

Abstract
Effective elimination of cancer cells by programmed cell death or apoptosis has been a mainstay and goal of clinical cancer therapy for over 3 decades. Clinical translation of effective proapoptotic agents includes drug discovery, bioavailability and stability, tumor penetration, acute or chronic normal tissue toxicity, drug interactions, off-target effects, tumor heterogeneity and evolution with emergence of resistant clones. The pathways of apoptosis include cell intrinsic and extrinsic processes triggered by cellular stress, DNA damage, and immune surveillance mechanisms. While tumor cell death can lead to therapy response, the selection, growth and dissemination of therapy-resistant cells makes tumors lethal, leading to patient mortality. We focus on apoptosis pathways, signaling pathways that impact on them as well as molecular targets, strategies, therapeutics in clinical translation and known resistance mechanisms.
Keywords: intrinsic pathway, BCL-2, BCL-XL, IAP, extrinsic pathway, TNF, TRAIL, death receptor, oncogene, growth factor, MCL1, mTOR, PI3K, EGFR
Introduction
The pathways of cell death are conserved with important insights coming from studying cell fate in lower organisms such as C. elegans work by Robert Horvitz that led to the 2002 Nobel Prize in Physiology or Medicine 1,2.
Early concepts of cancer development revolved around viral and cellular oncogenes 3, cellular proliferation and transformation. By the 1980’s with discovery of DNA fragmentation in lymphoid cells after glucocorticoid exposure 4 and subsequently in tumors undergoing chemotherapy 5, the induction of apoptosis as a goal for cancer therapy became a logical target and realistic goal. Discovery of anti-apoptotic oncogenes such as BCL-2 6 and later BCL-XL 7 provided insights into cell survival or the prevention of cell death 8,9 as a powerful mechanism for tumor growth 10 and drug resistance 11. These eventually would become therapeutic targets, and would lead to knowledge of secondary resistance mechanisms 12.
During the 1990’s and 2000’s there was much learned about the mechanisms leading to cell death from within a cell as well as through the immune system 13–15. In particular, features of apoptosis including the release of cytochrome c from the mitochondria became the focus of investigation for control of cell death 16. The balance of controlling families of proteins such the proapoptotic versus anti-apoptotic members of the BCL-2 family would regulate cytochrome c release from the mitochondria 17,18. Downstream of cytochrome c release would be formation of the apoptosome with caspase activation 19,20 that would be negatively regulated by the inhibitors of apoptosis (IAP) family of proteins 21. An overview of the current understanding of the apoptosis signaling pathways is shown in Figure 1, along with intersection of the major relevant survival pathways that impact on the cellular phenotype, including drug resistance.
Figure 1. Overview of apoptosis signaling pathways and pro-survival, immune, or tumor microenvironment influences that impact on them.
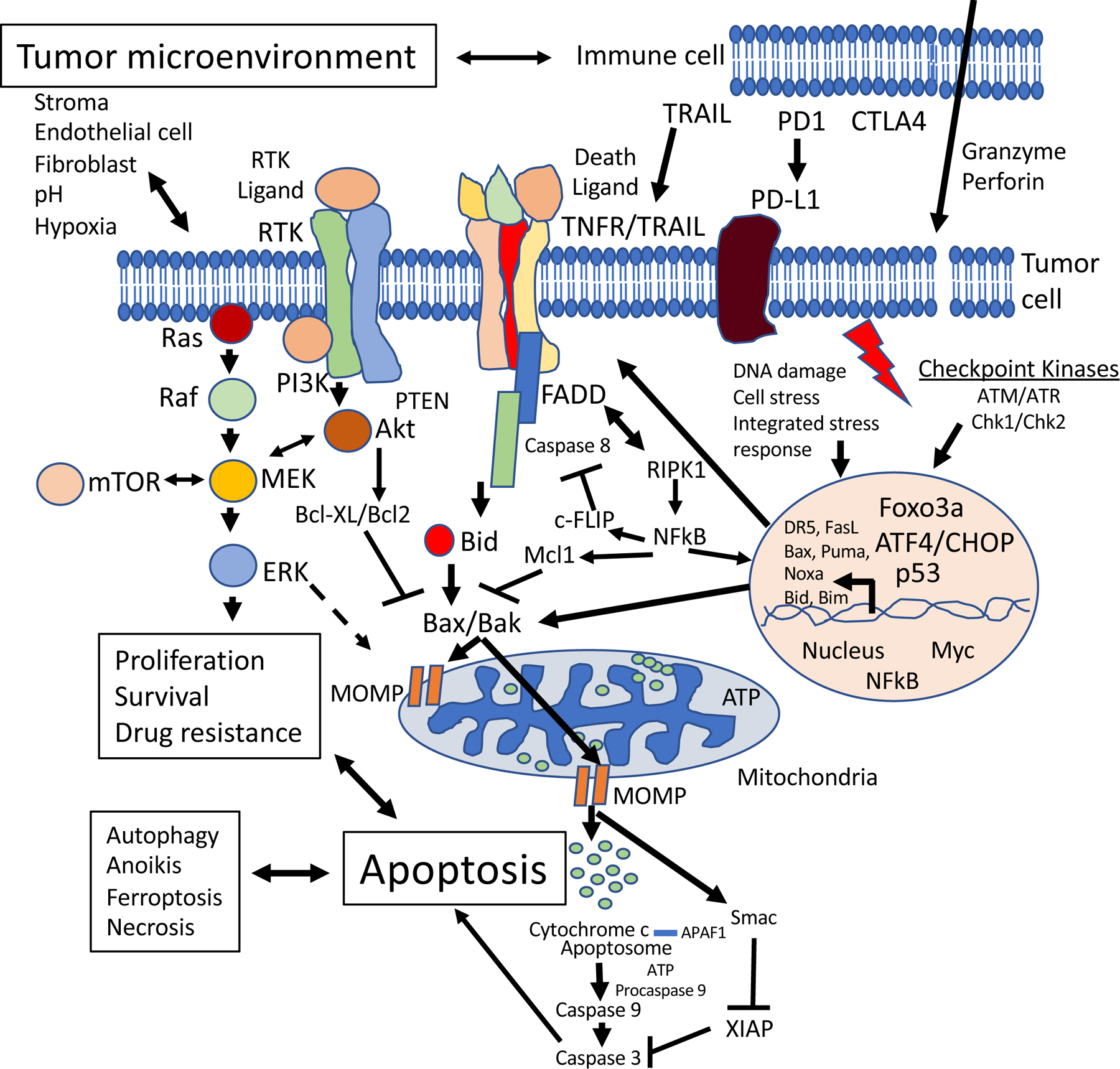
Intrinsic mitochondrial apoptosis involves proapoptotic BCL-2 family members (e.g. BAX or BAK) located in the mitochondrial outer membrane promoting mitochondrial outer membrane permeability (MOMP), cytochrome c release, caspase activation and apoptosis. Cytochrome c binds APAF1, and in an ATP-dependent process, the multimeric apoptosome is formed. Procaspase 9 binds to the apoptosome, gets cleaved and activated, and then activates caspase 3 20. Members of the IAP family including XIAP negatively regulate caspase activation and they can be inactivated by SMAC. Anti-apoptotic members of the BCL-2 family (e.g. BCL-2, BCL-XL, MCL1, Bcl-w, A1) inhibit the proapoptotic BCL-2 family members (e.g. BAX and BAK). BH3-only proteins such as BIM, BAD, BID, PUMA and NOXA can inhibit MCL1. It is important to note that within the intrinsic apoptosis pathway, mitochondrial permeabilization represents the point of commitment to cell death (irrespective of caspase activity). The extrinsic apoptosis pathway is depicted at the cell membrane as a trimeric TNF family receptor (Fas-R, TRAIL-R1, TRAIL-R2, TNF-R1, TNF-R2) that is bound by the respective ligand with proteins FADD and caspase 8 recruited to form the death-inducing signaling complex (DISC). Caspase 8 or 10 activation at the DISC results in Bid cleavage which provides cross-talk to the mitochondria in Type II cells which amplifies the death signal at the level of cytochrome c leading to apoptosis. c-FLIP binds to caspase 8 and prevents its activation. Prototypical growth factor activated receptor tyrosine kinases (RTKs) are shown in the cell membrane as dimeric proteins (e.g. members of the ERBB family such as EGFR, Her2, ERBB3) with downstream events involving PI3K and Akt activation. The RAS-MAPK pathway is also depicted. These pro-survival pathways also promote cell proliferation and negatively impact on the intrinsic cell death pathway, in part, through phosphorylation of BCL-2 family members. TNF and TRAIL receptors can activate NFkB which transcriptionally activates anti-apoptotic genes in the nucleus thereby attenuating cell death and promoting cell survival. Immune mechanisms can activate cell death through the extrinsic pathway, e.g. through TRAIL produced by Natural Killer (NK) cells in response to Interferon action, or through the blockade of cell death ligands, e.g. PD-L1. Immune cells such as cytotoxic T-cells or NK cells also produce granzyme which is a serine protease that induces apoptosis in target cells such as cancer cells. A protein called perforin forms pores in target cells to mediate granzyme-induced cell death. Various other components of the tumor microenvironment including stroma, fibroblasts and physical factors can impact cell death and cell survival pathways in ways that impact on drug responses. Drugs targeting endothelial cells can deprive tumor cells of nutrients leading to cell death. DNA damage response (e.g. those involving checkpoint kinases such as ATM/ATR, Chk1/Chk2) and cell stress response pathways (e.g. the integrated tress response leading to ATF4 and CHOP activation) ultimately activate proapoptotic genes that induce cell death, in addition to p53. Among the transcribed genes following p53 activation, Bax, Puma, Noxa, DR5, and FasL are noteworthy, but it should also be noted that many kinase inhibitors ultimately signal transcriptional activation of Bim. Additional cellular processes (e.g. autophagy, anoikis, ferroptosis, regulated necrosis) can cross-talk with apoptosis signaling pathways and are impacted by cancer therapeutics including those used in combinations with proapoptotic agents.
While progress over the last two decades has been made in targeted therapies for cancer through blockade of various kinases that promote tumor progression, in part through cell proliferation and survival, insights have been emerging regarding the connections between the cell death mechanisms and its contribution to efficacy of cancer targeted therapies 22,23. Ultimately, tumor cell death is impacted by the balance of these pathways as well as the cell extrinsic immune anti-tumor mechanisms.
Molecular markers of apoptosis
Apoptosis is a cellular death catalyzed by the proteolytic cleavage of thousands of proteins through the enzymatic activity of effector caspases such as caspases 3, 6 and 7 24. Apoptotic death can also be activated by granzyme contained within cytotoxic granules in T-cells or Natural Killer (NK) cells and through perforin-mediated pore formation in target cells 25. The apoptotic process involves nuclear membrane breakdown that occurs through lamin cleavage by caspase 6, membrane blebbing, and breakdown of genomic DNA into nucleosomal structures that classically was observed as a DNA ladder on agarose gels 26,27. A frequently used cellular protein marker for caspase 3 activation is the PARP protein with PARP cleavage being observed in cells undergoing apoptosis 28. M30 is a caspase 3 cleavage product of cytokeratin 18 detected in serum resulting from therapy-induced tumor apoptosis in patients 29,30. On tissue sections cleaved caspases are detectable by western blot as is cleavage of DNA through the TUNEL assay 31. Granzyme is detectable in cytolytic T-cells by immunohistochemistry 32. In cultured cells, apoptosis is detectable by flow cytometry though sub-G1 DNA content, externalization of phosphatidylserine, or various cleaved caspases 33. Caspase activation can also be seen by caspase enzymatic activity cell-based assays or even by imaging scans applied to humans 34,35. Granzyme B has been detected by PET scanning in preclinical studies as a marker of immunotherapy response 36. The detection of apoptosis has provided the tools for drug discovery as well as the experimental means for validation of drug action in clinical specimens. A nomenclature for cell death has been proposed to facilitate description of its various forms as well as use of appropriate assays for detection of the process in its various forms 37.
The core apoptotic signaling pathways in mammalian cells
Intrinsic cell death.
A final common pathway for cell-intrinsic apoptotic cell death involves mitochondrial outer membrane permeabilization (MOMP) 38 with the release of cytochrome c from the mitochondria, formation of the apoptosome with APAF1 and initiator caspase 9 followed by activation of executioner caspase 3 (Figure 1) 39. In most mammalian cells, MOMP and release of cytochrome c into the cytoplasm are critical requirements to trigger apoptosis, and it has been considered the point of no return in cellular commitment to cell death that can occur independent of caspase activation 16,40,41. Release of cytochrome c from the mitochondria is positively regulated by the proapoptotic BCL-2 family members such as BAX (BCL-2-associated X protein), BAK (BCL-2 antagonist killer 1), BIM, BID, PUMA and negatively regulated by the anti-apoptotic BCL-2 family members such as BCL-2, BCL-XL, Bcl-w, A1 and MCL1 42–44. BAX and BAK are central effectors of apoptosis as they form macropores in the mitochondria outer membrane causing MOMP . Following activation by proapoptotic proteins, BAX and BAK undergo conformational changes and oligomerization forming a pore in the mitochondrial membrane 45,46. Apoptosis-inducing BCL-2 members containing only one BCL-2 homology (BH) domain (BH3-only) can be grouped as activators and sensitizers 47,48. Activator proteins BIM, BID, and PUMA bind and directly activate BAX and/or BAK 49–57. Sensitizer proteins (i.e., BAD, NOXA, BIK, BMF, HRK) do not activate BAX and/or BAK directly, but provide important regulation by inhibiting anti-apoptotic proteins (i.e., BCL-2, BCL-XL, MCL1) and/or displacing activators such as BIM or monomers of BAX or BAK that could ultimately lead to apoptosis 41,58,59. Anti-apoptotic members have four BH domains forming a binding groove that sequesters proapoptotic activators or sensitizers BH3-only members as well as BAX and BAK 60. BH3-only proteins such as PUMA, NOXA, BID, or BIM have a strong affinity to and inhibit MCL1 47,60. Whereas BAD has a stronger affinity to BCL-2, BCL-W, BCL-XL and a relatively weak affinity to MCL1 47,61. The levels of BCL-2 proteins and their interactions modulate sensitivity and resistance to apoptosis and can be assessed by functional assays such as BH3 cellular profiling, an important tool to predict response to apoptosis targeted agents incorporated in recent clinical trials 62. For instance, cells with high levels of proapoptotic proteins are more susceptible to undergo apoptosis or “primed” as compared to cells with downregulation of BAX or BAK. BH3 profiling also reflects cellular dependence on specific anti-apoptotic proteins 63. In addition to transcriptional regulation, BCL-2 proteins are functionally modulated by cytoplasmic localization and post-translational modifications such as phosphorylation, ubiquitination, and proteasomal degradation 54,64–70. Cyclin-dependent kinases (CDK) and p53 mediate apoptosis by regulating transcription and phosphorylation of BCL-2 proteins 54. For example, pan CDK inhibitor flavopiridol blocks cell cycle progression and induces apoptosis by inhibiting MCL1 transcription and upregulating BIM, NOXA, and Bik/NBK in multiple myeloma cells 71,72. CDK5 regulates MCL1 through phosphorylation of its antagonist NOXA 73. CDK5 inhibition decreased the levels of MCL1 in pancreatic cancer cells and showed synergism with BCL2 inhibitor navitoclax inducing apoptosis 74. Following the release of cytochrome c from the mitochondria, caspase 3 activation can be negatively regulated by a family of Inhibitor of Apoptosis (IAP) proteins such as XIAP, c-IAP1 and c-IAP2 75,76. Peptides released from the mitochondria along with cytochrome c (e.g., SMAC/Diablo) can block XIAP and allow caspase activity to kill the cell 75,77. A number of drugs including small molecules 78–89 or stapled peptides 90–93 have been under various stages of development to block anti-apoptotic proteins such as BCL-2/BCL-XL, MCL1 or the IAP proteins, all of which can be overexpressed in human tumors. This includes SMAC mimetics 94,95. The balance of BCL-2 family of anti-apoptotic proteins versus the proapoptotic family members has long been considered a cellular rheostat that controls the threshold of cell death in mammalian cells 64,96,97. Proapoptotic members of the BCL-2 family such as BAX are classical targets for direct regulation by the p53 tumor suppressor protein 98. As such their levels increase after exposure of wild-type p53-expressing cells to various cytotoxic or DNA damaging agents 99. Increased expression of the proapoptotic BCL-2 family members can contribute to the cytotoxicity towards either tumor cells or normal cells. The molecular details of caspase processing and activation have been described 100.
Extrinsic cell death.
A second common pathway of cell death referred to as extrinsic cell death can initiate apoptosis from cell membrane proteins known as death receptors (DR). Proapoptotic death receptors include Fas, TNFR1, TNFR2, and the TRAIL receptors DR4 and DR5 13,39,101–111. Natural ligands such as FasL 112–116, TNF or TRAIL bind to Fas, the TNFR’s or the TRAIL receptors DR4 and DR5, respectively 117–120. The intracellular domains of the proapoptotic death receptors include a conserved protein-protein interaction domain referred to as the death domain 121,122. Thus, upon ligand binding, the death receptors trimerize and aggregate within the cell membrane (a phenomenon known as capping) and this is followed by recruitment of adaptor proteins such as FADD (Fas or other death receptor associated death domain protein) and initiator caspase proteins such as caspase 8 or caspase 10 found in humans (mice have no caspase 10) 120,123–125. Initiator caspase activation in the extrinsic pathway is negatively regulated by c-FLIP 126, while activation of caspases 8 and 10 can lead to activation of executioner caspases 3, 6, and 7 as well as BID cleavage. BID cleavage and myristoylation leads to its translocation to mitochondria and this can contribute to cytochrome c release 127. BID is regulated by p53 128. BID is the link between the extrinsic cell death pathway and the mitochondrial pathway and in so called Type II cells serves to amplify the cell death signal through the mitochondria leading to more efficient cell death that ultimately requires the mitochondrial events for cell execution. Type I cells efficiently activate downstream caspases such as caspase 3 leading to cell death without the need to amplify the signal through the mitochondria 129,130.
Signaling through death receptors can be attenuated through decoy receptors at the cell surface that bind to death ligands and compete for them, thereby reducing activation of the proapoptotic death receptors. A decoy receptor for Fas ligand is known to be amplified and overexpressed in some colorectal and lung tumor cells 131, and two TRAIL receptor decoys are known (DcR1 or TRID and DcRII or TRUND) to be overexpressed in normal cells thereby eliminating toxicity of TRAIL to normal tissues 132. Low caspase 8 expression occurs in normal cells, thereby also reducing toxicity of death ligands and receptors 133. Caspase 8 can also be actively degraded in tumor cells 134. The death receptors including to a large extent the TNF receptors and to a lesser extent the TRAIL receptors activate NFkB 135. Fas receptors do not activate NFkB, but their therapeutic activation leads to liver necrosis that was blocked by BID deletion in mice 136. The TNF receptors strongly activate NFkB and this limited the therapeutic development of TNF in cancer therapy due to systemic inflammatory responses in patients who were treated such that TNF is only used for regional therapy of bulky tumors such as sarcomas and melanomas combined with chemotherapy in limb perfusion protocols 137. While TNF does not have significant cytotoxic activity against tumor cells in vitro, its therapeutic benefit derives from the direct cytotoxicity on tumor vasculature promoting hemorrhagic necrosis, increased vascular permeability that enhances local accumulation of chemotherapy 138–140. The TRAIL receptors generally have minimal NFkB activation and this has been a key characteristic that favored therapeutics activating TRAIL receptors (TRAIL or TRAIL receptor agonists). However, in TRAIL-resistant tumors, activation of NFkB in the absence of cell death could promote tumor progression and metastasis and foster a tumor-supportive immune microenvironment by inducing secretion of CCL2 and increased accumulation of myeloid-derived suppressor cells (MDSCs) and M2-like macrophages 141,142. Furthermore, TRAIL activation promoted metastasis and invasion of KRAS-driven models of NSCLC and pancreatic adenocarcinoma via PI3K/Rac1, providing a biological rationale for these TRAIL-resistant tumors to overexpress TRAIL receptors 141. These results suggested a therapeutic benefit from blocking TRAIL receptors in KRAS-driven models with overexpression of TRAIL receptor. The p53 tumor suppressor directly transcriptionally regulates death receptors such as Fas or DR5, and thus therapeutics that stabilize p53 can induce cell death through both the extrinsic and intrinsic cell death pathways 143. Mice have only one proapoptotic TRAIL receptor that is regulated by p53, and the knockout mice have reduced apoptosis in vivo following exposure to whole-body irradiation 144,145.
Apoptosis is a stochastic process within a population of cells 146,147. Thus, there is a fractional cell killing within a population that has been attributed to variations in cell death-relevant protein concentrations 148. Recent evidence suggests the stochastic nature of apoptosis may be related to the cellular content of mitochondria and the distribution of BCL-2 family of proteins within the mitochondrial surface 149.
Extrinsic cell death can also occur through a process referred to as necroptosis 150 that involves loss of cell membrane integrity while caspase activity is inhibited. During the process of death receptor activation, RIPK1, cIAP1, and cIAP2 are recruited to the plasma membrane with subsequent activation of NFkB which together with its target c-FLIP inhibits apoptosis and caspase activation while allowing for plasma membrane disruption leading to the death known as necroptosis 150 that is RIPK1-dependent. The interplay between necroptosis and apoptosis, as well as other forms of regulated necrosis such as pyroptosis or ferroptosis are areas of active investigation 150, and ultimately effects of drugs on these interactions will be relevant to cancer therapy.
Survival pathways that impact on cell death.
In addition to the anti-apoptotic members of the BCL-2 family, the IAP proteins, and c-FLIP, a number of cellular pro-survival pathways impact on the cell death phenotype of cells treated with agents that activate the core apoptotic pathways 151–153. The pro-survival pathways are, in many if not all cases, oncogenic pathways and/or are otherwise activated by cellular oncogenes including growth factor receptors 22. While many of the pro-survival pathways have become established drug targets in oncology, it is important to recognize that ultimately the drugs induce apoptosis through the core apoptotic pathways 154–156. Examples include kinase signaling pathways involving AKT, ERK, Ras, Raf, MEK, or mTOR kinases 157–175. Inhibition of growth factor receptors, including EGFR, Her2/Neu, other ERBB family members, c-MET, or NTRK, among others, also leads to apoptosis 176–196.
These pathways to which tumor cells become addicted have cross-talk to the mediators of apoptosis in core cell death pathways to inactivate cell death. Various therapeutic inhibitors of these pathways relieve the blockade on cell death, thereby unleashing the cell death machinery in tumor cells. For instance, the anti-tumor efficacy of EGFR inhibitors is dependent on their ability to modulate the expression of BCL-2 family members and induce apoptosis. Expression of proapoptotic BIM (BCL-2 interacting mediator of cell death) in lung cancer cell lines predicts the apoptotic response to EGFR tyrosine kinase inhibitor (TKI) gefitinib, and downregulation of BIM attenuates the anti-tumor response in vivo 197. ERK1/2 signaling causes phosphorylation of BIM, leading to its proteasomal degradation and thereby promotes cell survival 198. Similarly, AKT and Ras-MAPK activation induce phosphorylation of BAD (BCL-2 agonist of cell death; a proapoptotic BCL-2 family member) and inhibit apoptosis; providing another intersection between cell survival signals and active inhibition of the cell death machinery 199,200. Intrinsic modulation of BCL-2 proteins has also been linked to the development of resistance to MEK and PI3K inhibitors providing a scientific rationale for combinations of BCL-2 family inhibitors and TKIs currently in clinical development 201,202.
Other forms of cells death.
Autophagy (‘self-eating’) is a regulated process that can promote cell survival (and tumor growth) when cells are deprived of nutrients, e.g. in the tumor microenvironment, but however, in its very late stages autophagy can cause cell death 203–206. The blockade of autophagy, for example with chloroquine is a strategy that has moved to the clinic and holds some promise 207. Current thinking is that the complex process of autophagy can involve other cellular functions including vesicle extrusion, cytokinesis, proliferation and autophagy-dependent cell death (ADCD) 208. The ultimate interplay between autophagic signaling and apoptosis pathways, and modulation of that cross-talk by cancer therapeutic agents is an important area of investigation.
Ferroptosis represents another distinct form of cell death triggered by the accumulation of cytosolic and lipid iron-dependent reactive oxygen species (ROS) 209–211. Glutathione peroxidase (GPX4) catalyzes the reduction of lipid peroxides utilizing reduced glutathione (GSH) and functions as a central switch to protect the cell against ferroptosis 212. Various cancer cell types are susceptible to ferroptosis, including melanoma, lymphoma, gastric cancer, and neuroblastoma 213–216. Dedifferentiation of melanoma cancer cells observed during the development of resistance to tyrosine kinase inhibitors occurs in tandem with increasing sensitivity to ferroptosis 213. Mesenchymal cellular features were associated with increased expression of ZEB1 (zinc finger E-box-binding homeobox 1) and changes in lipid metabolism creating a dependency on GPX4, which could be explored as a therapeutic vulnerability in treatment-resistant tumors and triggered to eradicate residual cancer cells and reduce recurrence 217. The GPX4 dependency of this mesenchymal state has sparked interest in strategies to induce ferroptosis for cancer therapeutics using direct inhibitors of GPX4 (i.e withaferin A, ML210, RSL3) 212,214,218,219, modulators of glutathione levels (i.e., buthionine sulfoximine) 212 or inhibitors of cystine-glutamate antiporter (i.e., erastin, sulfasalazine, sorafenib) 209,210,220. Naked nanoparticles or those carrying withaferin and other chemicals can induce ferroptosis 214,221,222. P53 also regulates sensitivity to ferroptosis, which seems to contribute to its tumor suppression function 223–225. P53 promotes ferroptosis in conditions of oxidative stress by downregulating SLC7A11 that reduces cystine cellular uptake 223. In other contexts, restoring wild-type P53 protected cancer cells against ferroptosis 226. Further progress in the understanding of molecular mechanisms of ferroptosis will promote novel therapeutic strategies targeting ferroptosis to control cancer growth 227.
Clinical translation of modulators of apoptosis
Monumental scientific efforts placed at translating the modulation of cell death mechanisms to therapeutic benefit of patients with cancer have led to FDA-approved BCL-2 targeting drugs with meaningful clinical benefits and spurred several strategies in clinical development.
Strategies targeting the intrinsic pathway
The discovery of the BCL-2 gene in patients with follicular lymphoma as a result of the chromosomal translocation t(14;18) ignited the interest in targeting this family of proteins to control cancer growth by promoting apoptosis 6,228. Initial efforts focused on identifying inhibitors of anti-apoptotic members of the family such as BCL-2. These approaches included oligonucleotides targeting BCL-2 expression and small molecules mimicking the binding of BH3-only proapoptotic members (BH3 mimetics) to anti-apoptotic BCL-2 members (Table 1).
Table 1.
Therapeutic approaches targeting the apoptosis intrinsic pathway
| Target | Active clinical trials | Phase | Histology focus | Clinicaltrial.gov ID |
|---|---|---|---|---|
| BH3 mimetics | ||||
| Dual BCL-2 and BCL-XL inhibitors | ||||
| Navitoclax | Yes | Phase I/II | CLL Advanced solid tumors Melanoma NSCLC |
NCT02079740 NCT02520778 NCT01989585 NCT02143401 |
| APG-1252 | Yes | Phase I/II | SCLC Advanced solid tumors |
NCT03387332 |
| AZD4320 | No | ALL NHL |
||
| S44563 | No | Melanoma SCLC |
||
| BCL2–32 | No | ALL NHL |
||
| BM-1197 | No | Colorectal cancer | ||
| Selective BCL-2 inhibitors | ||||
| Venetoclax # | Yes | Phase I-III | CLL NHL AML |
|
| S55746 (BCL201) | Yes | Phase I | CLL NHL Multiple myeloma |
NCT02920697 NCT02603445 |
| APG-2575 | Yes | Phase I | CLL NHL |
NCT03913949 NCT03537482 |
| BCL-XL inhibitors | ||||
| ABBV-155* | Yes | Phase I | Solid tumors | NCT03595059 |
| WEHI-539 | No | Breast cancer Ovarian cancer Chondrosarcoma Osteosarcoma |
||
| A-1155463 | No | AML | ||
| A-1331852 | No | Soft-tissue sarcoma | ||
| MCL1 inhibitors | ||||
| AMG 176 (**) | Yes | Phase I | AML NHL Multiple myeloma |
NCT03797261 NCT02675452 |
| MIK665 (S64315) (**) | Yes | Phase I | AML NHL Multiple myeloma |
NCT02992483 NCT02979366 NCT03672695 |
| AZD5991 | Yes | Phase I | NHL AML ALL Multiple myeloma |
NCT03218683 |
| S63845 | No | NHL AML |
||
| UMI-77 | No | Pancreatic cancer | ||
| A-1210477 | No | Esophageal carcinoma Head and neck carcinoma AML NHL Triple negative breast cancer |
||
| VU661013 | No | AML | ||
| Inhibitor of apoptosis (IAP) proteins and SMAC mimetics | ||||
| LCL161 | Yes | Phase I/II | Advanced solid tumors Breast cancer Colorectal NSCLC SCLC Multiple myeloma Polycythemia Vera Myelofibrosis |
NCT02649673 NCT03111992 NCT02098161 |
| Birinapant (TL32711) | Yes | Phase I/II | Advanced solid tumors NHL |
NCT02587962 NCT03803774 |
Venetoclax has been approved by the FDA for treatment of acute myeloid leukemia in combination with azacytidine, or decitabine, or low-dose cytarabine for elderly adults (> 75 years old) or adults with comorbidities that prevent the use of standard chemotherapy. Venetoclax was also approved for treatment of chronic lymphocytic leukemia (CCL) or small lymphocytic lymphoma (SLL).
antibody drug conjugate (ADC) based strategy
Clinical trials evaluating monotherapy and combinations with venetoclax
Administered as monotherapy or combined with radiation therapy or checkpoint inhibitors)
NSCLC: non-small cell lung carcinoma; SCLC: Small cell lung carcinoma; NHL: Non Hodgkin lymphoma; AML: acute myeloid leukemia; ALL: acute lymphoblastic leukemia; CLL: chronic lymphocytic leukemia
BH3 mimetics
ABT-737 was the first small molecule designed to selectively bind hydrophobic pockets within BCL-2, BCL-XL, and BCL-w where proapoptotic BH3-only proteins interact 229–231. ABT-737 demonstrated single-agent activity against lymphoma and small cell lung carcinoma cell lines as well as synergy with radiation and chemotherapy agents such as paclitaxel in other tumor types 229. These results provided proof-of-principle for this anti-cancer therapeutic strategy and supported the development of 2nd generation navitoclax (ABT-263) 232. Approximately 35% of patients with refractory chronic lymphocytic leukemia (CLL) achieved partial responses in the phase I trial with navitoclax 233. The response rates increased to 70% when it was combined with rituximab in untreated CLL 234. Development of navitoclax and dose escalation were hampered by (on-target) thrombocytopenia related to BCL-XL inhibition in platelets, but combinations of navitoclax with MEK inhibitors or other tyrosine kinase inhibitors continue to be explored in advanced solid tumors 235–238. BCL-2 selective inhibitor venetoclax (ABT-199) showed significant activity in preclinical models of CLL and non-Hodgkin lymphoma (NHL) overexpressing BCL2 without causing thrombocytopenia 239. The phase I study of venetoclax in heavily pretreated patients with refractory CLL showed tumor responses in all dose levels with an overall response rate of 79% and complete responses in 20% of the patients 240. Complete responses were also present among patients with chromosome 17p deletion – a group associated with poor outcomes. While thrombocytopenia was avoided by BCL-2 selectivity, the most relevant side effect observed with venetoclax was tumor lysis syndrome (TLS) related to rapid tumor responses, especially among patients with high tumor burden. This adverse event has been circumvented by an alternative dose schedule (5-week ramp-up) and preventative hydration and use of anti-hyperuricemic agents 241. Other side effects included mild nausea, diarrhea, and neutropenia. Progression-free survival (PFS) at 15 months was 66%, and the duration of response was longer among those achieving complete response. Aggregate results of four venetoclax early-phase trials in refractory CLL (347 patients) showed that patients with partial response had a median duration of response (DoR) of 24 months while a median DoR among patients with complete response patients was not reached 242. These results supported the first FDA approval for venetoclax for the treatment of patients with 17p deletion CLL 240,243. Subsequent studies with venetoclax combinations expanded the benefits of venetoclax in both refractory and treatment naïve CLL settings. A randomized phase 3 study showed that venetoclax combined with rituximab was superior to bendamustine/rituximab in refractory CLL with higher rates of response (92% vs. 72%) and progression-free survival (PFS) (2-year PFS 84% vs. 36%; P<0.001) 244. Venetoclax plus obinutuzumab (anti-CD20) was also incorporated to front line treatment of CLL, providing a chemotherapy-free option for these patients based its superior efficacy to chlorambucil/obinutuzumab in another phase 3 trial 245. The combination of venetoclax with ibrutinib - an inhibitor of Bruton tyrosine kinase - showed significant efficacy in the front line treatment of high-risk CLL representing another treatment option in this setting 246. Venetoclax plus standard chemoimmunotherapy regimen R-CHOP for treatment of B-cell NHL was safe and achieved 87% response rates in a phase I trial with subsequent phase 2 expansion cohort ongoing 247. In addition to transforming the treatment of lymphoid malignancies especially CLL, BCL-2 inhibition has had a significant impact on the treatment of elderly patients with acute myeloid leukemia (AML), a group of patients that frequently does not receive AML-directed treatment because of limited efficacy and toxicities with standard chemotherapy.
BCL-2 promotes survival of leukemic blasts, mediates resistance to chemotherapy, and its inhibition has significant activity in preclinical models of AML 248–250. Supporting the dependence on BCL-2 in AML cells, ABT-737 and venetoclax induced apoptosis of both AML myeloblasts and AML stem cells and demonstrated significant anti-tumor activity in AML xenograft models 248–250. Single-agent venetoclax was well tolerated with responses observed in 19% of patients with high risk and refractory AML but limited duration of responses fostered combination studies 251. BH3 profiling performed on pretreatment bone marrow biopsies and peripheral blood specimens showed the correlation between myeloblast dependence on BCL-XL or MCL-1 and shorter duration of therapy with venetoclax. Venetoclax also improved the clinical outcomes of elderly patients (> 75 years old) with AML when combined with low-dose cytarabine with 54% complete response rates and median overall survival (OS) of 10 months 252. These results were superior to low-dose cytarabine monotherapy (11–19% complete responses and OS 5.5 months), and compared favorably to intensive chemotherapy (45–57% remission rates and OS 5–12months) 253,254. Supported by synergistic effect between hypomethylating 5-azacytidine and BCL-2 inhibitors in preclinical models 231,255, a phase I trial showed that combination of venetoclax with azacytidine or decitabine displayed a favorable adverse profile with 60–70% complete remission rates and median OS of 17 months, a remarkable result for elderly patients with AML that led to FDA approval 256. The successful clinical translation and significant impact of the first BCL-2 inhibitor for the treatment of hematological malignancies fostered preclinical and clinical development of other selective and dual BCL-2 inhibitors 228,257–266.
Selective BCL-XL inhibitors have also been developed, and their clinical track has focused in solid tumors. A first-in-class BCL-XL inhibitor antibody-drug conjugate (ABBV-155) was recently disclosed and has reached the clinic in a phase I study investigating its safety and preliminary activity in refractory solid tumors as monotherapy or combined with taxanes 267,268. A BCL-XL-based vaccine is also being evaluated in a phase I trial enrolling patients with prostate cancer 269 (Table 1). Other compounds have demonstrated significant activity in preclinical models 270–277.
The success of BH3 mimetics has occurred in parallel to the advancement of strategies to target protein-protein interactions using stapled peptides, synthetic proteins “locked” in their α-helix secondary structure by a chemical “staple” 278,279. Stapled peptides carry the promise of targeting critical intracellular protein-protein interactions that have been considered undruggable and display chemical stability and resistance to proteolysis, increased cellular penetration and higher affinity to their target compared to small-molecules or biologics 278,279. The proof of concept and potential therapeutic application of hydrocarbon-stapled peptides was first demonstrated with peptides that mimic the α-helical BH3 segment of proapoptotic BID called SAHBA (stabilized alpha-helix of BCL-2 domains) 280. SAHBA penetrated leukemia cells, bound to BCL-XL, induced apoptosis and demonstrated anti-tumor activity in xenograft mice models of leukemia 280. Stapled-peptides targeting the p53-MDM2 interaction surface have been developed as an attempt to reactivate p53 and related compounds have transitioned to clinical trials and others are targeting MCL1, anti-apoptotic BFL-1 among other biological switches such as β-catenin 93,281–286.
Small molecules targeting a regulatory site at serine 184 of BAX which regulates its subcellular localization and ability to insert into the mitochondrial membrane have been shown to induce apoptosis in lung cancer models 287. The small molecule BAX agonists (SMBA1–3) selectively bind to BAX, inhibit S184 phosphorylation, promote oligomerization that leads to cytochrome c release, and trigger apoptosis. Significant activity of lead compound SMBA1 in models of lung cancer which is characterized by increased BAX expression fostered the development of optimized compounds with activity also in breast cancer cell lines such as CYD-4–61 and GL0385 288,289. Other BAX-activating compounds such as BAM-7 and BTSA1 have shown promising preclinical activity in glioblastoma and AML cell lines 290–292. Identification of small-molecule inhibitors of BAX called BAIs (BAX Activation Inhibitors) revealed a new binding site on inactive BAX and a mechanism for allosteric inhibition with significant therapeutic potential 293.
The proapoptotic protein BIM that preferentially activates BAX exemplifies the regulatory link between the intrinsic pathway and other kinases such as ERK1/2, PKB, and JNK-c-Jun pathways 198,294–297. The ERK1/2 and MAPK1 pathways promote cell survival in part by phosphorylating BIM that leads to its proteasomal degradation 198, an important mechanism for survival in kinase-driven tumors such as non-small cell lung cancer (NCSLC) and chronic myeloid leukemia (CML) 177,298. A germline deletion polymorphism in BCL2L11 encoding BIM without the BH3 domain results in resistance to EGFR TKIs in vitro and identifies patients with NSCLC that have inferior clinical outcomes when treated with gefitinib or crizotinib 299–301. These results coupled with the upregulation of BIM mediating chemotherapy-induced apoptosis and synergy between kinase inhibitors (i.e., cerdulatinib-SYK/JAK inhibitor) and venetoclax is fostering an interest in BIM as a biomarker and promoting strategies to modulate its expression for therapeutic applications 302,303.
MCL1 inhibitors
Significant efforts to develop inhibitors of anti-apoptotic MCL1 have reached an important milestone with novel compounds arriving in the clinic in first-in-human trials (Table 1). The therapeutic potential of these inhibitors is highlighted by the involvement of MCL1 in the pathogenesis of several malignancies, frequent amplification, and its association with resistance to chemotherapy or inhibitors of BCL-2/BCL-XL 304,305. Small molecule AM-8621, a novel class of spiromacrocyclic MCL1 inhibitor, binds to an MCL1 groove displacing BIM and inducing apoptosis of several cell lines with multiple myeloma, AML and lymphoma cells displaying the highest levels of sensitivity. AMG 176, a derivative of AM-8621 with a more favorable pharmacokinetic profile, showed synergy with chemotherapy and venetoclax paving the way to ongoing phase I trials of AMG 176 as monotherapy or combined with venetoclax in patients with refractory AML, lymphoma or multiple myeloma 85,306,307. AZD5991, another macrocyclic specific MCL1 inhibitor, is being investigated in patients with advanced hematologic malignancies 308,309. The MCL1 inhibitors VU661013 and S63845 showed synergy with venetoclax in preclinical models of hematological malignancies including venetoclax-resistant xenograft models of AML supporting the potential of combinatorial strategies to circumvent venetoclax resistance that might be relevant to other anti-BCL-2 compounds 310–316. The clinical investigation of these combinations will also inform the value of sequential treatments in settings where the temporal upregulation of these proteins determines response to treatment. BH3 profiling of cells and expression levels of BCL-2 could reveal anti-apoptotic addiction to BCL-2, BCL-XL, or MCL1 and help predict response to this growing number of apoptosis-inducing drugs 63,248,317–323.
B-cell lymphoma 2-related protein A1 (BCL2A1) also called BCL-2 related gene expressed in fetal liver (BFL-1) represents another anti-apoptotic BCL-2 protein overexpressed in hematological malignancies and solid tumors with specific inhibitors in development including stapled peptides 284,324–326. These molecules not only have significant therapeutic potential for BCL2A1/BFL-1 dependent malignancies (i.e., B cell malignancies) but recent results show that intrinsic resistance to inhibition of BCL-2, BCL-XL and MCL1 is mediated by BFL-1 and Bcl-w highlighting the importance of developing specific inhibitors of these relatively understudied members 318.
IAP inhibitors to target apoptosis
The inhibitor of apoptosis (IAP) proteins are overexpressed in several malignancies with a negative impact on prognosis through their promotion of cell survival by the prevention of caspase activation 327. Inhibitors of IAPs have long been sought as tools and potential cancer therapeutics that could induce cell death. Among the eight proteins in humans, the most relevant with anti-apoptotic activity are X-chromosome-linked IAP (XIAP), cellular IAP1 and IAP2 (c-IAP1 and c-IAP2), and melanoma IAP (ML-IAP). Second mitochondria-derived activator of caspase (SMAC) as well as Omi/HtrA2 are released from the mitochondria and can independently inhibit XIAP and prevent its negative regulation of the apoptosome 328. Cytochrome c released from the mitochondria together with cytoplasmic apoptotic protease-activating factor 1 (APAF1) and procaspase 9 form the apoptosome to drive caspase 9 activation 329. SMAC plays a central role binding to XIAP, c-IAP1, c-IAP2 and especially preventing XIAP from directly inhibiting caspases 3, 7 and 9. c-IAP1 and c-IAP2 are not potent inhibitors of caspases, but they can promote apoptosis by binding to SMAC and preventing its association with XIAP and subsequent inhibition of caspases 330,331. IAP proteins regulate cell survival at multiple levels and significant efforts have been devoted to develop inhibitors of IAP and SMAC mimetics as cancer therapeutics 21. Several IAP antagonists have transitioned to clinical trials including small molecules and oligonucleotides, but none has been FDA-approved thus far. Safety concerns have been raised with documented cytokine release syndrome and increased adverse events when combining the IAP antagonist LCL161 with chemotherapy 332,333. Interestingly, LCL161 displayed anti-tumor activity in patients with refractory multiple myeloma by upregulating IFN signaling and promoting an inflammatory response highlighting the potential therapeutic benefits independent of inducing cell death in this disease setting 334. LCL161 also displayed radiosensitization properties in head and neck squamous carcinoma models 335. Another compound birinapant (TL32711) which did not show significant activity as a single-agent is being evaluated in combination with anti-PD1 pembrolizumab in solid tumors and is being combined with radiation therapy in patients with head and neck cancer 336–339. Despite the challenges of these drugs with several trials without meaningful clinical results, the potential to enhance an anti-tumor immune response by combining SMAC mimetics with checkpoint inhibitors or radiation therapy remains high with ongoing clinical trials in advanced solid tumors including lung and colon cancers 340–343.
Mechanisms of resistance to BCL-2 targeting therapies
Several mechanisms of resistance to strategies targeting the BCL-2 family of proteins have been described including modulation of BCL-2 family members expression levels, mutations altering their binding site and cellular localization as well as modifications in mitochondrial function. Point mutations in the BCL-2 causing resistance to venetoclax were identified both in vitro and among patients with CLL progressing on venetoclax treatment 344,345. Continued exposure of lymphoma cell lines to venetoclax led to the acquisition of BCL-2 mutations in the BH3-binding groove interfering with drug binding 344. This in vitro result was supported by the identification of a novel mutation (Gly101Val) in the BCL-2 binding groove described using genomic analyses of paired specimens collected from patients with CLL treated with venetoclax and having progression of disease 345. This mutation was not detected in venetoclax naïve patients or cancer (COSMIC) or population databases (gnomAD). It reduced significantly BCL-2 binding affinity to venetoclax (~180-fold compared to wild type BCL-2) without altering the affinity to BAX and BIM, which allows the mutant protein to retain its pro-survival activities. Highly sensitive digital droplet PCR detected this mutation as early as 25 months prior to overt CLL clinical progression, potentially serving as an earlier biomarker of progression. While Gly101Val was not detected in other B-cell malignancies, another acquired BCL-2 mutation (Phe104Ile) also interfering with venetoclax binding was described in a patient with follicular lymphoma (FL) with secondary resistance to venetoclax 346. The crystal structures of venetoclax bound to wild type BCL-2 and mutants (G101V and F104L) were described, and restorage of venetoclax affinity was demonstrated with another BCL-2 mutation E152 in experimental models 347. Mutations in BTG1 and homozygous CDKN2A/B (p16Ink4a/p14Arf) deletions were also identified in patients with CLL and resistance to venetoclax 348. Other mechanisms of resistance have been described among the growing number of hematologic malignancies receiving treatment with inhibitors of BCL-2 family members.
Increased expression of BCL-XL induced by NFkB signaling was associated with venetoclax resistance in samples from patients with mantle cell lymphoma (MCL) and cell lines models 349,350. Furthermore, patients with MCL with primary resistance to the combination of venetoclax and ibrutinib 351 displayed loss of chromosome 9p and mutations in the SWI-SNF chromatin-remodeling complex that resulted in upregulation of BCL-XL 352. Resistance of AML cell lines and patient samples to venetoclax was also associated with overexpression of BCL-XL and lower levels of BCL-2 248. Genome-wide CRISPR/Cas 9 screen in human AML cell lines showed that inactivation of TP53, BAX and PMAIP1 genes leads to venetoclax resistance 353. The TP53 knockout cells had upregulation of BCL-XL and decreased expression of BCL-2 and MCL-1 making them sensitive to BCL-2/BCL-XL inhibitor AZD-4320 and less sensitive to MCL-1 inhibitor; BAX knockout cells did not have decreased levels of MCL-1 and were more sensitive to MCL-1 inhibitor. Venetoclax-resistant cells had distinctive changes in mitochondria metabolic profile characterized by increased anaerobic glycolysis and relative protection against mitochondria membrane depolarization in response to various stimuli. Another CRISPR/Cas 9 screen in AML cells uncovered the role of OPA1 (optic atrophy 1) protein and structural changes in mitochondria cristae in resistance 354. Mitochondria from venetoclax-resistant AML cells had tighter and increased number of cristae (inner membrane lamellae) compared to cells sensitive to venetoclax. These morphological changes were associated with increased expression of OPA1, which controls mitochondria fusion and protects the cell from apoptosis by regulating mitochondria cristae shape and cytochrome c release from the cristae to the intermembrane space, a crucial step prior to cytochrome c release into the cytosol 355–357. Venetoclax-resistance in AML cells was also mediated by CLPB protein (a mitochondrial AAA+ ATPase chaperonin) as demonstrated by its increased expression in resistant cells and resensitization following CLPB depletion using CRISPR sgRNA. This study expanded the known functions of CLPB beyond a protein chaperone by describing its anti-apoptotic function through direct interaction with proteins HAX1 and OPA1 modulating mitochondrial morphology 358–360. These results highlight the importance of molecular regulators of intrinsic mitochondrial function and structure as mediators of resistance to BCL-2-based therapeutic strategies.
While mutations in the proapoptotic BAX have well documented in various malignancies 361–363, a unique mutation in the C-terminal transmembrane domain of BAX (G179E) compromised its anchoring to mitochondria outer membrane causing resistance of lymphoma cell lines to venetoclax 344. Considering the importance of mitochondria size and membrane lipid composition to BAX-induced membrane permeabilization and sensitivity to apoptosis 364,365, it is conceivable that lipid composition might influence resistance to venetoclax. Further delineation of resistance mechanisms to BCL-2 inhibitors will be critical to guide patient selection, inform combinatorial treatment strategies and sequence of therapies.
Strategies targeting the extrinsic pathway
Death receptor agonists
Activation of the extrinsic pathway is triggered by extracellular signals activating the proapoptotic death receptors (DRs), transmembrane proteins members of the TNF receptor superfamily (TNFR) 14,27,39. The following DRs have been identified: TNFR1, Fas (CD95, APO-1), DR3, DR4 (TRAILR1), DR5 (TRAILR2) and DR6. Upon binding of their respective ligands, Fas, TNFR1, TNFR2, DR4 and DR5 recruit adaptor proteins such as FADD (Fas-associated death domain) to assemble the DISC (death-inducing signaling complex) that activates caspase-8 and −10 and the downstream apoptosis cascade. Cellular FLICE-inhibitory protein (c-FLIP) can also become recruited to the DISC and in some cellular settings competes with caspase-8 and −10 and inhibits initiator caspase activation and apoptosis (Figure 1).
Apo2L/TRAIL (TNF-Related Apoptosis-Inducing Ligand), the extracellular ligand of DR4 and DR5, is a transmembrane trimeric glycoprotein that be cleaved by metalloproteases and released as a soluble ligand 111,132. TRAIL is expressed on the surface of immune cells, including natural killer cells and cytotoxic T cells and regulates an innate immune response 366,367. The immune cells can also induce apoptosis in target cells using the granzyme-perforin pathway (Figure 1). TRAIL also binds to decoy receptors (DcR1, DcR2, osteoprotegerin) without intracellular caspase activation. Direct activation of the extrinsic pathway with agonists of DR4 and DR5 has provided a compelling rationale to induce apoptosis of cancer cells with therapeutic intent 368,369. While severe liver toxicity was observed with Fas ligands (CD95 agonists) in preclinical models and pro-inflammatory effects limited the application of TNF in the clinic, DR4 and DR5 agonists showed selective activity towards cancer cells without damage to normal tissues in preclinical models. Therapeutic approaches targeting DR4 and DR5 were brought to the clinic in the early 2000’s: recombinant human (rh)Apo2L/TRAIL (activating DR4 and DR5) and agonistic monoclonal antibodies against DR4 or DR5 (Figure 2) 370–373.
Figure 2. Cancer therapeutic approaches targeting apoptosis pathways.
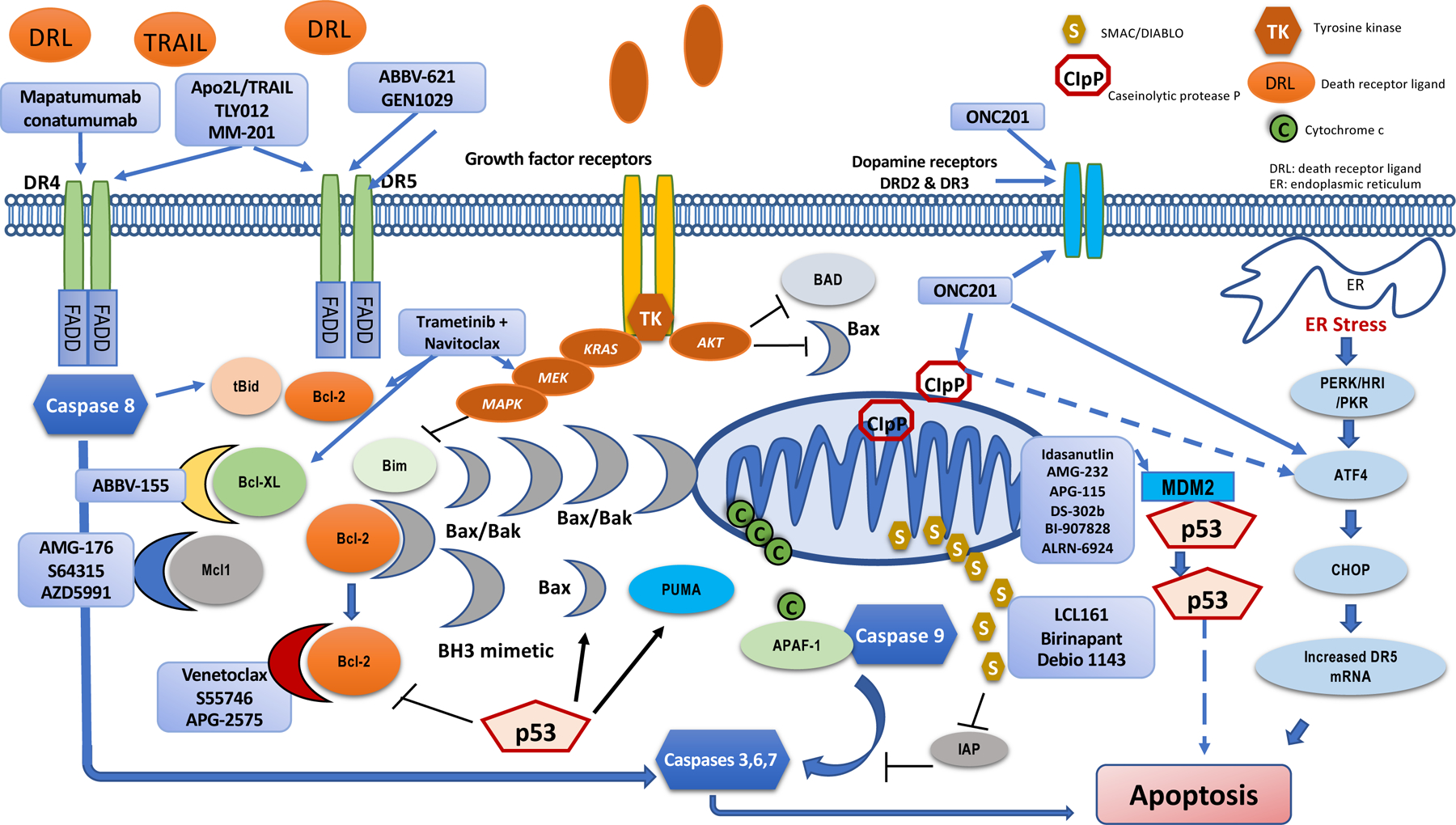
Death receptors 4 and 5 (DR4, DR5) can be activated by agonist antibodies such as mapatumumab, conatumumab, Apo2L/TRAIL, ABBV-621 and GEN1029, fusion protein of recombinant TRAIL with IgG1 (MM-201) or pegylated recombinant TRAIL (TLY012). BH3 mimetics bind to anti-apoptosis BCL-2 family members (BCL-2, BCL-XL, MCL1) leading to activation of BAX and BAK that form pores in the outer mitochondrial membrane resulting in release of cytochrome C. Selective inhibitors of BCL-2 (venetoclax, S55746, APG-2575), BCL-XL (ABBV-155) and MCL1 (AMG-176, S64315, AZD5991) have reached clinical trials. Modulation of growth signaling pathways also induces apoptosis and combinations of MEK inhibitors with navitoclax (BCL-2/BCL-XL inhibitor) are being investigated in clinical trials. Pro-survival pathways such as AKT and MEK inhibit apoptosis by phosphorylation of proapoptotic proteins such as BAD (BCL-2-associated agonist of cell death) and BIM (BCL-2-interacting mediator of cell death). Strategies targeting the p53 pathway to induce apoptosis include MDM2 inhibitors (i.e., idasanutlin, AMG-232, APG-115, DS-302b, BI-907828, ALRN-6924). The ONC201 which has a three-ring imipridone structure has been found to bind to dopamine receptors DRD2 and DRD3 415 as well as the mitochondrial caseinolytic protease P (CIpP) leading to activation of integrated stress response protein ATF4 416,417. ATF4 activation by ONC201 leads to CHOP-dependent DR5 upregulation and cell death 418,419. The relationship between ClpP and the other pathways engaged by ONC201 remains under study although the binding appears to be upstream of ATF4 activation (Lanlan Zhou and W.S.E-D., unpublished observations). SMAC (second mitochondria-derived activator of caspase) is released from mitochondria together with cytochrome c. Cytochrome s forms the apoptosome complex with caspase 9 and APAF-1 (apoptotic protease-activating factor 1). Inhibitors of apoptosis proteins (IAP) prevent caspase activation and this is blocked by SMAC or Omi/HtrA2 which is also released from mitochondria. SMAC mimetics and inhibitors of IAP (i.e., LCL161, birinapant, Debio 1143) have transitioned to clinical trials. tBid: truncated BH-3 interacting domain death agonist; BCL-XL: B cell lymphoma extra-large; MCL1: myeloid cell leukemia 1; BAX: BCL-2 associated X protein; BAK: BCL-2 antagonist/killer; MDM2: mouse double minute 2 protein; DRL: death receptor ligand; ER: endoplasmic reticulum. PERK: PKR-like ER kinase; ATF4: Activating transcription factor 4; CHOP: CCAAT-enhancer-binding protein homologous protein.
The recombinant APO2L/TRAIL (dulanermin) was extensively investigated in clinical trials as a single-agent and in combination with chemotherapy or rituximab in solid tumors and hematologic malignancies but clinically meaningful activity was not observed 374–377. These results have been attributed to its short half-life (30min), limited capacity to induce receptor clustering, binding to decoy DRs (i.e., DcR1, DcR2, osteoprotegerin) and lack of sufficient relevant biomarkers to inform sensitivity 369. Other potential mechanisms of resistance that have been proposed include epigenetic silencing of DR4, post-translational modifications of DR such as O-glycosylation, decreased expression and/or density of receptors on the cellular surface 378–383. Intracellular levels of BCL-2 family members such as BAX and MCL1 and other regulators of the intrinsic pathway (e.g., XIAP) can also mediate the resistance to TRAIL agonists in type II cells that depend on mitochondria for the induction of apoptosis 384–386. In colon cancer cell lines, the efficiency of BID cleavage by caspase influenced sensitivity to TRAIL 130. Targeting XIAP overcame TRAIL-resistance in cell lines deficient in BAX/BAK and those overexpressing BCL-2 385,387. Furthermore, the limited activity of soluble TRAIL can also be associated with its reduced ability to induce receptor clustering as suggested by higher responses with membrane-bound or oligomerized forms of TRAIL 388–390. Agonist monoclonal antibodies against DR4 and DR5 were promising given their longer half-lives, significant preclinical activity and ability to induce receptor clustering, but they also have shown limited clinical efficacy in various clinical settings particularly as single-agents. Mapatumumab, a DR4 agonist antibody, was well tolerated in clinical trials but showed limited clinical benefit when administered with chemotherapy in patients with NSCLC, colorectal cancer, and other solid tumors 391–394. Patients with untreated advanced NSCLC (stage IIIB or IV; 52% adenocarcinoma) were randomized to receive carboplatin and paclitaxel, or carboplatin, paclitaxel, and mapatumumab (10 mg/kg or 30 mg/kg) combination. Grade 3/4 lymphopenia occurred more frequently in the mapatumumab arms (22 and 19% vs. 3%), but there were no other significant toxicities attributed to mapatumumab and overall dose intensity was comparable among treatment arms. The addition of mapatumumab did not improve response rates, progression-free survival, or overall survival in this population. Lack of benefit was observed in all histological subgroups, and variable TRAIL-R1 receptor staining was not associated with response 391. Thirty-eight patients with colorectal cancer refractory to 5-fluorouracil (5-FU), oxaliplatin and irinotecan-based regimens were treated with single-agent mapatumumab which was relatively well tolerated without hepatotoxicity. There were no tumor responses and median PFS for this heavily pretreated population was only 1.2 months 393. Mapatumumab was also combined with gemcitabine in a phase I trial with 49 patients with advanced solid tumors (majority of patients with pancreatic cancer) 393. The combination was well tolerated with transient elevation of LFTs and 12 patients had confirmed partial responses that were associated with TRAIL-R1 staining. Single-agent mapatumumab showed encouraging activity in patients with refractory non-Hodgkin’s lymphoma, including 2 complete responses among 40 patients 395. It was also administered in combination with tyrosine kinase inhibitor sorafenib to patients with hepatocellular carcinoma without clinical benefit 396. The DR5 agonist lexatumumab was investigated as single-agent, and combined with chemotherapy in advanced solid tumors with some patients achieving durable stable disease including pediatric osteosarcoma and adult sarcomas 397–399. Thirty-seven patients with advanced solid tumors (32% soft tissue sarcoma, 19% osteogenic sarcoma, and 11% colorectal cancer) were treated with the most common adverse effects being constipation, fatigue, and mild nausea 397. Dose-limiting toxicities included grade 4 transaminitis. There were no confirmed responses, but 32% of patients had stable disease with 3 patients receiving > 10 treatment cycles. A second study evaluating lexatumumab every 2 weeks documented stable disease in 29% of patients and a mixed response in a patient with Hodgkin’s disease 400. Other DR5 agonists investigated in clinical trials included conatumumab (AMG655) 370,401–404, tigatuzumab 405,406, LBY135 407, drozitumab (PRO95780) 408,409. Approximately 190 treatment naïve patients with metastatic colorectal cancer were randomized to receive mFOLFOX6/bevacizumab, mFOLFOX6/bevacizumab with conatumumab or placebo. The combination was relatively well tolerated, but there were no significant differences in tumor responses, PFS or OS across the arms. Patients with high-affinity forms of the FCGR3A receptor (immunoglobulin gamma Fc region receptor III-A) had a non-significant improvement of PFS supporting previous results suggesting the importance of high affinity to promote receptor crosslinking 403. Recent efforts have explored combinations with drugs capable of sensitizing cells to TRAIL-induced apoptosis either by modulating the expression of TRAIL or inhibiting other anti-apoptotic mechanisms. A CDK9 inhibitor reversed the resistance of non-small cell lung cancer cell lines to TRAIL 410. Inhibition of MCL1 sensitized breast cancer cells to TRAIL through the upregulation of DR4 411. The combination of conatumumab and Apo2L/TRAIL enhanced the antitumor activity by promoting receptor crosslinking, oligomerization, and increasing the formation of death-inducing signaling complex (DISC) 412. This approach suggested that DR5 agonist-induced receptor crosslinking facilitated and enhanced the signaling of soluble Apo2L/TRAIL that could overcome the therapeutic limitations of monotherapy.
The first-in-class small molecule ONC201 was originally discovered as a TRAIL-inducing compound (TIC10) 413. TRAIL expression in numerous (but not all) cancer cell lines contributes to its potent anti-tumor activity, including against cancer stem cells 414 that has led to several ongoing clinical trials. The ONC201 which has a three-ring imipridone structure has been found to bind to dopamine receptors DRD2 and DRD3 415 as well as the mitochondrial caseinolytic protease P (CIpP) leading to activation of integrated stress response protein ATF4 416,417. ATF4 activation by ONC201 leads to CHOP-dependent DR5 upregulation and cell death 418,419. The anti-tumor activity of ONC201 was significantly enhanced by TRAIL or DR5 agonist in preclinical models of endometrial carcinoma and breast cancer 420,421. This appears to be due to priming of tumor cells treated with ONC201 to TRAIL-induced cell death, and represents one promising avenue for combination therapy targeting tumor cell death. ONC201 has immune stimulatory effects that trigger cell death of tumor cells 422,423 and this is currently being further tested in combination with immune checkpoint therapy 424. ONC201 has clinical activity against H3K27M-mutated midline gliomas such as DIPG with a reported 47% response rate 425,426. There is data to support the hypothesis that the interplay of dopamine receptor family members, as could be influenced by epigenetic alterations such as H3K27M mutation, may impact sensitivity of tumors to ONC201 427. The relationship between ClpP and the other pathways engaged by ONC201 remains under study although the binding appears to be upstream of ATF4 activation (Lanlan Zhou and W.S.E-D., unpublished observations). There is a literature that ER stress blockade 428 may induce apoptosis as a cancer therapeutic strategy including blockade of ATF4 in c-Myc-driven tumors 429. It remains unclear how or why some tumors respond and undergo apoptosis following ATF4 activation while others to ATF4 blockade. The HRI- and PKR-dependent activation of CHOP and DR5 by drugs such as ONC201 may be involved when ATF4 is therapeutically activated 418 while uncoupling of translation from bioenergetic demands may be involved in settings where PERK- and GCN2-dependent ATF4 activation is blocked 429.
A novel platform utilizing a mixture of two antibodies targeting distinct epitopes on DR5 using technology that promotes the formation of hexameters due to E430G mutations in the Fc domains of both antibodies and enhance antibody-dependent cellular cytotoxicity (ADCC) have also transitioned to phase I clinical trial in solid tumors 430–432 (GEN1029). Another TRAIL agonist fused with Fc-domain of a human immunoglobulin G1 (ABBV-621) that assembles to maximize receptor clustering is also undergoing a phase I trial in solid tumors and hematological malignancies with cohorts in colorectal and pancreatic cancers receiving treatment in combination with cytotoxic chemotherapy, and cohorts with AML and DLBCL treated with single-agent or combination with venetoclax 433–436. A fusion protein of IgG1 with single chain TRAIL has shown significant preclinical activity, particularly in PDX models of sarcoma combined with docetaxel leading to complete responses (MM-201) 437. Pegylated formulations of recombinant TRAIL with prolonged half-life (e.g., subcutaneously administered TLY012) and stability is undergoing development of treatment of liver fibrosis with significant potential to be explored in cancer treatment 438. These efforts have reinvigorated the interest and therapeutic potential of TRAIL-based strategies as a feasible anti-cancer therapy strategy. Combinatorial approaches utilizing TRAIL, DR agonists, and other modulators of apoptosis response such as BH3 mimetics have the potential to overcome the limitations of previous strategies.
Novel combinations and approaches
Targeting growth factor and oncogenic survival pathways to induce apoptosis
The therapeutic potential of targeting the interface between kinase signaling pathways and the apoptosis machinery with combinations of kinase inhibitors and inhibitors of the BCL-2 family has been demonstrated in preclinical models and is transitioning to clinical trials. Despite the upregulation of the MEK pathway in several malignancies including those with activating KRAS mutations, MEK inhibitors have limited activity as single-agents in part due to adaptive cell responses that include kinome reprogramming and upregulation of BH3 protein BIM 439–442. However, MEK inhibition upregulation of proapoptotic BIM primes cancer cells to respond to BCL-2/BCL-XL inhibitors. Xenograft models of lung cancers with KRAS mutations were exquisitely sensitive to the combination of MEK inhibitor and navitoclax (ABT-263; dual BCL-2/BCL-XL inhibitor) 443. The same combination also showed significant anti-tumor synergy in PDX models of serous ovarian cancer, and this response correlated with the degree of upregulation of BIM by MEK inhibitors 444. An ongoing phase Ib/II trial is investigating the combination of trametinib (MEK inhibitor) and navitoclax in advanced solid tumors with activating KRAS or NRAS mutations 235. A detailed analysis of KRAS-driven NSCLC cell lines showed that sensitivity to MEK inhibitors combined with BH3 mimetics (navitoclax, venetoclax and novel MCL1 inhibitors) is complex and dependent upon cancer cell dependency on MCL1or BCL-XL to neutralize upregulated BIM 202. Significant tumor regression of PDX models of KRAS-mutant lung adenocarcinoma was observed with trametinib plus AM-4907 (MCL1 inhibitor), but no activity was detected with trametinib plus navitoclax in these tumors where BIM was preferentially bound to MCL1. Nevertheless, pre-treatment with navitoclax shifted the subcellular localization of BIM to binding MCL1 and increased sensitivity to MEK plus MCL1 inhibitors. These results highlight that apoptotic responses of cancer cell lines have specific dependencies on BCL-2 proteins that could be challenging to prospectively identify in the clinic especially given the limitations of serial biopsies in patients with solid tumors. However, sequential treatment with BH3 mimetics offers the possibility to optimize the induction of apoptosis by targeted therapy combinations, an important strategy to be tested in the clinic as novel inhibitors enter clinical trials.
Ras and NFkB targeting to induce apoptosis
Intense effort has continued over three decades to target the Ras family of proteins, major oncogenic drivers, growth and pro-survival promoting proteins in multiple tumor types. Blockade of this signaling can lead to cell death, including apoptosis. Targeting farnesyl transferase activity was not an effective strategy in the clinic. In addition, targeting downstream effects such as through MEK inhibitor monotherapy has not led to sustained patient benefit in the clinic. An exciting development involves a specific KRAS mutant (G12C)-targeting drug that is now in clinical trials with recently reported promising early results in NSCLC and CRC 445.
NFkB activation leads to cell survival and pro-inflammatory signals that promote tumor progression and resistance to cancer therapeutics. There has been interest for many years in the therapeutic targeting of NFkB 446. Proteasome inhibitors have been developed for some hematological malignancies, and they may, in part, act through inhibition of NFkB 447. Much effort remains to target NFkB as a cancer therapeutic strategy that can lead to apoptotic cell death 448,449. Moreover, there is also much interest and effort to target downstream mediators of NFkB, such as IL6, as a cancer therapeutic strategy through cytotoxic and proapoptotic immune responses against cancer cells 450. IL-6 is regulated by another pro-survival pathway, STAT3, which is a cancer therapy target 451.
EGFR, Her2, c-MET, NTRK, BRAF, FLT3, BCR selective targeting induces apoptosis
Despite their dramatic clinical impact in EGFR-driven lung adenocarcinoma, resistance to EGFR tyrosine kinase inhibitors mediated by secondary EGFR mutations or MET gene amplification, among other mechanisms, have limited their benefit 452. Third-generation EGFR TKIs osimertinib can overcome EGFR T790M but succumb to modulation of BIM and MCL1 levels demonstrating the critical importance of apoptosis machinery to the efficacy of these drugs 453. Inhibitors of the c-Met/hepatocyte growth factor receptor (HGFR) such as crizotinib exert their anti-tumor activity by inducing apoptosis 191,192,454.
Tumors driven by NTRK-containing fusion proteins display constitutive activation of downstream pathways such as MAPK, PI3K, and STAT3 that can be effectively targeted with TRK inhibitors such as larotrectinib and entrectinib 194. Trk inhibition triggers apoptosis in several cancer models 455–457.
Targeting tumor suppressor pathways to induce apoptosis
A number of promising approaches targeting tumor suppressor pathways have been under development as anti-tumor agents. Blocking the unregulated oncogenic downstream effectors (PI3K, Akt, beta-catenin, Myc, mTOR, CDKs, VEGF) of mutated tumor suppressors has been an area with significant progress 458,459.
Multiple efforts have been directed at the p53 pathway, a major target for inactivation in human cancer 460–463. Among the most advanced, MDM2 (mouse double minute 2 protein) or MDMX inhibitors have been in clinical trials for several years 464,465. These therapeutics block the major pathway of wild-type p53 degradation without inducing DNA damage. Early results are promising, but there are on-target hematological toxicities 466,467. Numerous compounds targeting MDM2 have been developed since the nutlins, the first-in-class MDM2 inhibitors, including the nutlin-3a derivative RG7388 (idasanutlin) currently under investigation in trials for advanced hematologic malignancies and solid tumors as a single-agent or combined with chemotherapy, venetoclax or checkpoint inhibitors (atezolizumab) 468–473. Other MDM2 antagonists in clinical trials include AMG-232, HDM201 474, APG-115 (ongoing studies as single-agent and combination with pembrolizumab) 475, DS-3032b 476, BI 907828 477 and the stapled peptide ALRN-6924 478–486. Significant efforts have also been directed to combine MDM2 antagonists with other targeted agents (i.e., MEK 1/2, BCL-2/BCL-XL, PI3K, BRAF inhibitors among others) or developing bifunctional molecules such as PROTAC (Proteolysis Targeting Chimeric) compounds that unify a E3 ubiquitination ligase with a small molecule targeting MDM2, a compelling strategy especially when considering the reported emergence of p53 mutations during treatment with these agents and in vitro models describing the mechanisms of resistance 487–491.
Approaches targeting restoration of the p53 tumor suppressor pathway in tumors with mutant p53 are best exemplified by APR-246, which has been in multiple clinical trials 492. Boosting wild-type p53 or restoring the p53 signaling pathway in tumors with mutated p53 allows the endogenous growth arrest and apoptosis signaling in tumor cells to control tumor growth 493,494. Preliminary results of combination of APR-246 and azacytidine in patients with p53 mutant myelodysplastic syndromes (MDS) and AML showed a favorable toxicity profile and responses in all evaluable patients including complete responses. Pathway analysis after APR-246 treatment also showed transcriptional activation of p53 targets 495.
CDK4/6 inhibitors such as palbociclib, currently approved for treatment of breast cancer, induce cancer cell death by sensitizing cells to TRAIL-induced apoptosis and can overcome hypoxia-induced resistance in colon cancer cell lines and promotes apoptosis in other cell types by other mechanisms including inhibition of DNA repair following radiation therapy 496–498. Palbociclib also induces cell cycle arrest and senescence in models of glioblastoma and multiple myeloma 499,500. Palbociclib in combination with MEK inhibitor trametinib overcame resistance of KRAS-mutant lung adenocarcinoma cell lines to MEK inhibitor by inducing apoptosis 501. A preclinical approach involves micro-RNA targeting of CDK4/6 as an alternative strategy for therapeutic targeting of these kinases to induce tumor cell apoptosis 502.
Epigenetic approaches to targeting apoptosis
Epigenetic modulators such as histone deacetylases inhibitors (HDACi) and BET inhibitors (BETi) exert their anticancer activity through apoptosis with potential synergy with BCL-2 targeting agents. Combination BET inhibitor ABBV-075 with venetoclax has shown promising preclinical efficacy in models of AML, including in cell lines resistant to venetoclax and PDX models 503. This synergy seems to be mediated by decreased protein levels of MCL1, BCL-XL, BCL-2 in the AML cell lines resulting from BETi modulation of chromatin and gene expression. Similar results were also observed in patient-derived CD34+ blast progenitor cells. Synergy between BETi and venetoclax was also observed in patient-derived cutaneous T cell lymphoma (CTCL) cells 504.
HDAC inhibitors alter chromatin remodeling and induce apoptosis by several mechanisms including upregulation of death receptors and ligands TRAIL or Fas, increased expression of BH3-only proteins (i.e., Bid, BIM, Bmf and BAK), decreased expression of BCL-2, BCL-XL, BCL-W, MCL1, and elevation of reactive oxygen species that contribute to intrinsic pathway activation and provide rationale for combinations with BCL-2 family targeting agents 505–508. HDAC inhibitors prime rhabdomyosarcoma cells to venetoclax-induced apoptosis by increasing the expression of BIM 509. Similarly, panobinostat induces NOXA and decreases MCL1 in diffuse large B cell lymphoma (DLBCL) cell lines enhancing the sensitivity to BCL-2 inhibitor 510. HDAC inhibitors also potentiate the activity of MEK inhibitors and venetoclax in models of multiple myeloma 511. Clinical studies are investigating the combination of venetoclax and fimepinostat in refractory lymphoma 512,513. Hypomethylating agent azacytidine is also showing potent synergy with venetoclax and ABT-737. A clinical trial combining venetoclax with decitabine or azacytidine in patients with AML showed meaningful clinical efficacy with 67% of patients achieving complete remission including patients with high risk AML as described previously 231,248,255,256.
Targeting chaperones to induce apoptosis
Various strategies targeting cell stress including proteotoxic stress and ultimately leading to apoptosis continue to be explored in cancer therapy. Chaperones such as members of the heat shock protein family including HSP90 stabilize newly formed proteins which is very important in rapidly growing and dividing tumor cells that generate significant protein synthesis. HSP90 inhibitors including geldanamycin, ganatespib, onalespib, XL888, TAS116 and others have been under development 514–519. Endoplasmic reticulum (ER) stress caused by several stimuli lead to apoptosis mediated by activation of PERK (Protein Kinase R (PKR)-like Endoplasmic Reticulum Kinase or pancreatic EIF-2-alpha kinase), FADD, caspase 8 as well as upregulation of ER chaperone GRP78 in the cell surface which serves as a receptor of proapoptotic protein Par-4 (prostate apoptosis response-4) 520–523. Several lines of evidence highlight the crosstalk between ER stress response, ferroptosis, autophagy, and immune regulation of tumor microenvironment that collectively represent an active area of investigation 428,524,525. Further understanding of ER stress and the role of GRP78 in cancer pathogenesis coupled with the development of molecules inhibiting GRP78 are helping validate this chaperone as a relevant therapeutic target in various malignancies including colorectal and pancreatic cancers 526–528.
Intersection between programmed cell death and anti-tumor immune response
In contrast to apoptosis which is considered an immunogenically silent process, other forms of programmed cell death activate pro-inflammatory signaling and promote anti-tumor immune responses including pyroptosis, necroptosis, and caspase-independent cell death (CICD) 529. CICD triggered by mitochondrial outer membrane permeabilization (MOMP) 530 in the setting of caspase inhibition leads to the production of pro-inflammatory cytokines, immune cell tumor infiltration, and potent anti-tumor responses in vivo models 531. In cells with blocked mitochondria-dependent caspase activity (APAF-1 knockdown or caspase-9 CRISPR/Cas9 deletion), MOMP induced NF-kB activation and TNF production through degradation of IAP and upregulation of NF-kB inducing kinase (NIK). MOMP coupled with caspase inhibition allows translocation of mitochondrial DNA (mtDNA) through BAK/BAX macropores to the cytosol where it activates cGAS/STING, promotes type I interferon (IFN) production, and triggers an innate immune response 532–534. This mtDNA-driven immune response during cell death supports the role of caspases in suppressing the immune response during apoptosis. The mechanisms of these immunogenic forms of cell death could be explored therapeutically at intersection points to overcome apoptosis resistance and in conjunction with strategies in the clinic promoting an anti-tumor immune response such as with checkpoint inhibitors. In fact, the recent description that gasdermin E, an established executioner of pyroptosis cleaved by inflammatory caspases, can form pores in the mitochondria, activate caspases 3/7 and promote apoptosis, exemplifies the bridges between these pathways 535. Caspase 8 cleavage of gasdermin D and induction of pro-inflammatory lytic cell death provides another example of cross-talk between apoptosis and pyroptosis 536–538. One could envision interventions able to promote a switch between forms of cell death (i.e., apoptosis to CIDC) to enhance anti-tumor immune responses and/or overcome cell death resistance mechanisms.
Apoptotic priming as a road map to inform therapeutic index and predictive biomarkers
The sensitivity of healthy tissues to apoptosis varies according to histology and developmental stage, and is driven by the relationship between mitochondria and balance among BCl-2 family members. C-Myc-driven transcription of BCL-2 family members in healthy tissues during development plays a central role in the regulation of apoptosis priming 539. Tissues with high proliferation rates such as the developing brain display high levels of proapoptotic proteins such as BAX, BIM and BID making it highly sensitive to proapoptotic stimuli that might contribute to its vulnerability to toxins and drugs. This pattern changes over time with the downregulation of proapoptotic proteins in the post-natal period 540,541. In fact, the expression of BCL-2 family members is markedly reduced in adult mouse brain tissue 539. A similar state of apoptotic priming can be observed in the hematopoietic system, and other tissues such as the gastrointestinal tract have been described as unprimed given the cellular heterogeneity in terms of apoptosis sensitivity 47. These differences in tissue-specific patterns of apoptosis priming have been proposed as contributors of the enhanced susceptibility of pediatric cancer patients to chemotherapy and radiation toxicities as compared to adults 539. Functional assessment of apoptosis priming using BH3 profiling can also help predict the sensitivity of cancer cells (i.e. multiple myeloma, AML, CLL, ALL, and ovarian cancer) to chemotherapy drugs as well as to BH3 mimetics 542. Relevant differences in BH3 profiling between AML myeloblasts with greater BCL-2 dependency as compared to hematopoietic stem cells provided the therapeutic index of venetoclax that was successfully explored in the clinical trials 249,251,543. Distinct dependencies to BCL-2 proteins between cancer cells and normal counterparts have also been demonstrated in T cell development with early progenitor T cells (dependent on BCL-XL) and more mature pre-thymic T cell (dependent on BCL-2) 544. A change in overall dependency of T cell ALL on BCL-XL to BCL-2 in the early T cell progenitor ALL subtype identified the susceptibility of this aggressive subtype of ALL to venetoclax 542. While the BH3-profile and dependencies to specific BCL-2 family members has been well described in hematological malignancies and propelled the development of BCL-2 inhibitors, emerging results suggest strategies to optimize the efficacy of BH3 mimetics in solid tumors. A large screen of solid tumor cell lines revealed that approximately 50% of the cell lines tested displayed co-dependency on BCL-XL and MCL-1, and a marked synergistic efficacy of BCL-XL + MCL-1 inhibitors was observed 318. Relatively rare dependencies to BCL-XL in tumor types such as bladder cancer and pancreatic adenocarcinoma were associated with significant sensitivity to BCL-XL inhibition. Investigation of pathways associated with sensitivity patterns showed that epithelial signatures correlated with co-dependency to BCL-XL and MCL-1, and an EMT signature was associated with BCL-XL dependency in alignment with other reports 443,545. Some tumor types were resistant to all combinations of BCL-2, BCL-XL and MCL-1 inhibitors, but they were sensitive to BFL-1 and BCL-w inhibition. These results highlight potential therapeutic vulnerabilities in solid tumors that can inform the development of tissue-specific combinations of BH3 mimetics, and they demonstrate the value of expression profiling to determine BCL-2 gene dependencies. As novel BH3 mimetics enter the clinic, BH3 profiling assays and/or expression analyses will be critical to define not only cancer cell dependency and predict response, but also to guide the sensitivity of normal tissues to these agents 546. Further development of biomarkers refining the efficacy and safety of novel therapies promoting apoptosis need to incorporate the analysis of apoptosis priming state of healthy tissues for assessment of potential toxicities.
Prospects for advancing the field of apoptosis-inducing agents in cancer therapy
Targeting apoptotic pathways in tumor cells is a potent anti-cancer strategy as dead tumor cells contribute to clinical responses and have no chance to contribute to tumor relapse. While some drugs that directly target the intrinsic apoptosis pathway are approved such as venetoclax, most approved therapeutics that cause cell death target the pathways indirectly. Examples include inhibitors of growth factor signaling pathways, kinase inhibitors, mTOR inhibitors, proteasome inhibitors, or HDAC inhibitors. More direct targeting of the apoptotic pathways is under development with some promising agents having tumor selectivity.
The development of effective therapies targeting cell death pathways is exceedingly complex given the number of therapeutic targets, resistance mechanisms, different standard treatments for different tumor types, and toxicities of single agents or combinations. Some on-target or off-target toxicities are only discovered in clinical trials, and may necessitate development of secondary agents to avoid cardiotoxicity, or thrombocytopenia, by example. Additionally, it is important to bring agents into specific tumor types keeping in mind how those tumors are treated and which patients would be eligible and at what stage. It is typical for novel agents to be tested as salvage therapy for patients with advanced disease who are heavily pre-treated and then advanced in combinations to more front-line settings. Some agents that are promising in phase I trials are brought into phase II trials as combination therapies with an abbreviated phase I portion. A bias in the field has been that if certain agents are inactive in a particular tumor type, clinicians are somewhat reluctant to bring in promising combinations involving such agents that show preclinical synergy into therapeutic trials for that tumor type. Another bias is that some are reluctant to partner standard chemotherapy in novel combinations if the standard agent was used in a prior line of therapy. However, a rational approach would involve overcoming such biases and being more open to try, even on a small scale, promising therapeutic combinations as guided by available preclinical and clinical data.
It is essential to not miss the opportunity to learn from every patient in every clinical trial, and that includes biopsies of tissue, and liquid biopsies before treatment, during response and at progression. The history of clinical trials with apoptosis-inducing agents has missed opportunities to understand why certain agents appeared to fail even though the pathways (e.g., extrinsic cell death) are powerful tumor suppressor mechanisms. It is very important to incorporate candidate biomarkers, including BH3 profiling, as well as some open-ended molecular analysis (genomics, RNA-seq, proteomics, etc) of potential sensitivity and resistance mechanisms in order to continue to make progress. Analysis of tumor heterogeneity and evolution including at the single cell level has evolved to illuminate drug effects as well as the impact of the tumor microenvironment. Novel ways of targeting apoptosis and cell survival mechanisms (e.g. autophagy) in the hypoxic, low pH, nutrient-deprived tumor microenvironment are needed. These strategies must also unravel and target immune escape mechanisms within the tumor microenvironment. Additionally, a rational approach to novel therapy combination would go back to foundational concepts in oncology including using agents with largely non-cross-reacting mechanisms and non-overlapping toxicities. And to best help patients, clinicians need to be armed with back-up therapeutic strategies that exploit the common resistance mechanisms to cellular apoptosis, as well as patient tumor-specific changes that evolve with therapy. Monitoring tumor evolution and its heterogeneity using the latest available technologies such as liquid biopsy, single cell approaches, and imaging is expected to guide and improve cancer therapy targeting apoptotic pathways.
The future development of drugs that directly target apoptotic pathways could lead to tumor regression in difficult to treat tumors, and these drugs would be expected to be used in combination with agents targeting oncogenic survival pathways, immunotherapy, and classical chemotherapy and radiotherapy. Targeting apoptotic pathways remains a promising strategy for cancer therapy that will continue to evolve in future clinical practice.
Table 2.
Therapeutic approaches targeting the apoptosis extrinsic pathway and other cell death mechanisms
| Target | Active clinical trials | Phase | Histology focus | Clinicaltrials.gov ID |
|---|---|---|---|---|
| Death receptor agonists (DR4/5) | ||||
| GEN1029 | Yes | Phase I | Colorectal cancer NSCLC Triple negative breast cancer Renal carcinoma Gastric cancer Pancreatic cancer Urothelial carcinoma |
NCT03576131 |
| ABBV-621 | Yes | Phase I | AML NHL Pancreas cancer |
NCT03082209 |
| MM-201 | No | Sarcoma | ||
| TLY012 | No | Fibrosis | ||
| Strategies targeting p53 | ||||
| MDM2 inhibitors | ||||
| Idasanutlin (RG73882)* | Yes | Phase I | AML Multiple myeloma Breast cancer NHL |
NCT03850535 NCT02670044 NCT02545283 NCT02633059 NCT03566485 NCT03135262 |
| AMG-232 | Yes | Phase I/II | AML Sarcoma Multiple myeloma |
NCT03041688 NCT03217266 NCT03031730 |
| HDM201 ** | Yes | Phase I | AML | NCT03940352 |
| APG-115 | Yes | Phase I | Advanced solid tumors Melanoma NHL Salivary gland |
NCT02935907 NCT03611868 NCT03781986 |
| DS-3032b | Yes | Phase I | AML NHL Advanced solid tumors |
NCT03634228 NCT02319369 NCT01877382 |
| BI 907828 | Yes | Phase I | Advanced solid tumors | NCT03449381 |
| ALRN-6924 (stapled peptide) |
Yes | Phase I | Advanced solid tumors | NCT03725436 |
| Restoring wild type activity of mutant p53 | ||||
| APR246 | Yes | Phase I | AML Ovarian cancer Esophageal carcinoma Melanoma |
NCT02999893 NCT02098343 NCT03588078 NCT03391050 NCT03391050 |
| Other cell death mechanisms | ||||
| ONC201 (binds to dopamine receptors DRD2/DRD3 as well as the mitochondrial caseinolytic protease P (CIpP) leading to activation of integrated stress response protein ATF4 and cell death) | Yes | Phase I/II | Solid tumors Breast cancer Endometrial cancer Colorectal cancer H3K27M Gliomas Multiple myeloma NHL AML |
NCT03099499 NCT02863991 NCT03416530 NCT02420795 NCT03394027 NCT03295396 NCT03791398 NCT02392572 |
| Epigenetic modulators favoring activation of intrinsic pathway of apoptosis | ||||
| Fimepinostat +venetoclax | Yes | Phase I/II | NHL | NCT01742988 |
| Azacytidine or decitabine + venetoclax | Yes | Phase I-III | AML |
NCT03404193 NCT03941964 |
Administered as monotherapy or combined with checkpoint inhibitors)
Administered in combination with venetoclax
Key Points.
There are numerous ways of inducing apoptosis of cancer cells, some more direct than others, exploiting the various entry points into the final common pathway involving caspase-dependent proteolysis of 1000’s of cellular proteins, membrane blebbing and DNAase-dependent endonucleolytic cleavage of chromosomal DNA.
Few FDA-approved anti-cancer drugs act directly on the apoptotic pathway, near the point of execution, and these include small molecules that block anti-apoptotic BCL-2 family members to unleash the effect of proapoptotic BCL-2 family members on mitochondrial membrane permeability and cytochrome c release.
There remain many promising strategies for engaging cell death activation in cancer therapy including strategies that trigger the extrinsic cell death pathway, target tumor suppressor pathways, target the tumor microenvironment including stromal fibroblasts and combinatorial drug therapies.
Various FDA-approved drugs targeting cell survival and cell proliferation pathways in cells, including kinase inhibitors, growth factor targeting antibodies and small molecules such as MAPK pathway inhibitors such as MEK or BRAF inhibitors, mTOR inhibitors, proteasome and HDAC inhibitors, and anti-angiogenic agents, among others, impact less directly on cell death signaling pathways to achieve anti-tumor efficacy.
Immunotherapy whether cell-mediated or antibody-mediated induces apoptosis of cancer cells through granzyme or extrinsic cell death and is currently being combined with numerous other targeted therapies as well as classical chemotherapy or radiation.
Acknowledgements
The literature is vast and in several cases key reviews were cited to point readers to original articles. We apologize to colleagues for not including all possible high impact original references. W.S.E-D. is an American Cancer Society Research Professor, and the Mencoff Family University Professor at Brown University. This work was supported by NCI grants CA173453 and CA176289 to W.S.E-D.
Footnotes
Disclosures: B.A.C. has industry funding for clinical trials from AstraZeneca, Bayer, Astellas, Abbvie, MedImmune, Actuate Therapeutics. W.S.E-D. is the Scientific Founder and Shareholder of Oncoceutics, Inc., and p53-Therapeutics, Inc., and has received support for clinical trials from Bayer, Genentech, and BMS.
REFERENCES
- 1.Ellis HM & Horvitz HR Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829 (1986). [DOI] [PubMed] [Google Scholar]
- 2.Ellis RE, Yuan JY & Horvitz HR Mechanisms and functions of cell death. Annu Rev Cell Biol 7, 663–698, doi: 10.1146/annurev.cb.07.110191.003311 (1991). [DOI] [PubMed] [Google Scholar]
- 3.Varmus HE Nobel lecture. Retroviruses and oncogenes. I. Biosci Rep 10, 413–430, doi: 10.1007/bf01152288 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Wyllie AH Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 284, 555–556, doi: 10.1038/284555a0 (1980). [DOI] [PubMed] [Google Scholar]
- 5.Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE & Poirier GG Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res 53, 3976–3985 (1993). [PubMed] [Google Scholar]
- 6.Tsujimoto Y, Finger LR, Yunis J, Nowell PC & Croce CM Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226, 1097–1099, doi: 10.1126/science.6093263 (1984). [DOI] [PubMed] [Google Scholar]
- 7.Boise LH et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74, 597–608 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Hockenbery D, Nunez G, Milliman C, Schreiber RD & Korsmeyer SJ Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348, 334–336, doi: 10.1038/348334a0 (1990). [DOI] [PubMed] [Google Scholar]
- 9.Rathmell JC & Thompson CB Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell 109 Suppl, S97–107 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Fuchs Y & Steller H Programmed cell death in animal development and disease. Cell 147, 742–758, doi: 10.1016/j.cell.2011.10.033 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fulda S Tumor resistance to apoptosis. Int J Cancer 124, 511–515, doi: 10.1002/ijc.24064 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Thomas S et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther Targets 17, 61–75, doi: 10.1517/14728222.2013.733001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashkenazi A & Salvesen G Regulated cell death: signaling and mechanisms. Annu Rev Cell Dev Biol 30, 337–356, doi: 10.1146/annurev-cellbio-100913-013226 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Green DR & Llambi F Cell Death Signaling. Cold Spring Harb Perspect Biol 7, doi: 10.1101/cshperspect.a006080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagata S & Tanaka M Programmed cell death and the immune system. Nat Rev Immunol 17, 333–340, doi: 10.1038/nri.2016.153 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Li P et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Chao DT & Korsmeyer SJ BCL-2 family: regulators of cell death. Annu Rev Immunol 16, 395–419, doi: 10.1146/annurev.immunol.16.1.395 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Martinou JC & Youle RJ Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 21, 92–101, doi: 10.1016/j.devcel.2011.06.017 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schafer ZT & Kornbluth S The apoptosome: physiological, developmental, and pathological modes of regulation. Dev Cell 10, 549–561, doi: 10.1016/j.devcel.2006.04.008 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Ledgerwood EC & Morison IM Targeting the apoptosome for cancer therapy. Clin Cancer Res 15, 420–424, doi: 10.1158/1078-0432.CCR-08-1172 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Silke J & Meier P Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb Perspect Biol 5, doi: 10.1101/cshperspect.a008730 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhullar KS et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 17, 48, doi: 10.1186/s12943-018-0804-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurokawa M & Kornbluth S Caspases and kinases in a death grip. Cell 138, 838–854, doi: 10.1016/j.cell.2009.08.021 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McIlwain DR, Berger T & Mak TW Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 5, a008656, doi: 10.1101/cshperspect.a008656 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chowdhury D & Lieberman J Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol 26, 389–420, doi: 10.1146/annurev.immunol.26.021607.090404 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saraste A & Pulkki K Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res 45, 528–537, doi: 10.1016/s0008-6363(99)00384-3 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Elmore S Apoptosis: a review of programmed cell death. Toxicol Pathol 35, 495–516, doi: 10.1080/01926230701320337 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG & Earnshaw WC Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371, 346–347, doi: 10.1038/371346a0 (1994). [DOI] [PubMed] [Google Scholar]
- 29.Olofsson MH et al. Cytokeratin-18 is a useful serum biomarker for early determination of response of breast carcinomas to chemotherapy. Clin Cancer Res 13, 3198–3206, doi: 10.1158/1078-0432.CCR-07-0009 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Ulukaya E et al. Chemotherapy increases caspase-cleaved cytokeratin 18 in the serum of breast cancer patients. Radiol Oncol 45, 116–122, doi: 10.2478/v10019-011-0006-7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newbold A, Martin BP, Cullinane C & Bots M Detection of apoptotic cells using immunohistochemistry. Cold Spring Harb Protoc 2014, 1196–1201, doi: 10.1101/pdb.prot082537 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Hameed A, Truong LD, Price V, Kruhenbuhl O & Tschopp J Immunohistochemical localization of granzyme B antigen in cytotoxic cells in human tissues. Am J Pathol 138, 1069–1075 (1991). [PMC free article] [PubMed] [Google Scholar]
- 33.Wlodkowic D, Skommer J & Darzynkiewicz Z Flow cytometry-based apoptosis detection. Methods Mol Biol 559, 19–32, doi: 10.1007/978-1-60327-017-5_2 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu JJ, Wang W, Dicker DT & El-Deiry WS Bioluminescent imaging of TRAIL-induced apoptosis through detection of caspase activation following cleavage of DEVD-aminoluciferin. Cancer Biol Ther 4, 885–892, doi: 10.4161/cbt.4.8.2133 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Blankenberg FG et al. Imaging of apoptosis (programmed cell death) with 99mTc annexin V. J Nucl Med 40, 184–191 (1999). [PubMed] [Google Scholar]
- 36.Larimer BM et al. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res 77, 2318–2327, doi: 10.1158/0008-5472.CAN-16-3346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galluzzi L et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541, doi: 10.1038/s41418-017-0012-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalkavan H & Green DR MOMP, cell suicide as a BCL-2 family business. Cell Death Differ 25, 46–55, doi: 10.1038/cdd.2017.179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin Z & El-Deiry WS Overview of cell death signaling pathways. Cancer Biol Ther 4, 139–163, doi: 10.4161/cbt.4.2.1508 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Kim CN, Yang J, Jemmerson R & Wang X Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157, doi: 10.1016/s0092-8674(00)80085-9 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Bhola PD & Letai A Mitochondria-Judges and Executioners of Cell Death Sentences. Mol Cell 61, 695–704, doi: 10.1016/j.molcel.2016.02.019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132, doi: 10.1126/science.275.5303.1129 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Kharbanda S et al. Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc Natl Acad Sci U S A 94, 6939–6942, doi: 10.1073/pnas.94.13.6939 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nijhawan D et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev 17, 1475–1486, doi: 10.1101/gad.1093903 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bleicken S et al. Structural model of active Bax at the membrane. Mol Cell 56, 496–505, doi: 10.1016/j.molcel.2014.09.022 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borner C & Andrews DW The apoptotic pore on mitochondria: are we breaking through or still stuck? Cell Death Differ 21, 187–191, doi: 10.1038/cdd.2013.169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh R, Letai A & Sarosiek K Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol 20, 175–193, doi: 10.1038/s41580-018-0089-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zha H, Aime-Sempe C, Sato T & Reed JC Proapoptotic protein Bax heterodimerizes with Bcl-2 and homodimerizes with Bax via a novel domain (BH3) distinct from BH1 and BH2. J Biol Chem 271, 7440–7444, doi: 10.1074/jbc.271.13.7440 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Wang K, Yin XM, Chao DT, Milliman CL & Korsmeyer SJ BID: a novel BH3 domain-only death agonist. Genes Dev 10, 2859–2869, doi: 10.1101/gad.10.22.2859 (1996). [DOI] [PubMed] [Google Scholar]
- 50.O’Connor L et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J 17, 384–395, doi: 10.1093/emboj/17.2.384 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Letai A et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Cheng EH et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8, 705–711, doi: 10.1016/s1097-2765(01)00320-3 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Kuwana T et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331–342, doi: 10.1016/s0092-8674(02)01036-x (2002). [DOI] [PubMed] [Google Scholar]
- 54.Nakano K & Vousden KH PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7, 683–694, doi: 10.1016/s1097-2765(01)00214-3 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Yu J, Zhang L, Hwang PM, Kinzler KW & Vogelstein B PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7, 673–682, doi: 10.1016/s1097-2765(01)00213-1 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Dai H et al. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol 194, 39–48, doi: 10.1083/jcb.201102027 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim H et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 36, 487–499, doi: 10.1016/j.molcel.2009.09.030 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang E et al. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80, 285–291, doi: 10.1016/0092-8674(95)90411-5 (1995). [DOI] [PubMed] [Google Scholar]
- 59.Inohara N, Ding L, Chen S & Nunez G harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-X(L). EMBO J 16, 1686–1694, doi: 10.1093/emboj/16.7.1686 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Youle RJ & Strasser A The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9, 47–59, doi: 10.1038/nrm2308 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Czabotar PE, Lessene G, Strasser A & Adams JM Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15, 49–63, doi: 10.1038/nrm3722 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Del Gaizo Moore V & Letai A BH3 profiling--measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett 332, 202–205, doi: 10.1016/j.canlet.2011.12.021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Certo M et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365, doi: 10.1016/j.ccr.2006.03.027 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Kale J, Osterlund EJ & Andrews DW BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25, 65–80, doi: 10.1038/cdd.2017.186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cimmino A et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 102, 13944–13949, doi: 10.1073/pnas.0506654102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Inuzuka H et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471, 104–109, doi: 10.1038/nature09732 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Del Re DP et al. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol Cell 54, 639–650, doi: 10.1016/j.molcel.2014.04.007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hubner A, Barrett T, Flavell RA & Davis RJ Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell 30, 415–425, doi: 10.1016/j.molcel.2008.03.025 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gardai SJ et al. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem 279, 21085–21095, doi: 10.1074/jbc.M400063200 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Edlich F et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145, 104–116, doi: 10.1016/j.cell.2011.02.034 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gojo I, Zhang B & Fenton RG The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin Cancer Res 8, 3527–3538 (2002). [PubMed] [Google Scholar]
- 72.Chen S et al. CDK inhibitors upregulate BH3-only proteins to sensitize human myeloma cells to BH3 mimetic therapies. Cancer Res 72, 4225–4237, doi: 10.1158/0008-5472.CAN-12-1118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lowman XH et al. The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol Cell 40, 823–833, doi: 10.1016/j.molcel.2010.11.035 (2010). [DOI] [PubMed] [Google Scholar]
- 74.Kour S et al. CDK5 Inhibitor Downregulates Mcl-1 and Sensitizes Pancreatic Cancer Cell Lines to Navitoclax. Mol Pharmacol 96, 419–429, doi: 10.1124/mol.119.116855 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deveraux QL & Reed JC IAP family proteins--suppressors of apoptosis. Genes Dev. 13, 239–252 (1999). [DOI] [PubMed] [Google Scholar]
- 76.Liston P, Fong WG & Korneluk RG The inhibitors of apoptosis: there is more to life than Bcl2. Oncogene 22, 8568–8580, doi: 10.1038/sj.onc.1207101 (2003). [DOI] [PubMed] [Google Scholar]
- 77.van Loo G et al. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ 9, 1031–1042, doi: 10.1038/sj.cdd.4401088 (2002). [DOI] [PubMed] [Google Scholar]
- 78.Warr MR & Shore GC Small-molecule Bcl-2 antagonists as targeted therapy in oncology. Curr Oncol 15, 256–261, doi: 10.3747/co.v15i6.392 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vogler M, Dinsdale D, Dyer MJ & Cohen GM Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ 16, 360–367, doi: 10.1038/cdd.2008.137 (2009). [DOI] [PubMed] [Google Scholar]
- 80.Stauffer SR Small molecule inhibition of the Bcl-X(L)-BH3 protein-protein interaction: proof-of-concept of an in vivo chemopotentiator ABT-737. Curr Top Med Chem 7, 961–965 (2007). [DOI] [PubMed] [Google Scholar]
- 81.Manion MK, Fry J, Schwartz PS & Hockenbery DM Small-molecule inhibitors of Bcl-2. Curr Opin Investig Drugs 7, 1077–1084 (2006). [PubMed] [Google Scholar]
- 82.Park D et al. Novel small-molecule inhibitors of Bcl-XL to treat lung cancer. Cancer Res 73, 5485–5496, doi: 10.1158/0008-5472.CAN-12-2272 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Belmar J & Fesik SW Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther 145, 76–84, doi: 10.1016/j.pharmthera.2014.08.003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wan Y, Dai N, Tang Z & Fang H Small-molecule Mcl-1 inhibitors: Emerging anti-tumor agents. Eur J Med Chem 146, 471–482, doi: 10.1016/j.ejmech.2018.01.076 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Caenepeel S et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov 8, 1582–1597, doi: 10.1158/2159-8290.CD-18-0387 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Hunter AM, LaCasse EC & Korneluk RG The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 12, 1543–1568, doi: 10.1007/s10495-007-0087-3 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Rathore R, McCallum JE, Varghese E, Florea AM & Busselberg D Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 22, 898–919, doi: 10.1007/s10495-017-1375-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Derakhshan A, Chen Z & Van Waes C Therapeutic Small Molecules Target Inhibitor of Apoptosis Proteins in Cancers with Deregulation of Extrinsic and Intrinsic Cell Death Pathways. Clin Cancer Res 23, 1379–1387, doi: 10.1158/1078-0432.CCR-16-2172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Besbes S, Mirshahi M, Pocard M & Billard C New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget 6, 12862–12871, doi: 10.18632/oncotarget.3868 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Walensky LD & Bird GH Hydrocarbon-stapled peptides: principles, practice, and progress. J Med Chem 57, 6275–6288, doi: 10.1021/jm4011675 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee S, Braun CR, Bird GH & Walensky LD Photoreactive stapled peptides to identify and characterize BCL-2 family interaction sites by mass spectrometry. Methods Enzymol 544, 25–48, doi: 10.1016/B978-0-12-417158-9.00002-9 (2014). [DOI] [PubMed] [Google Scholar]
- 92.Adams JM Therapeutic potential of a peptide targeting BCL-2 cell guardians in cancer. J Clin Invest 122, 1965–1967, doi: 10.1172/JCI64120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rezaei Araghi R et al. Iterative optimization yields Mcl-1-targeting stapled peptides with selective cytotoxicity to Mcl-1-dependent cancer cells. Proc Natl Acad Sci U S A 115, E886–E895, doi: 10.1073/pnas.1712952115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Boddu P, Carter BZ, Verstovsek S & Pemmaraju N SMAC mimetics as potential cancer therapeutics in myeloid malignancies. Br J Haematol 185, 219–231, doi: 10.1111/bjh.15829 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Fulda S Promises and Challenges of Smac Mimetics as Cancer Therapeutics. Clin Cancer Res 21, 5030–5036, doi: 10.1158/1078-0432.CCR-15-0365 (2015). [DOI] [PubMed] [Google Scholar]
- 96.Adams JM & Cory S The Bcl-2 protein family: arbiters of cell survival. Science 281, 1322–1326 (1998). [DOI] [PubMed] [Google Scholar]
- 97.Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE & Oltvai ZN Bcl-2/Bax: a rheostat that regulates an anti-oxidant pathway and cell death. Semin Cancer Biol 4, 327–332 (1993). [PubMed] [Google Scholar]
- 98.Miyashita T & Reed JC Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80, 293–299 (1995). [DOI] [PubMed] [Google Scholar]
- 99.Kastenhuber ER & Lowe SW Putting p53 in Context. Cell 170, 1062–1078, doi: 10.1016/j.cell.2017.08.028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chang HY & Yang X Proteases for cell suicide: functions and regulation of caspases. Microbiol Mol Biol Rev 64, 821–846, doi: 10.1128/mmbr.64.4.821-846.2000 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.MacEwan DJ TNF ligands and receptors--a matter of life and death. Br J Pharmacol 135, 855–875, doi: 10.1038/sj.bjp.0704549 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Itoh N et al. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66, 233–243 (1991). [DOI] [PubMed] [Google Scholar]
- 103.Locksley RM, Killeen N & Lenardo MJ The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104, 487–501 (2001). [DOI] [PubMed] [Google Scholar]
- 104.Hehlgans T & Pfeffer K The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology 115, 1–20, doi: 10.1111/j.1365-2567.2005.02143.x (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pan G et al. The receptor for the cytotoxic ligand TRAIL. Science 276, 111–113, doi: 10.1126/science.276.5309.111 (1997). [DOI] [PubMed] [Google Scholar]
- 106.Schneider P et al. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 7, 831–836 (1997). [DOI] [PubMed] [Google Scholar]
- 107.Wu GS et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 17, 141–143, doi: 10.1038/ng1097-141 (1997). [DOI] [PubMed] [Google Scholar]
- 108.Walczak H et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J 16, 5386–5397, doi: 10.1093/emboj/16.17.5386 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chaudhary PM et al. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 7, 821–830 (1997). [DOI] [PubMed] [Google Scholar]
- 110.MacFarlane M et al. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem 272, 25417–25420, doi: 10.1074/jbc.272.41.25417 (1997). [DOI] [PubMed] [Google Scholar]
- 111.Hymowitz SG et al. Triggering cell death: the crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol Cell 4, 563–571 (1999). [DOI] [PubMed] [Google Scholar]
- 112.Strasser A, Jost PJ & Nagata S The many roles of FAS receptor signaling in the immune system. Immunity 30, 180–192, doi: 10.1016/j.immuni.2009.01.001 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Katagiri T, Cohen PL & Eisenberg RA The lpr gene causes an intrinsic T cell abnormality that is required for hyperproliferation. J Exp Med 167, 741–751, doi: 10.1084/jem.167.3.741 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA & Nagata S Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356, 314–317, doi: 10.1038/356314a0 (1992). [DOI] [PubMed] [Google Scholar]
- 115.Nagata S Apoptosis by death factor. Cell 88, 355–365, doi: 10.1016/s0092-8674(00)81874-7 (1997). [DOI] [PubMed] [Google Scholar]
- 116.Nagata S & Golstein P The Fas death factor. Science 267, 1449–1456, doi: 10.1126/science.7533326 (1995). [DOI] [PubMed] [Google Scholar]
- 117.Schneider P et al. Characterization of Fas (Apo-1, CD95)-Fas ligand interaction. J Biol Chem 272, 18827–18833, doi: 10.1074/jbc.272.30.18827 (1997). [DOI] [PubMed] [Google Scholar]
- 118.Pennica D et al. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature 312, 724–729, doi: 10.1038/312724a0 (1984). [DOI] [PubMed] [Google Scholar]
- 119.Wiley SR et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3, 673–682 (1995). [DOI] [PubMed] [Google Scholar]
- 120.Johnstone RW, Frew AJ & Smyth MJ The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer 8, 782–798, doi: 10.1038/nrc2465 (2008). [DOI] [PubMed] [Google Scholar]
- 121.Feinstein E, Kimchi A, Wallach D, Boldin M & Varfolomeev E The death domain: a module shared by proteins with diverse cellular functions. Trends Biochem Sci 20, 342–344 (1995). [DOI] [PubMed] [Google Scholar]
- 122.Huang B, Eberstadt M, Olejniczak ET, Meadows RP & Fesik SW NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 384, 638–641, doi: 10.1038/384638a0 (1996). [DOI] [PubMed] [Google Scholar]
- 123.Lavrik I, Golks A & Krammer PH Death receptor signaling. J Cell Sci 118, 265–267, doi: 10.1242/jcs.01610 (2005). [DOI] [PubMed] [Google Scholar]
- 124.Walczak H Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol 5, a008698, doi: 10.1101/cshperspect.a008698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guicciardi ME & Gores GJ Life and death by death receptors. FASEB J 23, 1625–1637, doi: 10.1096/fj.08-111005 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Irmler M et al. Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195, doi: 10.1038/40657 (1997). [DOI] [PubMed] [Google Scholar]
- 127.Billen LP, Shamas-Din A & Andrews DW Bid: a Bax-like BH3 protein. Oncogene 27 Suppl 1, S93–104, doi: 10.1038/onc.2009.47 (2008). [DOI] [PubMed] [Google Scholar]
- 128.Sax JK et al. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol 4, 842–849, doi: 10.1038/ncb866 (2002). [DOI] [PubMed] [Google Scholar]
- 129.Algeciras-Schimnich A et al. Molecular ordering of the initial signaling events of CD95. Mol Cell Biol 22, 207–220, doi: 10.1128/mcb.22.1.207-220.2002 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ozoren N & El-Deiry WS Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia 4, 551–557, doi: 10.1038/sj.neo.7900270 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pitti RM et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature 396, 699–703, doi: 10.1038/25387 (1998). [DOI] [PubMed] [Google Scholar]
- 132.LeBlanc HN & Ashkenazi A Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ 10, 66–75, doi: 10.1038/sj.cdd.4401187 (2003). [DOI] [PubMed] [Google Scholar]
- 133.Crowder RN, Dicker DT & El-Deiry WS The Deubiquitinase Inhibitor PR-619 Sensitizes Normal Human Fibroblasts to Tumor Necrosis Factor-related Apoptosis-inducing Ligand (TRAIL)-mediated Cell Death. J Biol Chem 291, 5960–5970, doi: 10.1074/jbc.M115.713545 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang L et al. Accelerated degradation of caspase-8 protein correlates with TRAIL resistance in a DLD1 human colon cancer cell line. Neoplasia 7, 594–602, doi: 10.1593/neo.04688 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hayden MS & Ghosh S Regulation of NF-kappaB by TNF family cytokines. Semin Immunol 26, 253–266, doi: 10.1016/j.smim.2014.05.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yin XM et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400, 886–891, doi: 10.1038/23730 (1999). [DOI] [PubMed] [Google Scholar]
- 137.van Horssen R, Ten Hagen TL & Eggermont AM TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist 11, 397–408, doi: 10.1634/theoncologist.11-4-397 (2006). [DOI] [PubMed] [Google Scholar]
- 138.Watanabe N et al. Synergistic cytotoxicity of recombinant human TNF and various anti-cancer drugs. Immunopharmacol Immunotoxicol 10, 117–127, doi: 10.3109/08923978809014406 (1988). [DOI] [PubMed] [Google Scholar]
- 139.Ruggiero V, Latham K & Baglioni C Cytostatic and cytotoxic activity of tumor necrosis factor on human cancer cells. J Immunol 138, 2711–2717 (1987). [PubMed] [Google Scholar]
- 140.Watanabe N et al. Toxic effect of tumor necrosis factor on tumor vasculature in mice. Cancer Res 48, 2179–2183 (1988). [PubMed] [Google Scholar]
- 141.von Karstedt S et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell 27, 561–573, doi: 10.1016/j.ccell.2015.02.014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hartwig T et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol Cell 65, 730–742 e735, doi: 10.1016/j.molcel.2017.01.021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.el-Deiry WS Regulation of p53 downstream genes. Semin Cancer Biol 8, 345–357 (1998). [DOI] [PubMed] [Google Scholar]
- 144.Finnberg N et al. DR5 knockout mice are compromised in radiation-induced apoptosis. Mol Cell Biol 25, 2000–2013, doi: 10.1128/MCB.25.5.2000-2013.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Finnberg N, Klein-Szanto AJ & El-Deiry WS TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J Clin Invest 118, 111–123, doi: 10.1172/JCI29900 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Raychaudhuri S, Willgohs E, Nguyen TN, Khan EM & Goldkorn T Monte Carlo simulation of cell death signaling predicts large cell-to-cell stochastic fluctuations through the type 2 pathway of apoptosis. Biophys J 95, 3559–3562, doi: 10.1529/biophysj.108.135483 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Spencer SL & Sorger PK Measuring and modeling apoptosis in single cells. Cell 144, 926–939, doi: 10.1016/j.cell.2011.03.002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Spencer SL, Gaudet S, Albeck JG, Burke JM & Sorger PK Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459, 428–432, doi: 10.1038/nature08012 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Santos LC et al. Mitochondrial origins of fractional control in regulated cell death. Nat Commun 10, 1313, doi: 10.1038/s41467-019-09275-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Green DR The Coming Decade of Cell Death Research: Five Riddles. Cell 177, 1094–1107, doi: 10.1016/j.cell.2019.04.024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dennis PA & Kastan MB Cellular survival pathways and resistance to cancer therapy. Drug Resist Updat 1, 301–309 (1998). [DOI] [PubMed] [Google Scholar]
- 152.Maddika S et al. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat 10, 13–29, doi: 10.1016/j.drup.2007.01.003 (2007). [DOI] [PubMed] [Google Scholar]
- 153.Boroughs LK & DeBerardinis RJ Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17, 351–359, doi: 10.1038/ncb3124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Baig S et al. Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand? Cell Death Dis 7, e2058, doi: 10.1038/cddis.2015.275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wong RS Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res 30, 87, doi: 10.1186/1756-9966-30-87 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hassan M, Watari H, AbuAlmaaty A, Ohba Y & Sakuragi N Apoptosis and molecular targeting therapy in cancer. Biomed Res Int 2014, 150845, doi: 10.1155/2014/150845 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 157.Levy DS, Kahana JA & Kumar R AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood 113, 1723–1729, doi: 10.1182/blood-2008-02-137737 (2009). [DOI] [PubMed] [Google Scholar]
- 158.de Frias M et al. Akt inhibitors induce apoptosis in chronic lymphocytic leukemia cells. Haematologica 94, 1698–1707, doi: 10.3324/haematol.2008.004028 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yang F et al. Feedback loops blockade potentiates apoptosis induction and antitumor activity of a novel AKT inhibitor DC120 in human liver cancer. Cell Death Dis 5, e1114, doi: 10.1038/cddis.2014.43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang X, Martindale JL & Holbrook NJ Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem 275, 39435–39443, doi: 10.1074/jbc.M004583200 (2000). [DOI] [PubMed] [Google Scholar]
- 161.Will M et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov 4, 334–347, doi: 10.1158/2159-8290.Cd-13-0611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lin L, Ding D, Jiang Y, Li Y & Li S MEK inhibitors induce apoptosis via FoxO3a-dependent PUMA induction in colorectal cancer cells. Oncogenesis 7, 67, doi: 10.1038/s41389-018-0078-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jasinski P, Zwolak P, Terai K & Dudek AZ Novel Ras pathway inhibitor induces apoptosis and growth inhibition of K-ras-mutated cancer cells in vitro and in vivo. Transl Res 152, 203–212, doi: 10.1016/j.trsl.2008.09.001 (2008). [DOI] [PubMed] [Google Scholar]
- 164.Lebowitz PF, Sakamuro D & Prendergast GC Farnesyl transferase inhibitors induce apoptosis of Ras-transformed cells denied substratum attachment. Cancer Res 57, 708–713 (1997). [PubMed] [Google Scholar]
- 165.Fujiwara D et al. Statins induce apoptosis through inhibition of Ras signaling pathways and enhancement of Bim and p27 expression in human hematopoietic tumor cells. Tumour Biol 39, 1010428317734947, doi: 10.1177/1010428317734947 (2017). [DOI] [PubMed] [Google Scholar]
- 166.Janes MR et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589 e517, doi: 10.1016/j.cell.2018.01.006 (2018). [DOI] [PubMed] [Google Scholar]
- 167.Jiang CC et al. Apoptosis of human melanoma cells induced by inhibition of B-RAFV600E involves preferential splicing of bimS. Cell Death Dis 1, e69, doi: 10.1038/cddis.2010.48 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Geserick P, Herlyn M & Leverkus M On the TRAIL to overcome BRAF-inhibitor resistance. J Invest Dermatol 134, 315–318, doi: 10.1038/jid.2013.348 (2014). [DOI] [PubMed] [Google Scholar]
- 169.Tsai J et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A 105, 3041–3046, doi: 10.1073/pnas.0711741105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Beck D et al. Vemurafenib potently induces endoplasmic reticulum stress-mediated apoptosis in BRAFV600E melanoma cells. Sci Signal 6, ra7, doi: 10.1126/scisignal.2003057 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Niessner H et al. BRAF Inhibitors Amplify the Proapoptotic Activity of MEK Inhibitors by Inducing ER Stress in NRAS-Mutant Melanoma. Clin Cancer Res 23, 6203–6214, doi: 10.1158/1078-0432.CCR-17-0098 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.He K, Zheng X, Li M, Zhang L & Yu J mTOR inhibitors induce apoptosis in colon cancer cells via CHOP-dependent DR5 induction on 4E-BP1 dephosphorylation. Oncogene 35, 148–157, doi: 10.1038/onc.2015.79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Blunt MD et al. The PI3K/mTOR inhibitor PF-04691502 induces apoptosis and inhibits microenvironmental signaling in CLL and the Emicro-TCL1 mouse model. Blood 125, 4032–4041, doi: 10.1182/blood-2014-11-610329 (2015). [DOI] [PubMed] [Google Scholar]
- 174.Zeng Z et al. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood 120, 2679–2689, doi: 10.1182/blood-2011-11-393934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Preuss E, Hugle M, Reimann R, Schlecht M & Fulda S Pan-mammalian target of rapamycin (mTOR) inhibitor AZD8055 primes rhabdomyosarcoma cells for ABT-737-induced apoptosis by down-regulating Mcl-1 protein. J Biol Chem 288, 35287–35296, doi: 10.1074/jbc.M113.495986 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Goel S, Hidalgo M & Perez-Soler R EGFR inhibitor-mediated apoptosis in solid tumors. J Exp Ther Oncol 6, 305–320 (2007). [PubMed] [Google Scholar]
- 177.Costa DB et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med 4, 1669–1679; discussion 1680, doi: 10.1371/journal.pmed.0040315 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ariyama H et al. Gefitinib, a selective EGFR tyrosine kinase inhibitor, induces apoptosis through activation of Bax in human gallbladder adenocarcinoma cells. J Cell Biochem 97, 724–734, doi: 10.1002/jcb.20678 (2006). [DOI] [PubMed] [Google Scholar]
- 179.Okamoto K et al. Role of survivin in EGFR inhibitor-induced apoptosis in non-small cell lung cancers positive for EGFR mutations. Cancer Res 70, 10402–10410, doi: 10.1158/0008-5472.CAN-10-2438 (2010). [DOI] [PubMed] [Google Scholar]
- 180.Sun Q et al. PUMA mediates EGFR tyrosine kinase inhibitor-induced apoptosis in head and neck cancer cells. Oncogene 28, 2348–2357, doi: 10.1038/onc.2009.108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hognason T et al. Epidermal growth factor receptor induced apoptosis: potentiation by inhibition of Ras signaling. FEBS Lett 491, 9–15, doi: 10.1016/s0014-5793(01)02166-4 (2001). [DOI] [PubMed] [Google Scholar]
- 182.Carpenter RL & Lo HW Regulation of Apoptosis by HER2 in Breast Cancer. J Carcinog Mutagen 2013, doi: 10.4172/2157-2518.S7-003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fink MY & Chipuk JE Survival of HER2-Positive Breast Cancer Cells: Receptor Signaling to Apoptotic Control Centers. Genes Cancer 4, 187–195, doi: 10.1177/1947601913488598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Faber AC et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A 106, 19503–19508, doi: 10.1073/pnas.0905056106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nahta R, Yuan LX, Du Y & Esteva FJ Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol Cancer Ther 6, 667–674, doi: 10.1158/1535-7163.MCT-06-0423 (2007). [DOI] [PubMed] [Google Scholar]
- 186.Henson ES, Hu X & Gibson SB Herceptin sensitizes ErbB2-overexpressing cells to apoptosis by reducing antiapoptotic Mcl-1 expression. Clin Cancer Res 12, 845–853, doi: 10.1158/1078-0432.CCR-05-0754 (2006). [DOI] [PubMed] [Google Scholar]
- 187.Fleming IN, Hogben M, Frame S, McClue SJ & Green SR Synergistic inhibition of ErbB signaling by combined treatment with seliciclib and ErbB-targeting agents. Clin Cancer Res 14, 4326–4335, doi: 10.1158/1078-0432.CCR-07-4633 (2008). [DOI] [PubMed] [Google Scholar]
- 188.Lee H, Lee H, Chin H, Kim K & Lee D ERBB3 knockdown induces cell cycle arrest and activation of Bak and Bax-dependent apoptosis in colon cancer cells. Oncotarget 5, 5138–5152, doi: 10.18632/oncotarget.2094 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jung HY, Joo HJ, Park JK & Kim YH The Blocking of c-Met Signaling Induces Apoptosis through the Increase of p53 Protein in Lung Cancer. Cancer Res Treat 44, 251–261, doi: 10.4143/crt.2012.44.4.251 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sattler M et al. A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res 63, 5462–5469 (2003). [PubMed] [Google Scholar]
- 191.Fuse MA et al. Combination Therapy with c-Met and Src Inhibitors Induces Caspase-Dependent Apoptosis of Merlin-Deficient Schwann Cells and Suppresses Growth of Schwannoma Cells. Mol Cancer Ther 16, 2387–2398, doi: 10.1158/1535-7163.MCT-17-0417 (2017). [DOI] [PubMed] [Google Scholar]
- 192.Dai L et al. Targeting HGF/c-MET induces cell cycle arrest, DNA damage, and apoptosis for primary effusion lymphoma. Blood 126, 2821–2831, doi: 10.1182/blood-2015-07-658823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liu Y et al. Inhibition of c-Met promoted apoptosis, autophagy and loss of the mitochondrial transmembrane potential in oridonin-induced A549 lung cancer cells. J Pharm Pharmacol 65, 1622–1642, doi: 10.1111/jphp.12140 (2013). [DOI] [PubMed] [Google Scholar]
- 194.Cocco E, Scaltriti M & Drilon A NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 15, 731–747, doi: 10.1038/s41571-018-0113-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ricciuti B, Genova C, Crino L, Libra M & Leonardi GC Antitumor activity of larotrectinib in tumors harboring NTRK gene fusions: a short review on the current evidence. Onco Targets Ther 12, 3171–3179, doi: 10.2147/OTT.S177051 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Smith KM et al. Antitumor Activity of Entrectinib, a Pan-TRK, ROS1, and ALK Inhibitor, in ETV6-NTRK3-Positive Acute Myeloid Leukemia. Mol Cancer Ther 17, 455–463, doi: 10.1158/1535-7163.MCT-17-0419 (2018). [DOI] [PubMed] [Google Scholar]
- 197.Faber AC et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov 1, 352–365, doi: 10.1158/2159-8290.CD-11-0106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ley R, Balmanno K, Hadfield K, Weston C & Cook SJ Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem 278, 18811–18816, doi: 10.1074/jbc.M301010200 (2003). [DOI] [PubMed] [Google Scholar]
- 199.Datta SR et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241 (1997). [DOI] [PubMed] [Google Scholar]
- 200.Bonni A et al. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286, 1358–1362 (1999). [DOI] [PubMed] [Google Scholar]
- 201.Hata AN et al. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers. Cancer Res 74, 3146–3156, doi: 10.1158/0008-5472.CAN-13-3728 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Nangia V et al. Exploiting MCL1 Dependency with Combination MEK + MCL1 Inhibitors Leads to Induction of Apoptosis and Tumor Regression in KRAS-Mutant Non-Small Cell Lung Cancer. Cancer Discov 8, 1598–1613, doi: 10.1158/2159-8290.CD-18-0277 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.White E The role for autophagy in cancer. J Clin Invest 125, 42–46, doi: 10.1172/jci73941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Levy JMM, Towers CG & Thorburn A Targeting autophagy in cancer. Nat Rev Cancer 17, 528–542, doi: 10.1038/nrc.2017.53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Thorburn A, Thamm DH & Gustafson DL Autophagy and cancer therapy. Mol Pharmacol 85, 830–838, doi: 10.1124/mol.114.091850 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Amaravadi R, Kimmelman AC & White E Recent insights into the function of autophagy in cancer. Genes Dev 30, 1913–1930, doi: 10.1101/gad.287524.116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chude CI & Amaravadi RK Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int J Mol Sci 18, doi: 10.3390/ijms18061279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Galluzzi L & Green DR Autophagy-Independent Functions of the Autophagy Machinery. Cell 177, 1682–1699, doi: 10.1016/j.cell.2019.05.026 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dixon SJ et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072, doi: 10.1016/j.cell.2012.03.042 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Stockwell BR et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285, doi: 10.1016/j.cell.2017.09.021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Friedmann Angeli JP, Krysko DV & Conrad M Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer 19, 405–414, doi: 10.1038/s41568-019-0149-1 (2019). [DOI] [PubMed] [Google Scholar]
- 212.Yang WS et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331, doi: 10.1016/j.cell.2013.12.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tsoi J et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904 e895, doi: 10.1016/j.ccell.2018.03.017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hassannia B et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest 128, 3341–3355, doi: 10.1172/jci99032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hao S et al. Cysteine Dioxygenase 1 Mediates Erastin-Induced Ferroptosis in Human Gastric Cancer Cells. Neoplasia 19, 1022–1032, doi: 10.1016/j.neo.2017.10.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Baumann C, Ullrich A & Torka R GAS6-expressing and self-sustaining cancer cells in 3D spheroids activate the PDK-RSK-mTOR pathway for survival and drug resistance. Mol Oncol 11, 1430–1447, doi: 10.1002/1878-0261.12109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hangauer MJ et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250, doi: 10.1038/nature24297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Shimada K et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol 12, 497–503, doi: 10.1038/nchembio.2079 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Viswanathan VS et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457, doi: 10.1038/nature23007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Guan J et al. The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: use of sulfasalazine. Cancer Chemother Pharmacol 64, 463–472, doi: 10.1007/s00280-008-0894-4 (2009). [DOI] [PubMed] [Google Scholar]
- 221.Kim SE et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol 11, 977–985, doi: 10.1038/nnano.2016.164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zhao Y, Zhao W, Lim YC & Liu T Salinomycin-Loaded Gold Nanoparticles for Treating Cancer Stem Cells by Ferroptosis-Induced Cell Death. Mol Pharm 16, 2532–2539, doi: 10.1021/acs.molpharmaceut.9b00132 (2019). [DOI] [PubMed] [Google Scholar]
- 223.Jiang L et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62, doi: 10.1038/nature14344 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Li T et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283, doi: 10.1016/j.cell.2012.04.026 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Wang SJ et al. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep 17, 366–373, doi: 10.1016/j.celrep.2016.09.022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tarangelo A et al. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep 22, 569–575, doi: 10.1016/j.celrep.2017.12.077 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hassannia B, Vandenabeele P & Vanden Berghe T Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 35, 830–849, doi: 10.1016/j.ccell.2019.04.002 (2019). [DOI] [PubMed] [Google Scholar]
- 228.Pekarsky Y, Balatti V & Croce CM BCL2 and miR-15/16: from gene discovery to treatment. Cell Death Differ 25, 21–26, doi: 10.1038/cdd.2017.159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Oltersdorf T et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681, doi: 10.1038/nature03579 (2005). [DOI] [PubMed] [Google Scholar]
- 230.Del Gaizo Moore V et al. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest 117, 112–121, doi: 10.1172/JCI28281 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tsao T et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann Hematol 91, 1861–1870, doi: 10.1007/s00277-012-1537-8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Tse C et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68, 3421–3428, doi: 10.1158/0008-5472.CAN-07-5836 (2008). [DOI] [PubMed] [Google Scholar]
- 233.Roberts AW et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol 30, 488–496, doi: 10.1200/JCO.2011.34.7898 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kipps TJ et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk Lymphoma 56, 2826–2833, doi: 10.3109/10428194.2015.1030638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02079740 (2019). [DOI] [PubMed]
- 236.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02520778 (2019). [DOI] [PubMed]
- 237.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT01989585 (2019). [DOI] [PubMed]
- 238.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02143401 (2019). [DOI] [PubMed]
- 239.Souers AJ et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19, 202–208, doi: 10.1038/nm.3048 (2013). [DOI] [PubMed] [Google Scholar]
- 240.Roberts AW et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med 374, 311–322, doi: 10.1056/NEJMoa1513257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Davids MS et al. Revised Dose Ramp-Up to Mitigate the Risk of Tumor Lysis Syndrome When Initiating Venetoclax in Patients With Mantle Cell Lymphoma. J Clin Oncol, JCO1800359, doi: 10.1200/JCO.18.00359 (2018). [DOI] [PubMed]
- 242.Roberts AW et al. Efficacy of venetoclax in relapsed chronic lymphocytic leukemia is influenced by disease and response variables. Blood 134, 111–122, doi: 10.1182/blood.2018882555 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Stilgenbauer S et al. Venetoclax for Patients With Chronic Lymphocytic Leukemia With 17p Deletion: Results From the Full Population of a Phase II Pivotal Trial. J Clin Oncol 36, 1973–1980, doi: 10.1200/JCO.2017.76.6840 (2018). [DOI] [PubMed] [Google Scholar]
- 244.Seymour JF et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N Engl J Med 378, 1107–1120, doi: 10.1056/NEJMoa1713976 (2018). [DOI] [PubMed] [Google Scholar]
- 245.Fischer K et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N Engl J Med 380, 2225–2236, doi: 10.1056/NEJMoa1815281 (2019). [DOI] [PubMed] [Google Scholar]
- 246.Jain N et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N Engl J Med 380, 2095–2103, doi: 10.1056/NEJMoa1900574 (2019). [DOI] [PubMed] [Google Scholar]
- 247.Zelenetz AD et al. Venetoclax plus R- or G-CHOP in non-Hodgkin lymphoma: results from the CAVALLI phase 1b trial. Blood 133, 1964–1976, doi: 10.1182/blood-2018-11-880526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Pan R et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 4, 362–375, doi: 10.1158/2159-8290.CD-13-0609 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Konopleva M et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 10, 375–388, doi: 10.1016/j.ccr.2006.10.006 (2006). [DOI] [PubMed] [Google Scholar]
- 250.Beurlet S et al. BCL-2 inhibition with ABT-737 prolongs survival in an NRAS/BCL-2 mouse model of AML by targeting primitive LSK and progenitor cells. Blood 122, 2864–2876, doi: 10.1182/blood-2012-07-445635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Konopleva M et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov 6, 1106–1117, doi: 10.1158/2159-8290.CD-16-0313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Wei AH et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J Clin Oncol 37, 1277–1284, doi: 10.1200/JCO.18.01600 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Kantarjian H et al. Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high-risk myelodysplastic syndrome: predictive prognostic models for outcome. Cancer 106, 1090–1098, doi: 10.1002/cncr.21723 (2006). [DOI] [PubMed] [Google Scholar]
- 254.Kantarjian H et al. Intensive chemotherapy does not benefit most older patients (age 70 years or older) with acute myeloid leukemia. Blood 116, 4422–4429, doi: 10.1182/blood-2010-03-276485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Bogenberger JM et al. Ex vivo activity of BCL-2 family inhibitors ABT-199 and ABT-737 combined with 5-azacytidine in myeloid malignancies. Leuk Lymphoma 56, 226–229, doi: 10.3109/10428194.2014.910657 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.DiNardo CD et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 133, 7–17, doi: 10.1182/blood-2018-08-868752 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Leverson JD et al. Found in Translation: How Preclinical Research Is Guiding the Clinical Development of the BCL2-Selective Inhibitor Venetoclax. Cancer Discov 7, 1376–1393, doi: 10.1158/2159-8290.CD-17-0797 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Lakhani NJ et al. A phase I study of novel dual Bcl-2/Bcl-xL inhibitor APG-1252 in patients with advanced small cell lung cancer (SCLC) or other solid tumor. Journal of Clinical Oncology 36, 2594–2594, doi: 10.1200/JCO.2018.36.15_suppl.2594 (2018). [DOI] [Google Scholar]
- 259.Ye L et al. The small-molecule compound BM-1197 inhibits the antiapoptotic regulators Bcl-2/Bcl-xL and triggers apoptotic cell death in human colorectal cancer cells. Tumour Biol 36, 3447–3455, doi: 10.1007/s13277-014-2980-z (2015). [DOI] [PubMed] [Google Scholar]
- 260.Nemati F et al. Targeting Bcl-2/Bcl-XL induces antitumor activity in uveal melanoma patient-derived xenografts. PLoS One 9, e80836, doi: 10.1371/journal.pone.0080836 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Loriot Y et al. Radiosensitization by a novel Bcl-2 and Bcl-XL inhibitor S44563 in small-cell lung cancer. Cell Death Dis 5, e1423, doi: 10.1038/cddis.2014.365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Adam A et al. A Dual Bcl-2/XL Inhibitor Induces Tumor Cell Apoptosis in a Hematopoietic Xenograft Model. Blood 124, 5304–5304 (2014). [Google Scholar]
- 263.Takimoto-Shimomura T et al. Dual targeting of bromodomain-containing 4 by AZD5153 and BCL2 by AZD4320 against B-cell lymphomas concomitantly overexpressing c-MYC and BCL2. Invest New Drugs 37, 210–222, doi: 10.1007/s10637-018-0623-8 (2019). [DOI] [PubMed] [Google Scholar]
- 264.Casara P et al. S55746 is a novel orally active BCL-2 selective and potent inhibitor that impairs hematological tumor growth. Oncotarget 9, 20075–20088, doi: 10.18632/oncotarget.24744 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Fang DD et al. BCL-2 selective inhibitor APG-2575 synergizes with BTK inhibitor in preclinical xenograft models of follicular lymphoma and diffuse large B-cell lymphoma. Cancer Research 79, 2058–2058, doi: 10.1158/1538-7445.Sabcs18-2058 (2019). [DOI] [Google Scholar]
- 266.Le Gouill S et al. A new Bcl-2 inhibitor (S55746/BCL201) as monotherapy in patients with relapsed or refractory Non-Hodgkin lymphoma: preliminary results of the first-in-human trial. Hematological Oncology 35, 47–48, doi: 10.1002/hon.2437_30 (2017). [DOI] [Google Scholar]
- 267.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03595059 (2019). [DOI] [PubMed]
- 268.Phillips AC in New Drugs on the Horizon: Part 2. Presentation at the 110th Annual Meeting of the American Association for Cancer Research; 2019 March 29 - April 3. [Google Scholar]
- 269.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03412786 (2019). [DOI] [PubMed]
- 270.Koehler MF et al. Structure-Guided Rescaffolding of Selective Antagonists of BCL-XL. ACS Med Chem Lett 5, 662–667, doi: 10.1021/ml500030p (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Tao ZF et al. Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity. ACS Med Chem Lett 5, 1088–1093, doi: 10.1021/ml5001867 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Baranski Z et al. Pharmacological inhibition of Bcl-xL sensitizes osteosarcoma to doxorubicin. Oncotarget 6, 36113–36125, doi: 10.18632/oncotarget.5333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Abed MN, Abdullah MI & Richardson A Antagonism of Bcl-XL is necessary for synergy between carboplatin and BH3 mimetics in ovarian cancer cells. J Ovarian Res 9, 25, doi: 10.1186/s13048-016-0234-y (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Lucantoni F, Lindner AU, O’Donovan N, Dussmann H & Prehn JHM Systems modeling accurately predicts responses to genotoxic agents and their synergism with BCL-2 inhibitors in triple negative breast cancer cells. Cell Death Dis 9, 42, doi: 10.1038/s41419-017-0039-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.de Jong Y et al. Bcl-xl as the most promising Bcl-2 family member in targeted treatment of chondrosarcoma. Oncogenesis 7, 74, doi: 10.1038/s41389-018-0084-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Lucantoni F, Dussmann H, Llorente-Folch I & Prehn JHM BCL2 and BCL(X)L selective inhibitors decrease mitochondrial ATP production in breast cancer cells and are synthetically lethal when combined with 2-deoxy-D-glucose. Oncotarget 9, 26046–26063, doi: 10.18632/oncotarget.25433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Faqar-Uz-Zaman SF, Heinicke U, Meister MT, Vogler M & Fulda S BCL-xL-selective BH3 mimetic sensitizes rhabdomyosarcoma cells to chemotherapeutics by activation of the mitochondrial pathway of apoptosis. Cancer Lett 412, 131–142, doi: 10.1016/j.canlet.2017.09.025 (2018). [DOI] [PubMed] [Google Scholar]
- 278.Ali AM, Atmaj J, Van Oosterwijk N, Groves MR & Domling A Stapled Peptides Inhibitors: A New Window for Target Drug Discovery. Comput Struct Biotechnol J 17, 263–281, doi: 10.1016/j.csbj.2019.01.012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Tsomaia N Peptide therapeutics: targeting the undruggable space. Eur J Med Chem 94, 459–470, doi: 10.1016/j.ejmech.2015.01.014 (2015). [DOI] [PubMed] [Google Scholar]
- 280.Walensky LD et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305, 1466–1470, doi: 10.1126/science.1099191 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Chang YS et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A 110, E3445–3454, doi: 10.1073/pnas.1303002110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kussie PH et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953, doi: 10.1126/science.274.5289.948 (1996). [DOI] [PubMed] [Google Scholar]
- 283.Baek S et al. Structure of the stapled p53 peptide bound to Mdm2. J Am Chem Soc 134, 103–106, doi: 10.1021/ja2090367 (2012). [DOI] [PubMed] [Google Scholar]
- 284.Harvey EP et al. Crystal Structures of Anti-apoptotic BFL-1 and Its Complex with a Covalent Stapled Peptide Inhibitor. Structure 26, 153–160 e154, doi: 10.1016/j.str.2017.11.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Stewart ML, Fire E, Keating AE & Walensky LD The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol 6, 595–601, doi: 10.1038/nchembio.391 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Grossmann TN et al. Inhibition of oncogenic Wnt signaling through direct targeting of beta-catenin. Proc Natl Acad Sci U S A 109, 17942–17947, doi: 10.1073/pnas.1208396109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Xin M et al. Small-molecule Bax agonists for cancer therapy. Nat Commun 5, 4935, doi: 10.1038/ncomms5935 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Li R et al. Modulation of Bax and mTOR for Cancer Therapeutics. Cancer Res 77, 3001–3012, doi: 10.1158/0008-5472.CAN-16-2356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Liu G et al. Discovery and optimization of small molecule Bax activators for cancer therapy. Cancer Research 79, 3-3, doi: 10.1158/1538-7445.Sabcs18-3 (2019).30602621 [DOI] [Google Scholar]
- 290.Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES & Walensky LD Direct and selective small-molecule activation of proapoptotic BAX. Nat Chem Biol 8, 639–645, doi: 10.1038/nchembio.995 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Daniele S et al. Bax Activation Blocks Self-Renewal and Induces Apoptosis of Human Glioblastoma Stem Cells. ACS Chem Neurosci 9, 85–99, doi: 10.1021/acschemneuro.7b00023 (2018). [DOI] [PubMed] [Google Scholar]
- 292.Reyna DE et al. Direct Activation of BAX by BTSA1 Overcomes Apoptosis Resistance in Acute Myeloid Leukemia. Cancer Cell 32, 490–505 e410, doi: 10.1016/j.ccell.2017.09.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Garner TP et al. Small-molecule allosteric inhibitors of BAX. Nat Chem Biol 15, 322–330, doi: 10.1038/s41589-018-0223-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Dijkers PF, Medema RH, Lammers JW, Koenderman L & Coffer PJ Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol 10, 1201–1204 (2000). [DOI] [PubMed] [Google Scholar]
- 295.Whitfield J, Neame SJ, Paquet L, Bernard O & Ham J Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29, 629–643 (2001). [DOI] [PubMed] [Google Scholar]
- 296.Weston CR et al. Activation of ERK1/2 by deltaRaf-1:ER* represses Bim expression independently of the JNK or PI3K pathways. Oncogene 22, 1281–1293, doi: 10.1038/sj.onc.1206261 (2003). [DOI] [PubMed] [Google Scholar]
- 297.Sarosiek KA et al. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell 51, 751–765, doi: 10.1016/j.molcel.2013.08.048 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Kuribara R et al. Roles of Bim in apoptosis of normal and Bcr-Abl-expressing hematopoietic progenitors. Mol Cell Biol 24, 6172–6183, doi: 10.1128/MCB.24.14.6172-6183.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Ng KP et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med 18, 521–528, doi: 10.1038/nm.2713 (2012). [DOI] [PubMed] [Google Scholar]
- 300.Yuan J et al. Clinical Implications of the BIM Deletion Polymorphism in Advanced Lung Adenocarcinoma Treated With Gefitinib. Clin Lung Cancer 19, e431–e438, doi: 10.1016/j.cllc.2018.02.007 (2018). [DOI] [PubMed] [Google Scholar]
- 301.Zhang L et al. Clinical features of Bim deletion polymorphism and its relation with crizotinib primary resistance in Chinese patients with ALK/ROS1 fusion-positive non-small cell lung cancer. Cancer 123, 2927–2935, doi: 10.1002/cncr.30677 (2017). [DOI] [PubMed] [Google Scholar]
- 302.Tan TT et al. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 7, 227–238, doi: 10.1016/j.ccr.2005.02.008 (2005). [DOI] [PubMed] [Google Scholar]
- 303.Steele AJ et al. Cerdulatinib induces Bim expression and synergistic cell kill in combination with venetoclax in follicular lymphoma cell lines. Cancer Res 78, Abstract nr 305 (2018). [Google Scholar]
- 304.Wertz IE et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471, 110–114, doi: 10.1038/nature09779 (2011). [DOI] [PubMed] [Google Scholar]
- 305.Zack TI et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet 45, 1134–1140, doi: 10.1038/ng.2760 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03797261 (2019). [DOI] [PubMed]
- 307.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02675452 (2019). [DOI] [PubMed]
- 308.Tron AE et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun 9, 5341, doi: 10.1038/s41467-018-07551-w (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03218683 (2019). [DOI] [PubMed]
- 310.Prukova D et al. Cotargeting of BCL2 with Venetoclax and MCL1 with S63845 Is Synthetically Lethal In Vivo in Relapsed Mantle Cell Lymphoma. Clin Cancer Res, doi: 10.1158/1078-0432.CCR-18-3275 (2019). [DOI] [PubMed]
- 311.Li Z, He S & Look AT The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia 33, 262–266, doi: 10.1038/s41375-018-0201-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Ramsey HE et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov 8, 1566–1581, doi: 10.1158/2159-8290.CD-18-0140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Moujalled DM et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia 33, 905–917, doi: 10.1038/s41375-018-0261-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Halilovic E et al. MIK665/S64315, a novel Mcl-1 inhibitor, in combination with Bcl-2 inhibitors exhibits strong synergistic antitumor activity in a range of hematologic malignancies. Cancer Research 79, 4477–4477, doi: 10.1158/1538-7445.Sabcs18-4477 (2019). [DOI] [Google Scholar]
- 315.Maragno AL et al. S64315 (MIK665) is a potent and selective Mcl1 inhibitor with strong antitumor activity across a diverse range of hematologic tumor models. Cancer Research 79, 4482–4482, doi: 10.1158/1538-7445.Sabcs18-4482 (2019). [DOI] [Google Scholar]
- 316.Lee T et al. Discovery and biological characterization of potent myeloid cell leukemia-1 inhibitors. FEBS Lett 591, 240–251, doi: 10.1002/1873-3468.12497 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Deng J et al. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12, 171–185, doi: 10.1016/j.ccr.2007.07.001 (2007). [DOI] [PubMed] [Google Scholar]
- 318.Soderquist RS et al. Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nat Commun 9, 3513, doi: 10.1038/s41467-018-05815-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Matulis SM et al. Preclinical Activity of Novel MCL1 Inhibitor AZD5991 in Multiple Myeloma. Blood 132, 952–952, doi: 10.1182/blood-2018-99-113504 (2018). [DOI] [Google Scholar]
- 320.Abulwerdi F et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther 13, 565–575, doi: 10.1158/1535-7163.MCT-12-0767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Ow TJ et al. Optimal targeting of BCL-family proteins in head and neck squamous cell carcinoma requires inhibition of both BCL-xL and MCL-1. Oncotarget 10, 494–510, doi: 10.18632/oncotarget.26563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Wang Q, Wan J, Zhang W & Hao S MCL-1 or BCL-xL-dependent resistance to the BCL-2 antagonist (ABT-199) can be overcome by specific inhibitor as single agents and in combination with ABT-199 in acute myeloid leukemia cells. Leuk Lymphoma, 1–11, doi: 10.1080/10428194.2018.1563694 (2019). [DOI] [PubMed]
- 323.Hird AW & Tron AE Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol Ther 198, 59–67, doi: 10.1016/j.pharmthera.2019.02.007 (2019). [DOI] [PubMed] [Google Scholar]
- 324.Guerra RM et al. Precision Targeting of BFL-1/A1 and an ATM Co-dependency in Human Cancer. Cell Rep 24, 3393–3403 e3395, doi: 10.1016/j.celrep.2018.08.089 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Vogler M BCL2A1: the underdog in the BCL2 family. Cell Death Differ 19, 67–74, doi: 10.1038/cdd.2011.158 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Brien G et al. Characterization of peptide aptamers targeting Bfl-1 anti-apoptotic protein. Biochemistry 50, 5120–5129, doi: 10.1021/bi101839p (2011). [DOI] [PubMed] [Google Scholar]
- 327.Fulda S & Vucic D Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov 11, 109–124, doi: 10.1038/nrd3627 (2012). [DOI] [PubMed] [Google Scholar]
- 328.Monian P & Jiang X Clearing the final hurdles to mitochondrial apoptosis: regulation post cytochrome C release. Exp Oncol 34, 185–191 (2012). [PubMed] [Google Scholar]
- 329.Zou H, Li Y, Liu X & Wang X An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274, 11549–11556, doi: 10.1074/jbc.274.17.11549 (1999). [DOI] [PubMed] [Google Scholar]
- 330.Eckelman BP, Salvesen GS & Scott FL Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep 7, 988–994, doi: 10.1038/sj.embor.7400795 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Vucic D et al. Engineering ML-IAP to produce an extraordinarily potent caspase 9 inhibitor: implications for Smac-dependent anti-apoptotic activity of ML-IAP. Biochem J 385, 11–20, doi: 10.1042/BJ20041108 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Infante JR et al. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol 32, 3103–3110, doi: 10.1200/JCO.2013.52.3993 (2014). [DOI] [PubMed] [Google Scholar]
- 333.Bardia A et al. Paclitaxel With Inhibitor of Apoptosis Antagonist, LCL161, for Localized Triple-Negative Breast Cancer, Prospectively Stratified by Gene Signature in a Biomarker-Driven Neoadjuvant Trial. J Clin Oncol 36, 3126–3133, doi: 10.1200/JCO.2017.74.8392 (2018). [DOI] [PubMed] [Google Scholar]
- 334.Chesi M et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat Med 22, 1411–1420, doi: 10.1038/nm.4229 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Yang L et al. LCL161, a SMAC-mimetic, Preferentially Radiosensitizes Human Papillomavirus-negative Head and Neck Squamous Cell Carcinoma. Mol Cancer Ther 18, 1025–1035, doi: 10.1158/1535-7163.MCT-18-1157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Amaravadi RK et al. A Phase I Study of the SMAC-Mimetic Birinapant in Adults with Refractory Solid Tumors or Lymphoma. Mol Cancer Ther 14, 2569–2575, doi: 10.1158/1535-7163.MCT-15-0475 (2015). [DOI] [PubMed] [Google Scholar]
- 337.Noonan AM et al. Pharmacodynamic markers and clinical results from the phase 2 study of the SMAC mimetic birinapant in women with relapsed platinum-resistant or -refractory epithelial ovarian cancer. Cancer 122, 588–597, doi: 10.1002/cncr.29783 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02587962 (2019). [DOI] [PubMed]
- 339.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03803774 (2019). [DOI] [PubMed]
- 340.Gomez-Roca C et al. Open-label, non-randomized, exploratory pre-operative window-of-opportunity trial to investigate the pharmacokinetics and pharmacodynamics of the smac mimetic Debio 1143 in patients with resectable squamous cell carcinoma of the head and neck. Cancer Research 79, 5001–5001, doi: 10.1158/1538-7445.Sabcs18-5001 (2019). [DOI] [Google Scholar]
- 341.Tao Z et al. SMAC Mimetic Debio 1143 and Ablative Radiation Therapy Synergize to Enhance Antitumor Immunity against Lung Cancer. Clin Cancer Res 25, 1113–1124, doi: 10.1158/1078-0432.CCR-17-3852 (2019). [DOI] [PubMed] [Google Scholar]
- 342.Juergens RA et al. A dose-finding study of the SMAC mimetic Debio 1143 when given in combination with avelumab to patients with advanced solid malignancies. Journal of Clinical Oncology 37, 2599–2599, doi: 10.1200/JCO.2019.37.15_suppl.2599 (2019). [DOI] [Google Scholar]
- 343.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03871959 (2019). [DOI] [PubMed]
- 344.Fresquet V, Rieger M, Carolis C, Garcia-Barchino MJ & Martinez-Climent JA Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 123, 4111–4119, doi: 10.1182/blood-2014-03-560284 (2014). [DOI] [PubMed] [Google Scholar]
- 345.Blombery P et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov 9, 342–353, doi: 10.1158/2159-8290.CD-18-1119 (2019). [DOI] [PubMed] [Google Scholar]
- 346.Blombery P et al. Characterization of a novel venetoclax resistance mutation (BCL2 Phe104Ile) observed in follicular lymphoma. Br J Haematol, doi: 10.1111/bjh.16069 (2019). [DOI] [PubMed]
- 347.Birkinshaw RW et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat Commun 10, 2385, doi: 10.1038/s41467-019-10363-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Herling CD et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat Commun 9, 727, doi: 10.1038/s41467-018-03170-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Jayappa KD et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv 1, 933–946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Chiron D et al. Rational targeted therapies to overcome microenvironment-dependent expansion of mantle cell lymphoma. Blood 128, 2808–2818, doi: 10.1182/blood-2016-06-720490 (2016). [DOI] [PubMed] [Google Scholar]
- 351.Tam CS et al. Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. N Engl J Med 378, 1211–1223, doi: 10.1056/NEJMoa1715519 (2018). [DOI] [PubMed] [Google Scholar]
- 352.Agarwal R et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med 25, 119–129, doi: 10.1038/s41591-018-0243-z (2019). [DOI] [PubMed] [Google Scholar]
- 353.Nechiporuk T et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov 9, 910–925, doi: 10.1158/2159-8290.CD-19-0125 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Chen X et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov 9, 890–909, doi: 10.1158/2159-8290.Cd-19-0117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Scorrano L et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2, 55–67 (2002). [DOI] [PubMed] [Google Scholar]
- 356.McQuibban GA, Saurya S & Freeman M Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 423, 537–541, doi: 10.1038/nature01633 (2003). [DOI] [PubMed] [Google Scholar]
- 357.Frezza C et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189, doi: 10.1016/j.cell.2006.06.025 (2006). [DOI] [PubMed] [Google Scholar]
- 358.Rottgers K, Zufall N, Guiard B & Voos W The ClpB homolog Hsp78 is required for the efficient degradation of proteins in the mitochondrial matrix. J Biol Chem 277, 45829–45837, doi: 10.1074/jbc.M207152200 (2002). [DOI] [PubMed] [Google Scholar]
- 359.Lee S et al. The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell 115, 229–240, doi: 10.1016/s0092-8674(03)00807-9 (2003). [DOI] [PubMed] [Google Scholar]
- 360.Wortmann SB et al. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am J Hum Genet 96, 245–257, doi: 10.1016/j.ajhg.2014.12.013 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Rampino N et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 275, 967–969, doi: 10.1126/science.275.5302.967 (1997). [DOI] [PubMed] [Google Scholar]
- 362.Brimmell M, Mendiola R, Mangion J & Packham G BAX frameshift mutations in cell lines derived from human haemopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene 16, 1803–1812, doi: 10.1038/sj.onc.1201704 (1998). [DOI] [PubMed] [Google Scholar]
- 363.Meijerink JP et al. Hematopoietic malignancies demonstrate loss-of-function mutations of BAX. Blood 91, 2991–2997 (1998). [PubMed] [Google Scholar]
- 364.Renault TT et al. Mitochondrial shape governs BAX-induced membrane permeabilization and apoptosis. Mol Cell 57, 69–82, doi: 10.1016/j.molcel.2014.10.028 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Shamas-Din A et al. Distinct lipid effects on tBid and Bim activation of membrane permeabilization by pro-apoptotic Bax. Biochem J 467, 495–505, doi: 10.1042/bj20141291 (2015). [DOI] [PubMed] [Google Scholar]
- 366.Almasan A & Ashkenazi A Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev 14, 337–348 (2003). [DOI] [PubMed] [Google Scholar]
- 367.Falschlehner C, Schaefer U & Walczak H Following TRAIL’s path in the immune system. Immunology 127, 145–154, doi: 10.1111/j.1365-2567.2009.03058.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Falschlehner C, Ganten TM, Koschny R, Schaefer U & Walczak H TRAIL and other TRAIL receptor agonists as novel cancer therapeutics. Adv Exp Med Biol 647, 195–206, doi: 10.1007/978-0-387-89520-8_14 (2009). [DOI] [PubMed] [Google Scholar]
- 369.Ashkenazi A Targeting the extrinsic apoptotic pathway in cancer: lessons learned and future directions. J Clin Invest 125, 487–489, doi: 10.1172/JCI80420 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Herbst RS et al. A first-in-human study of conatumumab in adult patients with advanced solid tumors. Clin Cancer Res 16, 5883–5891, doi: 10.1158/1078-0432.CCR-10-0631 (2010). [DOI] [PubMed] [Google Scholar]
- 371.Luster TA, Carrell JA, McCormick K, Sun D & Humphreys R Mapatumumab and lexatumumab induce apoptosis in TRAIL-R1 and TRAIL-R2 antibody-resistant NSCLC cell lines when treated in combination with bortezomib. Mol Cancer Ther 8, 292–302, doi: 10.1158/1535-7163.MCT-08-0918 (2009). [DOI] [PubMed] [Google Scholar]
- 372.Belyanskaya LL et al. Human agonistic TRAIL receptor antibodies Mapatumumab and Lexatumumab induce apoptosis in malignant mesothelioma and act synergistically with cisplatin. Mol Cancer 6, 66, doi: 10.1186/1476-4598-6-66 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Amm HM, Oliver PG, Lee CH, Li Y & Buchsbaum DJ Combined modality therapy with TRAIL or agonistic death receptor antibodies. Cancer Biol Ther 11, 431–449, doi: 10.4161/cbt.11.5.14671 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Herbst RS et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol 28, 2839–2846, doi: 10.1200/JCO.2009.25.1991 (2010). [DOI] [PubMed] [Google Scholar]
- 375.Soria JC et al. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J Clin Oncol 29, 4442–4451, doi: 10.1200/JCO.2011.37.2623 (2011). [DOI] [PubMed] [Google Scholar]
- 376.Cheah CY et al. Dulanermin with rituximab in patients with relapsed indolent B-cell lymphoma: an open-label phase 1b/2 randomised study. Lancet Haematol 2, e166–174, doi: 10.1016/S2352-3026(15)00026-5 (2015). [DOI] [PubMed] [Google Scholar]
- 377.Wainberg ZA et al. A phase 1B study of dulanermin in combination with modified FOLFOX6 plus bevacizumab in patients with metastatic colorectal cancer. Clin Colorectal Cancer 12, 248–254, doi: 10.1016/j.clcc.2013.06.002 (2013). [DOI] [PubMed] [Google Scholar]
- 378.Elias A et al. Epigenetic silencing of death receptor 4 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in gliomas. Clin Cancer Res 15, 5457–5465, doi: 10.1158/1078-0432.Ccr-09-1125 (2009). [DOI] [PubMed] [Google Scholar]
- 379.Horak P et al. Contribution of epigenetic silencing of tumor necrosis factor-related apoptosis inducing ligand receptor 1 (DR4) to TRAIL resistance and ovarian cancer. Mol Cancer Res 3, 335–343, doi: 10.1158/1541-7786.Mcr-04-0136 (2005). [DOI] [PubMed] [Google Scholar]
- 380.Wagner KW et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med 13, 1070–1077, doi: 10.1038/nm1627 (2007). [DOI] [PubMed] [Google Scholar]
- 381.Bae SI, Cheriyath V, Jacobs BS, Reu FJ & Borden EC Reversal of methylation silencing of Apo2L/TRAIL receptor 1 (DR4) expression overcomes resistance of SK-MEL-3 and SK-MEL-28 melanoma cells to interferons (IFNs) or Apo2L/TRAIL. Oncogene 27, 490–498, doi: 10.1038/sj.onc.1210655 (2008). [DOI] [PubMed] [Google Scholar]
- 382.Tang Z, Bauer JA, Morrison B & Lindner DJ Nitrosylcobalamin promotes cell death via S nitrosylation of Apo2L/TRAIL receptor DR4. Mol Cell Biol 26, 5588–5594, doi: 10.1128/mcb.00199-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Jin Z, McDonald ER 3rd, Dicker DT & El-Deiry WS Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J Biol Chem 279, 35829–35839, doi: 10.1074/jbc.M405538200 (2004). [DOI] [PubMed] [Google Scholar]
- 384.Gillissen B et al. Endogenous Bak inhibitors Mcl-1 and Bcl-xL: differential impact on TRAIL resistance in Bax-deficient carcinoma. J Cell Biol 188, 851–862, doi: 10.1083/jcb.200912070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Vogler M et al. Targeting XIAP bypasses Bcl-2-mediated resistance to TRAIL and cooperates with TRAIL to suppress pancreatic cancer growth in vitro and in vivo. Cancer Res 68, 7956–7965, doi: 10.1158/0008-5472.Can-08-1296 (2008). [DOI] [PubMed] [Google Scholar]
- 386.Zhang L & Fang B Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther 12, 228–237, doi: 10.1038/sj.cgt.7700792 (2005). [DOI] [PubMed] [Google Scholar]
- 387.Gillissen B et al. Targeted therapy of the XIAP/proteasome pathway overcomes TRAIL-resistance in carcinoma by switching apoptosis signaling to a Bax/Bak-independent ‘type I’ mode. Cell Death Dis 4, e643, doi: 10.1038/cddis.2013.67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Muhlenbeck F et al. The tumor necrosis factor-related apoptosis-inducing ligand receptors TRAIL-R1 and TRAIL-R2 have distinct cross-linking requirements for initiation of apoptosis and are non-redundant in JNK activation. J Biol Chem 275, 32208–32213, doi: 10.1074/jbc.M000482200 (2000). [DOI] [PubMed] [Google Scholar]
- 389.Wajant H et al. Differential activation of TRAIL-R1 and −2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRAIL-R2 by a soluble TRAIL derivative. Oncogene 20, 4101–4106, doi: 10.1038/sj.onc.1204558 (2001). [DOI] [PubMed] [Google Scholar]
- 390.Berg D et al. Enforced covalent trimerization increases the activity of the TNF ligand family members TRAIL and CD95L. Cell Death Differ 14, 2021–2034, doi: 10.1038/sj.cdd.4402213 (2007). [DOI] [PubMed] [Google Scholar]
- 391.von Pawel J et al. Phase II trial of mapatumumab, a fully human agonist monoclonal antibody to tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1), in combination with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Clin Lung Cancer 15, 188–196 e182, doi: 10.1016/j.cllc.2013.12.005 (2014). [DOI] [PubMed] [Google Scholar]
- 392.Hotte SJ et al. A phase 1 study of mapatumumab (fully human monoclonal antibody to TRAIL-R1) in patients with advanced solid malignancies. Clin Cancer Res 14, 3450–3455, doi: 10.1158/1078-0432.CCR-07-1416 (2008). [DOI] [PubMed] [Google Scholar]
- 393.Trarbach T et al. Phase II trial of mapatumumab, a fully human agonistic monoclonal antibody that targets and activates the tumour necrosis factor apoptosis-inducing ligand receptor-1 (TRAIL-R1), in patients with refractory colorectal cancer. Br J Cancer 102, 506–512, doi: 10.1038/sj.bjc.6605507 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Mom CH et al. Mapatumumab, a fully human agonistic monoclonal antibody that targets TRAIL-R1, in combination with gemcitabine and cisplatin: a phase I study. Clin Cancer Res 15, 5584–5590, doi: 10.1158/1078-0432.CCR-09-0996 (2009). [DOI] [PubMed] [Google Scholar]
- 395.Younes A et al. A Phase 1b/2 trial of mapatumumab in patients with relapsed/refractory non-Hodgkin’s lymphoma. Br J Cancer 103, 1783–1787, doi: 10.1038/sj.bjc.6605987 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Ciuleanu T et al. A randomized, double-blind, placebo-controlled phase II study to assess the efficacy and safety of mapatumumab with sorafenib in patients with advanced hepatocellular carcinoma. Ann Oncol 27, 680–687, doi: 10.1093/annonc/mdw004 (2016). [DOI] [PubMed] [Google Scholar]
- 397.Plummer R et al. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin Cancer Res 13, 6187–6194, doi: 10.1158/1078-0432.CCR-07-0950 (2007). [DOI] [PubMed] [Google Scholar]
- 398.Merchant MS et al. Phase I trial and pharmacokinetic study of lexatumumab in pediatric patients with solid tumors. J Clin Oncol 30, 4141–4147, doi: 10.1200/JCO.2012.44.1055 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Sikic BI et al. A phase Ib study to assess the safety of lexatumumab, a human monoclonal antibody that activates TRAIL-R2, in combination with gemcitabine, pemetrexed, doxorubicin or FOLFIRI. Journal of Clinical Oncology 25, 14006–14006 (2007). [Google Scholar]
- 400.Wakelee HA et al. Phase I and pharmacokinetic study of lexatumumab (HGS-ETR2) given every 2 weeks in patients with advanced solid tumors. Ann Oncol 21, 376–381, doi: 10.1093/annonc/mdp292 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Fuchs CS et al. TRAIL receptor agonist conatumumab with modified FOLFOX6 plus bevacizumab for first-line treatment of metastatic colorectal cancer: A randomized phase 1b/2 trial. Cancer 119, 4290–4298, doi: 10.1002/cncr.28353 (2013). [DOI] [PubMed] [Google Scholar]
- 402.Cohn AL et al. A randomized, placebo-controlled phase 2 study of ganitumab or conatumumab in combination with FOLFIRI for second-line treatment of mutant KRAS metastatic colorectal cancer. Ann Oncol 24, 1777–1785, doi: 10.1093/annonc/mdt057 (2013). [DOI] [PubMed] [Google Scholar]
- 403.Paz-Ares L et al. A randomized phase 2 study of paclitaxel and carboplatin with or without conatumumab for first-line treatment of advanced non-small-cell lung cancer. J Thorac Oncol 8, 329–337, doi: 10.1097/JTO.0b013e31827ce554 (2013). [DOI] [PubMed] [Google Scholar]
- 404.Kindler HL et al. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann Oncol 23, 2834–2842, doi: 10.1093/annonc/mds142 (2012). [DOI] [PubMed] [Google Scholar]
- 405.Forero-Torres A et al. TBCRC 019: A Phase II Trial of Nanoparticle Albumin-Bound Paclitaxel with or without the Anti-Death Receptor 5 Monoclonal Antibody Tigatuzumab in Patients with Triple-Negative Breast Cancer. Clin Cancer Res 21, 2722–2729, doi: 10.1158/1078-0432.CCR-14-2780 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Forero-Torres A et al. Phase 2, multicenter, open-label study of tigatuzumab (CS-1008), a humanized monoclonal antibody targeting death receptor 5, in combination with gemcitabine in chemotherapy-naive patients with unresectable or metastatic pancreatic cancer. Cancer Med 2, 925–932, doi: 10.1002/cam4.137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Sharma S et al. Safety, pharmacokinetics, and pharmacodynamics of the DR5 antibody LBY135 alone and in combination with capecitabine in patients with advanced solid tumors. Invest New Drugs 32, 135–144, doi: 10.1007/s10637-013-9952-9 (2014). [DOI] [PubMed] [Google Scholar]
- 408.Camidge DR et al. A phase I safety and pharmacokinetic study of the death receptor 5 agonistic antibody PRO95780 in patients with advanced malignancies. Clin Cancer Res 16, 1256–1263, doi: 10.1158/1078-0432.CCR-09-1267 (2010). [DOI] [PubMed] [Google Scholar]
- 409.Ashkenazi A Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov 7, 1001–1012, doi: 10.1038/nrd2637 (2008). [DOI] [PubMed] [Google Scholar]
- 410.Lemke J et al. Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl-1. Cell Death Differ 21, 491–502, doi: 10.1038/cdd.2013.179 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.De Blasio A et al. Loss of MCL1 function sensitizes the MDA-MB-231 breast cancer cells to rh-TRAIL by increasing DR4 levels. J Cell Physiol 234, 18432–18447, doi: 10.1002/jcp.28479 (2019). [DOI] [PubMed] [Google Scholar]
- 412.Graves JD et al. Apo2L/TRAIL and the death receptor 5 agonist antibody AMG 655 cooperate to promote receptor clustering and antitumor activity. Cancer Cell 26, 177–189, doi: 10.1016/j.ccr.2014.04.028 (2014). [DOI] [PubMed] [Google Scholar]
- 413.Allen JE et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med 5, 171ra117, doi: 10.1126/scitranslmed.3004828 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Prabhu VV, Allen JE, Dicker DT & El-Deiry WS Small-Molecule ONC201/TIC10 Targets Chemotherapy-Resistant Colorectal Cancer Stem-like Cells in an Akt/Foxo3a/TRAIL-Dependent Manner. Cancer Res 75, 1423–1432, doi: 10.1158/0008-5472.CAN-13-3451 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Kline CLB et al. Role of Dopamine Receptors in the Anticancer Activity of ONC201. Neoplasia 20, 80–91, doi: 10.1016/j.neo.2017.10.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Graves PR et al. Mitochondrial Protease ClpP is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem Biol 14, 1020–1029, doi: 10.1021/acschembio.9b00222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Ishizawa J et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 35, 721–737 e729, doi: 10.1016/j.ccell.2019.03.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Kline CL et al. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Sci Signal 9, ra18, doi: 10.1126/scisignal.aac4374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Ishizawa J et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal 9, ra17, doi: 10.1126/scisignal.aac4380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Ralff MD et al. Recombinant human TRAIL or a DR5 agonistic antibody convert the response of non-triple negative breast cancer cells to ONC201 from anti-proliferative to apoptotic. Cancer Research 79, 258–258, doi: 10.1158/1538-7445.Sabcs18-258 (2019). [DOI] [Google Scholar]
- 421.Ray J, Ralff M, Dicker D & El-Deiry W Anti-tumorigenic effect of ONC201 is enhanced by combination treatment with TRAIL or a DR5 agonist in endometrial cancer in vitro. Cancer Research 79, 262-262, doi: 10.1158/1538-7445.Sabcs18-262 (2019). [DOI] [Google Scholar]
- 422.Wagner J et al. Dose intensification of TRAIL-inducing ONC201 inhibits metastasis and promotes intratumoral NK cell recruitment. J Clin Invest 128, 2325–2338, doi: 10.1172/JCI96711 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Stein MN et al. Safety and enhanced immunostimulatory activity of the DRD2 antagonist ONC201 in advanced solid tumor patients with weekly oral administration. J Immunother Cancer 7, 136, doi: 10.1186/s40425-019-0599-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03791398 (2019). [DOI] [PubMed]
- 425.Arrillaga I et al. Single agent ONC201 in adult recurrent H3 K27M-mutant glioma. J Clin Oncol 37, abstr 3005 (2019). [Google Scholar]
- 426.Hall MD et al. First clinical experience with DRD2/3 antagonist ONC201 in H3 K27M-mutant pediatric diffuse intrinsic pontine glioma: a case report. J Neurosurg Pediatr, 1–7, doi: 10.3171/2019.2.PEDS18480 (2019). [DOI] [PubMed]
- 427.Prabhu VV et al. Dopamine Receptor D5 is a Modulator of Tumor Response to Dopamine Receptor D2 Antagonism. Clin Cancer Res 25, 2305–2313, doi: 10.1158/1078-0432.CCR-18-2572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Cubillos-Ruiz JR, Bettigole SE & Glimcher LH Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 168, 692–706, doi: 10.1016/j.cell.2016.12.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Tameire F et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat Cell Biol 21, 889–899, doi: 10.1038/s41556-019-0347-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.de Jong RN et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol 14, e1002344, doi: 10.1371/journal.pbio.1002344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03576131 (2019). [DOI] [PubMed]
- 432.Overdijk MB et al. DR5 agonist activity of HexaBody DR5/DR5 (GEN1029) is potentiated by C1q and independent of Fc-gamma receptor binding in preclinical tumor models. Cancer Research 79, 2391–2391, doi: 10.1158/1538-7445.Sabcs18-2391 (2019). [DOI] [Google Scholar]
- 433.Tahir SK et al. ABBV-621 Is a Novel and Potent TRAIL Receptor Agonist Fusion Protein That Induces Apoptosis Alone and in Combination with Navitoclax and Venetoclax in Hematological Tumors. Blood 130, 2812–2812 (2017). [Google Scholar]
- 434.Morgan-Lappe SE ABBV-621: A best-in-class TRAIL-receptor agonist fusion protein that enhances optimal clustering for the treatment of solid and hematologic tumors. Cancer Research 77, Abstract DDT01–03, doi: 10.1158/1538-7445.Am2017-ddt01-03 (2017). [DOI] [Google Scholar]
- 435.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03082209 (2019). [DOI] [PubMed]
- 436.Ratain MJ et al. Phase 1, first-in-human study of TRAIL receptor agonist fusion protein ABBV-621. Journal of Clinical Oncology 37, 3013–3013, doi: 10.1200/JCO.2019.37.15_suppl.3013 (2019). [DOI] [Google Scholar]
- 437.Sawyer AJ et al. Engineering and preclinical activity of MM-201, a best-in-class TRAIL receptor agonist. Cancer Research 79, 2491–2491, doi: 10.1158/1538-7445.Sabcs18-2491 (2019). [DOI] [Google Scholar]
- 438.Park JS et al. Targeting of dermal myofibroblasts through death receptor 5 arrests fibrosis in mouse models of scleroderma. Nat Commun 10, 1128, doi: 10.1038/s41467-019-09101-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Blumenschein GR Jr. et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Ann Oncol 26, 894–901, doi: 10.1093/annonc/mdv072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Kurimchak AM et al. Intrinsic Resistance to MEK Inhibition through BET Protein-Mediated Kinome Reprogramming in NF1-Deficient Ovarian Cancer. Mol Cancer Res, doi: 10.1158/1541-7786.MCR-18-1332 (2019). [DOI] [PMC free article] [PubMed]
- 441.Johnson GL, Stuhlmiller TJ, Angus SP, Zawistowski JS & Graves LM Molecular pathways: adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin Cancer Res 20, 2516–2522, doi: 10.1158/1078-0432.CCR-13-1081 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Meng J et al. Apoptosis induction by MEK inhibition in human lung cancer cells is mediated by Bim. PLoS One 5, e13026, doi: 10.1371/journal.pone.0013026 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Corcoran RB et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell 23, 121–128, doi: 10.1016/j.ccr.2012.11.007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Iavarone C et al. Combined MEK and BCL-2/XL Inhibition Is Effective in High-Grade Serous Ovarian Cancer Patient-Derived Xenograft Models and BIM Levels Are Predictive of Responsiveness. Mol Cancer Ther 18, 642–655, doi: 10.1158/1535-7163.MCT-18-0413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Fakih M et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRASG12C inhibitor, in advanced solid tumors. J Clin Oncol 37, abstract 3003 (2019). [Google Scholar]
- 446.Baud V & Karin M Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov 8, 33–40, doi: 10.1038/nrd2781 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Crawford LJ, Walker B & Irvine AE Proteasome inhibitors in cancer therapy. J Cell Commun Signal 5, 101–110, doi: 10.1007/s12079-011-0121-7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Hoesel B & Schmid JA The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer 12, 86, doi: 10.1186/1476-4598-12-86 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Luo JL, Kamata H & Karin M IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest 115, 2625–2632, doi: 10.1172/JCI26322 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Guo Y, Xu F, Lu T, Duan Z & Zhang Z Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev 38, 904–910, doi: 10.1016/j.ctrv.2012.04.007 (2012). [DOI] [PubMed] [Google Scholar]
- 451.Johnson DE, O’Keefe RA & Grandis JR Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 15, 234–248, doi: 10.1038/nrclinonc.2018.8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Rotow J & Bivona TG Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer 17, 637–658, doi: 10.1038/nrc.2017.84 (2017). [DOI] [PubMed] [Google Scholar]
- 453.Shi P et al. Overcoming Acquired Resistance to AZD9291, A Third-Generation EGFR Inhibitor, through Modulation of MEK/ERK-Dependent Bim and Mcl-1 Degradation. Clin Cancer Res 23, 6567–6579, doi: 10.1158/1078-0432.CCR-17-1574 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Hamedani FS et al. Crizotinib (PF-2341066) induces apoptosis due to downregulation of pSTAT3 and BCL-2 family proteins in NPM-ALK(+) anaplastic large cell lymphoma. Leuk Res 38, 503–508, doi: 10.1016/j.leukres.2013.12.027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Pinski J et al. Trk receptor inhibition induces apoptosis of proliferating but not quiescent human osteoblasts. Cancer Res 62, 986–989 (2002). [PubMed] [Google Scholar]
- 456.Weeraratna AT et al. Pan-trk inhibition decreases metastasis and enhances host survival in experimental models as a result of its selective induction of apoptosis of prostate cancer cells. Clin Cancer Res 7, 2237–2245 (2001). [PubMed] [Google Scholar]
- 457.Lavoie JF et al. TrkA induces apoptosis of neuroblastoma cells and does so via a p53-dependent mechanism. J Biol Chem 280, 29199–29207, doi: 10.1074/jbc.M502364200 (2005). [DOI] [PubMed] [Google Scholar]
- 458.Amin A et al. Evasion of anti-growth signaling: A key step in tumorigenesis and potential target for treatment and prophylaxis by natural compounds. Semin Cancer Biol 35 Suppl, S55–S77, doi: 10.1016/j.semcancer.2015.02.005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Sever R & Brugge JS Signal transduction in cancer. Cold Spring Harb Perspect Med 5, doi: 10.1101/cshperspect.a006098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Bykov VJN, Eriksson SE, Bianchi J & Wiman KG Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer 18, 89–102, doi: 10.1038/nrc.2017.109 (2018). [DOI] [PubMed] [Google Scholar]
- 461.Blandino G & Di Agostino S New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J Exp Clin Cancer Res 37, 30, doi: 10.1186/s13046-018-0705-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Parrales A & Iwakuma T Targeting Oncogenic Mutant p53 for Cancer Therapy. Front Oncol 5, 288, doi: 10.3389/fonc.2015.00288 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Levine AJ Targeting Therapies for the p53 Protein in Cancer Treatments. Annual Review of Cancer Biology 3, 21–34, doi: 10.1146/annurev-cancerbio-030518-055455 (2019). [DOI] [Google Scholar]
- 464.Wang S, Zhao Y, Aguilar A, Bernard D & Yang CY Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb Perspect Med 7, doi: 10.1101/cshperspect.a026245 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Burgess A et al. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol 6, 7, doi: 10.3389/fonc.2016.00007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Tisato V, Voltan R, Gonelli A, Secchiero P & Zauli G MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol 10, 133, doi: 10.1186/s13045-017-0500-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Kocik J et al. Helping the Released Guardian: Drug Combinations for Supporting the Anticancer Activity of HDM2 (MDM2) Antagonists. Cancers (Basel) 11, doi: 10.3390/cancers11071014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Ding Q et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem 56, 5979–5983, doi: 10.1021/jm400487c (2013). [DOI] [PubMed] [Google Scholar]
- 469.Reis B et al. Acute myeloid leukemia patients’ clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blasts. Haematologica 101, e185–188, doi: 10.3324/haematol.2015.139717 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Blotner S, Chen LC, Ferlini C & Zhi J Phase 1 summary of plasma concentration-QTc analysis for idasanutlin, an MDM2 antagonist, in patients with advanced solid tumors and AML. Cancer Chemother Pharmacol 81, 597–607, doi: 10.1007/s00280-018-3534-7 (2018). [DOI] [PubMed] [Google Scholar]
- 471.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03850535 (2019). [DOI] [PubMed]
- 472.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03135262 (2019). [DOI] [PubMed]
- 473.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03566485 (2019). [DOI] [PubMed]
- 474.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03940352 (2019). [DOI] [PubMed]
- 475.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02935907 (2019). [DOI] [PubMed]
- 476.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03634228 (2019). [DOI] [PubMed]
- 477.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03449381 (2019). [DOI] [PubMed]
- 478.Liao G et al. The development of piperidinones as potent MDM2-P53 protein-protein interaction inhibitors for cancer therapy. Eur J Med Chem 159, 1–9, doi: 10.1016/j.ejmech.2018.09.044 (2018). [DOI] [PubMed] [Google Scholar]
- 479.Furet P et al. Discovery of a novel class of highly potent inhibitors of the p53-MDM2 interaction by structure-based design starting from a conformational argument. Bioorg Med Chem Lett 26, 4837–4841, doi: 10.1016/j.bmcl.2016.08.010 (2016). [DOI] [PubMed] [Google Scholar]
- 480.Erba HP et al. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv 3, 1939–1949, doi: 10.1182/bloodadvances.2019030916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Tolcher AW et al. A phase Ib/II study of APG-115 in combination with pembrolizumab in patients with unresectable or metastatic melanomas or advanced solid tumors. Ann Oncol 30, doi: 10.1093/annonc/mdz027 (2019). [DOI] [Google Scholar]
- 482.Aguilar A et al. Discovery of 4-((3’R,4’S,5’R)-6’’-Chloro-4’-(3-chloro-2-fluorophenyl)-1’-ethyl-2’’-oxodispiro[ cyclohexane-1,2’-pyrrolidine-3’,3’’-indoline]-5’-carboxamido)bicyclo[2.2.2]octane −1-carboxylic Acid (AA-115/APG-115): A Potent and Orally Active Murine Double Minute 2 (MDM2) Inhibitor in Clinical Development. J Med Chem 60, 2819–2839, doi: 10.1021/acs.jmedchem.6b01665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Arnhold V et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 9, 2304–2319, doi: 10.18632/oncotarget.23409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Cornillie J et al. Anti-tumor activity of the MDM2-TP53 inhibitor BI-907828 in dedifferentiated liposarcoma patient-derived xenograft models harboring MDM2 amplification. Clin Transl Oncol, doi: 10.1007/s12094-019-02158-z (2019). [DOI] [PubMed]
- 485.Shustov AR et al. Preliminary Results of the Stapled Peptide ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Two Phase IIa Dose Expansion Cohorts in Relapsed/Refractory TP53 Wild-Type Peripheral T-Cell Lymphoma. Blood 132, 1623-1623, doi: 10.1182/blood-2018-99-116780 (2018). [DOI] [Google Scholar]
- 486.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03725436 (2019). [DOI] [PubMed]
- 487.Skalniak L, Surmiak E & Holak TA A therapeutic patent overview of MDM2/X-targeted therapies (2014–2018). Expert Opin Ther Pat 29, 151–170, doi: 10.1080/13543776.2019.1582645 (2019). [DOI] [PubMed] [Google Scholar]
- 488.Jung J et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun 7, 12609, doi: 10.1038/ncomms12609 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.de Jonge M et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur J Cancer 76, 144–151, doi: 10.1016/j.ejca.2017.02.005 (2017). [DOI] [PubMed] [Google Scholar]
- 490.Skalniak L et al. Prolonged Idasanutlin (RG7388) Treatment Leads to the Generation of p53-Mutated Cells. Cancers (Basel) 10, doi: 10.3390/cancers10110396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Aziz MH, Shen H & Maki CG Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene 30, 4678–4686, doi: 10.1038/onc.2011.185 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Zhang Q, Bykov VJN, Wiman KG & Zawacka-Pankau J APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis 9, 439, doi: 10.1038/s41419-018-0463-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Bassett EA, Wang W, Rastinejad F & El-Deiry WS Structural and functional basis for therapeutic modulation of p53 signaling. Clin Cancer Res 14, 6376–6386, doi: 10.1158/1078-0432.CCR-08-1526 (2008). [DOI] [PubMed] [Google Scholar]
- 494.Chen F, Wang W & El-Deiry WS Current strategies to target p53 in cancer. Biochem Pharmacol 80, 724–730, doi: 10.1016/j.bcp.2010.04.031 (2010). [DOI] [PubMed] [Google Scholar]
- 495.Sallman DA et al. Phase 1b/2 Combination Study of APR-246 and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Acute Myeloid Leukemia (AML). Blood 132, 3091 (2018). [Google Scholar]
- 496.Zhang J, Zhou L, Zhao S, Dicker DT & El-Deiry WS The CDK4/6 inhibitor palbociclib synergizes with irinotecan to promote colorectal cancer cell death under hypoxia. Cell Cycle 16, 1193–1200, doi: 10.1080/15384101.2017.1320005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Hashizume R et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neuro Oncol 18, 1519–1528, doi: 10.1093/neuonc/now106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Zhang G et al. Palbociclib triggers apoptosis in bladder cancer cells by Cdk2-induced Rad9-mediated reorganization of the Bak.Bcl-xl complex. Biochem Pharmacol 163, 133–141, doi: 10.1016/j.bcp.2019.02.017 (2019). [DOI] [PubMed] [Google Scholar]
- 499.Michaud K et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res 70, 3228–3238, doi: 10.1158/0008-5472.CAN-09-4559 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Baughn LB et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res 66, 7661–7667, doi: 10.1158/0008-5472.CAN-06-1098 (2006). [DOI] [PubMed] [Google Scholar]
- 501.Tao Z et al. Coadministration of Trametinib and Palbociclib Radiosensitizes KRAS-Mutant Non-Small Cell Lung Cancers In Vitro and In Vivo. Clin Cancer Res 22, 122–133, doi: 10.1158/1078-0432.CCR-15-0589 (2016). [DOI] [PubMed] [Google Scholar]
- 502.Lulla AR et al. miR-6883 Family miRNAs Target CDK4/6 to Induce G1 Phase Cell-Cycle Arrest in Colon Cancer Cells. Cancer Res 77, 6902–6913, doi: 10.1158/0008-5472.CAN-17-1767 (2017). [DOI] [PubMed] [Google Scholar]
- 503.Fiskus W et al. Superior efficacy of cotreatment with BET protein inhibitor and BCL2 or MCL1 inhibitor against AML blast progenitor cells. Blood Cancer J 9, 4, doi: 10.1038/s41408-018-0165-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Kim SR et al. BET inhibition in advanced cutaneous T cell lymphoma is synergistically potentiated by BCL2 inhibition or HDAC inhibition. Oncotarget 9, 29193–29207, doi: 10.18632/oncotarget.25670 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Bolden JE, Peart MJ & Johnstone RW Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5, 769–784, doi: 10.1038/nrd2133 (2006). [DOI] [PubMed] [Google Scholar]
- 506.Zhao Y et al. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc Natl Acad Sci U S A 102, 16090–16095, doi: 10.1073/pnas.0505585102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Ruefli AA et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A 98, 10833–10838, doi: 10.1073/pnas.191208598 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Zhang Y, Adachi M, Kawamura R & Imai K Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ 13, 129–140, doi: 10.1038/sj.cdd.4401686 (2006). [DOI] [PubMed] [Google Scholar]
- 509.Heinicke U, Haydn T, Kehr S, Vogler M & Fulda S BCL-2 selective inhibitor ABT-199 primes rhabdomyosarcoma cells to histone deacetylase inhibitor-induced apoptosis. Oncogene 37, 5325–5339, doi: 10.1038/s41388-018-0212-5 (2018). [DOI] [PubMed] [Google Scholar]
- 510.Liu Y et al. NOXA genetic amplification or pharmacologic induction primes lymphoma cells to BCL2 inhibitor-induced cell death. Proc Natl Acad Sci U S A 115, 12034–12039, doi: 10.1073/pnas.1806928115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Ramakrishnan VG et al. Histone deacetylase inhibition in combination with MEK or BCL-2 inhibition in multiple myeloma. Haematologica, doi: 10.3324/haematol.2018.211110 (2019). [DOI] [PMC free article] [PubMed]
- 512.Sun K et al. The Combination of Venetoclax and CUDC-907 Exhibits Synergistic Activity in Venetoclax-Refractory DLBCL. Blood 128, 4184–4184 (2016). [Google Scholar]
- 513.US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT01742988 (2019). [DOI] [PubMed]
- 514.Neckers L & Workman P Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18, 64–76, doi: 10.1158/1078-0432.CCR-11-1000 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Scaltriti M, Dawood S & Cortes J Molecular pathways: targeting hsp90--who benefits and who does not. Clin Cancer Res 18, 4508–4513, doi: 10.1158/1078-0432.CCR-11-2138 (2012). [DOI] [PubMed] [Google Scholar]
- 516.Centenera MM et al. Evidence for efficacy of new Hsp90 inhibitors revealed by ex vivo culture of human prostate tumors. Clin Cancer Res 18, 3562–3570, doi: 10.1158/1078-0432.CCR-12-0782 (2012). [DOI] [PubMed] [Google Scholar]
- 517.Shah S et al. Results from phase II trial of HSP90 inhibitor, STA-9090 (ganetespib), in metastatic uveal melanoma. Melanoma Res 28, 605–610, doi: 10.1097/CMR.0000000000000509 (2018). [DOI] [PubMed] [Google Scholar]
- 518.Shapiro GI et al. First-in-human phase I dose escalation study of a second-generation non-ansamycin HSP90 inhibitor, AT13387, in patients with advanced solid tumors. Clin Cancer Res 21, 87–97, doi: 10.1158/1078-0432.CCR-14-0979 (2015). [DOI] [PubMed] [Google Scholar]
- 519.Bussenius J et al. Discovery of XL888: a novel tropane-derived small molecule inhibitor of HSP90. Bioorg Med Chem Lett 22, 5396–5404, doi: 10.1016/j.bmcl.2012.07.052 (2012). [DOI] [PubMed] [Google Scholar]
- 520.Park MA et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol Ther 7, 1648–1662, doi: 10.4161/cbt.7.10.6623 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Pootrakul L et al. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin Cancer Res 12, 5987–5993, doi: 10.1158/1078-0432.CCR-06-0133 (2006). [DOI] [PubMed] [Google Scholar]
- 522.Burikhanov R et al. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell 138, 377–388, doi: 10.1016/j.cell.2009.05.022 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Hart LS & El-Deiry WS Cell death: a new Par-4 the TRAIL. Cell 138, 220–222, doi: 10.1016/j.cell.2009.07.007 (2009). [DOI] [PubMed] [Google Scholar]
- 524.Lee YS, Lee DH, Choudry HA, Bartlett DL & Lee YJ Ferroptosis-Induced Endoplasmic Reticulum Stress: Cross-talk between Ferroptosis and Apoptosis. Mol Cancer Res 16, 1073–1076, doi: 10.1158/1541-7786.MCR-18-0055 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Luhr M et al. The kinase PERK and the transcription factor ATF4 play distinct and essential roles in autophagy resulting from tunicamycin-induced ER stress. J Biol Chem 294, 8197–8217, doi: 10.1074/jbc.RA118.002829 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Bailly C & Waring MJ Pharmacological effectors of GRP78 chaperone in cancers. Biochem Pharmacol 163, 269–278, doi: 10.1016/j.bcp.2019.02.038 (2019). [DOI] [PubMed] [Google Scholar]
- 527.Chern YJ et al. The interaction between SPARC and GRP78 interferes with ER stress signaling and potentiates apoptosis via PERK/eIF2alpha and IRE1alpha/XBP-1 in colorectal cancer. Cell Death Dis 10, 504, doi: 10.1038/s41419-019-1687-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Dauer P et al. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis 10, 132, doi: 10.1038/s41419-019-1408-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Tait SW, Ichim G & Green DR Die another way--non-apoptotic mechanisms of cell death. J Cell Sci 127, 2135–2144, doi: 10.1242/jcs.093575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Lartigue L et al. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell 20, 4871–4884, doi: 10.1091/mbc.E09-07-0649 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Giampazolias E et al. Mitochondrial permeabilization engages NF-kappaB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol 19, 1116–1129, doi: 10.1038/ncb3596 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.White MJ et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562, doi: 10.1016/j.cell.2014.11.036 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Rongvaux A et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577, doi: 10.1016/j.cell.2014.11.037 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.McArthur K et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359, doi: 10.1126/science.aao6047 (2018). [DOI] [PubMed] [Google Scholar]
- 535.Rogers C et al. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10, 1689, doi: 10.1038/s41467-019-09397-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Orning P et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069, doi: 10.1126/science.aau2818 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Sarhan J et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A 115, E10888–E10897, doi: 10.1073/pnas.1809548115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Chen KW et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J 38, doi: 10.15252/embj.2019101638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Sarosiek KA et al. Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell 31, 142–156, doi: 10.1016/j.ccell.2016.11.011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Madden SD, Donovan M & Cotter TG Key apoptosis regulating proteins are down-regulated during postnatal tissue development. Int J Dev Biol 51, 415–423, doi: 10.1387/ijdb.062263sm (2007). [DOI] [PubMed] [Google Scholar]
- 541.Nakaya K et al. Sensitivity to radiation-induced apoptosis and neuron loss declines rapidly in the postnatal mouse neocortex. Int J Radiat Biol 81, 545–554, doi: 10.1080/09553000500280492 (2005). [DOI] [PubMed] [Google Scholar]
- 542.Chonghaile TN et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov 4, 1074–1087, doi: 10.1158/2159-8290.CD-14-0353 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Vo TT et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 151, 344–355, doi: 10.1016/j.cell.2012.08.038 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Ryan JA, Brunelle JK & Letai A Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci U S A 107, 12895–12900, doi: 10.1073/pnas.0914878107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Gupta PB et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138, 645–659, doi: 10.1016/j.cell.2009.06.034 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Ryan J & Letai A BH3 profiling in whole cells by fluorimeter or FACS. Methods 61, 156–164, doi: 10.1016/j.ymeth.2013.04.006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]