

Introduction
This white paper provides updated ITC recommendations on transporters that are important in drug development following the 3rd ITC workshop. New additions include prospective evaluation of OCT1 and retrospective evaluation of OATP2B1 because of their important roles in drug absorption, disposition and effects. For the first time, the ITC underscores importance of transporters involved in drug-induced vitamin deficiency (THTR2) and those involved in the disposition of biomarkers of organ function (OAT2 and bile acid transporters).
The third International Transporter Consortium (ITC) transporter workshop was held with the purpose of updating recommendations on transporters in drug development. A key goal was to discuss transporters of emerging clinical relevance, and reach consensus on whether the clinical evidence supports incorporation of these transporters into the practice of drug development. Authors of this manuscript formed a working group to reach consensus on emerging drug transporters of clinical relevance. Previously, ITC recommendations were largely focused on transporters important in mediating drug-drug interactions (DDIs) or associated with liver toxicity. In this white paper, ITC recommendations extend beyond transporters important in DDIs to include transporters that are potentially important in mediating drug-induced vitamin deficiencies and those that are involved in the disposition of biomarkers associated with drug-induced organ toxicities. Updated ITC recommendations on transporters in drug development are summarized in this revised ITC transporter figure (Figure 1). Below, new recommendations are summarized under each transporter along with the evidence supporting those recommendations.
Figure 1.
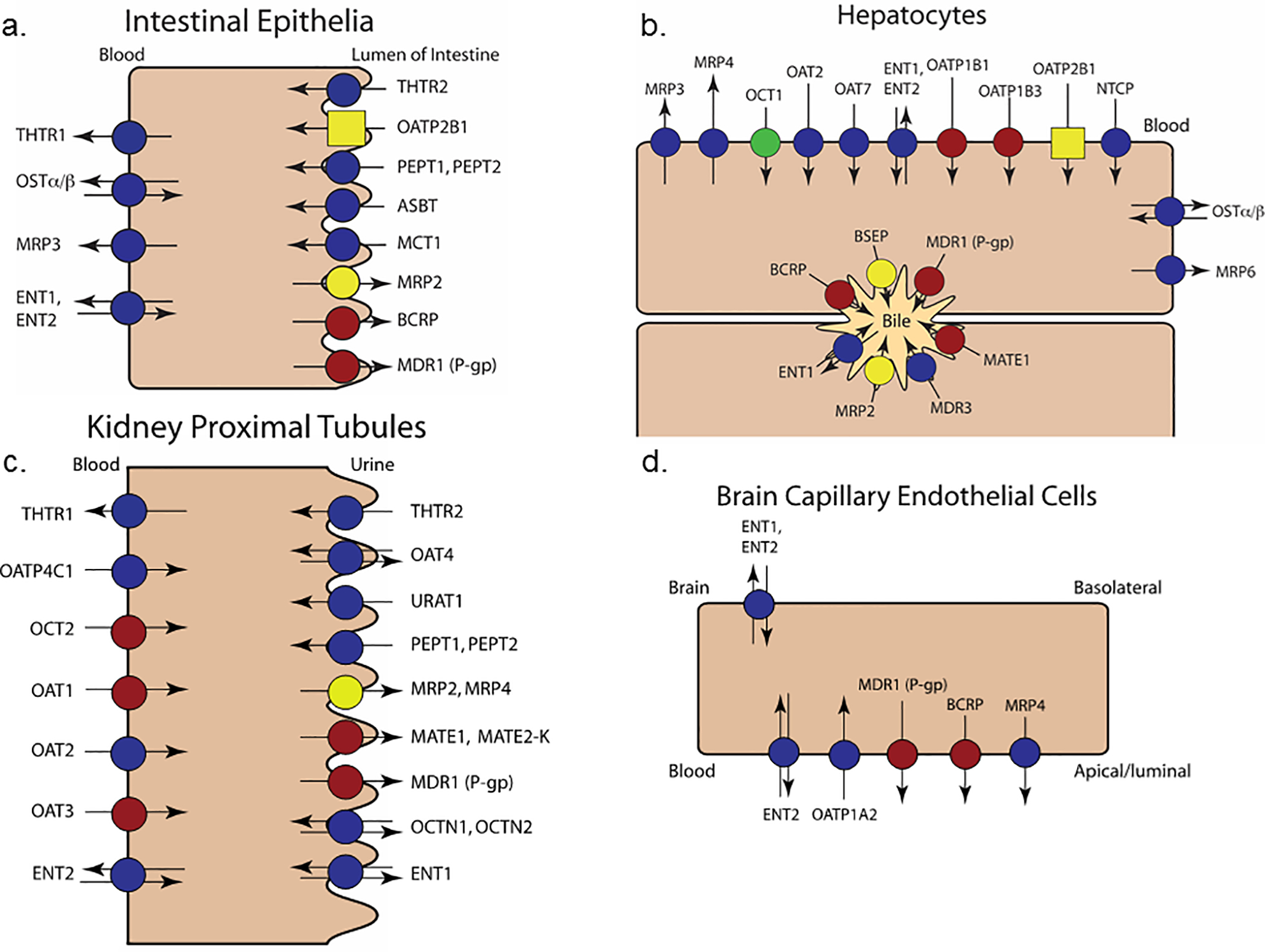
Transporters proposed for evaluation during drug development in the intestine (a), liver (b), kidney (c), and brain (d). The transporters previously recommended for evaluation by the ITC are marked with red circles; transporters previously recommended for retrospective mechanistic explanation of clinical observations are marked with yellow circles. New transporters were highlighted on the basis of broad clinical relevance to drug interactions. OCT1 (green circle) is proposed for evaluation during drug development both for transport of substrates and inhibitory potential. Retrospective study of OATP2B1 (yellow square) is warranted in limited specific instances of DDIs or disposition otherwise unexplained by more common mechanisms. THTRs are new transporters added to the figure; however, drug inhibition of this transporter is likely relevant in the context of pre-existing thiamine deficiency and, therefore, should only be considered for drugs used chronically in sensitive populations or when evidence of drug-induced thiamine deficiency is observed. Symbols for the other discussed transporters (OATP1A2, OATP4C1, OAT2, ASBT [a.k.a. IBAT], OSTα/β, NTCP, MDR3) remain as dark blue circles, because evidence is lacking for specific drug development recommendations.
I. New Recommendations for Transporters as Mediators of Clinical DDIs
In this section, evidence is presented for several transporters, as mediators of clinical DDIs in the liver and intestine. In particular, recent clinical studies support the roles of the hepatic transporter, OCT1, in mediating clinically important DDIs. The possible involvement of the organic anion transporting polypeptide transporter, OATP2B1, in mediating both intestinal and hepatic DDIs, needs to be considered to understand observed DDIs that cannot be attributed to more common mechanisms (e.g., intestinal P-gp/BCRP, hepatic OATP1B1/1B3). Brief comments on other transporters, for which clinical evidence is sparse, are included.
Organic Cation Transporter 1 (OCT1)
Human OCT1 (SLC22A1) is mainly expressed in the liver, localized on the sinusoidal side of hepatocytes. Hepatic OCT1 was discussed in the 2010 ITC white paper (1), and evaluation of inhibitors of this transporter was first recommended in European Medicines Agency’s 2010 draft DDI guideline and remains in their final DDI guideline for understanding potential alterations in metformin distribution to the liver, the primary site of metformin pharmacology. Practical implementation of this recommendation has been challenging, because systemic exposure is the major endpoint of DDI studies in drug development. Notably, OCT1 is not a determinant of metformin plasma levels, because this drug is eliminated almost exclusively by the kidney (2). Metformin clinical DDI studies, to date, have been primarily triggered by in vitro studies of investigational drugs’ interactions with the renal transporters, OCT2, MATE1, and MATE2-K, and have focused on changes in systemic metformin exposure, an endpoint that could be inadequate to inform rational metformin dose adjustment in DDIs (3–6). For example, selective impairment of OCT1 can reduce hepatic metformin exposure and subsequently its anti-hyperglycemic effects, with no apparent changes in systemic pharmacokinetics (6, 7). Furthermore, nonspecific inhibition of OCTs can reduce the renal clearance of metformin through OCT2 inhibition, while simultaneously reducing the hepatic uptake of the drug (through OCT1 inhibition); thus resulting in little net change in hepatic drug exposure and drug response despite increased systemic exposure as demonstrated directly in Oct1/2-knockout mice (4). Many OCT/MATE inhibitors are nonspecific for renal and hepatic metformin transporters, and may alter metformin hepatic distribution and pharmacodynamics, in addition to renal clearance and systemic pharmacokinetics (8; also see UCSF-FDA TransPortal http://transportal.compbio.ucsf.edu/). As such, metformin DDI studies may require pharmacodynamic endpoints (e.g. oral glucose tolerance test) to understand the true effects of concomitant drugs on metformin pharmacodynamics (3, 4, 6; metformin clinical DDI study design commentary in this issue). OCT1 inhibitory potential is critical, in addition to inhibitory potential to OCT2/MATE, to understand whether the pharmacologic effect of metformin may be altered in the presence of a DDI.
OCT1 inhibition is important to consider for metformin dose adjustments in DDI studies that are conducted for drugs that modulate metformin renal elimination. However, for reasons outlined above, metformin is not a sensitive pharmacokinetic probe for OCT1 inhibition. As detailed below, recent clinical studies identified several OCT1-substrate drugs whose systemic exposure is sensitive to OCT1 modulation and may be considered as OCT1 systemic probe substrates. Collectively these studies demonstrated that OCT1 can act as the rate-determining step in hepatic drug clearance of these drugs in a manner conceptually analogous to hepatic OATPs.
Recent clinical evidence from pharmacogenetic studies supports hepatic OCT1 as a potential target for systemic DDIs with notable pharmacokinetic and pharmacodynamic consequences (see accompanying white paper of clinically-relevant transporter polymorphisms in this issue of CPT). For example, the OCT1-substrate fenoterol, a narrow therapeutic index beta2 agonist, exhibited a 2-fold elevated systemic exposure in homozygous carriers of OCT1 null alleles, which elicited clinically concerning 1.5-fold and 3.4-fold greater increases in heart rate and blood glucose, respectively (9). Similarly, the active O-desmethyl metabolite of tramadol, which is a substrate of OCT1, exhibited a 2-fold greater exposure, resulting in enhanced opioid pharmacodynamic activity in healthy subjects, and in clinical practice, decreased drug self-administration for postoperative pain (10, 11). Further, OCT1 reduced function polymorphisms were associated with greater systemic exposure (2–4-fold) and enhanced efficacy (up to 10-fold reduction in vomiting) of the serotonin receptor (5-HT3) antagonists, ondansetron and tropisetron in over 200 patients (12). In people harboring OCT1 reduced function polymorphisms, the systemic exposure of the antimigraine medication, sumatriptan, was increased 2-fold (13). OCT1 reduced function polymorphisms have been associated with the disposition and pharmacologic effects of the opioid, morphine; however, these effects are inconsistent between studies and may be confounded by oral administration of the prodrug versus parenteral administration of morphine directly (14). DDIs with specific and strong inhibitors may phenocopy PK and possible PD changes of these OCT1-substrate drugs associated with genetic polymorphisms, and as such, studies with genetic polymorphisms provide clinical support for OCT1 in mediating DDIs.
Based on clinical evidence, evaluation of substrate and inhibition potential for OCT1 is recommended during drug development. As a hepatic uptake transporter, existing OATP substrate and inhibitor decision trees can be extended to OCT1 (Figure 2). Note the cut-off values shown in Figure 2 are consistent with OATP decision trees proposed in the 2017 FDA in vitro DDI draft guidance, and these may change based on future data. The conduct of in vitro studies is generally preferred for the clinically-relevant substrate-inhibitor pairs that will be studied in the clinic; therefore, choice of the substrate to use in assays will depend on concomitant medications that are most relevant for the new molecular entity (NME) under study and, if warranted based on decision tree analysis, would be the subject of a subsequent clinical DDI study. For example, for NMEs used in cancer patients, the sponsor may wish to consider interactions with 5-HT3 receptor antagonists that appear to be sensitive substrates based on pharmacogenomics studies discussed above. For drugs predicted to be OCT1 substrates in vivo, as noted, there are no available specific clinical inhibitors of the transporter. In this case, the high prevalence of homozygous carriers of OCT1 null alleles (e.g. up to 9% of Caucasians) enables the use of pharmacogenetic studies as a substitute for a traditional DDI evaluation. Alternatively, a DDI study can be conducted with a nonspecific OCT1 inhibitor with careful consideration of the effects that inhibition of other pathways may have on the victim NME.
Figure 2.
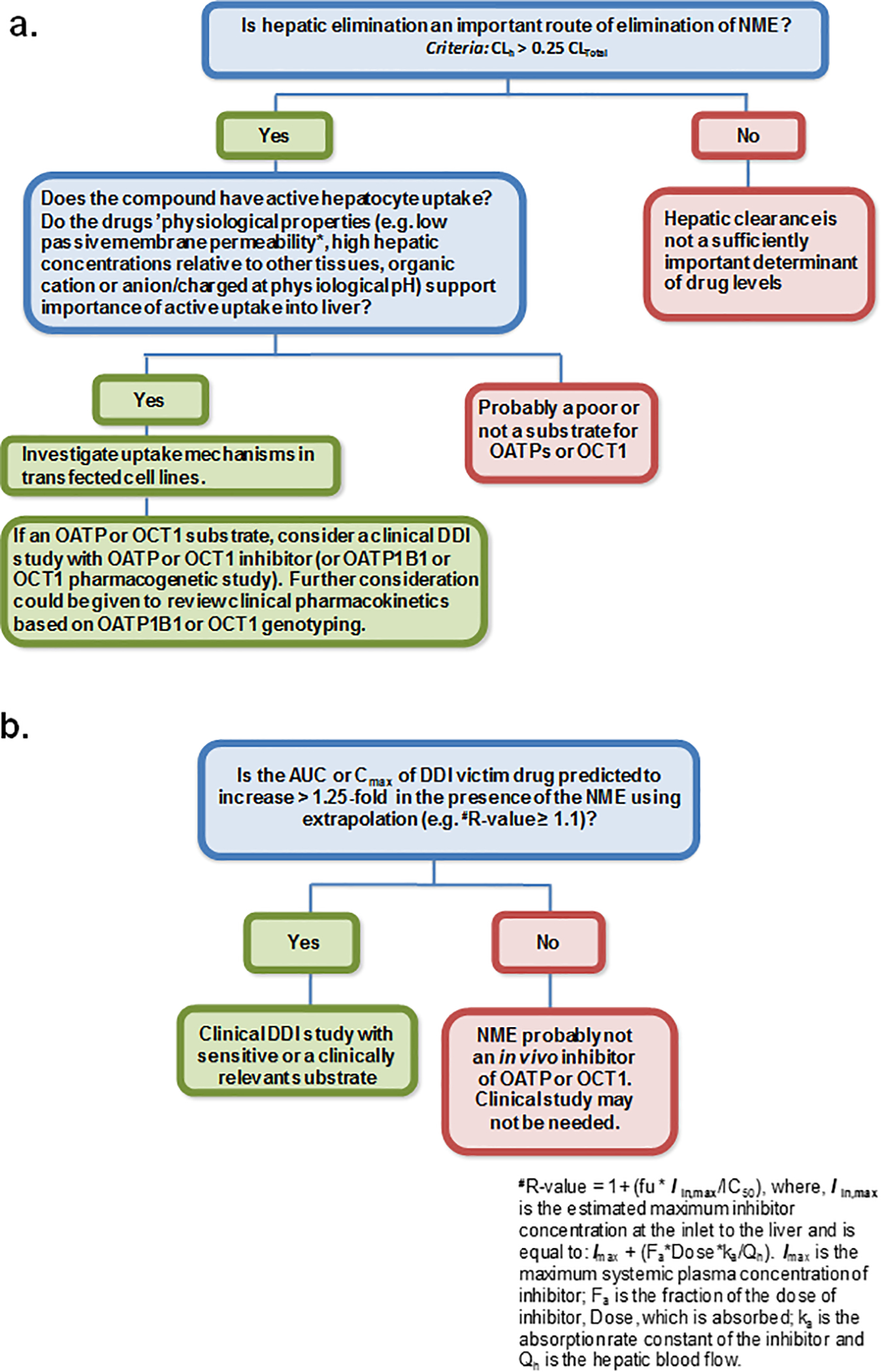
Proposed addition of OCT1 to hepatic OATP substrate (a) and inhibitor (b) decision trees. Note the cut-off values are consistent with OATP decision trees in the 2017 FDA in vitro DDI draft guidance. Cut-off values may change based on future data, so the emphasis is placed not on any specific threshold, but on the concept that OCT1 be considered in a manner analogous to hepatic OATPs.
Organic Anion Transporting Polypeptide 2B1 (OATP2B1)
OATP2B1 (SLCO2B1) exhibits broad tissue expression relative to OATP1B1/1B3 (15). Particularly, OATP2B1 is the main intestinal OATP isoform (16, 17) and is responsible for DDIs previously attributed to OATP1A2 (18). Figure 1 shows OATP2B1 as an intestinal apical uptake mechanism, although apical/basolateral localization in enterocytes remains controversial (19, 20). Additionally, recent proteomic data indicate that OATP2B1 has similar expression as OATP1B3 in the liver (21–23), suggesting that its importance in hepatic uptake may have been underestimated.
OATP2B1 has broad substrate specificity, transporting many clinically used drugs, including atorvastatin and rosuvastatin (15, 24). Detailed analysis of the transport kinetics of OATP2B1 revealed low- and high-affinity binding sites (25). These binding sites show distinct Km and Vmax values for typical probe substrates, such as estrone-3-sulfate, pravastatin, and fexofenadine; likewise, many inhibitors bind preferentially to either the high- or low-affinity binding site (18, 25). OATP2B1 exhibits pH-dependent transport (26), which can be clinically relevant in the intestine. Therefore, in addition to standard in vitro studies at neutral pH, mechanistic evaluation at acidic conditions (e.g. pH 5.5) could potentially improve in vitro to in vivo extrapolation. Several polymorphisms of OATP2B1 have been described (27). SLCO2B1*3 (c.1457C>T) is the most studied variant, but clinical relevance is controversial, even for the same substrate drug [e.g., fexofenadine (15)].
Drugs such as the tyrosine kinase inhibitors gefitinib and erlotinib (28), and natural products, such as naringin and hesperidin in fruit juice (29) are potent inhibitors of OATP2B1. However, to date, known inhibitors are not specific to OATP2B1 and may interact with other transporters and drug metabolizing enzymes.
In vivo evidence in support of intestinal OATP2B1 as a mediator of DDIs comes largely from drug-fruit juice interactions. In particular, the Cmax and AUC of several OATP2B1 substrates (including fexofenadine, aliskiren, and celiprolol) were decreased 1.5- to 2-fold by grapefruit, apple, and orange juice (15, 30–32). Inhibition of intestinal OATP2B1 can decrease systemic exposure of OATP2B1 substrate drugs without increasing the terminal half-life. Recent studies showed that ronacaleret, an inhibitor of OATP2B1, decreased rosuvastain plasma exposure by 50% without altering its terminal half-life (33).
Hepatic OATP2B1 may play an important role in drug clearance. However, many OATP2B1 substrates and inhibitors interact with other hepatic transporters and a clear clinical example of a hepatic OATP2B1 DDI remains elusive (34). For example, asunaprevir, a potent inhibitor of OATP1B1 and OATP2B1, increased the plasma AUC of rosuvastatin 1.9-fold (35). In addition to OATP1B1, the asunaprevir/rosuvastatin DDI appears to require additional consideration of the role of hepatic OATP2B1 for mechanistic explanation. The relative contribution of OATP2B1 to overall hepatic uptake merits assessment when this process cannot be otherwise explained.
In summary, OATP2B1 can play a role in intestinal absorption and hepatic clearance of drugs. Present clinical evidence is insufficient to recommend prospective evaluation of OATP2B1 in drug development. As an emerging transporter, the role of OATP2B1 should be explored based on evidence of in vivo transport in the intestine or liver that cannot be attributed to more common mechanisms (e.g., intestinal P-gp/BCRP, hepatic OATP1B1/1B3). Intestinal OATP2B1 should be considered as a mechanistic explanation of DDIs or drug-fruit juice interactions upon observation of apparently decreased absorption of the substrate drug.
Organic Anion Transporting Polypeptide 4C1 (OATP4C1)
Human OATP4C1 (SLCO4C1) is localized to the basolateral membrane of the renal proximal tubule (36). Substrates of OATP4C1 include digoxin, ouabain, and sitagliptin (37). To-date, the contribution of OATP4C1 to renal elimination of drugs and DDIs has not been well established beyond tubular secretion of digoxin (38). OATP4C1 has been challenging to study due to inconsistent ability to attain functional expression in vitro. A recent OATP4C1 in vitro inhibition screening of 53 drugs only identified ritonavir as a clinically-relevant inhibitor (39). An unexpected DDI between bupropion and digoxin was reported, where bupropion increased renal clearance of digoxin by 80% (40). Activation of OATP4C1-mediated digoxin tubular secretion by bupropion and its metabolites was proposed as the mechanistic explanation based on in vitro studies (41). Overall, the clinical significance of OATP4C1 is not well established, and therefore, no recommendations can be made for drug development at present.
Organic Anion Transporting Polypeptide 1A2 (OATP1A2)
OATP1A2 (SLCO1A2) is expressed in the brain (42). Notably OATP1A2 is not expressed in enterocytes, despite initial reports misattributing intestinal OATP transport to OATP1A2. Although not present in hepatocytes, cholangiocytes lining the bile ducts express OATP1A2 (43). OATP1A2 transports opioid agonists and hydrophilic antimigraine tryptans (42, 44). There is no direct clinical evidence to link OATP1A2 to enhanced drug entry into the central nervous system (CNS), but knockout mice lacking the murine ortholog suggest this to be the case (45). To date, there is no clear evidence for clinical DDIs due to brain OATP1A2 inhibition, and successful targeting of OATP1A2 for CNS drug delivery has yet to be demonstrated. As such, no recommendations can be made for drug development.
II. New Recommendations for Transporters as Mediators of Biomarker Disposition and Drug-Vitamin Interactions
In this section, evidence is presented for the organic anion transporter, OAT2, as a mediator of the disposition of creatinine, and for the thiamine transporter, THTR2, as a mediator of drug-induced vitamin deficiency in certain populations. Finally, an update is presented on key bile acid transporters.
Organic Anion Transporter 2 (OAT2)
Considerably less attention has been given to the clinical significance of OAT2 (SLC22A7) relative to its SLC22A counterparts, OAT1/3 (SLC22A6/8) and OCT2 (SLC22A2). Nevertheless, OAT2 has a broad substrate specificity and is highly expressed in the liver and kidney making it a likely candidate to be a mediator of clinically relevant DDIs. Particularly intriguing is that OAT2 is the only SLC22A member highly expressed in the liver with a bias towards anionic substrates. While suffering from some contradictory findings (46), the main reason OAT2 has not been routinely studied during the drug development is likely due to the lack of an established role as the molecular mechanism for clinically significant effects. Although creatinine tubular secretion has been accepted to be mediated by OCT2 and MATE1/2-K (47), recent studies suggest that OAT2 may be an important contributor to creatinine uptake into the renal proximal tubule (48). OAT2 therefore has the potential to play an important role in the determination of creatinine clearance, a commonly used clinical biomarker of renal function.
Substrates of OAT2 include structurally diverse physiologic metabolites, signaling molecules, and drugs. Notably, OAT2 is the most efficient SLC transporter identified to date for acyclic guanosine nucleoside analogs (acyclovir, ganciclovir and penciclovir) (46, 49–52), cyclic GMP (46, 48, 49, 53), creatinine (46, 48), and diclofenac (52). OAT2 may play a role in the hepatic clearance of tolbutamide (54).
Recently, in vitro studies have identified OAT2 as the most efficient transporter of creatinine, approximately 40-fold more efficient than OCT2 (46, 48). Further, the protein expression of OAT2 in the kidney is within 10-fold of OCT2 expression (55, 56). Taken together, these data suggest that OAT2 may contribute to the active tubular secretion of creatinine. Indeed, OAT2 demonstrated predictive utility for clinically changes in serum creatinine with the experimental Janus Kinase 1 inhibitor, itacitinib (57). Inhibition of OAT2 has also been suggested to play a role in the serum creatinine elevations observed with indomethacin (46). However, a study of creatinine excursion mechanisms for 15 drugs suggested that clinical OAT2 inhibition may be uncommon (58).
Because there is no selective in vivo inhibitor of OAT2, it has been difficult to truly establish the in vivo role of OAT2 in creatinine disposition. While the broad assessment of OAT2 transport and inhibition for drug candidates cannot be recommended at this time, it is a promising candidate for future mechanistic, pharmacogenomic, and pharmacologic research.
Thiamine Transporter 2 (THTR2)
The thiamine transporters (THTR) 1 (SLC19A2) and 2 (SLC19A3) are responsible for the oral absorption and tissue distribution of thiamine (vitamin B1). They are expressed in many organs and important physiologic barriers including the intestine, blood-brain barrier (BBB) and the kidney proximal tubule (59–61). While THTR2 is localized on the apical side of enterocytes, and kidney proximal tubules, the localization of THTR1 is mainly on the basolateral side (59–61). The tissue distribution and localization makes THTRs potentially important in thiamine intestinal absorption, brain penetration, and renal reabsorption. THTRs have been thought of as highly selective for thiamine until recent studies demonstrated interactions of THTRs with commonly prescribed drugs including metform and trimethoprim and suggested potentially clinically relevant consequences due to their inhibition by the drug candidate fedratinib (62–64).
Fedratinib, a Janus Kinase 2 inhibitor, was terminated in phase 3 due to the observation of neurological adverse events consistent with Wernicke’s encephalopathy (WE) in 8 out of approximately 600 patients (65). The WE reported with fedratinib use may have been due to thiamine deficiency (TD), and were ameliorated in at least one patient upon thiamine supplementation (65). Fedratinib, which shares a common moiety of 4-aminopyrimidine with thiamine, is a potent inhibitor of THTR2 but not THTR1, and may have interfered with the oral absorption of thiamine resulting in TD (62). Subsequent studies have identified several inhibitors of THTRs (63, 64). In particular, the commonly prescribed antibiotic trimethoprim, which has been associated with TD (66), was found to potently inhibit both THTR1 and THTR2 at clinically relevant concentrations (63). In addition to the identification of inhibitors, several drugs have now been identified to be substrates for THTRs. While fedratinib is a substrate of THTR2 only, trimethoprim is transported by both THTR1 and THTR2 (63). Although more limited in scope, some substrate overlap has been observed between THTR2 and other xenobiotic cationic transporters, such as OCT1, OCT2, MATE1 and MATE2-K. For example prototypical OCT/MATE substrates, metformin, 1-methyl-4-phenylpyridinium (MPP+), and famotidine are also transported by THTR2 (64).
Inhibition of THTR2 may have significant impact on thiamine intestinal absorption and renal reabsorption resulting in TD. However, in addition to THTR2 inhibition, many other factors including malnutrition, reduced gastrointestinal absorption due to gastric bypass surgery, prolonged vomiting or diarrhea and chronic alcohol use can cause TD (67). In addition, many reported cases of drug-induced TD, which ultimately caused WE, were due to interruption of the thiamine metabolism pathway. For example, tolazamide, a glucose lowering drug used for the treatment of type 2 diabetes, and isofamide, a chemotherapeutic agent, have been associated with WE likely caused by their inhibition of thiamine conversion to thiamine pyrophosphate (68) (69).
While many drug-induced TD mechanisms are related to thiamine metabolism, either directly or indirectly, cases that involve the inhibition of THTR2 resulting in disruption of thiamine absorption are rather sparse. Although both the [I2]/IC50 and [I1]/IC50 values of fedratinib for THTR2 and trimethoprim for both THTR1 and THTR2 exceed the cutoff for a clinical DDI study (i.e., [I2]/IC50 > 10 and [I1]/IC50 > 1.25, where I2 is the intestinal maximal concentration [dose/250 mL] and I1 is the unbound systemic Cmax), WE has been observed only with fedratinib treatment. Fedratinib was tested in patients suffering from myelofibrosis, a rare cancer of the bone marrow, while trimethoprim is often used for treating bladder and urinary tract infections. Therefore, certain patients who are more vulnerable to vitamin deficiency due to impaired absorption and malnutrition may be more prone to develop WE than other disease populations following treatment with drugs which can affect thiamine absorption and distribution. For example, thiamine deficiency has been reported to be common in the elderly, individuals who abuse alcohol, and in patients who have undergone bariatric surgery (70, 71). Duration of treatment with a THTR2-inhibitor drug also is a likely risk factor for TD. In addition, a recent study reported a high rate of thiamine deficiency in patients with myeloproliferative disorders including an increased incidence of WE (72) perhaps making this population more sensitive to an inhibitor of thiamine transport like fedratinib. Therefore, monitoring thiamine concentrations in those vulnerable patient populations during the treatment with THTR2 inhibitors may help identify patients at increased risk, and therefore, prevent the onset of drug-induced WE.
The incidence of drug-induced vitamin deficiencies may be underappreciated. Factors affecting the observation of effects on nutrients include the lack of specific monitoring and that severe clinical events may only manifest in sensitive populations with underlying vitamin deficiency. THTR2 is important in the absorption and the distribution of thiamine, as well as various drugs and xenobiotics. Overall, assessing drug interactions with THTR2 in vitro may aid drug developers in predicting which drugs should be carefully monitored in susceptible populations for drug-induced thiamine deficiency.
Hepatic Bile Acid Transporters
Several hepatic transporters contribute to the disposition of bile acids, and drug interactions involving some of these transporters may cause cholestasis and liver injury. In addition to bile acids, phosphatidylcholine and cholesterol also are important constituents of bile. Phosphatidylcholine forms mixed micelles with cholesterol and bile acids, protecting bile ducts from toxic bile acids. This section briefly highlights the key hepatobiliary transporters.
Biliary excretion of bile acids is predominantly mediated by BSEP (ABCB11), and a separate white paper dedicated to BSEP inhibition as a mechanism of drug-induced liver injury (DILI) is published in this issue of CPT. Clinically, rare mutations that result in farnesoid X receptor (FXR) deficiency and undetectable BSEP expression can cause rapidly progressive cholestatic liver injury (73). Although BSEP inhibition has been linked to intrahepatic accumulation of bile acids, which can trigger clinical cholestasis and may lead to DILI, it is difficult to prospectively link in vitro inhibition of BSEP with clinical hepatotoxicity. In addition to BSEP, other hepatic uptake and efflux transporters contribute to bile acid hepatobiliary disposition. Sodium taurocholate co-transporting polypeptide (NTCP), a solute carrier protein (SLC10A1) expressed on the basolateral membrane of hepatocytes, is primarily responsible for hepatic uptake of bile acids in humans. Many BSEP inhibitors can also inhibit NTCP-/OATP-mediated hepatic uptake of bile acids (74), which may result in increased systemic concentrations of bile acids. This overlap in bile acid uptake/efflux inhibition may serve as a protective mechanism to prevent potentially toxic accumulation of bile acids in the liver (75).
Multidrug resistance proteins 3 and 4 (MRP3, MRP4) are ATP-dependent efflux pumps localized to the sinusoidal membrane that mediate the transport of substrates from the hepatocyte into blood. MRP3 (ABCC3) and MRP4 (ABCC4) are induced under cholestatic conditions, leading to the hypothesis that these transporters can serve as a compensatory mechanism to alleviate increases in intracellular bile acid concentrations when biliary excretion is impaired (76, 77). Increases in liver bile acids and liver toxicity were observed in bile duct ligated Mrp4-knockout mice, but not Mrp3-knockout mice (78, 79). Direct human translation of data obtained from pre-clinical bile acid disposition studies can be challenging due to species differences in the bile acid pool, the relative contribution of various mechanisms to overall bile acid hepatobiliary disposition, and regulation of those mechanisms (76, 80). Several groups have attempted to assess whether MRP inhibition in addition to BSEP inhibition increases DILI predictability; however, this resulted in conflicting conclusions (81–83). In addition to bile acids, MRP2, MRP3, and MRP4 are important determinants of hepatobiliary disposition of polar drug metabolites (e.g. glucuronide conjugates), which can contribute to overall victim and perpetrator DDI potential (84).
The organic solute transporter alpha/beta (OSTα/β), which consists of two gene products encoded by SLC51A and SLC51B, is a bidirectional transporter expressed on the sinusoidal membrane of human hepatocytes, cholangiocytes and enterocytes (85). Heterodimerization of the two subunits and movement of the complex to the basolateral membrane is required for OSTα/β to transport substrates by facilitated diffusion (86). OSTα/β substrates include bile acids, sulfate conjugates of steroid hormones, and some drugs (85, 87). Hepatic expression of OSTα and OSTβ is significantly increased in obstructive cholestasis (88) and primary biliary cirrhosis (89), a response that is dependent on regulation by FXR. Adaptive upregulation of OSTα/β may protect the liver from hepatotoxic bile acids that accumulate in cholestatic conditions (90). Recent studies revealed that troglitazone sulfate, a potent BSEP inhibitor, also inhibits taurocholate transport by OSTα/β (Malinen in press 2018). OSTα/β is an emerging compensatory basolateral efflux transporter that functions much like a “safety-valve” to protect the liver from toxic concentrations of bile acids. Inhibition of OSTα/β is a potential, but understudied, mechanism that may contribute to bile acid-mediated DILI.
Multidrug resistance-associated protein 2 (MRP2, ABCC2) mediates the biliary excretion of glucuronide and sulfate conjugates of bile acids (e.g., sulfated tauro- or glycolithocholate), endogenous compounds (e.g., bilirubin glucuronides), and xenobiotics. Dubin–Johnson syndrome, genetic deficiency of MRP2, is characterized by hyperbilirubinaemia. Although total serum bile acid concentrations are unaffected in Dubin-Johnson patients (91), hepatocyte and systemic concentrations of individual bile acid species may be altered. Inhibition of MRP2 may result in hepatocellular accumulation of toxic metabolites, which may contribute to DILI (92). Studies (93) suggest that genetic variations in MRP2 may increase susceptibility to herb- and drug-induced liver injury. Alterations in MRP2 expression or function may impact the hepatobiliary disposition of bile acids and hepatotoxic drugs, and could be a contributing factor in DILI.
Biliary secretion of phosphatidylcholine is mediated by multidrug resistance protein 3 (MDR3, ABCB4) (94). Mice lacking Mdr2, the mouse ortholog of MDR3, exhibit impaired biliary secretion of phosphatidylcholine and develop hepatobiliary disease (95). In humans, genetic polymorphisms resulting in MDR3 deficiency lead to cholestatic disorders (96, 97). Inhibitory effects of drugs on MDR3 have not been extensively investigated, but several studies support the potential role of MDR3 inhibition in DILI. Itraconazole, which induced cholestatic liver injury in patients, inhibits MDR3 (98). Screening of 125 drugs demonstrated that MDR3 clinical inhibition potency may help differentiate hepatotoxic drugs and non-hepatotoxic drugs (99). In addition, combined analysis of BSEP and MDR3 inhibition data showed that 9 compounds potently inhibited both MDR3 and BSEP (IC50 < 20 μM), all of which were classified as drugs with the highest DILI incidence (99).
As reviewed in this section, the physiology of bile acid disposition is complex with involvement of multiple hepatic transporters in addition to BSEP (ABCB11). Moreover, bile acids undergo extensive enterohepatic recirculation and are metabolized by enzymes in hepatocytes, intestinal epithelial cells, and by the gut microbiome. The first step in bile acid re-absorption is mediated by intestinal apical sodium-dependent bile transporter (ASBT, a.k.a. IBAT), which is not specifically discussed, as it is not a mechanism of cholestasis, but whose inhibition may warrant consideration upon observation of impaired bile acid absorption. Hepatic bile acid levels are regulated through the by farnesoid X receptor through FXR-SHP and FXR-FGF19 pathways, which control multiple adaptation mechanisms (e.g., decrease in CYP7A1, the rate-limiting step in bile acid synthesis, induction of sinusoidal efflux transporters, induction of conjugating enzymes and BSEP) (100, 101). Systematic understanding of a drug’s effects on multiple mechanisms involved in bile acid homeostasis, beyond direct BSEP inhibition, should be considered for mechanistic understanding of altered bile acid disposition.
III. Concluding remarks
Clinical evidence has emerged to justify evaluation of hepatic OCT1 in drug development in addition to renal transporters OCT2 and MATEs. The proposed evaluation approach for OCT1 substrates and inhibitors follows principles established for hepatic OATP1B1 and OATP1B3 (Figure 2), although specific cut-off values necessitate further study and refinement. Both intestinal and hepatic drug uptake via OATP2B1 should be evaluated retrospectively in instances of DDIs, drug-food interactions, or disposition otherwise unexplained by more common mechanisms. Evidence to justify the evaluation of brain OATP1A2 and renal OATP4C1 with a high level of concern from a DDI perspective is lacking. THTR2 may be relevant in the context of pre-existing thiamine deficiency, and therefore, should be considered for drugs used in sensitive populations or when evidence of drug-induced thiamine deficiency is observed. The manuscript highlights the emerging importance of OAT2 as a mechanism of creatinine renal secretion, but relevance to drug development is not sufficiently well established to support specific recommendations. Transporters involved in bile acid disposition are not specifically recommended for evaluation with a high level of concern from a DDI perspective, but may be considered in cholestasis/DILI.
Acknowledgement
We acknowledge Drs. Aleksandra Galetin, Shiew-Mei Huang, Vikram Arya, and Xinning Yang for their review of this manuscript prior to submission.
Funding
No funding was received for this work.
Abbreviations:
- (ITC)
International Transporter Consortium
- (DDI)
drug-drug interaction
- (NME)
new molecular entity
- OCT
(organic cation transporter)
- (MATE)
multidrug and toxin extrusion protein
- (OATP)
organic anion transporting polypeptide
- (OAT)
organic anion transporter
- (THTR)
thiamine transporter
- (MRP)
multidrug resistance protein
- (MDR)
multidrug resistance
- (DILI)
drug-induced liver injury
- (NTCP)
sodium taurocholate co-transporting polypeptide
- (BSEP)
bile salt export pump
- (ASBT, a.k.a. IBAT)
apical sodium-dependent bile transporter
- (OSTα/β)
organic solute transporter alpha/beta
- (CYP)
cytochrome P450
- (FXR)
farnesoid-X-receptor
- (ICP)
intrahepatic cholestasis of pregnancy
- (LPAC)
low phospholipid-associated cholelithiasis
Footnotes
Conflict of Interest
The authors declared no competing interests for this work.
Disclaimer
The contents of this manuscript reflect the views of the authors and should not be construed to represent the FDA’s views or policies. No official support or endorsement by the FDA is intended or should be inferred. The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the FDA.
References
- (1).International Transporter C et al. Membrane transporters in drug development. Nat Rev Drug Discov 9, 215–36 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zamek-Gliszczynski MJ, Giacomini KM & Zhang L Emerging Clinical Importance of Hepatic Organic Cation Transporter 1 (OCT1) in Drug Pharmacokinetics, Dynamics, Pharmacogenetic Variability, and Drug Interactions. Clin Pharmacol Ther, (2017). [DOI] [PubMed] [Google Scholar]
- (3).Hibma JE et al. The Effect of Famotidine, a MATE1-Selective Inhibitor, on the Pharmacokinetics and Pharmacodynamics of Metformin. Clin Pharmacokinet 55, 711–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Higgins JW, Bedwell DW & Zamek-Gliszczynski MJ Ablation of both organic cation transporter (OCT)1 and OCT2 alters metformin pharmacokinetics but has no effect on tissue drug exposure and pharmacodynamics. Drug Metab Dispos 40, 1170–7 (2012). [DOI] [PubMed] [Google Scholar]
- (5).Song IH et al. The Effect of Dolutegravir on the Pharmacokinetics of Metformin in Healthy Subjects. J Acquir Immune Defic Syndr 72, 400–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sundelin E et al. Genetic Polymorphisms in Organic Cation Transporter 1 Attenuates Hepatic Metformin Exposure in Humans. Clin Pharmacol Ther 102, 841–8 (2017). [DOI] [PubMed] [Google Scholar]
- (7).Cho SK, Kim CO, Park ES & Chung JY Verapamil decreases the glucose-lowering effect of metformin in healthy volunteers. Br J Clin Pharmacol 78, 1426–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wittwer MB et al. Discovery of potent, selective multidrug and toxin extrusion transporter 1 (MATE1, SLC47A1) inhibitors through prescription drug profiling and computational modeling. J Med Chem 56, 781–95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tzvetkov MV et al. Increased systemic exposure and stronger cardiovascular and metabolic adverse reactions to fenoterol in individuals with heritable OCT1 deficiency. Clin Pharmacol Ther, (2017). [DOI] [PubMed] [Google Scholar]
- (10).Stamer UM, Musshoff F, Stuber F, Brockmoller J, Steffens M & Tzvetkov MV Loss-of-function polymorphisms in the organic cation transporter OCT1 are associated with reduced postoperative tramadol consumption. Pain 157, 2467–75 (2016). [DOI] [PubMed] [Google Scholar]
- (11).Tzvetkov MV, Saadatmand AR, Lotsch J, Tegeder I, Stingl JC & Brockmoller J Genetically polymorphic OCT1: another piece in the puzzle of the variable pharmacokinetics and pharmacodynamics of the opioidergic drug tramadol. Clin Pharmacol Ther 90, 143–50 (2011). [DOI] [PubMed] [Google Scholar]
- (12).Tzvetkov MV, Saadatmand AR, Bokelmann K, Meineke I, Kaiser R & Brockmoller J Effects of OCT1 polymorphisms on the cellular uptake, plasma concentrations and efficacy of the 5-HT(3) antagonists tropisetron and ondansetron. Pharmacogenomics J 12, 22–9 (2012). [DOI] [PubMed] [Google Scholar]
- (13).Matthaei J et al. OCT1 mediates hepatic uptake of sumatriptan and loss-of-function OCT1 polymorphisms affect sumatriptan pharmacokinetics. Clin Pharmacol Ther 99, 633–41 (2016). [DOI] [PubMed] [Google Scholar]
- (14).Tzvetkov MV OCT1 pharmacogenetics in pain management: is a clinical application within reach? Pharmacogenomics, (2017). [DOI] [PubMed] [Google Scholar]
- (15).Yu J, Zhou Z, Tay-Sontheimer J, Levy RH & Ragueneau-Majlessi I Intestinal Drug Interactions Mediated by OATPs: A Systematic Review of Preclinical and Clinical Findings. J Pharm Sci 106, 2312–25 (2017). [DOI] [PubMed] [Google Scholar]
- (16).Drozdzik M et al. Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine. Mol Pharm 11, 3547–55 (2014). [DOI] [PubMed] [Google Scholar]
- (17).Groer C et al. LC-MS/MS-based quantification of clinically relevant intestinal uptake and efflux transporter proteins. J Pharm Biomed Anal 85, 253–61 (2013). [DOI] [PubMed] [Google Scholar]
- (18).Shirasaka Y, Mori T, Murata Y, Nakanishi T & Tamai I Substrate- and dose-dependent drug interactions with grapefruit juice caused by multiple binding sites on OATP2B1. Pharm Res 31, 2035–43 (2014). [DOI] [PubMed] [Google Scholar]
- (19).Keiser M et al. The Organic Anion-Transporting Peptide 2B1 Is Localized in the Basolateral Membrane of the Human Jejunum and Caco-2 Monolayers. J Pharm Sci 106, 2657–63 (2017). [DOI] [PubMed] [Google Scholar]
- (20).Muller J, Keiser M, Drozdzik M & Oswald S Expression, regulation and function of intestinal drug transporters: an update. Biol Chem 398, 175–92 (2017). [DOI] [PubMed] [Google Scholar]
- (21).Prasad B et al. Ontogeny of Hepatic Drug Transporters as Quantified by LC-MS/MS Proteomics. Clin Pharmacol Ther 100, 362–70 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Vildhede A, Wisniewski JR, Noren A, Karlgren M & Artursson P Comparative Proteomic Analysis of Human Liver Tissue and Isolated Hepatocytes with a Focus on Proteins Determining Drug Exposure. J Proteome Res 14, 3305–14 (2015). [DOI] [PubMed] [Google Scholar]
- (23).Badee J, Achour B, Rostami-Hodjegan A & Galetin A Meta-analysis of expression of hepatic organic anion-transporting polypeptide (OATP) transporters in cellular systems relative to human liver tissue. Drug Metab Dispos 43, 424–32 (2015). [DOI] [PubMed] [Google Scholar]
- (24).Vildhede A et al. Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug-drug interactions. Drug Metab Dispos 42, 1210–8 (2014). [DOI] [PubMed] [Google Scholar]
- (25).Shirasaka Y, Mori T, Shichiri M, Nakanishi T & Tamai I Functional pleiotropy of organic anion transporting polypeptide OATP2B1 due to multiple binding sites. Drug Metab Pharmacokinet 27, 360–4 (2012). [DOI] [PubMed] [Google Scholar]
- (26).Kobayashi D, Nozawa T, Imai K, Nezu J, Tsuji A & Tamai I Involvement of human organic anion transporting polypeptide OATP-B (SLC21A9) in pH-dependent transport across intestinal apical membrane. J Pharmacol Exp Ther 306, 703–8 (2003). [DOI] [PubMed] [Google Scholar]
- (27).Nozawa T et al. Genetic polymorphisms of human organic anion transporters OATP-C (SLC21A6) and OATP-B (SLC21A9): allele frequencies in the Japanese population and functional analysis. J Pharmacol Exp Ther 302, 804–13 (2002). [DOI] [PubMed] [Google Scholar]
- (28).Johnston RA, Rawling T, Chan T, Zhou F & Murray M Selective inhibition of human solute carrier transporters by multikinase inhibitors. Drug Metab Dispos 42, 1851–7 (2014). [DOI] [PubMed] [Google Scholar]
- (29).Shirasaka Y, Shichiri M, Mori T, Nakanishi T & Tamai I Major active components in grapefruit, orange, and apple juices responsible for OATP2B1-mediated drug interactions. J Pharm Sci 102, 280–8 (2013). [DOI] [PubMed] [Google Scholar]
- (30).Tapaninen T, Neuvonen PJ & Niemi M Orange and apple juice greatly reduce the plasma concentrations of the OATP2B1 substrate aliskiren. Br J Clin Pharmacol 71, 718–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ieiri I et al. Microdosing clinical study: pharmacokinetic, pharmacogenomic (SLCO2B1), and interaction (grapefruit juice) profiles of celiprolol following the oral microdose and therapeutic dose. J Clin Pharmacol 52, 1078–89 (2012). [DOI] [PubMed] [Google Scholar]
- (32).Imanaga J et al. The effects of the SLCO2B1 c.1457C > T polymorphism and apple juice on the pharmacokinetics of fexofenadine and midazolam in humans. Pharmacogenet Genomics 21, 84–93 (2011). [DOI] [PubMed] [Google Scholar]
- (33).Johnson M, Patel D, Matheny C, Ho M, Chen L & Ellens H Inhibition of Intestinal OATP2B1 by the Calcium Receptor Antagonist Ronacaleret Results in a Significant Drug-Drug Interaction by Causing a 2-Fold Decrease in Exposure of Rosuvastatin. Drug Metab Dispos 45, 27–34 (2017). [DOI] [PubMed] [Google Scholar]
- (34).Kitamura S, Maeda K, Wang Y & Sugiyama Y Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos 36, 2014–23 (2008). [DOI] [PubMed] [Google Scholar]
- (35).Eley T et al. Organic anion transporting polypeptide-mediated transport of, and inhibition by, asunaprevir, an inhibitor of hepatitis C virus NS3 protease. Clin Pharmacol Ther 97, 159–66 (2015). [DOI] [PubMed] [Google Scholar]
- (36).Mikkaichi T et al. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc Natl Acad Sci U S A 101, 3569–74 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chu XY et al. Transport of the dipeptidyl peptidase-4 inhibitor sitagliptin by human organic anion transporter 3, organic anion transporting polypeptide 4C1, and multidrug resistance P-glycoprotein. J Pharmacol Exp Ther 321, 673–83 (2007). [DOI] [PubMed] [Google Scholar]
- (38).Scotcher D, Jones CR, Galetin A & Rostami-Hodjegan A Delineating the Role of Various Factors in Renal Disposition of Digoxin through Application of Physiologically Based Kidney Model to Renal Impairment Populations. J Pharmacol Exp Ther 360, 484–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sato T, Mishima E, Mano N, Abe T & Yamaguchi H Potential Drug Interactions Mediated by Renal Organic Anion Transporter OATP4C1. J Pharmacol Exp Ther 362, 271–7 (2017). [DOI] [PubMed] [Google Scholar]
- (40).Kirby BJ, Collier AC, Kharasch ED, Whittington D, Thummel KE & Unadkat JD Complex drug interactions of the HIV protease inhibitors 3: effect of simultaneous or staggered dosing of digoxin and ritonavir, nelfinavir, rifampin, or bupropion. Drug Metab Dispos 40, 610–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).He J, Yu Y, Prasad B, Chen X & Unadkat JD Mechanism of an unusual, but clinically significant, digoxin-bupropion drug interaction. Biopharm Drug Dispos 35, 253–63 (2014). [DOI] [PubMed] [Google Scholar]
- (42).Gao B, Vavricka SR, Meier PJ & Stieger B Differential cellular expression of organic anion transporting peptides OATP1A2 and OATP2B1 in the human retina and brain: implications for carrier-mediated transport of neuropeptides and neurosteriods in the CNS. Pflugers Arch 467, 1481–93 (2015). [DOI] [PubMed] [Google Scholar]
- (43).Glaser SS & Alpini G Activation of the cholehepatic shunt as a potential therapy for primary sclerosing cholangitis. Hepatology 49, 1795–7 (2009). [DOI] [PubMed] [Google Scholar]
- (44).Cheng Z et al. Hydrophilic anti-migraine triptans are substrates for OATP1A2, a transporter expressed at human blood-brain barrier. Xenobiotica 42, 880–90 (2012). [DOI] [PubMed] [Google Scholar]
- (45).Higgins JW et al. Utility of Oatp1a/1b-knockout and OATP1B1/3-humanized mice in the study of OATP-mediated pharmacokinetics and tissue distribution: case studies with pravastatin, atorvastatin, simvastatin, and carboxydichlorofluorescein. Drug Metab Dispos 42, 182–92 (2014). [DOI] [PubMed] [Google Scholar]
- (46).Shen H, Lai Y & Rodrigues AD Organic Anion Transporter 2: An Enigmatic Human Solute Carrier. Drug Metab Dispos 45, 228–36 (2017). [DOI] [PubMed] [Google Scholar]
- (47).Imamura Y et al. Prediction of fluoroquinolone-induced elevation in serum creatinine levels: a case of drug-endogenous substance interaction involving the inhibition of renal secretion. Clin Pharmacol Ther 89, 81–8 (2011). [DOI] [PubMed] [Google Scholar]
- (48).Lepist EI et al. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int 86, 350–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Cropp CD et al. Organic anion transporter 2 (SLC22A7) is a facilitative transporter of cGMP. Mol Pharmacol 73, 1151–8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Cheng Y, Vapurcuyan A, Shahidullah M, Aleksunes LM & Pelis RM Expression of organic anion transporter 2 in the human kidney and its potential role in the tubular secretion of guanine-containing antiviral drugs. Drug Metab Dispos 40, 617–24 (2012). [DOI] [PubMed] [Google Scholar]
- (51).Dahlin A et al. Gene expression profiling of transporters in the solute carrier and ATP-binding cassette superfamilies in human eye substructures. Mol Pharm 10, 650–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zhang Y et al. Diclofenac and Its Acyl Glucuronide: Determination of In Vivo Exposure in Human Subjects and Characterization as Human Drug Transporter Substrates In Vitro. Drug Metab Dispos 44, 320–8 (2016). [DOI] [PubMed] [Google Scholar]
- (53).Marada VV, Florl S, Kuhne A, Muller J, Burckhardt G & Hagos Y Interaction of human organic anion transporter 2 (OAT2) and sodium taurocholate cotransporting polypeptide (NTCP) with antineoplastic drugs. Pharmacol Res 91, 78–87 (2015). [DOI] [PubMed] [Google Scholar]
- (54).Bi YA et al. Organic Anion Transporter 2 mediates hepatic uptake of tolbutamide, a Cytochrome P450 2C9 probe drug. J Pharmacol Exp Ther, (2018). [DOI] [PubMed] [Google Scholar]
- (55).Prasad B et al. Abundance of Drug Transporters in the Human Kidney Cortex as Quantified by Quantitative Targeted Proteomics. Drug Metab Dispos 44, 1920–4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Nakamura K et al. Large-scale multiplex absolute protein quantification of drug-metabolizing enzymes and transporters in human intestine, liver, and kidney microsomes by SWATH-MS: Comparison with MRM/SRM and HR-MRM/PRM. Proteomics 16, 2106–17 (2016). [DOI] [PubMed] [Google Scholar]
- (57).Zhang Y et al. Impact on creatinine renal clearance by the interplay of multiple renal transporters: a case study with INCB039110. Drug Metab Dispos 43, 485–9 (2015). [DOI] [PubMed] [Google Scholar]
- (58).Mathialagan S, Rodrigues AD & Feng B Evaluation of Renal Transporter Inhibition Using Creatinine as a Substrate In Vitro to Assess the Clinical Risk of Elevated Serum Creatinine. J Pharm Sci 106, 2535–41 (2017). [DOI] [PubMed] [Google Scholar]
- (59).Said HM, Balamurugan K, Subramanian VS & Marchant JS Expression and functional contribution of hTHTR-2 in thiamin absorption in human intestine. Am J Physiol Gastrointest Liver Physiol 286, G491–8 (2004). [DOI] [PubMed] [Google Scholar]
- (60).Boulware MJ, Subramanian VS, Said HM & Marchant JS Polarized expression of members of the solute carrier SLC19A gene family of water-soluble multivitamin transporters: implications for physiological function. Biochem J 376, 43–8 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Larkin JR et al. Glucose-induced down regulation of thiamine transporters in the kidney proximal tubular epithelium produces thiamine insufficiency in diabetes. PLoS One 7, e53175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhang Q et al. The Janus kinase 2 inhibitor fedratinib inhibits thiamine uptake: a putative mechanism for the onset of Wernicke’s encephalopathy. Drug Metab Dispos 42, 1656–62 (2014). [DOI] [PubMed] [Google Scholar]
- (63).Giacomini MM et al. Interaction of 2,4-Diaminopyrimidine-Containing Drugs Including Fedratinib and Trimethoprim with Thiamine Transporters. Drug Metab Dispos 45, 76–85 (2017). [DOI] [PubMed] [Google Scholar]
- (64).Liang X et al. Metformin Is a Substrate and Inhibitor of the Human Thiamine Transporter, THTR-2 (SLC19A3). Mol Pharm 12, 4301–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Pardanani A et al. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J 5, e335 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Thiamine. Monograph. Altern Med Rev 8, 59–62 (2003). [PubMed] [Google Scholar]
- (67).Sechi G & Serra A Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol 6, 442–55 (2007). [DOI] [PubMed] [Google Scholar]
- (68).Kwee IL & Nakada T Wernicke’s encephalopathy induced by tolazamide. N Engl J Med 309, 599–600 (1983). [DOI] [PubMed] [Google Scholar]
- (69).Buesa JM, Garcia-Teijido P, Losa R & Fra J Treatment of ifosfamide encephalopathy with intravenous thiamin. Clin Cancer Res 9, 4636–7 (2003). [PubMed] [Google Scholar]
- (70).O’Keeffe ST, Tormey WP, Glasgow R & Lavan JN Thiamine deficiency in hospitalized elderly patients. Gerontology 40, 18–24 (1994). [DOI] [PubMed] [Google Scholar]
- (71).Wilkinson TJ, Hanger HC, George PM & Sainsbury R Is thiamine deficiency in elderly people related to age or co-morbidity? Age Ageing 29, 111–6 (2000). [DOI] [PubMed] [Google Scholar]
- (72).Wu J, Zhang L, Vaze A, Lin S & Juhaeri J Risk of Wernicke’s encephalopathy and cardiac disorders in patients with myeloproliferative neoplasm. Cancer Epidemiol 39, 242–9 (2015). [DOI] [PubMed] [Google Scholar]
- (73).Gomez-Ospina N et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat Commun 7, 10713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Lepist EI et al. Evaluation of the endothelin receptor antagonists ambrisentan, bosentan, macitentan, and sitaxsentan as hepatobiliary transporter inhibitors and substrates in sandwich-cultured human hepatocytes. PLoS One 9, e87548 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Leslie EM, Watkins PB, Kim RB & Brouwer KL Differential inhibition of rat and human Na+-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1)by bosentan: a mechanism for species differences in hepatotoxicity. J Pharmacol Exp Ther 321, 1170–8 (2007). [DOI] [PubMed] [Google Scholar]
- (76).Keppler D Progress in the Molecular Characterization of Hepatobiliary Transporters. Dig Dis 35, 197–202 (2017). [DOI] [PubMed] [Google Scholar]
- (77).Kock K & Brouwer KL A perspective on efflux transport proteins in the liver. Clin Pharmacol Ther 92, 599–612 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Mennone A et al. Mrp4−/− mice have an impaired cytoprotective response in obstructive cholestasis. Hepatology 43, 1013–21 (2006). [DOI] [PubMed] [Google Scholar]
- (79).Zelcer N et al. Mice lacking Mrp3 (Abcc3) have normal bile salt transport, but altered hepatic transport of endogenous glucuronides. J Hepatol 44, 768–75 (2006). [DOI] [PubMed] [Google Scholar]
- (80).Yang K, Pfeifer ND, Kock K & Brouwer KL Species differences in hepatobiliary disposition of taurocholic acid in human and rat sandwich-cultured hepatocytes: implications for drug-induced liver injury. J Pharmacol Exp Ther 353, 415–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Kock K et al. Risk factors for development of cholestatic drug-induced liver injury: inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3 and 4. Drug Metab Dispos 42, 665–74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Morgan RE et al. A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol Sci 136, 216–41 (2013). [DOI] [PubMed] [Google Scholar]
- (83).Yucha RW et al. In Vitro Drug-Induced Liver Injury Prediction: Criteria Optimization of Efflux Transporter IC50 and Physicochemical Properties. Toxicol Sci 157, 487–99 (2017). [DOI] [PubMed] [Google Scholar]
- (84).Zamek-Gliszczynski MJ, Chu X, Polli JW, Paine MF & Galetin A Understanding the transport properties of metabolites: case studies and considerations for drug development. Drug Metab Dispos 42, 650–64 (2014). [DOI] [PubMed] [Google Scholar]
- (85).Ballatori N et al. OSTalpha-OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 42, 1270–9 (2005). [DOI] [PubMed] [Google Scholar]
- (86).Li N, Cui Z, Fang F, Lee JY & Ballatori N Heterodimerization, trafficking and membrane topology of the two proteins, Ost alpha and Ost beta, that constitute the organic solute and steroid transporter. Biochem J 407, 363–72 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Wang W, Seward DJ, Li L, Boyer JL & Ballatori N Expression cloning of two genes that together mediate organic solute and steroid transport in the liver of a marine vertebrate. Proc Natl Acad Sci U S A 98, 9431–6 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Chai J et al. Hepatic expression of detoxification enzymes is decreased in human obstructive cholestasis due to gallstone biliary obstruction. PLoS One 10, e0120055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Boyer JL et al. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am J Physiol Gastrointest Liver Physiol 290, G1124–30 (2006). [DOI] [PubMed] [Google Scholar]
- (90).Zhang Y, Jackson JP, St Claire RL 3rd, Freeman K, Brouwer KR & Edwards JE Obeticholic acid, a selective farnesoid X receptor agonist, regulates bile acid homeostasis in sandwich-cultured human hepatocytes. Pharmacol Res Perspect 5, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Iyanagi T, Emi Y & Ikushiro S Biochemical and molecular aspects of genetic disorders of bilirubin metabolism. Biochim Biophys Acta 1407, 173–84 (1998). [DOI] [PubMed] [Google Scholar]
- (92).Tang W Drug metabolite profiling and elucidation of drug-induced hepatotoxicity. Expert Opin Drug Metab Toxicol 3, 407–20 (2007). [DOI] [PubMed] [Google Scholar]
- (93).Choi JH et al. MRP2 haplotypes confer differential susceptibility to toxic liver injury. Pharmacogenet Genomics 17, 403–15 (2007). [DOI] [PubMed] [Google Scholar]
- (94).Van der Bliek AM, Baas F, Ten Houte de Lange, T., Kooiman, P.M., Van der Velde-Koerts, T. & Borst, P. The human mdr3 gene encodes a novel P-glycoprotein homologue and gives rise to alternatively spliced mRNAs in liver. EMBO J 6, 3325–31 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Smit JJ et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 75, 451–62 (1993). [DOI] [PubMed] [Google Scholar]
- (96).Davit-Spraul A, Gonzales E, Baussan C & Jacquemin E The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis 30, 134–46 (2010). [DOI] [PubMed] [Google Scholar]
- (97).Erlinger S Low phospholipid-associated cholestasis and cholelithiasis. Clin Res Hepatol Gastroenterol 36 Suppl 1, S36–40 (2012). [DOI] [PubMed] [Google Scholar]
- (98).Mahdi ZM, Synal-Hermanns U, Yoker A, Locher KP & Stieger B Role of Multidrug Resistance Protein 3 in Antifungal-Induced Cholestasis. Mol Pharmacol 90, 23–34 (2016). [DOI] [PubMed] [Google Scholar]
- (99).Aleo MD, Shah F, He K, Bonin PD & Rodrigues AD Evaluating the Role of Multidrug Resistance Protein 3 (MDR3) Inhibition in Predicting Drug-Induced Liver Injury Using 125 Pharmaceuticals. Chem Res Toxicol 30, 1219–29 (2017). [DOI] [PubMed] [Google Scholar]
- (100).Chiang JY Bile acids: regulation of synthesis. J Lipid Res 50, 1955–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Liu J et al. Potency of individual bile acids to regulate bile acid synthesis and transport genes in primary human hepatocyte cultures. Toxicol Sci 141, 538–46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]