is out of the scope of the current study. Finally, further external validations of the classification models and derived formulas need to be performed on an additional cohort with same inclusion criteria as the current cohort.
In sum, utilizing this well-characterized cohort and leveraging machine learning tools, we proposed an effective computational framework to quantify the role of the intricate interactions between gut microbiota and host immune markers in CDI pathogenesis. We believe this framework offers the potential for microbiome-immune marker derived CDI diagnosis, as well as therapeutic strategies for its prevention and treatment.
Material and methods
Study cohort
The background and design of this cohort has been described in detail previously67. Basically, our clinical cohort consists of 243 well-characterized recruited participants, who were divided into four groups associated with different C. difficile infection/colonization statuses: (1) CDI (n = 112): Eligible patients were inpatients 18 years old with new-onset diarrhea, positive clinical stool NAAT result, and a decision to treat for CDI. The stool sample was captured as a discarded sample, and a discarded serum sample collected within 24 hours of that stool sample also captured. Patients were excluded if the diagnostic stool specimen was 72 hours old, if they had received CDI treatment for 24 hours prior to stool collection, or if they had a colostomy. Assessment for the presence of diarrhea included review of nursing input/output logs for number and consistency of stools, consultation with treating clinicians, and detailed chart review (requiring mention of “diarrhea”, “loose stools”, and/or increased frequency, in notes written by multiple providers). Patients for whom there was any doubt about the presence of diarrhea, or who had chronic diarrhea, were excluded. (2) Asymptomatic Carriage (n = 40): Eligible patients were inpatients 18 years old, admitted for at least 72 hours, who had received at least one dose of an antibiotics within the past 7 days, and did not have diarrhea in the 48 hours prior to stool specimen submission. Patients with 2 or more loose stools within 24 hours were excluded; patients who had 1 loose stool were included only if they had recently received a laxative. Patients were excluded if they had a colostomy; received oral or intravenous metronidazole, oral vancomycin, oral rifaximin, and/or oral fidaxomicin for 24 hours within the prior 7 days; had been diagnosed with CDI in the past 6 months; or had tested negative for C. difficile within the past 7 days. Stool specimen were collected prospectively under verbal informed consent. A discarded serum sample from within 24 hours of the stool specimen were also captured. NAAT (Xpert C. difficile/Epi) was performed on all samples, and positive samples retained as the Asymptomatic Carriage cohort. (3) Non-CDI Diarrhea (n = 44): patients with diarrhea (confirmed using the same definition used for the CDI cohort) but had NAAT-negative stool on clinical C. difficile testing; (4) Control (n = 47): patients without diarrhea who had screened as eligible for the Asymptomatic Carriage cohort but were NAAT-negative on research stool testing. In our previous study36, the four groups were named as (1) CDI-NAAT = CDI; (2) Carrier-NAAT = Asymptomatic Carriage; (3) diarrhea NAAT-negative = Non-CDI Diarrhea and (4) no Diarrhea NAAT-Negative = Control. In this work, for simplicity we used the simpler and more clearly descriptive titles.
Serum immune marker measurement
The measurement of host serum cytokines concentrations of IL-2, IL-4, IL-6, IL-8, IL-10, IL-13, IL-15, IL-1β, G-CSF, MCP-1, VEGF-A, and TNF-α was performed using a Milliplex magnetic bead kit and Luminex analyzer (MAGPIX) (Millipore Sigma, Inc., Burlington, MA) as per the manufacturer’s instructions. Purified toxin A and B were separately prepared from C. difficile strain VPI 10463 (American Type Culture Collection 43255-FZ, Manassas, VA). Serum antibody (IgA, IgG, and IgM) levels against C. difficile toxins A and B were measured by semi-quantitative enzyme-linked immunosorbent assay (ELISA). All the experimental details have been reported previously36,67.
Fecal DNA extraction and bacterial 16S rRNA sequencing data analysis
Stool DNA was extracted using the DNeasy PowerSoil Pro Kit (Qiagen, cat# 12888–100) in a QiaCube-automated DNA extraction system (Qiagen) according to instructions. Briefly, 250 mg stool was transferred into a PowerBead Pro Tube provided with the kit and 200 ug RNaseA and 800 μl of CD1 solution were added. Tubes were vortexed briefly, transferred into an adapter, and then vortexed at maximum speed for 10 min. Tubes were centrifuged at 15,000 xg for 1 min and about 500–600 μl supernatant was used for DNA extraction according to instructions. DNA were eluted in 70 μl elution solution C6 and stored at −80°C until use. 16S rRNA microbiome characterization was performed by sequencing the V4 region of the 16S rRNA gene using the Illumina MiSeq68. Each sample was amplified using a barcoded primer, which yielded a unique sequence identifier tagged onto each individual sample library. Illumina-based sequencing yielded greater than 15,000 reads per sample. CLC Genomics Workbench version 12 (Qiagen) was used for OTU clustering and generation of abundance tables. Analyses were performed using the tutorial “OTU Clustering Step by Step” updated September 2, 2019 and available at: https://resources.qiagenbioinformatics.com/tutorials/OTU_Clustering_Steps.pdf
Microbial diversity and differential abundance analysis
The diversity measures and permutational multivariate analysis of variance (PERMANOVA, compare groups of objects and test the null hypothesis that the centroids and dispersion of the groups as defined by measure space are equivalent for all groups) were calculated using the vegan package in R (see Supporting methods for details). For differential abundance analysis, we used ANCOM37 (analysis of composition of microbiomes), with a Benjamini–Hochberg correction at 5% level of significance, and adjusted for age and sex. The Mann–Whitney U test was used to compare the difference of immune marker levels between different groups.
Microbial correlation network and microbiome-immune association analysis
The microbial correlation networks were constructed using SparCC38 (sparse correlations for compositional data, https://github.com/luispedro/sparcc) (see Supporting methods for details). We also used the NetShift39 (https://web.rniapps.net/netshift) to identify potential “driver” taxa that are responsible for the differences of microbial correlations between the CDI and Asymptomatic Carriage (or between Non-CDI Diarrhea and Non-CDI) networks. The driver taxa were identified based on their neighbor shift (NESH) scores, and betweenness centrality (BC) measures in the two networks. Associations between the gut microbiota and host immune markers were quantified by Spearman correlation coefficients in combination with Benjamini-Hochberg FDR correction to account for multiple hypothesis testing (significance threshold α ≤ 0.05). All included genera were required to be detected in ≥50% of all samples in each group.
Classification with Random Forests model
To build a classification model capable of testing the overall contribution of immunological or microbial data in distinguishing the CDI status, we developed a multi-class random forest (RF) classifier. The data is split into a training set and a test set, with 70% of the data forming the training data and the remaining 30% forming the test set. The performance of the multi-class model was measured by micro-average (aggregate the contributions of all classes to compute the average metric) and macro-average (compute the metric independently for each class and then take the average) AUC (the area under the receiver operating characteristic (ROC) curve, the measure of the ability of a classifier to distinguish between classes and is used as a summary of the ROC curve). To determine whether more specific host immune markers or gut microbial taxa could differentiate CDI subjects from Asymptomatic Carriage, Non-CDI Diarrhea and Non-CDI groups, we constructed the binary classifiers based on RF models with integrated immune markers and microbiome data (see Supporting methods for details).
Symbolic classification with genetic programming
We employed Karoo GP69, a genetic programming application suite written in Python that support both symbolic regression (SR) and symbolic classification (SC) analysis, to derive simple formulas for CDI diagnosis. Due to the different training sets, SC will derive different formulas, but their classification performances are quite comparable (S8 Table). The formulas shown in Table 2 were derived based on the whole dataset (for details see supplementary methods). To demonstrate the advantage of SC, for each classification task (i.e., CDI vs. Asymptomatic Carriage, CDI vs. Non-CDI Diarrhea, and CDI vs. Non-CDI), we also performed logistic regression (LR) using the same set of selected features as used in SC (Table 2) (see Supporting methods for details).
Supplementary Material
Acknowledgments
The authors thank all patients who participated in this study, as well as Carolyn Alonso, Javier Villafuerte Gálvez, and the technologists in the Beth Israel Deaconess Medical Center Clinical Microbiology Laboratory for their help with sample collection. The authors thank Zheng Sun for valuable discussion on the microbiome data analysis. S.K. would like to acknowledge the support and help from Professor Lusheng Huang (Jiangxi Agricultural University).
Funding Statement
Y.-Y.L. acknowledged grants from National Institutes of Health (R01AI141529, RF1AG067744, R01HD093761, UH3OD023268, U19AI095219 and U01HL089856). N.R.P. and C.P.K. acknowledged grants from National Institutes of Health (R01AI116596) and Institut Mérieux.
Authors’ contributions
Y.-Y.L, N.R.P., X.C., and C.P.K. conceived and designed the project. C.P.K., N.R.P., X.C. and K.D. performed the clinical study. X.C., H.X., and Q.L. contributed to the serum immune marker measurement. K.W.G. and A.J.G. performed fecal DNA extraction and bacterial 16S rRNA sequencing. S.K., X.-W.W., and Y.-Y.L. performed all the data analysis and wrote the manuscript. N.R.P., K.W.G., C.P.K., and K.D. edited the manuscript.
Ethics approval and consent to participate
Approval of this study was given by the Beth Israel Deaconess Medical Center. All human subjects provided informed consent for participation in the study and collection and analysis of data.
Consent for publication
Not applicable.
Data availability statement
The sequencing data that support the findings of this study have been deposited in the National Center for Biotechnology Information Sequence Read Archive with the BioProject ID: PRJNA668194.
Competing interests
C.P.K. has acted as a paid consultant to Artugen, Facile Therapeutics, First Light Biosciences, Finch, Matrivax, Merck, Seres Health, and Vedanta and has received grant support from Merck. X. C. has acted as a paid consultant to Artugen. All other authors report no potential conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, et al. Burden of Clostridium difficile Infection in the United States. N Engl J Med. 2015;372(9):825–18. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Depestel DD, Aronoff DM.. Epidemiology of Clostridium difficile Infection. J Pharm Pract. 2013;26(464–475):464–475. doi: 10.1177/0897190013499521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDonald LC, Gerding DN, Johnson S, Bakken JS, Carroll KC, Coffin SE, Dubberke ER, Garey KW, Gould CV, Kelly C, et al. Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis. 2018;66:e1–e48. doi: 10.1093/cid/cix1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rupnik M, Wilcox MH, Gerding DN.. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7(7):526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 5.Schaffler H, Breitruck A. Clostridium difficile – from Colonization to Infection. Front Microbiol. 2018;9(646). doi: 10.3389/fmicb.2018.00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tenover FC, Baron EJ, Peterson LR, Persing DH. Laboratory diagnosis of Clostridium difficile infection can molecular amplification methods move us out of uncertainty? J Mol Diagn. 2011;13(6):573–582. doi: 10.1016/j.jmoldx.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burnham CA, Carroll KC. Diagnosis of Clostridium difficile infection: an ongoing conundrum for clinicians and for clinical laboratories. Clin Microbiol Rev. 2013;26(604–630). doi: 10.1128/CMR.00016-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musher DM, Manhas A, Jain P, Nuila F, Waqar A, Logan N, Marino B, Graviss EA. Detection of Clostridium difficile toxin: comparison of enzyme immunoassay results with results obtained by cytotoxicity assay. J Clin Microbiol. 2007;45(8):2737–2739. doi: 10.1128/JCM.00686-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bagdasarian N, Rao K, Malani PN. Diagnosis and Treatment of Clostridium difficile in Adults. JAMA. 2015;313(4):398. doi: 10.1001/jama.2014.17103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rineh A, Kelso MJ, Vatansever F, Tegos GP, Hamblin MR. Clostridium difficile infection: molecular pathogenesis and novel therapeutics. Expert Rev Anti Infect Ther. 2014;12(131–150). doi: 10.1586/14787210.2014.866515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevens V, Dumyati G, Fine LS, Fisher SG, van Wijngaarden E. Cumulative antibiotic exposures over time and the risk of Clostridium difficile infection. Clin Infect Dis. 2011;53(42–48). doi: 10.1093/cid/cir301. [DOI] [PubMed] [Google Scholar]
- 12.Slimings C, Riley TV. Antibiotics and hospital-acquired Clostridium difficile infection: update of systematic review and meta-analysis. J Antimicrob Chemother. 2014;69(881–891). doi: 10.1093/jac/dkt477. [DOI] [PubMed] [Google Scholar]
- 13.Lewis BB, Buffie CG, Carter RA, Leiner I, Toussaint NC, Miller LC, Gobourne A, Ling L, Pamer EG. Loss of Microbiota-Mediated Colonization Resistance to Clostridium difficile Infection With Oral Vancomycin Compared With Metronidazole. J Infect Dis. 2015;212(1656–1665). doi: 10.1093/infdis/jiv256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becattini S, Taur Y, Pamer EG. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol Med. 2016;22(458–478). doi: 10.1016/j.molmed.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, Pamer EG. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun. 2012;80(1):62–73. doi: 10.1128/IAI.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517(7533):205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rea MC, Sit CS, Clayton E, O'Connor PM, Whittal RM, Zheng J, Vederas JC, Ross RP, Hill C. Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc Natl Acad Sci U S A. 2010;107(9352–9357). doi: 10.1073/pnas.0913554107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leffler DA, Lamont JT. Clostridium difficile Infection. N Engl J Med. 2015;373(287–288). doi: 10.1056/NEJMc1506004. [DOI] [PubMed] [Google Scholar]
- 19.Genth H, Dreger SC, Huelsenbeck J, Just I. Clostridium difficile toxins: more than mere inhibitors of Rho proteins. Int J Biochem Cell Biol. 2008;40(592–597). doi: 10.1016/j.biocel.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 20.Sun X, He X, Tzipori S, Gerhard R, Feng H. Essential role of the glucosyltransferase activity in Clostridium difficile toxin-induced secretion of TNF-alpha by macrophages. Microb Pathog. 2009;46(298–305). doi: 10.1016/j.micpath.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riegler M, Sedivy R, Pothoulakis C, Hamilton G, Zacherl J, Bischof G, Cosentini E, Feil W, Schiessel R, LaMont JT, et al. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J Clin Invest. 1995;95(2004–2011). doi: 10.1172/JCI117885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun X, Hirota SA. The roles of host and pathogen factors and the innate immune response in the pathogenesis of Clostridium difficile infection. Mol Immunol. 2015;63(193–202). doi: 10.1016/j.molimm.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly CP, Kyne L. The host immune response to Clostridium difficile. J Med Microbiol. 2011;60(8):1070–1079. doi: 10.1099/jmm.0.030015-0. [DOI] [PubMed] [Google Scholar]
- 24.Bibbo S, Lopetuso LR, Ianiro G, Di Rienzo T, Gasbarrini A, Cammarota G. Role of Microbiota and Innate Immunity in Recurrent Clostridium difficile Infection. J Immunol Res. 2014;2014(462740):1–8. doi: 10.1155/2014/462740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iacob S, Iacob DG, Luminos LM. Intestinal Microbiota as a Host Defense Mechanism to Infectious Threats. Front Microbiol. 2018;9(3328). doi: 10.3389/fmicb.2018.03328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madan R, Jr WA Jr.. Immune responses to Clostridium difficile infection. Trends Mol Med. 2012;18(11):658–666. doi: 10.1016/j.molmed.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun X, Savidge T, Feng H. The enterotoxicity of Clostridium difficile toxins. Toxins (Basel). 2010;2(7):1848–1880. doi: 10.3390/toxins2071848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kyne L, Warny M, Qamar A, Kelly CP. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet. 2001;357(189–193). doi: 10.1016/S0140-6736(00)03592-3. [DOI] [PubMed] [Google Scholar]
- 29.Wilcox MH, Gerding DN, Poxton IR, Kelly C, Nathan R, Birch T, Cornely OA, Rahav G, Bouza E, Lee C, et al. Bezlotoxumab for Prevention of Recurrent Clostridium difficile Infection. N Engl J Med. 2017;376(4):305–317. doi: 10.1056/NEJMoa1602615. [DOI] [PubMed] [Google Scholar]
- 30.Giannasca PJ, Zhang ZX, Lei WD, Boden JA, Giel MA, Monath TP, Thomas WD Jr. Serum antitoxin antibodies mediate systemic and mucosal protection from Clostridium difficile disease in hamsters. Infect Immun. 1999;67(527–538). doi: 10.1128/IAI.67.2.527-538.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnston PF, Gerding DN, Knight KL. Protection from Clostridium difficile infection in CD4 T Cell- and polymeric immunoglobulin receptor-deficient mice. Infect Immun. 2014;82(522–531). doi: 10.1128/IAI.01273-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abos A, Baggio HC, Segura B, García-Díaz AI, Compta Y, Martí MJ, Valldeoriola F, Junqué C. Discriminating cognitive status in Parkinson’s disease through functional connectomics and machine learning. Sci Rep. 2017;7(1). doi: 10.1038/srep45347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dagliati A, Marini S, Sacchi L, Cogni G, Teliti M, Tibollo V, De Cata P, Chiovato L, Bellazzi R. Machine Learning Methods to Predict Diabetes Complications. J Diabetes Sci Technol. 2018;12(2):295–302. doi: 10.1177/1932296817706375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mossotto E, Ashton JJ, Coelho T, Beattie RM, MacArthur BD, Ennis S. Classification of Paediatric Inflammatory Bowel Disease using Machine Learning. Sci Rep. 2011;53(1). doi: 10.1093/cid/cir301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schubert AM, Rogers MAM, Ring C, Mogle J, Petrosino JP, Young VB, Aronoff DM, Schloss PD. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. mBio. 2014;5(3):e01021–01014. doi: 10.1128/mBio.01021-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelly CP, Chen X, Williams D, Xu H, Cuddemi CA, Daugherty K, Barrett C, Miller M, Foussadier A, Lantz A, et al. Host Immune Markers Distinguish Clostridioides difficile Infection From Asymptomatic Carriage and Non–C. difficile Diarrhea. Clin Infect Dis. 2019. doi: 10.1093/cid/ciz330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis. 2015;26(27663). doi: 10.3402/mehd.v26.27663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedman J, Alm EJ, von Mering C. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8(9):e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuntal BK, Chandrakar P, Sadhu S, Mande SS. ‘NetShift’: a methodology for understanding ‘driver microbes’ from healthy and disease microbiome datasets. ISME J. 2019;13(442–454). doi: 10.1038/s41396-018-0291-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bannister CA, Halcox JP, Currie CJ, Preece A, Spasic I, Pławiak P. A genetic programming approach to development of clinical prediction models: a case study in symptomatic cardiovascular disease. PLoS One. 2018;13(9):e0202685. doi: 10.1371/journal.pone.0202685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt M, Lipson H. Distilling free-form natural laws from experimental data. Science. 2009;324(5923):81–85. doi: 10.1126/science.1165893. [DOI] [PubMed] [Google Scholar]
- 42.Song Y, Garg S, Girotra M, Maddox C, von Rosenvinge EC, Dutta A, Dutta S, Fricke WF. Microbiota dynamics in patients treated with fecal microbiota transplantation for recurrent Clostridium difficile infection. PLoS One. 2013;8(11):e81330. doi: 10.1371/journal.pone.0081330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milani C, Ticinesi A, Gerritsen J, Nouvenne A, Lugli GA, Mancabelli L, Turroni F, Duranti S, Mangifesta M, Viappiani A, et al. Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Sci Rep. 2016;6(1). doi: 10.1038/srep25945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang ZD, Ajami NJ, Petrosino JF, Jun G, Hanis CL, Shah M, Hochman L, Ankoma-Sey V, DuPont AW, Wong MC, et al. Randomised clinical trial: faecal microbiota transplantation for recurrent Clostridum difficile infection - fresh, or frozen, or lyophilised microbiota from a small pool of healthy donors delivered by colonoscopy. Aliment Pharmacol Ther. 2017;45(7):899–908. doi: 10.1111/apt.13969. [DOI] [PubMed] [Google Scholar]
- 45.Shankar V, Hamilton MJ, Khoruts A, Kilburn A, Unno T, Paliy O, Sadowsky MJ. Species and genus level resolution analysis of gut microbiota in Clostridium difficile patients following fecal microbiota transplantation. Microbiome. 2014;2(1):13. doi: 10.1186/2049-2618-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaneveld JR, McMinds R, Vega Thurber R. Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat Microbiol. 2017;2(9). doi: 10.1038/nmicrobiol.2017.121. [DOI] [PubMed] [Google Scholar]
- 47.Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, Drew JC, Ilonen J, Knip M, Hyöty H, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5(1):82–91. doi: 10.1038/ismej.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caussy C, Tripathi A, Humphrey G, Bassirian S, Singh S, Faulkner C, Bettencourt R, Rizo E, Richards L, Xu ZZ, et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat Commun. 2019;10(1). doi: 10.1038/s41467-019-09455-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kolling GL, Wu M, Warren CA, Durmaz E, Klaenhammer TR, Guerrant RL. Lactic acid production by Streptococcus thermophilus alters Clostridium difficile infection and in vitro Toxin A production. Gut Microbes. 2012;3(6):523–529. doi: 10.4161/gmic.21757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rowan F, Docherty NG, Murphy M, Murphy B, Coffey JC, O‘Connell PR. Desulfovibrio bacterial species are increased in ulcerative colitis. Dis Colon Rectum. 2010;53(11):1530–1536. doi: 10.1007/DCR.0b013e3181f1e620. [DOI] [PubMed] [Google Scholar]
- 51.Arnold RS, Thom KA, Sharma S, Phillips M, Kristie Johnson J, Morgan DJ. Emergence of Klebsiella pneumoniae carbapenemase-producing bacteria. South Med J. 2011;104(1):40–45. doi: 10.1097/SMJ.0b013e3181fd7d5a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Navon-Venezia S, Kondratyeva K, Carattoli A. Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol Rev. 2017;41(3):252–275. doi: 10.1093/femsre/fux013. [DOI] [PubMed] [Google Scholar]
- 53.Antharam VC, McEwen DC, Garrett TJ, Dossey AT, Li EC, Kozlov AN, Mesbah Z, Wang GP. An Integrated Metabolomic and Microbiome Analysis Identified Specific Gut Microbiota Associated with Fecal Cholesterol and Coprostanol in Clostridium difficile Infection. PLoS One. 2016;11(2):e0148824. doi: 10.1371/journal.pone.0148824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khanna S, Montassier E, Schmidt B, Patel R, Knights D, Pardi DS, Kashyap PC. Gut microbiome predictors of treatment response and recurrence in primary Clostridium difficile infection. Aliment Pharmacol Ther. 2016;44(7):715–727. doi: 10.1111/apt.13750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han SH, Yi J, Kim JH, Lee S, Moon HW. Composition of gut microbiota in patients with toxigenic Clostridioides (Clostridium) difficile: comparison between subgroups according to clinical criteria and toxin gene load. PLoS One. 2019;14(e0212626). doi: 10.1371/journal.pone.0212626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daquigan N, Seekatz AM, Greathouse KL, Young VB, White JR. High-resolution profiling of the gut microbiome reveals the extent of Clostridium difficile burden. NPJ Biofilms Microbiomes. 2017;3(1). doi: 10.1038/s41522-017-0043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hudson LE, Anderson SE, Corbett AH, Lamb TJ. Gleaning Insights from Fecal Microbiota Transplantation and Probiotic Studies for the Rational Design of Combination Microbial Therapies. Clin Microbiol Rev. 2017;30(191–231). doi: 10.1128/CMR.00049-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujitani S, George WL, Morgan MA, Nichols S, Murthy AR. Implications for vancomycin-resistant Enterococcus colonization associated with Clostridium difficile infections. Am J Infect Control. 2011;39(3):188–193. doi: 10.1016/j.ajic.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 59.Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, Wang GP. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol. 2013;51(9):2884–2892. doi: 10.1128/JCM.00845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sokol H, Jegou S, McQuitty C, Straub M, Leducq V, Landman C, Kirchgesner J, Le Gall G, Bourrier A, Nion-Larmurier I, et al. Specificities of the intestinal microbiota in patients with inflammatory bowel disease and Clostridium difficile infection. Gut Microbes. 2018;9(1):55–60. doi: 10.1080/19490976.2017.1361092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mezzatesta ML, Gona F, Stefani S. Enterobacter cloacae complex: clinical impact and emerging antibiotic resistance. Future Microbiology. 2013;26(5):887–902. doi: 10.1177/0897190013499521. [DOI] [PubMed] [Google Scholar]
- 62.Umana A, Sanders BE, Yoo CC, Casasanta MA, Udayasuryan B, Verbridge SS, Slade DJ. Utilizing Whole Fusobacterium Genomes To Identify, Correct, and Characterize Potential Virulence Protein Families. J Bacteriol. 2019;201(23). doi: 10.1128/JB.00273-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng H. Interpreting tree ensembles with inTrees. International Journal of Data Science and Analytics. 2019;7(4):277–287. doi: 10.1007/s41060-018-0144-8. [DOI] [Google Scholar]
- 64.Tollenaar N, van der Heijden PGM, Stiglic G. Optimizing predictive performance of criminal recidivism models using registration data with binary and survival outcomes. PLoS One. 2019;14(3):e0213245. doi: 10.1371/journal.pone.0213245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Norton EC, Dowd BE. Log Odds and the Interpretation of Logit Models. Health Serv Res. 2018;53(2):859–878. doi: 10.1111/1475-6773.12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu K-H, Xu C-G. A genetic programming-based approach to the classification of multiclass microarray datasets. Bioinformatics. 2009;25(3):331–337. doi: 10.1093/bioinformatics/btn644. [DOI] [PubMed] [Google Scholar]
- 67.Pollock NR, Banz A, Chen X, Williams D, Xu H, Cuddemi CA, Cui AX, Perrotta M, Alhassan E, Riou B, et al. Comparison of Clostridioides difficile Stool Toxin Concentrations in Adults With Symptomatic Infection and Asymptomatic Carriage Using an Ultrasensitive Quantitative Immunoassay. Clin Infect Dis. 2019;68(78–86). doi: 10.1093/cid/ciy415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2(1):6. doi: 10.1186/2049-2618-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Staats K, Pantridge E, Cavaglia M, Milovanov I, Aniyan A. TensorFlow Enabled Genetic Programming. arXiv E-prints, arXiv. 2017. 1708.03157. <https://ui.adsabs.harvard.edu/abs/2017arXiv170803157S>. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data that support the findings of this study have been deposited in the National Center for Biotechnology Information Sequence Read Archive with the BioProject ID: PRJNA668194.