

Abstract
Stable isotope labeling is a leading strategy for mass-spectrometry-based peptide quantification. Whereas TMTpro isobaric tagging can quantify up to 16 multiplexed samples in a single experiment, nonisobaric, yet chromatographically indistinguishable, variants of TMTpro reagents can be used in conjunction with the isobaric tag series for various peptide-targeting applications. Here we test the performance of two nonisobaric TMTpro variants, a stable-isotope-free TMTproZero tag and a nearly fully isotope-labeled “super-heavy” variant, shTMTpro, in a targeted assay for peptides of charge state 4+. We label each peptide with TMTproZero or Super Heavy TMTpro reagents and separately spike each peptide into a TMTpro16-labeled background (equal amount of peptide across all 16 channels). We observe that the expected 1:1 reporter ion ratio is distorted when a TMTproZero-labeled peptide is used; however, we note no such interference when shTMTpro substitutes the TMTproZero tag. Our data suggest that using the Super Heavy TMTpro reagent is an improvement over the TMTproZero reagent for the accurate quantification of high-charge-state peptides for trigger-based multiplexed assays.
Graphical Abstract
Stable isotope labeling of peptides is the foundation for numerous mass-spectrometry-based quantification strategies. Developments in quantitative proteomics have yielded many innovative and streamlined methodologies for targeting low-abundance peptides. Techniques such as TOMAHAQ1 and TOMAHTO2 use relatively high amounts (at times, 100× or greater) of trigger peptides to quantify low amounts of target peptides. These trigger peptides are labeled with chromatographically indistinguishable, isotopologue variants of isobaric tags, such as TMTzero and Super Heavy TMT reagents, collectively known as mTMT,3,4 which are variants of the original TMT molecule.5 Here we introduce the Super Heavy TMTpro (shTMTpro) reagent, aptly named as it incorporates nine more stable isotopes than the TMTpro16 reagents that are used in isobaric tagging experiments.
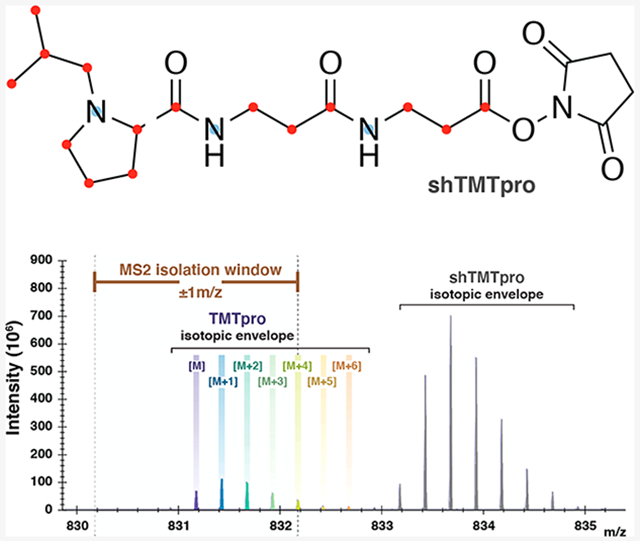
TMTpro reagents encompass a set of 16 isobaric, stable-isotope-labeled, NHS-ester-reactive chemical-labeling reagents used for mass-spectrometry-based quantitative protein profiling. The TMTpro molecule itself is incorporated with a total of nine stable isotopes (seven 13C and two 15N) that are distributed between a reporter ion and a mass balancer region.6 This isobaric set of tags has a peptide modification mass of 304.2011 Da and produces reporter ions that range between 126.1277 and 134.1482 Da (Figure 1A, left). Analogous to the mTMT variants of the classic TMT molecule, TMTpro variants include a stable-isotope-free (TMTproZero reagent) and a nearly fully 13C- and 15N-isotope-labeled (Super Heavy TMTpro reagent) version of the molecule, all of which are chromatographically indistinguishable. The lighter version of the TMTpro reagent, TMTproZero, has a modification mass of 295.1896 Da and generates a reporter ion with a mass of 126.1277 Da (Figure 1A, middle). In contrast, the heavier version of the TMTpro reagent, shTMTpro, has a total of 18 stable isotopes (15 13C and 3 15N). These stable isotopes impart the shTMTpro tag with a modification mass of 313.2310 Da and a reporter ion mass of 135.1516 Da (Figure 1A, right). As such, the mass difference between the TMTpro16 reagent set and either variant is ±9 Da. In general, a lysine-terminating, fully tryptic peptide can accept two TMTpro labels, one at the N-terminus and another at the C-terminal lysine, which creates a mass difference of ±18 Da relative to TMTpro16 whether TMTproZero or shTMTpro reagent is used. However, an arginine-terminating peptide (with the TMTpro tag present only at the N-terminus) will have a mass difference of only ±9 Da. Because tryptic peptides are generally of charge state 2+ or greater, this mass difference will decrease with increasing charge state. As such, an arginine-terminating peptide (barring any internal lysine residues) that is of charge state 4+ will have a mass difference of only 2.25 Da between the nonisobaric labels. This small mass difference between monoisotopic peaks allows higher order isotope peaks from one isotopic envelope (e.g., the high-abundance trigger peptide) to overlap with the envelope of a second peptide (e.g., the low-abundance target peptide), which ultimately results in reporter ion ratios that are distorted from those expected.
Figure 1.

Showcasing the interference of coeluting, nonisobaric TMTpro variants. (A) Chemical structures, modification masses, and reporter ion masses of TMTpro (left), TMTproZero (middle), and shTMTpro (right) reagents. Reporter ion fragments are represented on the left of the HCD fragmentation site. (B) Theoretical spectrum depicting isotopic envelopes of the three TMTpro nonisobaric variants for a peptide of charge state 4+. (C) Experimental overview in which a peptide labeled with the TMTpro16 tag series is mixed with five times as much peptide labeled with either TMTproZero or shTMTpro reagent, separated on a 30 min gradient, and analyzed on an Orbitrap Eclipse mass spectrometer by MS/MS. (D) Chromatogram showing the coelution of TMTproZero (left) and shTMTpro (right) tagged with TMTpro16 reagents for the Parkin peptide (residues 52–75) with sequence NDWTVQNCDLDQQSIVHIVQRPWR. The peptide is alkylated at the cysteine residue, and the first arginine residue is heavy-isotope-labeled (+10.0082 Da). (E) MS1-stage spectrum depicting the TMTproZero-labeled Parkin peptide and the overlap of isotopic envelopes (top). The TMTpro16 reporter ion profile shows interference due to TMTproZero-labeled peptides (bottom left). The relative peak areas of TMTpro reporter ions across the 16 channels indicate interference (bottom right). (F) MS1-stage spectrum depicting the shTMTpro-labeled Parkin peptide and showing no overlap of isotopic envelopes (top). The TMTpro16 reporter ion profile indicates no measurable interference due to shTMTpro-labeled peptides (bottom left). The relative peak areas of TMTpro reporter ions across the 16 channels show equal peak areas for each reporter ion (bottom right).
Targeted assays must balance signal intensity and specificity. Naturally occurring isotopes are distributed across the peptide molecule and generally form an isotopic envelope on a mass spectrum. For tandem MS analysis, wider isolation windows (e.g., ±1 Th) that are centered around the monoisotopic peak are often recommended to maximize the signal for low-abundance species. It is conceivable that when targeting a given peptide, the isolation window may capture the tail of an overlapping isotopic envelope from a coeluting peptide. While aiming for high sensitivity, wide windows facilitate the coisolation, cofragmentation, and thus coanalysis of interfering species. The effect of interference is exacerbated in the case of TOMAHAQ-style assays in which a high-abundance trigger peptide permits the isolation and fragmentation of a low-abundance, coeluting target of interest using an a priori mass offset.1 The caveat of interference is generally observed when using classic TMTzero triggering of TMT10plex samples. In this case, only a 5 Da difference separates these two coeluting, nonisobaric tags such that peptides not containing lysine residues and that are triply charged (or greater) have a mass difference of <2 Da. The isotopic envelopes of the target and trigger overlap and thereby limit the use of TMTzero for triggering lysine-residue-deficient peptide targets. As such, the TMTzero reagent has been supplanted by Super Heavy TMT reagent in the TOMAHTO2 workflow. Because the mass difference of TMTproZero from TMTpro is larger (9 Da), the effect of interference on lysine-free peptides with charge states of 2+ and 3+ is unlikely. However, we suspect that higher charge states (i.e., z = 4+ and above, Figure 1B) may be affected and suggest that the shTMTpro reagent could alleviate any possible interference. We recognize that 4+ peptides often make up just 4 to 5% of the total peptide-spectrum matches (PSMs) in a complex mixture; however, the utility of shTMTpro is for targeting a peptide of interest that may be rarely observed at a lower charge state (i.e., z = 2+ and 3+), where using TMTproZero would result in interference.
To showcase the benefits of shTMTpro over TMTproZero, we used an AQUA peptide7 with the sequence NDWTVQNCDLDQQSIVHIVQRPWR (representing residues 52–75 of the Parkin protein). We tested for potential interference when spiking this 4+ charge-state TMTproZero-labeled trigger peptide into a ratio-defined TMTpro16-labeled multiplexed sample. We labeled the Parkin peptide with TMTpro16 reagents and mixed the 16 differentially tagged peptides at a 1:1 ratio (details in the Supplemental Methods). We also labeled the peptide with TMTproZero or Super Heavy TMTpro tags following standard protocols.5 We then spiked five times the amount of the TMTproZero-labeled peptide into the TMTpro16 multiplexed sample. The labeled peptides (total of 50 pmol on-column) were analyzed across a 30 min gradient on an EASY-nLC 1200 liquid chromatograph that was coupled to an Orbitrap Eclipse mass spectrometer. We used higher-energy collisional dissociation (HCD) for fragmentation and high-resolution MS2 analysis (hrMS2). Similarly, we spiked five times the amount of shTMTpro-labeled peptide into the TMTpro16 multiplexed sample and analyzed this sample using the same method (Figure 1C). As expected, we noted that the retention time was similar (~13.5 min) for the peptides labeled with TMTproZero and TMTpro16 reagents (Figure 1D, left) as well as for shTMTpro and TMTpro16 reagents (Figure 1D, right).
When the TMTproZero-labeled peptide was spiked into the TMTpro16-labeled sample, we observed an area of isotopic interference where the M+5 and greater isotope peaks of the trigger peptide were coisolated and cofragmented with the target precursor of interest (Figure 1E, top). As previously noted, the reporter ion masses from TMTpro16 reagents range between 126.1277 and 134.1482 Da. TMTproZero produces a reporter ion of 126.1277 Da that overlaps perfectly with the first channel of TMTpro16 (Figure 1E, bottom left). As a prototypical example of interference, the area under the peak of the 126 reporter ion was approximately five times greater than the expected 1:1 ratio across the remaining TMTpro16 reporter ions (Figure 1E, bottom right), as determined using Skyline.8 Moreover, the naturally occurring 13C isotope of the TMTproZero’s 126 reporter ion also influenced the 127c reporter ion of TMTpro16, distorting this ratio to approximately twice of what was expected. These data using the TMTproZero tag showed that overlapping isotopic envelopes can compromise accurate peptide quantification, in particular when applying isobaric labeling techniques. In contrast, no area of isotopic interference is observed when the Super Heavy TMTpro tag is used to label peptides spiked into the TMTpro16 sample (Figure 1F, top). It is also noteworthy that the fragment ions of peptides labeled with the shTMTpro reagent have a reporter ion mass of 135.1516 Da, which is beyond the mass of the TMTpro16 reporter ion set (Figure 1F, bottom left). As such, the TMTpro16 reporter ions retained the expected 1:1 ratio across all 16 channels when the shTMTpro-labeled peptide was used (Figure 1F, bottom right).
We also verified the effect of interference for both of the TMTpro variants for a second peptide, which is a phosphorylated version of the Parkin peptide on Serine 65, a key residue for its enzymatic activation (again representing residues 52–75), NDWTVQNCDLDQQpSIVHIVQRPWR.12 The labeled peptides were analyzed over a 30 min gradient. Agreeing with previous data, we noted that retention time was similar (~14.7 min) for the peptides labeled with TMTproZero and TMTpro16 reagents (Figure S1A, left) as well as for Super Heavy TMTpro and TMTpro16 reagents (Figure S1A, right). As expected, we observed an area of isotopic interference when we spiked five times the amount of the TMTproZero-labeled pS65-Parkin into the TMTpro16-labeled pS65-Parkin sample (Figure S1B, top). Like the unphosphorylated version of the peptide, the intensity and peak area of the 126 and 127c reporter ions from TMTpro16 reagent set also increased, respectively, due to the coisolation and cofragmentation of the 126-reporter ion and its associated stable-isotope (M + 1) peak from the TMTproZero-labeled peptide (Figure S1B, bottom). It follows that this interference was not observed when we spiked five times the shTMTpro-labeled pS65-Parkin peptide into another aliquot of the TMTpro16-labeled pS65-Parkin sample (Figure S1C), as was true for the unphosphorylated peptide. We noted that decreasing the width of the MS2 isolation window does decrease (but does not eliminate) interference due to the TMTproZero reagent (Figure S1D and E).
We have shown that the Super Heavy TMTpro reagent can alleviate the effect of interference for high-charge-state peptides when used in lieu of the TMTproZero tag for multiplexed targeted assays. As such, the Super Heavy TMTpro reagent has evident advantages over the TMTproZero reagent in applications such as TOMAHAQ1 and TOMAHTO2 that use relatively high amounts of trigger peptides to quantify low amounts of target peptides. To generalize the advantages of using shTMTpro over TMTproZero for peptides other than the two highlighted here, we labeled yeast (S. cerevisiae) tryptic peptides at a 1:1 ratio across all channels using TMTpro16. We then labeled yeast peptides with shTMTpro and TMTproZero, each of which we mixed with TMTpro16 at a 1:1 ratio (Figure S2A). The TMTpro16 sample with TMTproZero had a predominant 126 peak (Figure S2B), whereas the TMTpro16 sample with shTMTpro approximated a 1:1 ratio across all channels (Figure S2C). We expected the distribution of the coefficients of variation (CV) of the first four channels (replicate strains) to approach unity. We plotted the kernel density distribution of the CVs for the first four channels for all charge states (Figure S2D) and for individual charge states z = 4+ (Figure S2E), z = 5+ (Figure S2F), and z = 6+ (Figure S2G). For all charge states, the distribution of CVs was substantially broader and shifted to the right for the TMTproZero compared with shTMTpro. These data further support that using the TMTproZero reagent can distort ratios due to the 126-reporter ion that is shared with the TMTpro16 isobaric set and that the shTMTpro reagent is compatible with more complex samples, including charge states >4+. However, if the TMTproZero reagent must be used, then we recommend narrowing the isolation window or using an m/z window offset, which may concomitantly decrease the signal and thereby be detrimental to the quantification of lower abundance peptide species. As an alternative application, Super Heavy TMTpro may be used in conjunction with its nonisobaric and chromatographically indistinguishable counterparts, TMTproZero and a TMTpro16 tag, for MS1-based quantification that is akin to SILAC,9 reductive dimethylation,10 mTRAQ,11 or mTMT.3 In summary, the Super Heavy TMTpro tag is a promising and highly versatile chemical-labeling reagent for quantitative mass-spectrometry applications.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant nos. R01GM132129 (J.A.P), GM67945 (S.P.G.), and HG006673-07 and NS083524-11 (J.W.H).
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c01056.
Supplemental Methods: sample preparation, liquid chromatography and tandem mass spectrometry, and data analysis. Figure S1: Showcasing interference of coeluting, nonisobaric TMTpro variants using a phosphopeptide. Figure S2: TMTpro variant-labeled proteomes in a TMTpro16 background (PDF)
Contributor Information
Alban Ordureau, Department of Cell Biology, Blavatnik Institute of Harvard Medical School, Boston, Massachusetts 02115, United States.
Qing Yu, Department of Cell Biology, Blavatnik Institute of Harvard Medical School, Boston, Massachusetts 02115, United States.
Ryan D. Bomgarden, Thermo Fisher Scientific, Rockford, Illinois 61105, United States
John C. Rogers, Thermo Fisher Scientific, Rockford, Illinois 61105, United States
J. Wade Harper, Department of Cell Biology, Blavatnik Institute of Harvard Medical School, Boston, Massachusetts 02115, United States.
Steven P. Gygi, Department of Cell Biology, Blavatnik Institute of Harvard Medical School, Boston, Massachusetts 02115, United States.
Joao A. Paulo, Department of Cell Biology, Blavatnik Institute of Harvard Medical School, Boston, Massachusetts 02115, United States.
REFERENCES
- (1).Erickson BK; Rose CM; Braun CR; Erickson AR; Knott J; McAlister GC; Wuhr M; Paulo JA; Everley RA; Gygi SP A Strategy to Combine Sample Multiplexing with Targeted Proteomics Assays for High-Throughput Protein Signature Characterization. Mol. Cell 2017, 65 (2), 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Yu Q; Xiao H; Jedrychowski MP; Schweppe DK; Navarrete-Perea J; Knott J; Rogers J; Chouchani ET; Gygi SP Sample multiplexing for targeted pathway proteomics in aging mice. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (18), 9723–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Paulo JA; Gygi SP mTMT: An Alternative, Nonisobaric, Tandem Mass Tag Allowing for Precursor-Based Quantification. Anal. Chem 2019, 91 (19), 12167–12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Paulo JA; Navarrete-Perea J; Erickson AR; Knott J; Gygi SP An Internal Standard for Assessing Phosphopeptide Recovery from Metal Ion/Oxide Enrichment Strategies. J. Am. Soc. Mass Spectrom 2018, 29 (7), 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Thompson A; Schafer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem 2003, 75 (8), 1895–1904. [DOI] [PubMed] [Google Scholar]
- (6).Li J; Van Vranken JG; Pontano Vaites L; Schweppe DK; Huttlin EL; Etienne C; Nandhikonda P; Viner R; Robitaille AM; Thompson AH; Kuhn K; Pike I; Bomgarden RD; Rogers JC; Gygi SP; Paulo JA TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 2020, 17 (4), 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gerber SA; Rush J; Stemman O; Kirschner MW; Gygi SP Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (12), 6940–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26 (7), 966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lundberg E; Fagerberg L; Klevebring D; Matic I; Geiger T; Cox J; Algenas C; Lundeberg J; Mann M; Uhlen M Defining the transcriptome and proteome in three functionally different human cell lines. Mol. Syst. Biol 2010, 6, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Boersema PJ; Raijmakers R; Lemeer S; Mohammed S; Heck AJ Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc 2009, 4 (4), 484–94. [DOI] [PubMed] [Google Scholar]
- (11).Oppermann FS; Klammer M; Bobe C; Cox J; Schaab C; Tebbe A; Daub H Comparison of SILAC and mTRAQ quantification for phosphoproteomics on a quadrupole orbitrap mass spectrometer. J. Proteome Res 2013, 12 (9), 4089–100. [DOI] [PubMed] [Google Scholar]
- (12).Ordureau A; Sarraf SA; Duda DM; Heo JM; Jedrychowski MP; Sviderskiy VO; Olszewski JL; Koerber JT; Xie T; Beausoleil SA; Wells JA; Gygi SP; Schulman BA; Harper JW Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56 (3), 360–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.