

Abstract
Protein kinase CK2 plays multiple roles in cell function in normal and disease states. CK2 is elevated in numerous types of cancer cells, and CK2 suppression of apoptosis represents a key link to the cancer cell phenotype. CK2 regulation of cell survival and death involves diverse processes, and our previous work suggested that mitochondrial machinery is a key locus of this function. One of the earliest responses of prostate cells to inhibition of CK2 is a change in mitochondrial membrane potential, possibly associated with Ca2+ signaling. Thus, in the present work, we investigated early impact of CK2 on intracellular Ca2+ dynamics. Three prostate cancer (PCa) cell lines, PC3-LN4, C4–2B, and 22Rv1, were studied. PCa cells were treated with the CK2 small molecule inhibitors 4,5,6,7-tetrabrombenzotriazole and CX-4945 followed by analysis of Ca2+ levels in various cellular compartments over time. The results showed dose-dependent loss in cytosolic Ca2+ levels starting within 2 min and reaching maximal loss within 5–10 min. There was a concomitant increase in Ca2+ in the endoplasmic reticulum (ER) and mitochondrial compartments. The results suggest that inhibition of CK2 activity results in a rapid movement of Ca2+ out of the cytosol and into the ER and mitochondria, which may be among the earliest contributory factors for induction of apoptosis in cells subjected to inhibition of CK2. In cells with death-inducing levels of CK2 inhibition, total cellular Ca2+ levels dropped at 2 h post-treatment. These novel observations represent a potential mechanism underlying regulation of cell survival and death by CK2 activity.
Keywords: CK2, Calcium, Mitochondria, Endoplasmic reticulum, Cytoplasm, Cell death, Apoptosis, Prostate cancer
Introduction
Protein kinase CK2 (official acronym for former name casein kinase 2 or II) is a ubiquitous and highly conserved protein Ser/Thr kinase consisting of two catalytic subunits (α and α′) linked through two regulatory subunits (β). CK2 is essential for cell survival, and much work has contributed to the recognition that CK2 is a master regulator of numerous cell functions in both normal and abnormal states [1–6]. The kinase subunits are localized to nuclear and cytoplasmic compartments where protein and activity levels may be dynamically regulated contributing to the execution of its functions [7–9]. Originally, the role of elevated CK2 in cancer was not clear since it is also increased (albeit transiently) in normal proliferating cells (see, e.g., [10]). However, our discovery that, in addition to roles in cell growth and proliferation, CK2 has a potent effect on suppression of cell death provided its functional link to the cancer cell phenotype where both proliferation and cell death are dysregulated [11–14]. Thus, the impact of CK2 on cell death has been recognized as a strikingly important function, but the underlying mechanisms of this function remain to be fully elucidated.
In previous studies, we demonstrated that one mode of CK2 regulation of apoptotic activity is mediated by its action on mitochondrial machinery. For example, downregulation of CK2 results in upregulation of Bax and downregulation of Bcl-2 and Bcl-xL accompanied with release of cytochrome c. On the other hand, elevation of CK2α by transient ectopic expression produces an opposite effect on these signals [15, 16]. Production of hydrogen peroxide (H2O2) in PCa cells subjected to CK2 downregulation for 6–24 h (h) is another trigger for apoptotic circuitry [17]. However, the underlying initiating mechanism of these observations remained unclear. In our subsequent work to identify the earliest triggering signals for mediation of CK2 regulated apoptosis, we reported that loss of CK2 expression and activity resulted in dramatically decreased mitochondrial membrane potential (Δψm) that was apparent within 2 h of CK2 inhibition, hinting at a proximal event to activation of apoptosis. Our results also suggested that alterations in Ca2+ signaling were involved in the CK2-mediated loss of Δψm and mitochondrial permeability [18].
Ca2+ is essential for cell function (e.g., [19]). Ca2+ homeostasis is strictly maintained to regulate its roles in life and death functions, and both loss and gain in cell Ca2+ levels can result in cell death [20]. In general, cancer cells have the ability to tolerate higher levels of Ca2+ relative to normal cells (see, e.g., [21]). Since our previous results suggested alterations in Ca2+ signaling in connection with Δψm and mitochondrial permeability [18], we undertook a detailed examination of the regulation of Ca2+ levels in PCa cells caused by decreased CK2 activity. Here we show that CK2 inhibition caused a rapid loss of cytosolic Ca2+ levels and coincident increase in mitochondrial and endoplasmic reticulum levels of Ca2+. Our results are the first to demonstrate that rapid impact on Ca2+ homeostasis is one of the earliest events underlying the initiation of apoptotic machinery in response to reduction in CK2 activity, and thus represents a mechanism for CK2 mediated regulation of cell death.
Materials and methods
Cell lines and culture
Acquisition of cells was as follows: PC3-LN4 cells were selected by orthotopic implantation within the prostate of athymic nude mice and maintained thereafter by monolayer culture [22]; C4–2B cells were a kind gift from Dr. Allen Gao (University of California at Davis, Davis, CA, USA) [23]; 22Rv1 cells were purchased from ATCC (Manassas, VA, USA) and the original freeze back of these cells was used for this work. PC3-LN4 cells were authenticated by Johns Hopkins University genetics core facility using a 9 marker STR profile (Baltimore, MD, USA). 22Rv1 cells were authenticated by IDEXX Bioresearch using a 9 marker STR profile (Columbia, MO, USA). All cells were grown in RPMI-1640 with 25 mM HEPES and L-glutamine (catalog # SH30255.01; Lot #AB216533, 0.42 mM Ca2+, HyClone Laboratories, Logan, UT, USA) with 10% fetal bovine serum (catalog # S11150, Lot #D14038; Atlanta Biologicals, Minneapolis, MN, USA) and 1% Pen/Strep. All cell lines were grown in monolayer culture in an incubator at 37 °C with 5% CO2. All cells had undetectable levels of mycoplasma when thawed, and were maintained in culture for no more than 2 months.
CK2 inhibitors
TBB was made up as a 30 mM stock in DMSO, stored at − 20 °C, and discarded after 30 days (14,453, Cayman Chemicals, Ann Arbor, MI, USA) CX-4945 was made up as a 30 mM stock in DMSO, stored at − 20 °C, and discarded after 30 days (A11060, AdooQ Bioscience, Irving, CA, USA).
Ca2+ monitoring: all compartments
For cytosolic, endoplasmic reticulum, and mitochondrial calcium monitoring, dye charged cells under growth conditions were brought to the plate reader or microscope, baseline readings were recorded, cells were immediately treated with the respective pre-warmed drugs (using a multichannel pipette whenever possible), and sequential fluorescence recordings initiated using a pre-set program on the plate reader or microscope.
Cytosolic Ca2+ monitoring
A FluoForte Ca2+ assay kit (Enz 51,017, Enzo Life Sciences, Farmingdale, NY, USA) was used for monitoring intracellular cytosolic free Ca2+ over time after inhibition of CK2 activity. PC3-LN4 (10 × 103), C4–2B and 22Rv1 (15 × 103, each) cells were plated in 100 μL total volume per well onto black-walled clear bottom 96-well tissue culture plates and grown overnight. When cells reached 70–80% confluency, they were loaded with Fluoforte dye which had been prepared in kit assay buffer [Hanks’ buffer with 20 mM HEPES (HHBS)] containing dye efflux inhibitor, and incubated for 45 min (min) at 37 °C. The dye charged cells were treated with CK2 inhibitor drugs which had been prepared in kit assay buffer; (TBB from 8 to 80 μM, and CX-4945 at 10 μM); DMSO was added for baseline negative control and ATP (150 μM) for positive control. Ca+2 changes were detected by recording fluorescent signals at excitation 490 nm/emission 525 nm with fluorometric plate reader (SpectraMax, Molecular Devices, San Jose, Ca, USA) in kinetic mode for 15 min.
Endoplasmic reticulum Ca2+ monitoring
We employed a low affinity Fluo-5N/AM dye (F14204, Molecular Probes, Thermo Fisher Scientific, Walter, MA, USA) to monitor the free Ca2+ changes in the endoplasmic reticulum (ER) in the three cell lines. PC3-LN4, C4–2B and 22Rv1 cells at 70–80% density in black-walled clear bottom 96-well plates were loaded with optimized 2 μM Fluo-5N dye which had been prepared in live cell imaging solution (A14291DJ, Molecular Probes) containing 1.8 mM CaCl2 and 10 mM glucose. They were incubated for 30 min at 25 °C under growth conditions of 5% CO2 and 20% O2. Fluo-5N dye loading solution was replaced with imaging solution followed by incubation for 15 min at 37 °C under growth conditions to allow complete de-esterification of intracellular AM esters. Imaging solution was replaced with Ca2+/Mg2+-free HBSS buffer (Molecular Probes) for baseline fluorescence recoding and then CK2 inhibitors which had been prepared in 2 mM CaCl2 HBSS buffer (TBB 80, 40, 20, 8 μM, and CX-4945 at 10 μM) were added directly onto wells. DMSO and 150 μM ATP were employed for baseline and positive controls, respectively. Intra-ER Ca+2 changes were detected by recording fluorescence signals at excitation 494 nm/emission 536 nm using plate reader (Molecular Devices).
Mitochondrial Ca2+ monitoring
We used Rhod-2/AM dye (R1244, Molecular Probes) to monitor the mitochondrial Ca2+ influx. PC3-LN4, C4–2B and 22Rv1 cells at 70–80% density in black-walled clear bottom 96-well plates were loaded with optimized 2 μM Rhod-2/AM dye which had been prepared in live cell imaging solution containing 1.8 mM CaCl2 and 10 mM glucose, and were incubated for 30 min at 25 °C under growth conditions of 5% CO2 and 20% O2. Rhod-2/AM dye loading solution was replaced with imaging solution and cells were incubated for 10–15 min at 37 °C under growth conditions to allow complete de-esterification of intracellular AM esters. Imaging solution was replaced with Ca2+/Mg2+-free HBSS buffer (Molecular Probes, Invitrogen) for baseline fluorescence recoding and then CK2 inhibitors (TBB at 80, 40, 20, 8 μM, and CX-4945 at 10 μM) which had been prepared in 2 mM CaCl2 HBSS buffer were added directly into the wells. DMSO served as control for baseline fluorescence detection, and 10 μM RU360 was used as inhibitory control to inhibit Ca+2 entry through mitochondrial Ca+2 uniporter complex to confirm no change in florescence were included. Mitochondrial Ca+2 flux was measured by recording fluorescence signals at excitation 552 nm/emission 581 nm with Fluorometric Plate Reader (Molecular Devices).
Fluorescence microscopy
Microscopy was performed for all three cell lines to qualitatively monitor the intensity of cytosolic Ca2+ signals. The cells were prepared and treated similarly as for Fluoforte plate reader assays, and then immediately imaged with fluorescence microscope (EVOS® FL Auto, Thermo Fisher Scientific, Walter, MA, USA) over 15 min using the GFP filter cube. Qualitative fluorescence imaging was also performed on cells to visually monitor intra-organelle Ca+2. The cells were loaded with low affinity 2 μM Fluo-5N/AM green dye for ER Ca2+ and ER-ID Red dye for ER network (Enz 51,026, Enzo Life Sciences) for detection of co-localization of ER and Ca2+. The Rhod-2/AM red dye (2 μM) was employed for monitoring mitochondrial Ca2+, and MitoTracker green-FM (M7514, Molecular Probes) to stain mitochondria networks for co-localization detection of Ca+2 in mitochondria. Stains were directly added and incubated at 25 °C for 15 min in growth chamber. Cell were then placed in microscope-attached incubator chamber (EVOS® Onstage Incubator) under growth conditions of 30 °C, 5% CO2 and 20% O2. The cells loaded with dyes were then treated with CK2 inhibitors (at 80 μM TBB or CX-4945 at 10 μM) and fluorescence images at 20X were taken using RFP and GFP filter cubes.
The following image adjustments were made using Adobe Photoshop: All cytosolic images shown in Fig. 1b were sharpened to improve focus using the options filter/other/high pass 10 px with blending to hard light at 100% opacity. Contrast was increased to 50% for all cytosolic images and for ER images in the PC3-LN4 cell line treated with DMSO, CX-4945 and ATP.
Fig. 1.

Change in the intracellular cytosolic Ca2+ levels in response to inhibition of CK2 activity. a Kinetic monitoring of intracellular cytosolic Ca2+ in PC3-LN4, C4–2B, and 22Rv1 cells loaded with Fluoforte and treated with 80 μM TBB or 10 μM CX-4945 was carried out over a period of 15 min. Appropriate concentration of DMSO was used in the controls, and 150 μM ATP was used as a positive control. The data represent means of 3 independent experiments. The p values represent *p < 0.05, **p < 0.01, and ***p < 001. b Fluorescence imaging of cytosolic Ca2+ using Fluoforte dye (green) after inhibition of CK2 activity. Cells treated with appropriate volume of DMSO served as baseline controls while those with 150 μM ATP served as positive control. All other details are described under Materials and Methods. Cell lines, treatments, time points and scale bars are indicated. CX4945 in the figure represents CX-4945.
Total cellular calcium measurement
For all total Ca2+ assays, cells were grown on 15 cm plates, 2 plates per final cell pellet. Cells were treated for 2 h with TBB, equivalent concentration of DMSO, or A23187 in media containing 10 mM Ca2+. Timing was carefully controlled by staggering treatments to allow for the first step of cell pellet collection. Cells were collected into pre-weighed polystyrene tubes in growth media using a cell lifter, and centrifuged at 180×g for 5 min at 4 °C. The media was poured off, and the cell pellet placed on ice until all cell pellets were collected. Each pellet was resuspended gently with 10 mL of Ca2+/Mg2+-free phosphate buffered saline (PBS), and centrifuged as before. The PBS was poured off, the cell pellet washed by pulse vortexing in 1 mL Mg2+/Ca2+-free PBS, and centrifuged as before. The PBS was removed completely, and pellet weights were obtained. Cell pellets were stored at − 20 °C.
The Cayman Calcium Assay kit was used to determine total cellular levels of Ca2+ in cell lysates as directed by the manufacturer with minor modifications (701,220; Cayman Chemical, Ann Arbor, MI). PCa cell lysates were tested at various concentrations to determine the best lysis conditions for detection of Ca2+ within the standard curve, incorporating 0.25 mg/dL into the standard curve. Cell pellets were lysed using 100 μL of RIPA (without glycerol [24]) per 80 mg of pellet mass. After 5 min incubation on ice, the lysate was vortexed at high speed for 5 s (s) and centrifuged 2800×g for 10 min at room temperature. The supernatant was transferred to a fresh polystyrene tube and both experimental samples and standard curve samples were measured in triplicate. Calcium concentrations were calculated from the standard curve.
The procedure described by Lamboley et al., using BAPTA (1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid), was also used to verify changes in total cellular Ca2+ [25]. Briefly, cell pellets were resuspended in MeS buffer without BAPTA (MeS/no-BAPTA) to final concentration of 4 mg/mL. To this suspension, 20% sodium dodecyl sulfate was added at a ratio of 1:40. A 0.4 mL aliquot of this mixture was removed, diluted to 2 mg/mL with MeS/no BAPTA containing 1:40 20% SDS, and centrifuged at 17,000×g for 45 min at room temperature. The supernatant was used for background control measures. To the remaining suspension, an equal volume of MeS containing 0.3 mM BAPTA was added (final BAPTA 0.15 mM). Measurements were taken as described [25]. Background lysate (no BAPTA; 292 nm) values were subtracted from the BAPTA-containing lysate values, and Ca2+ amounts were calculated from the standard curve.
For both Cayman- (o-Cresolphthalein Complexone) and BAPTA-based Ca2+ measures, comparison of cells treated for 2 h with and without additional 10 mM Ca2+ in the media showed more reproducible measurement of Ca2+ levels when 10 mM Ca2+ supplementation was used.
Statistical analysis
The IBM SPSS statistics program (version 21.0, IBM. Chicago, IL, USA) was used for statistical analysis of cytosolic, ER, and mitochondrial assays. Independent experiments for each cell line were repeated 3 times with 3 to 6 replications within each experiment. Data were represented as Mean ± SD, and statistical significance was determined by Student t test to compare the means between two groups, one-way and two-way ANOVA to compare mean differences among drug groups, and employing Tukey’s honest significance difference (HSD) post hoc test to identify which specific cell line or drug group was significant. Mean differences values were considered as significant at p < 0.05. Total cellular Ca2+ analysis was performed by GraphPad Prism 6, using one sample t-test to compare drug treatment with TBB to control DMSO.
Results
Effect of CK2 inhibition on cellular cytosolic Ca2+ levels
Our previous work suggested that CK2 activity influenced cellular Ca2+ dynamics [18]. Here, we undertook a systematic investigation of the intracellular Ca2+ response subsequent to treatment of PCa cells with small molecule inhibitors of CK2. Using the Fluoforte calcium assay system, we examined free cytosolic Ca2+ levels in PC3-LN4, C4–2B, and 22Rv1 cell lines in response to CK2 inhibition over time from 0 to 15 min. Treatment with 80 μM TBB resulted in an initial increased cytosolic Ca2+ detection followed by significant and progressive loss of cytosolic Ca2+ levels that was apparent within 5 min in the three cell lines (Fig. 1a). By 10 min the loss in cytosolic Ca2+ was from 15 to 23% compared with the pre-treatment levels. The results in 22Rv1 cells showed an apparent recovery of Ca2+ at 15 min post-treatment with 80 μM TBB, but not with 40 or 20 μM TBB (Table 1). Although treatment with TBB resulted in decreased cytosolic Ca2+ levels in the three cell lines, we noted that decreased cytosolic Ca2+ levels after CX-4945 treatment was only observed in 22Rv1 cells at 5 and 10 min. Moreover, at 15 min the three cell lines had elevated cytosolic Ca2+ levels due to CX-4945 treatment (Table 1); the possible basis of this observation is considered subsequently.
Table 1.
Effect of CK2 inhibition on cytosolic Ca2+ levels
| Time in min | Drug (concentration in μM) | Cytosolic Ca2+ level relative to pre-treatment Mean ± SD (p) | ||
|---|---|---|---|---|
| PC3-LN4 | C4–2B | 22Rv1 | ||
| 0 | DMSO | 1.11 ± 0.02 | 1.04 ± 0.01 | 1.00 ± 0.02 |
| TBB (80) | 1.39 ± 0.05* | 1.06 ± 0.00 | 1.13 ± 0.07* | |
| TBB (40) | 1.16 ± 0.04 | 1.02 ± 0.00 | 1.04 ± 0.03 | |
| TBB (20) | 1.17 ± 0.01 | 1.09 ± 0.01*** | 1.06 ± 0.01 | |
| CX-4945 (10) | 1.02 ± 0.01 | 0.97 ± 0.02*** | 1.11 ± 0.08# | |
| ATP (150) | 1.80 ± 0.22*** | 1.18 ± 0.01*** | 0.85 ± 0.01* | |
| 5 | DMSO | 1.15 ± 0.04 | 1.07 ± 0.04 | 1.08 ± 0.03 |
| TBB (80) | 1.01 ± 0.03* | 0.88 ± 0.03*** | 0.80 ± 0.02*** | |
| TBB (40) | 0.99 ± 0.03*** | 0.79 ± 0.00*** | 0.90 ± 0.02*** | |
| TBB (20) | 1.03 ± 0.02* | 0.98 ± 0.03* | 0.84 ± 0.01*** | |
| CX-4945 (10) | 1.08 ± 0.07 | 1.10 ± 0.02 | 0.83 ± 0.03*** | |
| ATP (150) | 1.05 ± 0.03# | 0.88 ± 0.03*** | 0.78 ± 0.01*** | |
| 10 | DMSO | 1.16 ± 0.02 | 1.08 ± 0.04 | 1.09 ± 0.01 |
| TBB (80) | 0.91 ± 0.01*** | 0.85 ± 0.03*** | 0.77 ± 0.06*** | |
| TBB (40) | 0.90 ± 0.04*** | 1.03 ± 0.04 | 0.78 ± 0.02*** | |
| TBB (20) | 1.00 ± 0.01*** | 0.95 ± 0.02*** | 0.70 ± 0.01*** | |
| CX-4945 (10) | 1.09 ± 0.03* | 1.19 ± 0.01* | 0.89 ± 0.04*** | |
| ATP (150) | 1.03 ± 0.04*** | 0.83 ± 0.02*** | 0.66 ± 0.03*** | |
| 15 | DMSO | 1.16 ± 0.02 | 1.07 ± 0.04 | 1.02 ± 0.03 |
| TBB (80) | 0.85 ± 0.02*** | 0.89 ± 0.03*** | 1.05 ± 0.10 | |
| TBB (40) | 0.87 ± 0.03*** | 0.90 ± 0.03*** | 0.84 ± 0.04* | |
| TBB (20) | 0.94 ± 0.03*** | 0.94 ± 0.03*** | 0.83 ± 0.04* | |
| CX-4945 (10) | 1.09 ± 0.03 | 1.22 ± 0.03*** | 1.12 ± 0.04 | |
| ATP (150) | 1.09 ± 0.07 | 0.71 ± 0.02*** | 0.70 ± 0.04*** | |
Ca2+ levels represent the change within the treatment condition relative to pre-treatment of cells with the various agents shown in the drug column. DMSO served as vehicle control while ATP served as positive control. Mean and standard deviation are listed with p values included in parentheses:
p < 0.1;
p < 0.05;
p < 0.001
The above described observations were confirmed by fluorescence image analysis of the three cell lines loaded with Fluoforte and treated with 80 μM TBB or 10 μM CX-4945. The changes in green fluorescence in response to TBB were subtly apparent in cells treated with 80 μM TBB at 10 or 15 min (Fig. 1b).
Effect of CK2 inhibition on ER Ca2+ levels
The notable early loss of cytosolic Ca2+ levels after inhibition of CK2 suggested a possible translocation of cytosolic Ca2+ to intracellular organelles such as endoplasmic reticulum and mitochondria, which are the major sites of cellular Ca2+ sequestration. Thus, we employed Fluo-5N/AM dye to examine the effects of TBB and CX-4945 on ER Ca2+ levels in PC3-LN4, C4–2B, and 22Rv1 cells over a period of 15 min. The results in Fig. 2a demonstrate that strong inhibition of CK2 activity in each of the cell lines resulted in a rapid increase in Ca2+ levels in the ER occurring within 5 min post-treatment. The increases over 5 to 15 min varied from 8 to 72% due to inhibition by either TBB or CX-4945, with significance reached for each PCa cell type. The results are also presented in tabulated form in Table 2.
Fig. 2.

Change in the intracellular endoplasmic reticulum Ca2+ levels in response to inhibition of CK2 activity. a Kinetic monitoring of Ca2+ in the ER in PC3-LN4, C4–2B, and 22Rv1 cells loaded with Fluo-5N dye and treated with 80 μM TBB or 10 μM CX-4945 over the indicated time. Ca2+ was detected by recording the fluorescent signal as described under Materials and Methods. Baseline controls used corresponding volume of DMSO, and 150 μM ATP served as a positive control. The data represent means of 3 independent experiments; p values are indicated by *p < 0.05, **p < 0.01. b Fluorescence imaging of ER Ca2+ after inhibition of CK2 activity. Fluo-5N/AM green dye indicates Ca2+ binding and ER-ID red dye depicts the ER network. Co-localization of Ca2+ (green) and ER (red) signals is indicted by orange-yellow fluorescence. Baseline control is represented with DMSO and positive control with 150 μM ATP. Cell lines, treatments, time points and scale bars are indicated. CX4945 in the figure represents CX-4945.
Table 2.
Effect of CK2 inhibition on endoplasmic reticulum (ER) Ca2+ levels
| Time in min | Drug (concentration in μM) | ER Ca2+ level relative to pre-treatment Mean ± SD (p) | ||
|---|---|---|---|---|
| PC3-LN4 | C4–2B | 22Rv1 | ||
| 0 | DMSO | 1.05 ± 0.08 | 0.89 ± 0.03 | 1.00 ± 0.05 |
| TBB (80) | 1.00 ± 0.04 | 0.95 ± 0.06 | 1.04 ± 0.10 | |
| TBB (40) | 0.98 ± 0.12 | 0.89 ± 0.04 | 1.03 ± 0.01 | |
| TBB (20) | 1.02 ± 0.08 | 0.82 ± 0.08 | 1.05 ± 0.08 | |
| CX-4945 (10) | 0.91 ± 0.03 | 0.95 ± 0.06 | 0.99 ± 0.01 | |
| ATP (150) | 0.95 ± 0.04 | 0.91 ± 0.04 | 1.02 ± 0.04 | |
| 5 | DMSO | 0.99 ± 0.06 | 0.95 ± 0.08 | 1.04 ± 0.03 |
| TBB (80) | 1.46 ± 0.11*** | 1.08 ± 0.05# | 1.40 ± 0.03*** | |
| TBB (40) | 1.04 ± 0.12 | 1.13 ± 0.05* | 1.25 ± 0.13# | |
| TBB (20) | 1.21 ± 0.07 | 1.06 ± 0.04 | 1.17 ± 0.08 | |
| CX-4945 (10) | 1.33 ± 0.06* | 1.12 ± 0.05* | 1.25 ± 0.03* | |
| ATP (150) | 1.31 ± 0.06* | 1.22 ± 0.05*** | 1.28 ± 0.09* | |
| 10 | DMSO | 1.06 ± 0.12 | 0.95 ± 0.09 | 0.98 ± 0.06 |
| TBB (80) | 1.48 ± 0.18# | 1.22 ± 0.01*** | 1.43 ± 0.01*** | |
| TBB (40) | 1.26 ± 0.17 | 1.14 ± 0.07* | 1.22 ± 0.08 | |
| TBB (20) | 1.55 ± 0.09* | 1.16 ± 0.02* | 1.38 ± 0.17*** | |
| CX-4945 (10) | 1.49 ± 0.06# | 1.15 ± 0.03* | 1.26 ± 0.07# | |
| ATP (150) | 1.65 ± 0.20* | 1.28 ± 0.06*** | 1.33 ± 0.13* | |
| 15 | DMSO | 1.14 ± 0.04 | 0.97 ± 0.06 | 0.98 ± 0.06 |
| TBB (80) | 1.72 ± 0.24* | 1.23 ± 0.05*** | 1.24 ± 0.13# | |
| TBB (40) | 1.28 ± 0.12 | 1.15 ± 0.04* | 1.13 ± 0.09 | |
| TBB (20) | 1.43 ± 0.11 | 1.17 ± 0.04* | 1.29 ± 0.10* | |
| CX-4945 (10) | 1.38 ± 0.15 | 1.13 ± 0.03* | 1.20 ± 0.10 | |
| ATP (150) | 1.74 ± 0.16* | 1.30 ± 0.08*** | 1.39 ± 0.03*** | |
Ca2+ levels represent the change within the treatment condition relative to pre-treatment of cells with the various agents shown in the drug column. DMSO served as vehicle control while ATP served as positive control. Mean and standard deviation are listed with p values included in parentheses:
p < 0.1;
p < 0.05;
p < 0.001
The corresponding fluorescence imaging for ER Ca2+ in TBB and CX-4945 treated cells at 15 min was also determined. Cells were loaded with Fluo-5N/AM green dye (for Ca2+) and ER-ID red dye (for ER network identification). The results in Fig. 2b in which the Ca2+ and ER signals are overlaid demonstrate a shift from predominantly green signal in control DMSO treated cells to reddish/yellow signal in TBB, CX-4945 and positive control ATP-treated cells, indicating increased Ca2+ presence in ER due to CK2 inhibition.
Effect of CK2 inhibition on mitochondrial Ca2+ levels
We investigated the response of mitochondrial Ca2+ levels to inhibition of CK2 activity. Cells were loaded with Rhod-2/FM dye and subjected to treatment with various drugs over time. As shown in Fig. 3a, analogous to the results for ER, we observed a rapid increase in levels of Ca2+ by up to 30% in mitochondria of PC3-LN4, C4–2B, and 22Rv1 cells treated with inhibitors of CK2 for 5 min. At 15 min, mitochondrial Ca2+ increased by 29 to 50% after TBB treatment of 80 μM. Again, both CK2 inhibitors caused movement of Ca2+ into the mitochondria. A more detailed presentation of these data is given in Table 3, incorporating all doses of the inhibitors tested.
Fig. 3.

Change in the intracellular mitochondrial Ca2+ levels in response to inhibition of CK2 activity. a Kinetic monitoring of Ca2+ in mitochondria of PC3-LN4, C4–2B, and 22Rv1 cells loaded with 2 μM Rhod-2 dye and treated with 80 μM TBB or 10 μM CX-4945 over the indicated time. Ca2+ movement was monitored by fluorescent signals as described in Materials and Methods. DMSO was used for baseline control, and 10 μM RU360 was used as mitochondrial Ca2+ uptake inhibitor control. The data represent means of 3 independent experiments, and p values are indicated by *p < 0.05, **p < 0.01, and ***p < 0001. b Fluorescence imaging of Ca2+ in mitochondria after CK2 inhibition. Rhod-2/AM red dye indicates Ca2+ binding and MitoTracker green dye depicts the mitochondrial network. Co-localization of Ca2+ (red) and mitochondria (green) signals is indicted by orange-yellow fluorescence. DMSO and RU360 controls were as indicated above. Cell lines, treatments, time points and scale bars are indicated. CX4945 in the figure represents CX-4945.
Table 3.
Effect of CK2 inhibition on mitochondrial Ca2+ levels
| Time in min | Drug (concentration in μM) | Mitochondrial Ca2+ level relative to pre-treatment Mean ± SD (p) | ||
|---|---|---|---|---|
| PC3-LN4 | C4–2B | 22Rv1 | ||
| 0 | DMSO | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 |
| TBB (80) | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | |
| TBB (40) | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | |
| TBB (20) | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | |
| CX-4945 (10) | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | |
| RU360 (10) | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | |
| 5 | DMSO | 0.97 ± 0.04 | 1.03 ± 0.04 | 1.00 ± 0.00 |
| TBB (80) | 1.30 ± 0.02** | 1.14 ± 0.03* | 1.30 ± 0.06*** | |
| TBB (40) | 1.15 ± 0.16 | 1.04 ± 0.02 | 1.22 ± 0.07*** | |
| TBB (20) | 1.20 ± 0.13 | 1.09 ± 0.05 | 1.11 ± 0.03# | |
| CX-4945 (10) | 1.06 ± 0.13 | 1.11 ± 0.03# | 1.07 ± 0.02 | |
| RU360 (10) | 1.05 ± 0.03 | 1.00 ± 0.01 | 1.00 ± 0.01 | |
| 10 | DMSO | 0.90 ± 0.02 | 1.04 ± 0.05 | 0.96 ± 0.05 |
| TBB (80) | 1.34 ± 0.11*** | 1.28 ± 0.03*** | 1.23 ± 0.03*** | |
| TBB (40) | 1.22 ± 0.09# | 1.16 ± 0.03* | 1.22 ± 0.05*** | |
| TBB (20) | 1.39 ± 0.04*** | 1.12 ± 0.04 | 1.21 ± 0.03*** | |
| CX-4945 (10) | 1.10 ± 0.10* | 1.15 ± 0.04* | 1.15 ± 0.04*** | |
| RU360 (10) | 1.04 ± 0.01 | 1.00 ± 0.01 | 1.01 ± 0.01 | |
| 15 | DMSO | 0.97 ± 0.03 | 1.03 ± 0.02 | 0.99 ± 0.01 |
| TBB (80) | 1.50 ± 0.09*** | 1.29 ± 0.04*** | 1.30 ± 0.06*** | |
| TBB (40) | 1.29 ± 0.07*** | 1.14 ± 0.06# | 1.20 ± 0.03*** | |
| TBB (20) | 1.37 ± 0.10*** | 1.19 ± 0.03** | 1.12 ± 0.05* | |
| CX-4945 (10) | 1.17 ± 0.11# | 1.19 ± 0.04** | 1.07 ± 0.04 | |
| RU360 (10) | 1.06 ± 0.03 | 1.02 ± 0.04 | 1.03 ± 0.01 | |
Ca2+ levels represent the change within the treatment condition relative to pre-treatment of cells with the various agents shown in the drug column. DMSO served as vehicle control while RU360 served as control inhibiting Ca2+ import into mitochondria. Mean and standard deviation are listed with p values included in parentheses:
p < 0.1;
p < 0.05;
p < 0.01;
p < 0.001
Fluorescence imaging analysis of Ca2+ in mitochondria was also carried out in cells loaded with Rhod-2/AM red dye (for Ca2+) and MitoTracker green dye (for mitochondrial network) followed by treatment with various agents for 15 min. As shown in Fig. 3b, prostate cancer cells PC3-LN4, C4–2B, and 22Rv1 treated with inhibitors of CK2 compared with controls demonstrated strongly increased yellow/orange fluorescence intensity representing increased Ca2+ presence in mitochondria relative to controls (treated with DMSO or 10 μM RU360).
Effect of CK2 inhibition on total cellular Ca2+ levels
Given the dramatic changes observed in the intracellular dynamics of Ca2+ in response to altered CK2 activity, we wanted to determine the effect of CK2 inhibition on total cellular Ca2+ levels using the Cayman assay or the method developed by Lamboley et al. for total tissue Ca2+ analysis [25]. Total cellular Ca2+ in cell lysates of PC3-LN4 and C4–2B cells treated with 8 or 80 μM TBB for 2 h was carried out using the Cayman assay. We chose these two concentrations of TBB based on our published data demonstrating no impact on cell viability at 2 or 24 h (h) using 8 μM TBB, but strong negative impact using 80 μM TBB. The results in Fig. 4a show the effect of TBB on total Ca2+ levels in PC3-LN4 and C4–2B cells. Strong CK2 inhibition using 80 μM TBB caused 11–16% loss of total cellular Ca2+ in the two cell lines. Treatment with 8 μM TBB did not cause reduced total cellular Ca2+, in fact levels increased slightly but this change was not significant. Because the variation in data for C4–2B cells resulted in lack of significance, we measured the effect of 80 μM TBB on C4–2B total cellular Ca2+ levels using the method of Lamboley et al. In this experiment, we also used a calcium ionophore, A23187, to induce calcium uptake in the PCa cells. These results demonstrated a significant 26% decreased Ca2+ level caused by TBB, in contrast to the 1.8-fold increased Ca2+ level due to A23187 treatment. In sum, these experiments suggested that alteration in CK2 activity caused an early (5–15 min) response in intracellular Ca2+ distribution and total cellular Ca2+ levels (2 h) which is significantly prior to the appearance of other markers of cell death detectable starting around 6 h following inhibition of CK2 activity [18] (Fig. 5).
Fig. 4.
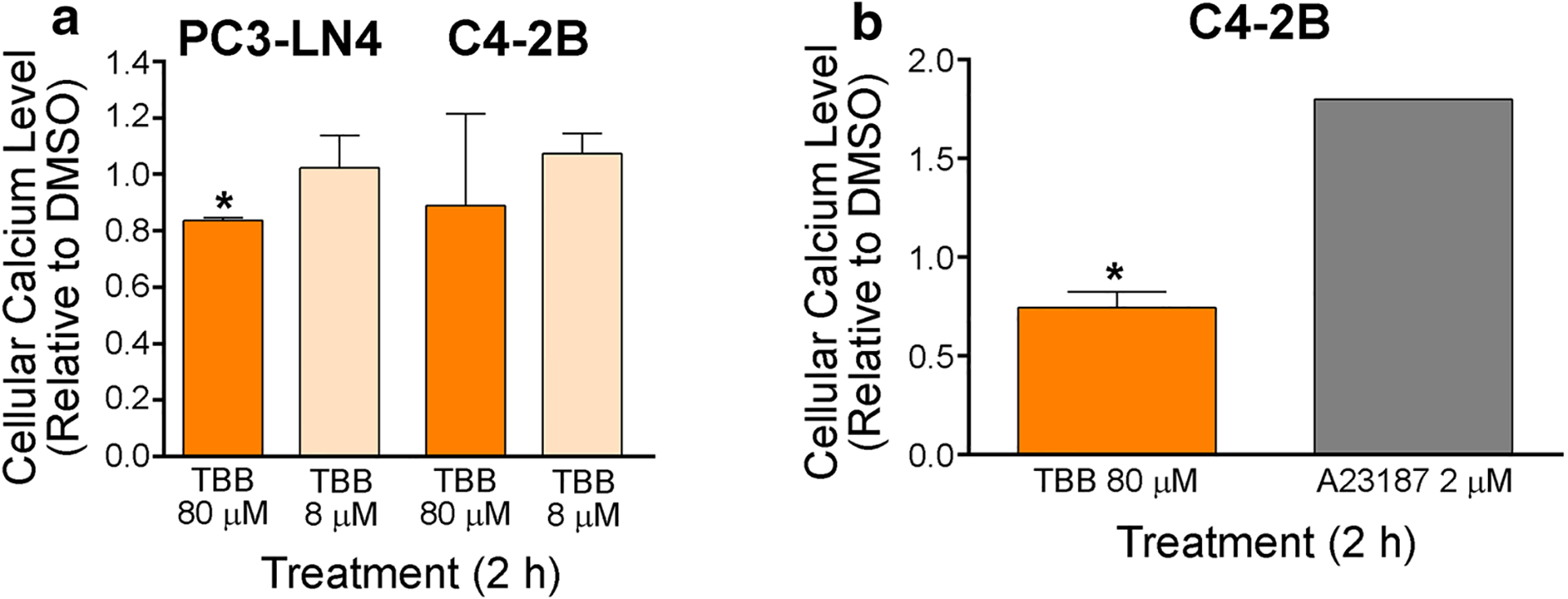
Effect of inhibition of CK2 activity on total intracellular Ca2+ levels. a PC3-LN4 and C4–2B cells in culture were treated with TBB (8 or 80 μM) for 2 h. Cell lysates were analyzed for Ca2+ levels using the Cayman assay. The response of total cellular Ca2+ to CK2 inhibition relative to DMSO control is shown. b The effect of 80 μM TBB (expressed relative to DMSO control) on total cellular Ca2+ level in C4–2B cells measured by the BAPTA assay. Treatment with A23187 was included as a positive control for Ca2+ uptake. *p < 0.05. CX4945 in the figure represents CX-4945
Fig. 5.

The temporal sequence of events leading to apoptosis in response to CK2 inhibition. We propose that inhibition of CK2 results in intracellular Ca2+ flux so that there is rapid loss of cytosolic Ca2+ with a concomitant increase in ER and mitochondrial Ca2+. These changes are reflected in the loss of Δψm within 2 h. If the cells are treated with 80 μM TBB (high CK2i), they follow a path that results in decreased total cellular Ca2+ and induction of the mitochondrial apoptotic pathway and cell death. If the cells are treated with 8 μM TBB (low CK2i), there is no change in total cellular Ca2+ and concomitant recovery of Δψm, with the result that cells survive
Discussion
In this work, we have identified cellular Ca2+ homeostasis as the earliest target of CK2 impact in regulation of cell death. Protein kinase CK2 is well recognized to play diverse roles in cell function, including cell proliferation and decisions for cell survival. In the latter context, one of its major functions is to serve as a suppressor of apoptosis [13, 14, 26]. In previous work, we demonstrated that alteration in CK2 activity or level influences the production of H2O2, suggesting generation of reactive oxygen species (ROS) as an early mediator of cell death via the mitochondrial apoptotic machinery [16, 17]. However, our subsequent work hinted at this production of ROS not being the earliest mediator of cell death in response to CK2 inhibition. For example, we documented that CK2 inhibition in cells causes rapid loss in mitochondrial membrane potential (Δψm), apparent within 2 h after treatment of cells with CK2 inhibitors TBB or TBCA (tetrabromocinnamic acid); further, mitochondrial membrane permeability transition was induced immediately after CK2 inhibition in purified mitochondria. We showed that chelation of Ca2+ abrogated the impact of blocking CK2 activity on Δψm and mitochondrial permeability. These results accorded with cell viability and clonal survival data in response to 80 μM TBB, which indicated that loss of cell survival occurred after only 6–8 h of CK2 inhibitor treatment [18, 26]. Inhibition of CK2 has been suggested to induce ER stress [27]. We have also observed the expression of ER stress response signals in PCa cells on downregulation of CK2; however, the appearance of these signals was not a rapid event [28]. These cumulative observations suggest that the very early changes in Ca2+ signaling occurring in response to inhibition of CK2 orchestrate the induction of cellular apoptosis.
The present work has provided experimental data showing that various compartments of the cell (cytosol, mitochondria, and ER) demonstrate profound rapid changes in Ca2+ dynamics in response to altered CK2 activity. There is considerable evidence that ER and mitochondria are the major organelles involved in physiological sequestration of Ca2+ in the cell, and the level of Ca2+ in these organelles is tightly regulated for their physiological functions. If the level of Ca2+ exceeds these levels, there is an onset of ER stress and a juxtaposition of ER with mitochondria, resulting in increased mitochondrial Ca2+ levels. This elevation of Ca2+ in the mitochondria leads to breakdown of permeability regulation and loss of Δψm, instigating a further choreography of events leading to cell death (see, e.g., [19, 29–32]). In cells treated with CK2 inhibitor, there is a loss of Ca2+ in the cytosol and a corresponding gain in Ca2+ in the mitochondrial and ER compartments that occurs within 5 min. In light of the foregoing discussion regarding transfer of Ca2+ from ER to mitochondria, our results do not indicate whether increased Ca2+ in the mitochondria follows that in ER, since the temporal change in Ca2+ levels in the two compartments appears analogous. Regardless, our results represent the earliest observed effects on intracellular Ca2+ homeostasis caused by inhibition of CK2 activity, and represent an important addition to the mechanism of CK2 regulation of cell death.
CK2 control over intracellular Ca2+ levels and dynamics has many possible pathways. The results presented here on an immediate impact of CK2 inhibition suggest future focus on protein/protein interactions and CK2 substrate proteins involved in Ca2+ binding or shuttling. Calmodulin phosphorylation by CK2 can modulate its structure as well as the binding of calmodulin to interacting partner proteins such as the M-type potassium channel subunit KCNQ2 (see, e.g., [33, 34]). Various Bcl-2 pro-and anti-apoptotic family member proteins play pivotal roles in mediating ER and mitochondrial Ca2+ flux and cell death programs. Further, CK2 may have an effect on various plasma membrane-associated pumps and channels involved in Ca2+ transport, as well as an effect on the mitochondrial membrane transport mechanisms. These various possibilities remain to be investigated in future studies.
Our data on the effect of CK2 inhibition on total cellular Ca2+ are intriguing. It has been observed that, in general, an increase in cellular free Ca2+ promotes cell death, the most notable example being that of necrotic cell death (e.g., [35]). It is also noteworthy that Ca2+ levels in cells are strictly regulated such that cells cannot tolerate an increase or a decrease in the physiological levels of Ca2+ (see, e.g., [20, 36, 37]). Thus, our observation that a death-inducing level of CK2 inhibition causes the total cellular Ca2+ amount to drop within 2 h represents an example of lethally perturbed Ca2+ homeostasis. Cancer cells have the ability to tolerate higher levels of Ca2+ relative to corresponding non-malignant cells (e.g., [38]). Further, the role of ER and mitochondrial Ca2+ crosstalk in cell death has been discussed in detail (see, e.g., [30, 39–41]. Our observations accord with the well-described changes in mitochondrial and ER associated Ca2+ pools and the consequence of interaction of these organelles in altering cell viability [29, 31, 39, 42]. Based on our previous data [18] and the present results, a suggested temporal sequence of events leading to apoptosis in response to CK2 inhibition is presented in Fig. 5. We propose that acute inhibition of CK2 induces Ca2+ entry into both the ER and mitochondria with consequent initiation of apoptotic activity via the mitochondrial machinery.
In summary, CK2 control of cell survival exploits control of Ca2+ dynamics and signaling in the cell. CK2 inhibition is associated with immediate loss of cytosolic Ca2+ and dramatic increase in Ca2+ levels in the ER and mitochondrial compartments, suggesting these changes to be the triggering event for eventual induction of apoptosis. These events are followed by decreased total cellular Ca2+. A number of transient receptor potential channels have been described, and in particular, TRPV6 has been considered to be particularly involved in regulation of Ca2+ in cells [38, 43–45]. At present, it is unclear as to the specific roles of various pumps or channels that regulate Ca2+ in response to altered CK2 activity, and this will be the subject of our future studies. Our observations are the first to link CK2 impact on intracellular Ca2+ homeostasis as the earliest event related to CK2 in regulation of cell death.
Acknowledgements
KA holds the title of Senior Research Career Scientist awarded by the U.S. Department of Veterans Affairs. M.A. was recipient of a scholarship awarded by the Higher Education Commission of Pakistan.
Funding
This work was supported by Merit Review research funds BX003282 awarded by the Department of Veterans Affairs (K.A.), and research grant R01CA150182 awarded by the National Cancer Institute, NIH, Department of Health and Human Services (K.A.).
Footnotes
Publisher's Disclaimer: Disclaimer The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the U.S. Department of Veterans Affairs or the U.S. government.
Conflict of interest The authors declare no conflict of interest or competing interests.
References
- 1.Ahmed K (1999) Nuclear matrix and protein kinase CK2 signaling. Crit Rev Eukaryot Gene Expr 9(3–4):329–336 [DOI] [PubMed] [Google Scholar]
- 2.Tawfic S, Yu S, Wang H, Faust R, Davis A, Ahmed K (2001) Protein kinase CK2 signal in neoplasia. Histol Histopathol 16(2):573–582 [DOI] [PubMed] [Google Scholar]
- 3.Pinna LA (2002) Protein kinase CK2: a challenge to canons. J Cell Sci 115(Pt 20):3873–3878 [DOI] [PubMed] [Google Scholar]
- 4.Meggio F, Pinna LA (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J 17(3):349–368. 10.1096/fj.02-0473rev [DOI] [PubMed] [Google Scholar]
- 5.Guerra B, Issinger O-G (2008) Protein kinase CK2 in human diseases. Curr Med Chem 15(19):1870–1886. 10.1007/s00018-009-9148-9 [DOI] [PubMed] [Google Scholar]
- 6.St-Denis NA, Litchfield DW (2009) Protein kinase CK2 in health and disease: from birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci 66(11–12):1817–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmed K, Yenice S, Davis A, Goueli SA (1993) Association of casein kinase 2 with nuclear chromatin in relation to androgenic regulation of rat prostate. Proc Natl Acad Sci USA 90(10):4426–4430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faust RA, Niehans G, Gapany M, Hoistad D, Knapp D, Cherwitz D, Davis A, Adams GL, Ahmed K (1999) Subcellular immunolocalization of protein kinase CK2 in normal and carcinoma cells. Int J Biochem Cell Biol 31(9):941–949. 10.1016/S1357-2725(99)00050-3 [DOI] [PubMed] [Google Scholar]
- 9.Faust M, Montenarh M (2000) Subcellular localization of protein kinase CK2. A key to its function? Cell Tissue Res 301(3):329–340 [DOI] [PubMed] [Google Scholar]
- 10.Guerra B, Issinger O-G (1999) Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis 20(2):391–408. [DOI] [PubMed] [Google Scholar]
- 11.Guo C, Yu S, Davis AT, Wang H, Green JE, Ahmed K (2001) A potential role of nuclear matrix-associated protein kinase CK2 in protection against drug-induced apoptosis in cancer cells. J Biol Chem 276(8):5992–5999. 10.1074/jbc.M004862200 [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Davis A, Yu S, Ahmed K (2001) Response of cancer cells to molecular interruption of the CK2 signal. Mol Cell Biochem 227(1–2):167–174. 10.1023/A:1013112908734 [DOI] [PubMed] [Google Scholar]
- 13.Ahmed K, Gerber DA, Cochet C (2002) Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol 12(5):226–230 [DOI] [PubMed] [Google Scholar]
- 14.Ahmad KA, Wang G, Unger G, Slaton J, Ahmed K (2008) Protein kinase CK2–a key suppressor of apoptosis. Adv Enzyme Regul 48:179–187. 10.1016/j.advenzreg.2008.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang G, Ahmad KA, Ahmed K (2005) Modulation of death receptor-mediated apoptosis by CK2. Mol Cell Biochem 274(1–2):201–205 [DOI] [PubMed] [Google Scholar]
- 16.Wang G, Ahmad KA, Ahmed K (2006) Role of protein kinase CK2 in the regulation of tumor necrosis factor-related apoptosis inducing ligand-induced apoptosis in prostate cancer cells. Cancer Res 66(4):2242–2249. 10.1158/0008-5472.CAN-05-2772 [DOI] [PubMed] [Google Scholar]
- 17.Ahmad KA, Wang G, Ahmed K (2006) Intracellular hydrogen peroxide production is an upstream event in apoptosis induced by down-regulation of casein kinase 2 in prostate cancer cells. Mol Cancer Res 4(5):331–338. 10.1158/1541-7786.MCR-06-0073 [DOI] [PubMed] [Google Scholar]
- 18.Qaiser F, Trembley JH, Kren BT, Wu JJ, Naveed AK, Ahmed K (2014) Protein kinase CK2 inhibition induces cell death via early impact on mitochondrial function. J Cell Biochem 115(12):2103–2115. 10.1002/jcb.24887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clapham DE (2007) Calcium signaling. Cell 131(6):1047–1058. 10.1016/j.cell.2007.11.028 [DOI] [PubMed] [Google Scholar]
- 20.Brini M, Ottolini D, Cali T, Carafoli E (2013) Calcium in health and disease. Metal ions in life sciences 13:81–137. 10.1007/978-94-007-7500-8_4 [DOI] [PubMed] [Google Scholar]
- 21.Ritaine A, Shapovalov G, Prevarskaya N (2017) Metabolic disorders and cancer: store-operated Ca(2+) entry in cancer: focus on IP3R-mediated Ca(2+) release from intracellular stores and its role in migration and invasion. Adv Exp Med Biol 993:623–637. 10.1007/978-3-319-57732-6_31 [DOI] [PubMed] [Google Scholar]
- 22.Slaton JW, Unger GM, Sloper DT, Davis AT, Ahmed K (2004) Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol Cancer Res 2(12):712–721 [PubMed] [Google Scholar]
- 23.Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, Gao AC (2014) Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castrationresistant prostate cancer. Clin Cancer Res 20(12):3198–3210. 10.1158/1078-0432.Ccr-13-3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trembley JH, Unger GM, Tobolt DK, Korman VL, Wang G, Ahmad KA, Slaton JW, Kren BT, Ahmed K (2011) Systemic administration of antisense oligonucleotides simultaneously targeting CK2alpha and alpha’ subunits reduces orthotopic xenograft prostate tumors in mice. Mol Cell Biochem 356(1–2):21–35. 10.1007/s11010-011-0943-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamboley CR, Kake Guena SA, Toure F, Hebert C, Yaddaden L, Nadeau S, Bouchard P, Wei-LaPierre L, Laine J, Rousseau EC, Frenette J, Protasi F, Dirksen RT, Pape PC (2015) New method for determining total calcium content in tissue applied to skeletal muscle with and without calsequestrin. J Gen Physiol 145(2):127–153. 10.1085/jgp.201411250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trembley JH, Qaiser F, Kren BT, Ahmed K (2015) CK2—a global regulator of cell death. In: Ahmed K, Issinger O-G, Szyszka R (eds) Protein kinase CK2 cellular function in normal and disease states, Advances in Biochemistry in Health and Disease, vol 12. Springer, Switzerland, pp 159–181. 10.1007/978-3-319-14544-0_10 [DOI] [Google Scholar]
- 27.Hessenauer A, Schneider CC, Gotz C, Montenarh M (2011) CK2 inhibition induces apoptosis via the ER stress response. Cell Signal 23(1):145–151. 10.1016/j.cellsig.2010.08.014 [DOI] [PubMed] [Google Scholar]
- 28.Trembley JH, Kren BT, Abedin MJ, Shaughnessy DP, Li Y, Dehm SM, Ahmed K (2019) CK2 Pro-survival role in prostate cancer is mediated via maintenance and promotion of androgen receptor and NFkappaB p65 expression. Pharmaceuticals 12(2):89. 10.3390/ph12020089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174(7):915–921. 10.1083/jcb.200604016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orrenius S, Gogvadze V, Zhivotovsky B (2015) Calcium and mitochondria in the regulation of cell death. Biochem Biophys Res Commun 460(1):72–81. 10.1016/j.bbrc.2015.01.137 [DOI] [PubMed] [Google Scholar]
- 31.Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, Giorgi C, Pinton P (2018) Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69:62–72. 10.1016/j.ceca.2017.05.003 [DOI] [PubMed] [Google Scholar]
- 32.Fliniaux I, Germain E, Farfariello V, Prevarskaya N (2018) TRPs and Ca2+ in cell death and survival. Cell Calcium 69:4–18. 10.1016/j.ceca.2017.07.002 [DOI] [PubMed] [Google Scholar]
- 33.Arrigoni G, Marin O, Pagano MA, Settimo L, Paolin B, Meggio F, Pinna LA (2004) Phosphorylation of calmodulin fragments by protein kinase CK2. Mech Aspects Struct Conseq Biochem 43(40):12788–12798. 10.1021/bi049365c [DOI] [PubMed] [Google Scholar]
- 34.Pan J, Zhang S, Borchers CH (2016) Protein species-specific characterization of conformational change induced by multisite phosphorylation. J Proteomics 134:138–143. 10.1016/j.jprot.2015.12.002 [DOI] [PubMed] [Google Scholar]
- 35.Zhivotovsky B, Orrenius S (2011) Calcium and cell death mechanisms: a perspective from the cell death community. Cell Calcium 50(3):211–221. 10.1016/j.ceca.2011.03.003 [DOI] [PubMed] [Google Scholar]
- 36.Carafoli E, Krebs J (2016) Why calcium? How calcium became the best communicator. J Biol Chem 291(40):20849–20857. 10.1074/jbc.R116.735894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cali T, Brini M, Carafoli E (2017) Regulation of cell calcium and role of plasma membrane calcium ATPases. Int Rev Cell Mol Biol 332:259–296. 10.1016/bs.ircmb.2017.01.002 [DOI] [PubMed] [Google Scholar]
- 38.Monteith GR, Prevarskaya N, Roberts-Thomson SJ (2017) The calcium-cancer signalling nexus. Nat Rev Cancer 17(6):367–380. 10.1038/nrc.2017.18 [DOI] [PubMed] [Google Scholar]
- 39.Grimm S (2012) The ER-mitochondria interface: the social network of cell death. Biochim Biophys Acta 1823(2):327–334. 10.1016/j.bbamcr.2011.11.018 [DOI] [PubMed] [Google Scholar]
- 40.Ivanova H, Kerkhofs M, La Rovere RM, Bultynck G (2017) Endoplasmic reticulum-mitochondrial Ca2+ fluxes underlying cancer cell survival. Front Oncol 7:70. 10.3389/fonc.2017.00070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart TA, Yapa KTDS (1848) Monteith GR (2015) Altered calcium signaling in cancer cells. Biochimica et Biophysica Acta (BBA) 10:2502–2511. 10.1016/j.bbamem.2014.08.016 [DOI] [PubMed] [Google Scholar]
- 42.Naon D (1843) Scorrano L (2014) At the right distance: ER-mitochondria juxtaposition in cell life and death. Bba-Mol Cell Res 10:2184–2194. 10.1016/j.bbamcr.2014.05.011 [DOI] [PubMed] [Google Scholar]
- 43.Stewart JM (2020) TRPV6 as a target for cancer therapy. J Cancer 11(2):374–387. 10.7150/jca.31640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lehen’kyi V, Flourakis M, Skryma R, Prevarskaya N (2007) TRPV6 channel controls prostate cancer cell proliferation via Ca(2+)/NFAT-dependent pathways. Oncogene 26(52):7380–7385. 10.1038/sj.onc.1210545 [DOI] [PubMed] [Google Scholar]
- 45.Prevarskaya N, Skryma R, Shuba Y (2018) Ion channels in cancer: are cancer hallmarks oncochannelopathies? Physiol Rev 98(2):559–621. 10.1152/physrev.00044.2016 [DOI] [PubMed] [Google Scholar]