

Abstract
This study provides a comprehensive review of the published research on the association between early life adversity and markers of inflammation in children and adolescents. We conducted a systematic review of the published literature on the association between early life adversity and markers of inflammation in pediatric populations. To date, 27 studies have been published in this area representing a wide range of global populations and diverse methods of which nearly half were prospective, longitudinal studies. Of these 27, only 12 studies shared an inflammatory outcome with 4 or more other studies; 9 for CRP, and 6 for IL-6. The association between early life adversity and both CRP, z = .07 [.04, .10], and IL-6, z = .17 [−.07, .42], were small and only significant for CRP although comparable in magnitude to the effects observed in adult samples. Descriptively, the association between early life adversity and CRP appeared to be stronger in studies conducted in infants and adolescents compared with middle childhood. There was minimal evidence of publication bias for studies measuring CRP, but evidence of publication bias for studies using IL-6. Eight studies have looked at the association between early life adversity and stimulated inflammatory cytokines in vitro, and both the methods and results of these studies were mixed; the majority observed exaggerated production of inflammatory cytokines despite mixed methodological approaches that make comparisons across studies difficult. In summary, the evidence supporting an association between early life adversity and inflammation in pediatric samples is limited so far by the number of studies and their heterogeneous methodological approaches. More research that is grounded in a developmental framework and informed by the complexity of the innate immune system is needed in this area.
Keywords: Early life adversity, Childhood, Adolescents, Inflammation, Meta-analysis
1. Introduction
Approximately 12% of youth, from infancy to age 18, will be exposed to early life adversity to such an extent that they will experience lifelong mental and physical health disparities (Felitti et al., 1998; Kalmakis and Chandler, 2015). Early life adversity encompasses abuse by a caregiver, parent psychopathology, poverty, parent separation, household dysfunction, natural disasters, assault, and other unpredictable, potentially traumatic events (Felitti et al., 1998; Kessler et al., 2010; Kuhlman et al., 2018b). This minority of youth will account for nearly half of all psychiatric disorders (Kessler et al., 2010), experience at least a 44% increase in heart attacks and strokes (Korkeila et al., 2010), and an increased risk of all-cause mortality by age 50 (Chen et al., 2016; Kelly-Irving et al., 2013). Yet, the developmental pathways through which early life adversity places youth on a trajectory of lifelong health disparity is not well understood, thus impeding prevention and the development of targeted interventions that could mitigate these lifelong sequelae. One pathway through which early life adversity may presage lifelong psychiatric and cardiovascular health disparities is through alterations to the immune system, particularly by increasing inflammation. The purpose of this review was to synthesize the published research on the association between early life adversity and markers of inflammation measured during childhood and adolescence.
One hypothesized pathway through which early life adversity may lead to these health disparities is through alterations to the innate immune system, which is responsible for inflammation or the body’s initial defense against pathogens and tissue injury. In the short term, inflammation is protective against a wide range of bacterial and viral infections (See Mogensen, 2009 for review). However, chronic inflammation can also contribute to the pathogenesis of other diseases. For example, inflammation reorganizes human behavior to promote healing and recovery, which can mimic the behavioral phenotype observed in depression (Dantzer et al., 2008; Eisenberger et al., 2010), and has been prospectively linked to the pathogenesis of depression (Gimeno et al., 2009; Miller and Raison, 2016). This phenomenon has been the topic of investigation for the past three decades (Dantzer and Kelley, 2007). Chronic inflammation is also a well-established biological marker of cardiovascular disease risk (Koenig et al., 1999; Ridker, 2003; Ridker et al., 1997), has been implicated in the development and progression of cancer (Harris, 2007; Harris et al., 2014), and can lead to a number of serious health conditions commonly observed in older adults (Franceschi and Campisi, 2014).
There are several methods of assessing inflammatory activity. The most common is to measure circulating concentrations of inflammatory cytokines and markers of inflammation under resting/basal conditions in peripheral tissues (plasma, serum, dried blood spots, and saliva). This approach provides an index of the amount of inflammatory signaling occurring in the body at any given time. Another approach is to stimulate immune cells in vitro with bacterial agents, such as lipopolysaccharide (LPS), or other antigens that activate inflammatory processes. In this method, the amount of pathogen exposure in vitro is tightly controlled, and provides an index of the phenotypic potential for inflammatory reactivity within an individual’s immune system.
The association between early life adversity and markers of inflammation in adulthood is well-established (Baumeister et al., 2016; Danese et al., 2009, 2007; Matthews et al., 2014). Indeed, a meta-analysis of the 25 studies investigating the association between childhood trauma exposure and circulating inflammatory markers in adulthood was published only 2 years ago showing small, yet significant, effect sizes overall; z = 0.10 for CRP, z = 0.08 for IL-6, and z = 0.23 for TNF-α (Baumeister et al., 2016). Yet, it remains unclear whether relatively healthy youth exposed to early life adversity exhibit a similar exaggerated inflammatory profile to that observed in adults (Slopen et al., 2012).
Inflammation is carefully regulated throughout childhood and adolescence, and accumulates with age through biological aging, behavioral, psychosocial, and environmental pathways that impede effective regulation (Franceschi and Campisi, 2014; Miller et al., 2011). Thus, it is possible that the association between adversity exposure and inflammatory biomarkers does not emerge before adulthood or may be masked by regulatory processes. That being said, if inflammation is a pathway through which early life adversity contributes to lifelong health disparities, identifying the earliest signs of this pathway will allow early detection of those at greatest risk.
In the present study, we conducted a systematic review of the published literature on the association between early life adversity and inflammation in pediatric populations (ages birth to 18 years) with particular interest in characterizing the methodological landscape of the field to date; a meta-analysis of the studies that have indexed inflammation using circulating CRP and IL-6; and a qualitative analysis of studies looking at inflammatory responses of immune cells following in vitro stimulation. We hypothesized that youth exposed to early life adversity would demonstrate greater circulating markers of inflammation, and an emerging pro-inflammatory phenotype as measured in vitro by stimulated production of inflammatory cytokines.
2. Methods
2.1. Search strategy and inclusion criteria
The literature reviewed and subjected to systematic review and meta-analytic analyses represent a subset of published articles identified for a larger project within our research laboratory (Horn et al., 2017; Kuhlman et al., 2017). For the larger project, we conducted a systematic review of the literature on early life adversity and peripheral physiological stress systems using the PUBMED database according to the guidelines established in the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) (Moher et al., 2009). We identified relevant studies conducted in humans using cross-referenced search terms related to adversity (i.e., “child adversity”, “child trauma”, “child maltreatment”, “early life stress”, “child poverty”, “early life adversity”, “childhood socioeconomic status”) with search terms related to functioning of the HPA-axis or inflammatory arm of the immune system (i.e., “inflammation”, “inflammatory”, “innate immunity”, “cytokine”, “interleukin”, “cortisol”, “neuroendocrine”, “HPA-axis”). All duplicate articles were removed. Eligibility for article inclusion in the larger project was independently determined by two members of the research staff. Reliability for inclusion across raters was good, α = 0.88, and articles where raters were discrepant on inclusion were reviewed individually by the first author to determine eligibility. Criteria for inclusion were full peer-reviewed journal articles, available in English, which examined the association between exposure to adversity during childhood and functioning of either the hypothalamic-pituitary-adrenal (HPA)-axis or the inflammatory arm of the immune system. Articles that were relevant to the scope of the project but had used non-human animals, were reviews, or were retracted were excluded.
For each article, pertinent methodological details regarding the sample, study design, measurement of early life adversity, and measurement of the biological marker were extracted by trained research assistants. Data extracted from each article included: year of publication, journal of publication, study design (longitudinal or cross-sectional), sample size, the percent of participants that were female, percent of participants that were Caucasian, whether the study excluded participants for taking steroid or anti-depressant medication, the early life adversity measure used, the mean age of the sample at the time of the early life adversity assessment, whether early life adversity was assessed retrospectively, who reported the early life adversity exposure (caregiver, victim, 3rd party), the method of assessing early life adversity (self-report questionnaire, clinical interview, record review), whether the biological outcome was related to the HPA-axis or inflammation, the type of tissue used to measure the biomarker (blood, saliva, hair, urine), the mean age of the sample at the biomarker assessment, and whether the study looked at functioning of these physiological systems under resting/basal conditions or following a stressor/stimulation.
Articles from this larger database selected for inclusion in the present systematic review and meta-analysis were those studies looking specifically at the association between early life adversity and 1) a marker of inflammatory function that was 2) measured during childhood or adolescence. Given the nascent state of this field, inflammatory function was broadly defined and in some cases included markers that are non-specifically linked to both innate and adaptive immune function. For each of these included articles, two of the authors extracted which cytokines were measured in the study, the reported association between early life adversity and each inflammatory marker, and the covariates included in the models used to compute these associations. If results were reported by stratified groups, such as by sex or presence of a physical or mental illness, the coefficient for each group was recorded. Relevant tests of this association are Pearson correlations (r), t-tests (t), F-tests (F), odds ratios (OR), Cohen’s Ds (d), standardized betas (ß), unstandardized betas (b), Mann Whitney U (z), or mean differences between groups. If multiple statistics could be extracted from the manuscript, a statistic reflecting the association between ELS and the inflammatory marker corrected for known confounds, such as BMI, was preferred and the covariates noted over an uncorrected coefficient. The effect size extracted from the article and the test used to compute it was recorded by two authors. Discrepancies between authors were discussed until a consensus was reached. For articles where the association between early life adversity and the study’s chosen inflammatory marker were not clear or complete, the corresponding author of that manuscript was emailed to provide the pertinent information or to clarify the details of the model presented in the published article.
2.2. Meta-analysis
Meta-analyses were conducted separately for any inflammatory biomarker with 5 or more studies. This only applied to two inflammatory markers: C-reactive protein (CRP) and interleukin-6 (IL-6). To conduct our meta-analysis, we employed a random-effects model, which is a more conservative meta-analytic approach that accounts for within- and between-study variation. Random-effects models assume a distribution of effect sizes across studies and produce wider confidence intervals compared to the fixed-model approach (Egger et al., 1997; Hedges and Vevea, 1998). All effect sizes were transformed to r-values, with positive r-values signifying a positive relationship between early life adversity and the inflammatory marker. R-values are comparable across studies with different designs (e.g., predictor type) and were easily computable from information extracted from the manuscripts. In the event that studies stratified results by group (e.g., sex, weight), the results were treated as separate analyses. The metafor and user-friendlyscience packages on RStudio 1.0.136 were utilized to compute and aggregate effect sizes. All results are reported as Fisher’s r-to-z transformed correlation coefficients.
The Q and I2 statistics were calculated to assess heterogeneity among effect sizes. The Q statistic, distributed as χ2, specifies if variability among study outcomes is large enough to reject the null hypothesis that they are drawn from a common population. The I2 value describes the percentage of variation across studies due to heterogeneity. Moderator analyses were conducted if there was evidence of statistically significant heterogeneity (Higgins and Thompson, 2002). The following moderators were evaluated: 1) average age of participants at the time of the inflammatory measure (i.e., infancy = 0–2 years, childhood = age 3 to 9, late childhood/adolescent = 10 years of age or older) (Canadian Paediatric Society, 2003), 2) statistical adjustment for body mass index or waist circumference, 3) statistical adjustment for sex, 4) statistical adjustment for age, 5) tissue type (inflammatory marker measured in blood, or saliva, 6) type of early life adversity measure (self-report, clinical interview, record review), and 7) reporter of early life adversity (i.e., victim or caregiver/third party). See O’Connor and colleagues (2009) for a review on why moderators such as BMI, sex, and age should be considered in studies of inflammatory markers.
Forest plots were created to display the effect sizes for each study included in the meta-analysis with associated 95%CIs. To explore potential publication bias, funnel plots that illustrate the distribution of effect sizes by their standard error were created (Sterne and Egger, 2001). Studies falling outside the funnels depicting increasing magnitude effect sizes with decreasing standard errors are considered evidence of publication bias (Sterne and Egger, 2001).
3. Results
We identified 27 published, empirical studies that examined the association between early life adversity and an inflammatory marker in pediatric samples. Fig. 1 contains the PRISMA diagram depicting the article selection process that resulted in these 27 studies published between 1995 and 2018.
Fig. 1.
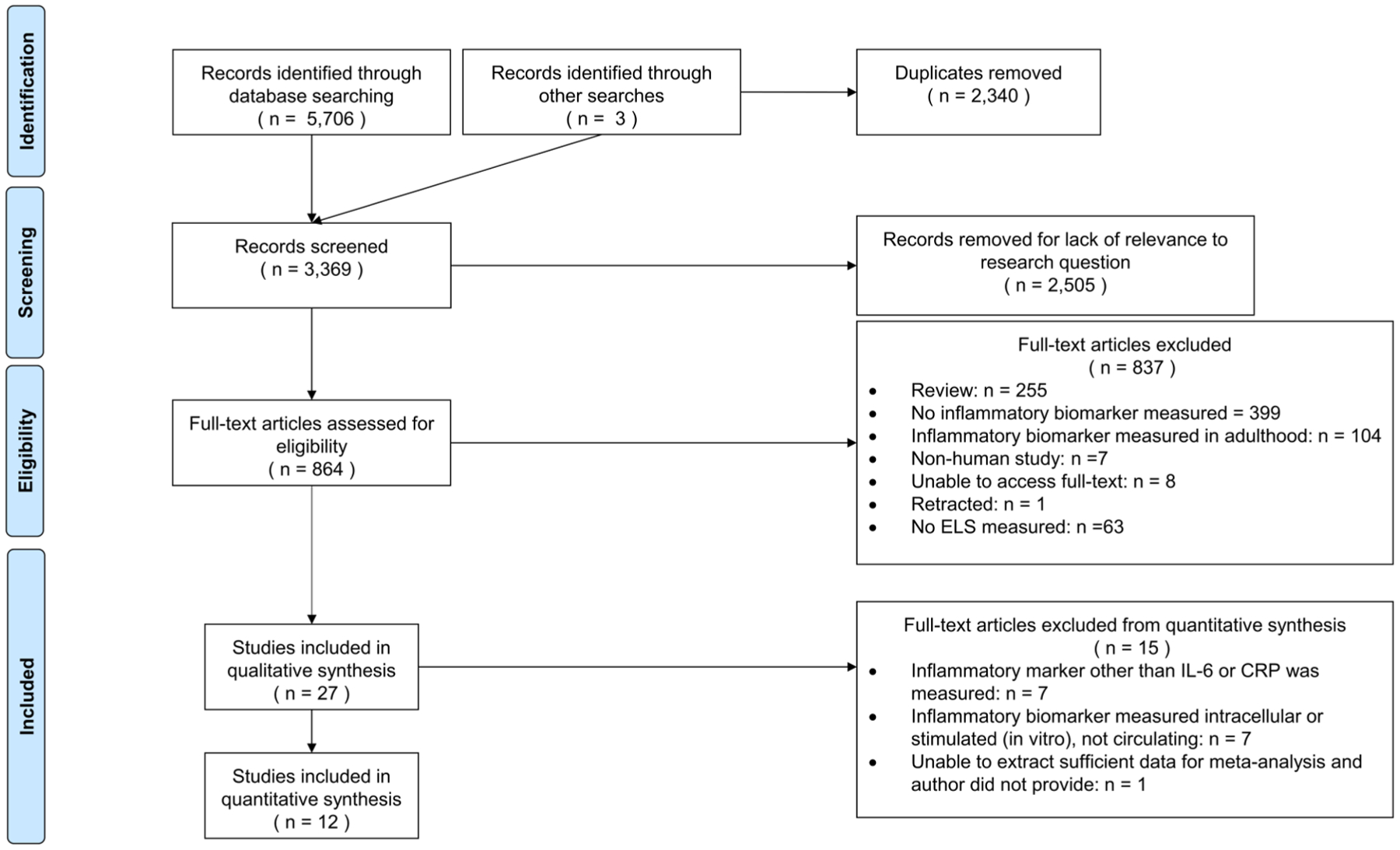
PRISMA diagram depicting articles identification.
First, we provide descriptive analysis of the 27 studies identified in our systematic review. Table 1 provides descriptive characteristics of these 27 studies. This information is organized into information pertaining to the sample, assessment of early life adversity, and measurement of the inflammatory biomarker. Despite the somewhat small number of studies published in this area, they reflect a wide range of methodologies, in diverse samples across the globe. First, these studies used samples that ranged from 27 to 2232 participants in youth from birth to age 19 and represented multiple ethnic groups. Notably, almost half used a prospective longitudinal design, where early life adversity was measured before the inflammatory markers; and one third of studies reported that participants were excluded from participating for steroid or antidepressant medication use.
Table 1.
Methodological and descriptive characteristics of published studies on association between childhood adversity and inflammatory biomarkers in pediatric samples (n = 27).
| M (SD) | Range | % (n) | |
|---|---|---|---|
| Sample characteristics | |||
| Sample size | 267.33 (474.20) | 27–2,232 | |
| Percent female | 58.11 (23.65) | 0–100 | |
| Percent Caucasian | 40.42 (30.49) | 0–100 | |
| Age of participants at adversity assessment | 9.59 (1.17) | 0–17.80 | |
| Age of participant at inflammatory assessment | 10.60 (5.88) | 1.42–19.50 | |
| Longitudinal study design | 37.0 (10) | ||
| Length of follow-up (n = 14) | 2.85 (4.11) | 0–13 | |
| Participants were excluded for steroid medication use | 29.6 (8) | ||
| Participants were excluded for anti-depressant medication use | 29.6 (8) | ||
| Childhood adversity measurement characteristics | |||
| Adversity was retrospectively reported | 40.7 (11) | ||
| Adversity reporter | |||
| Victim/child | 37.0 (10) | ||
| Caregiver | 40.7 (11) | ||
| Third party/record review | 18.5 (5) | ||
| Combination | 3.7 (1) | ||
| Adversity assessment format | |||
| Self-report questionnaire | 48.1 (13) | ||
| Clinical interview | 22.2 (6) | ||
| Record review | 22.2 (6) | ||
| Multiple | 7.4 (2) | ||
| Inflammatory biomarker measurement characteristics | |||
| Type of tissue used for inflammatory biomarker1 | |||
| Blood | 70.4 (19) | ||
| Saliva | 33.3 (9) | ||
| Included measure of HPA-axis function | 29.6 (8) | ||
| Circulating inflammation at rest | 74.1 (20) | ||
| Inflammatory response to challenge | 44.4 (12) | ||
Groups not mutually exclusive.
Early life adversity was measured using a number of methods in these studies. Given that the vast majority of studies in adult samples use retrospective self-report of early life adversity, it is notable that only 41% of studies used retrospective reports of adversity, and 37% of studies used victim reported adversity exposure. The remaining studies used adversity exposure that was reported by a caregiver, a third party, or used multiple informants. The distribution of methods used to assess early life adversity was similarly varied such that the most common method was via a questionnaire, yet this only accounted for 48% of studies. A similarly large proportion of studies used clinical interviews or third party record reviews.
The inflammatory biomarkers measured were heterogeneous across these studies, including studies of both circulating and stimulated cytokines. The most commonly measured inflammatory markers were IL-6 (n = 7), TNF-α (n = 2), CRP (n = 9), IFN-γ (n = 1), IL-10 (n = 2), and IL-1β (n = 3). To avoid spurious results, only studies examining CRP and IL-6 were subjected to meta-analysis because they have been examined in 5 or more studies. Finally, 1/3 of the studies also reported measures of neuroendocrine functioning, such as salivary cortisol or glucocorticoid receptor sensitivity.
3.1. Early life adversity and inflammation using circulating markers
Table 2 summarizes the studies measuring any circulating markers of inflammation, their methods, and results. A total of 12 studies, with k = 19 analyses, were included in the meta-analysis. Nine studies examined associations between early life adversity and CRP (Baldwin et al., 2018; Chen et al., 2013; Cicchetti et al., 2015; David et al., 2017; Goosby et al., 2015; Hadley and Decaro, 2014; Miller and Cole, 2012; Tyrka et al., 2015; Walsh et al., 2016) and six examined associations between early life adversity and IL-6 (Bücker et al., 2015; Chen et al., 2013; Kılıç et al., 2017; Miller and Chen, 2010; Miller and Cole, 2012; Riis et al., 2016; Walsh et al., 2016).
Table 2.
Childhood adversity and inflammation using circulating markers of inflammation.
| Study | Included in meta-analysis | Sample size | Mean Age of sample (years) | Sample characteristics1 | Adversity exposure | Inflammatory outcomes2 | Main result |
|---|---|---|---|---|---|---|---|
| David et al. (2017) | Yes | 49 | 1.42 | Healthy, low income community sample of mother-infant dyads participating in a larger longitudinal sample | Socioeconomic disadvantage and maternal psychosocial stress | Salivary C-reactive protein |
|
| Measelle and Ablow (2018) | No | 49 | 1.42 | Healthy, low income community sample of mother-infant dyads participating in a larger longitudinal sample | Maternal depression, socioeconomic disadvantage, and familial stress | Composite measure of pro-inflammatory cytokines (IL-1β, IL-6, IL-8, and TNF-α) and CRP measured via saliva |
|
| Phillips et al. (2004) | No | 130 | 2.67 | Rural children in Nigeria | Measles exposure | Th1 and Th2 cytokines, IL-12, IL-6 |
|
|
|||||||
| Hadley and Decaro (2014) | Yes | 1,387 | 3.0 | Nationally representative study of children in Tanzania | Household wealth rank | CRP via dried blood spots |
|
| Tyrka et al. (2015) | Yes | 40 | 4.16 | Maltreated children identified through child welfare, and community controls recruited from low-income pediatric clinic | Past-month contextual stressors, lifetime contextual stressors, and traumatic life events | Salivary IL-1ß and CRP |
|
|
|||||||
| Riis et al. (2016) | No | 125 | 5.45 | Healthy, community sample of mother-child pairs | Maternal psychological distress | Salivary IL-1β, IL-6, IL-8, TNF-α |
|
| Hagel et al. (1995) | No | 85 | 8.50 | Ascaris-infected children living in an urban slum area of Caracas, Venezuela | Malnutrition | IgE and IL-4 |
|
| Bücker et al., (2015) | Yes | 61 | 9.24 | Children (ages 3–12) drawn from a Child Protection Programme / foster care system or primary care in south Brazil | Met Criteria A for PTSD in DSM-IV via clinical interview | TNF-α, IL-12p70, IL-6, IL-8, IL-10, and IL-1β |
|
| Cicchetti et al. (2015) | Yes | 489 | 9.72 | Youth participating in a summer day camp for low-income children | Maltreatment determined via Department of Human Services records | Salivary CRP |
|
| Ulmer-Yaniv et al. (2018) | No | 177 | 10 | Israeli families enrolled in a longitudinal study living in comparable towns either near or far from Gaza border. | Exposure to continuous wartime trauma | Salivary IgA |
|
| Worthman and Panter-Brick (2008) | No | 107 | 11.80 | Rural, homeless, and urban Nepali children | Homelessness | Alpha 1-antichymotrypsin |
|
| Goosby et al. (2015) | Yes | 40 | 12.32 | Low-income African American and Caucasian mothers with a focal child between the ages of 10 and 15 were recruited through health fairs. | Perceived discrimination | CRP via dried blood spots |
|
| Falcone et al. (2015) | No | 115 | 14.20 | Youth admitted to child and adolescent psychiatric unit with mood disorders or psychosis | Childhood trauma using the Life Events Checklist and the Adverse Childhood Experiences scale | S100B |
|
| Chen et al. (2013) | Yes | 163 | 14.53 | Community sample of families from Vancouver, British Columbia, Canada | Low childhood socioeconomic status | IL-6 and CRP |
|
| Kılıç et al. (2017) | Yes | 27 | 15 | Treatment seeking population in hospital within 72 hours of sexual abuse experience in Bursa, Turkey | Sexual abuse | IL-6 and IL-10 |
|
| Bick et al. (2015) | No | 206 | 15.30 | Community sample | History of childhood neglect via Childhood Trauma Questionnaire (CTQ) | Macrophage Migration Inhibitory Factor (MIF) |
|
|
|||||||
| Chen et al. (2003) | No | 30 | 14.65 | Adolescents with persistent asthma | Low SES and cumulative life stress | IL-4, IL-5, IFN-γ |
|
| Walsh et al. (2016) | Yes | 124 (T2), 109 (T3) | 16.50 | Pregnant adolescent girls | Exposure to child abuse via the CTQ | IL-6 and CRP |
|
| Baldwin et al. (2018) | Yes | 1,732 | 18 | Longitudinal birth cohort of British twins in the United Kingdom | Cumulative exposure to several types of victimization (domestic violence between the mother and her partner; frequent bullying by peers; physical maltreatment by an adult; sexual abuse; emotional abuse and neglect; and physical neglect) assessed at ages 5, 7, 10, and 12 years | CRP |
|
| Miller and Chen (2010) | Yes | 135 | 17.0 | Young women at risk for affective disorders in Vancouver, British Columbia, Canada | Harsh family climate and episodic recent stressors | IL-6 |
|
| Miller and Cole (2012) | Yes | 147 | 19.5 | All female community sample in Vancouver, British Columbia, Canada | Childhood adversity was indexed by parental separation, low socioeconomic status, and familial psychopathology | CRP and IL-6 |
|
All studies conducted in the United States of America unless otherwise noted.
Measured in blood unless otherwise noted.
C-reactive protein.
A total of 9 studies, representing data from 4692 youth, with k = 12 analyses, were included in the meta-analysis between CRP and early life adversity. Five of these studies examined multiple types of adversity (i.e., physical and sexual abuse, emotional abuse, neglect, family psychopathology, domestic violence, bullying, low SES, or parental separation). One study examined discrimination and three studies examined socioeconomic or poverty status. One study (David et al., 2017) examined two separate types of ELS (SES and maternal stress) and both analyses were included1. The aggregated effect size was small but significant, z = .07, SE = .02, 95%CI = .04 – .10, p < .0001. Little heterogeneity was observed, Q(df = 11) = 13.52, p = .26, I2 = 0.10% suggesting no moderators of this effect. There was no evidence of publication. Fig. 2 depicts the forest plot of studies looking at early life adversity sorted by the average participant age at the time of the CRP assessment and CRP and Fig. 3 depicts the funnel plot illustrating minimal publication bias across these studies.
Fig. 2.
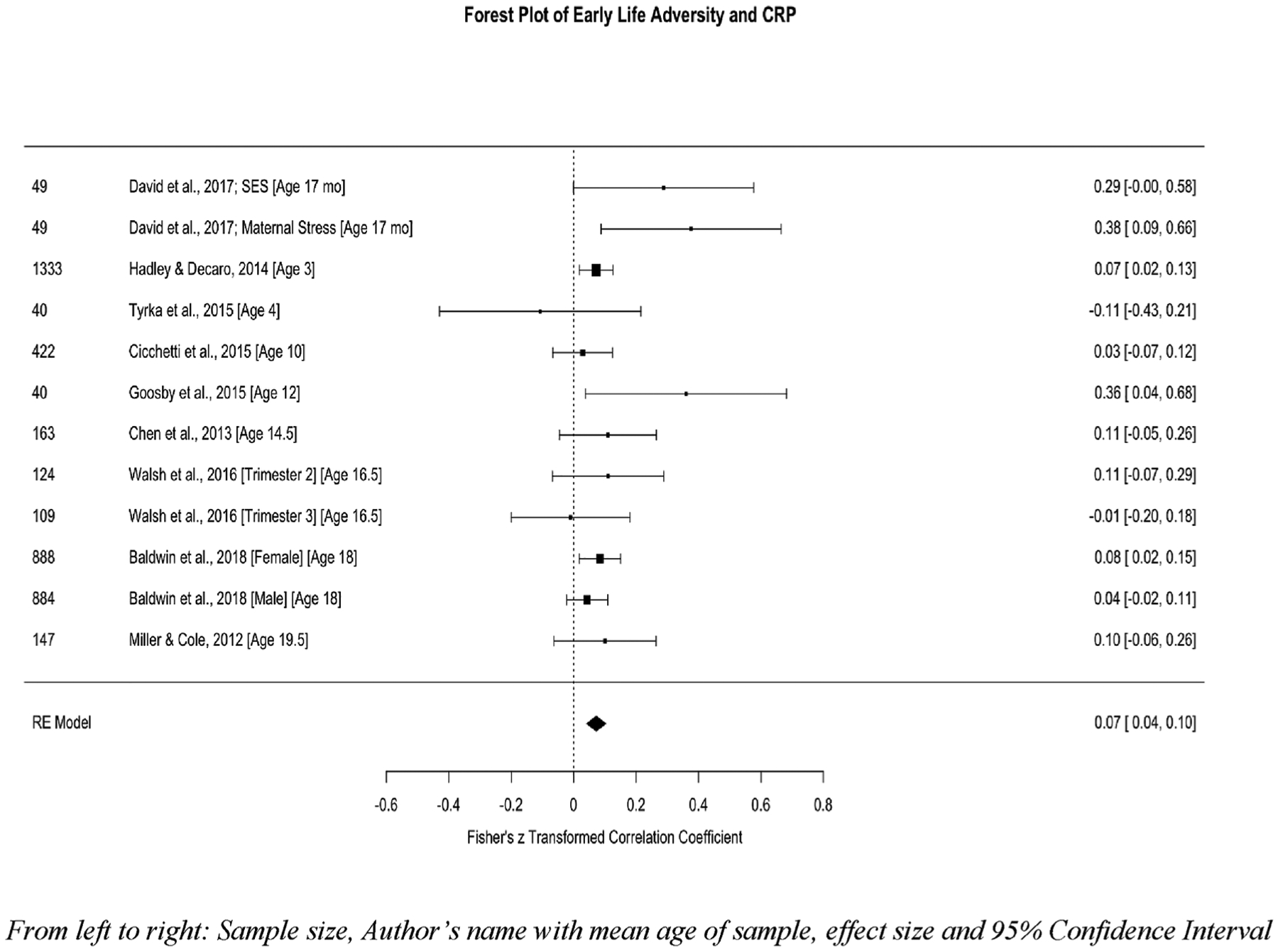
Forest plot of childhood adversity and CRP.
Fig. 3.
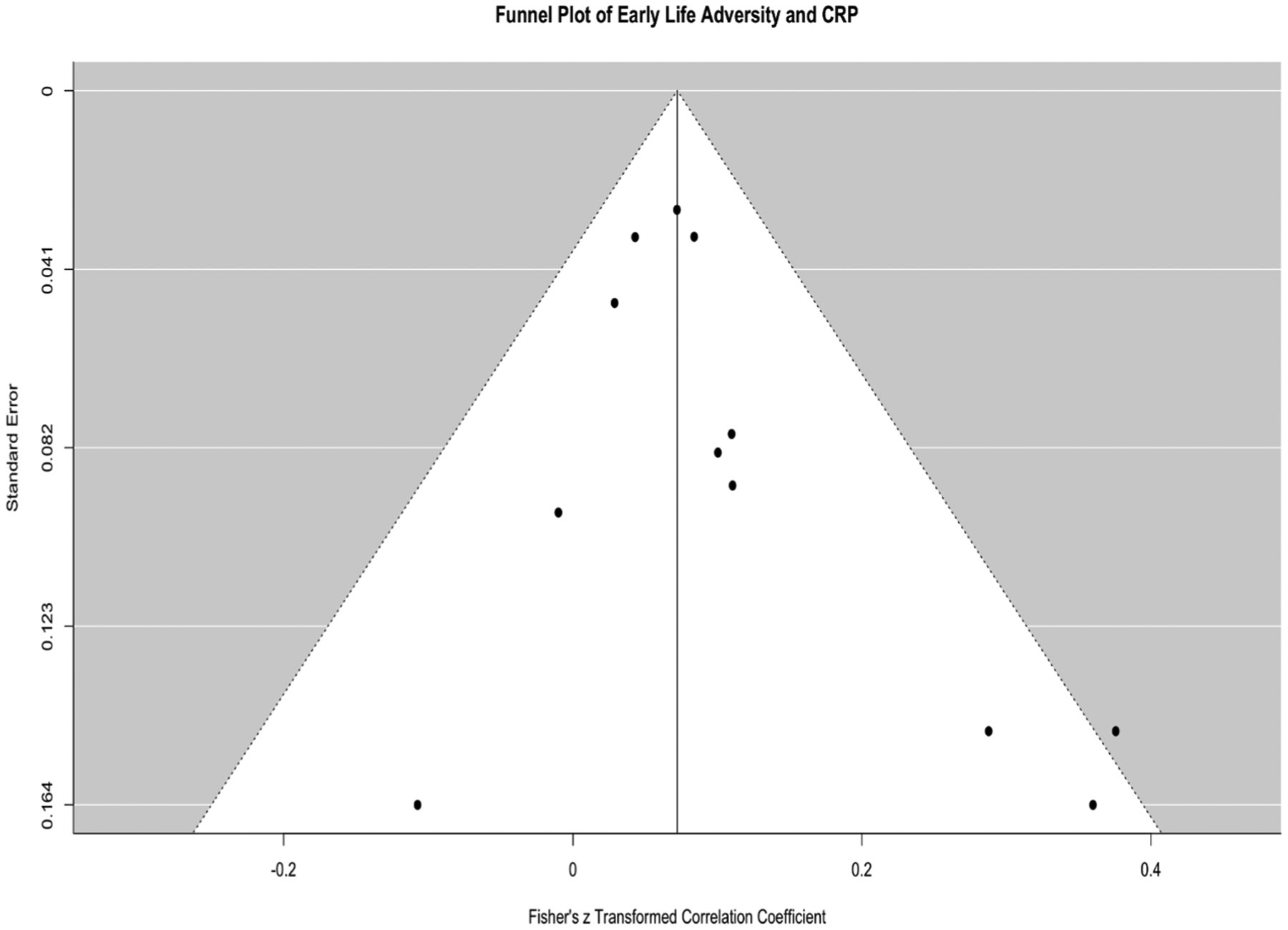
Funnel plot for studies examining childhood adversity and CRP.
Moderators of the association between early life adversity and CRP.
All proposed moderators were explored. Type of trauma exposure, reporter of trauma, tissue type, age of participant, and statistical adjustment for age, sex, and BMI were all non-significant moderators. However, the moderation analyses revealed a qualitatively interesting pattern of results by age that this analysis is likely underpowered to detect given the small sample size, QM = 1.43, p = 23. Specifically, the association between adversity and CRP appears to be significant and positive during infancy and adolescence, z = 0.33, SE = 0.10, p = .002, 95%CI[0.13,0.54] and z = .07, SE = .02, p = .0004, 95%CI[.03, .10] respectively, and non-significant during early childhood, z = .06, SE = .05, p = .28, 95%CI[−.05, .16]. Fig. 4 uses a violin plot to illustrate the association between adversity and CRP by age group.
Fig. 4.

Violin plot illustrating association between early life adversity and CRP by age groups (infancy, early childhood, adolescence).
Interleukin-6.
A total of 6 studies, representing data from 522 youth, with k = 7 analyses, were included in the meta-analysis between early life adversity and IL-6. Fig. 5 depicts a forest plot of studies looking at early life adversity and IL-6 sorted by the average participant age at the time of the IL-6 assessment. Three studies examined multiple types of adversity (e.g., index trauma, parental separation, SES, family psychopathology, physical, sexual, and emotional abuse, neglect). Two studies examined family dynamics (i.e., parenting stress and harsh family environment), one study examined low SES, and one study examined sexual abuse. The aggregated effect size was non-significant, z = .17, SE = .12, 95%CI[−.07, .42], p = .16. High levels of heterogeneity were observed, Q (df = 6) = 46.12, p < .0001, I2 = 90.57%, suggesting that there may be important moderators to consider in this effect. There was some evidence of publication bias. Fig. 6 depicts the funnel plot illustrating publication bias in studies reporting IL-6 outcomes.
Fig. 5.
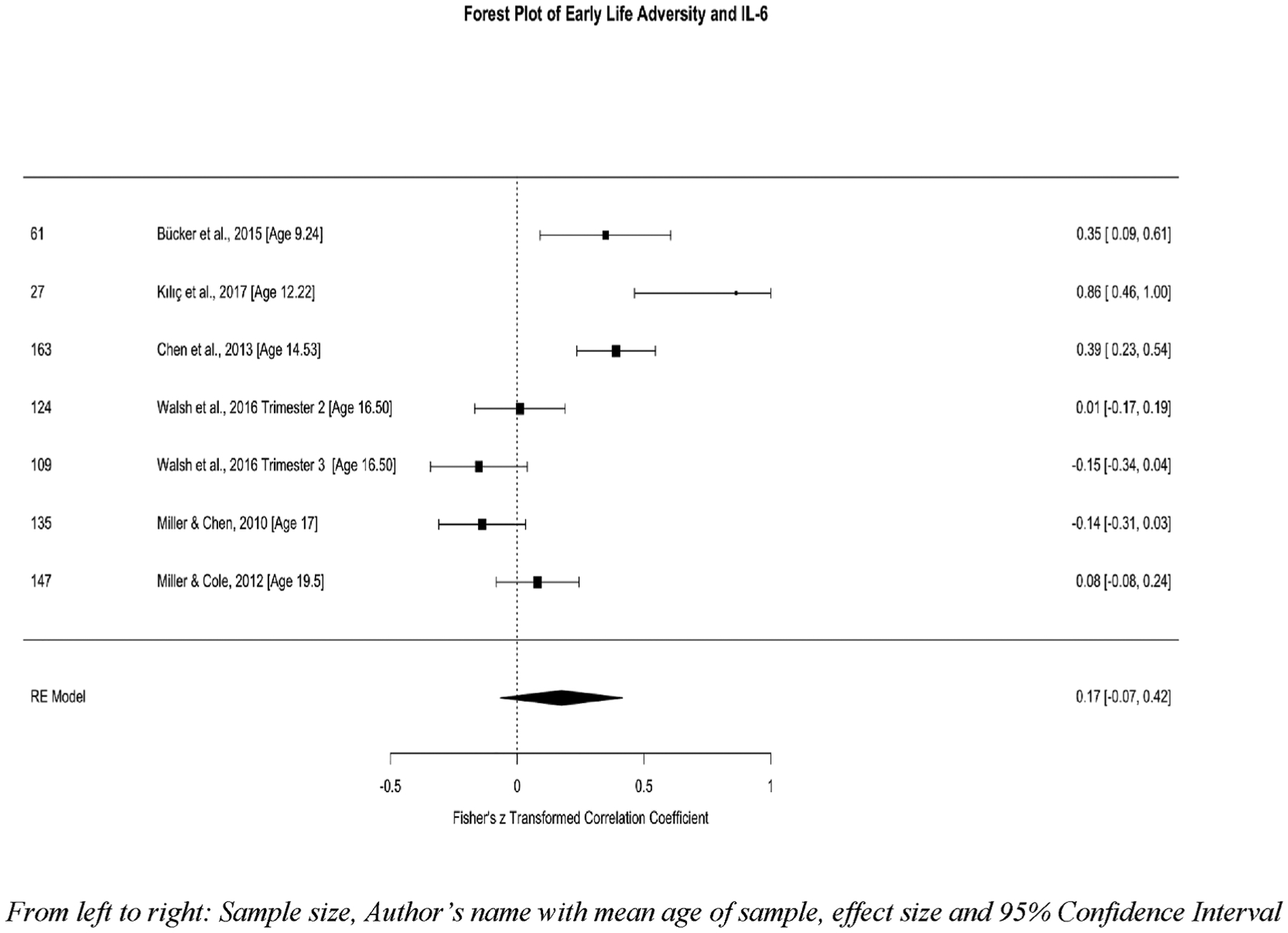
Forest plot of childhood adversity and circulating IL-6.
Fig. 6.

Funnel plot for studies examining childhood adversity and IL-6.
Moderators of the association between early life adversity and IL-6.
All proposed moderators were explored given the high degree of heterogeneity observed. All moderators were non-significant (i.e., age of participant, statistical adjustment for age, sex, and BMI, type of trauma exposure, and tissue type), with the exception of reporter of trauma, QM = 35.48, p < .0001, r = .43 (SE = .07), 95%CI[.29, .58]. Specifically, in studies where the ELS reporter was the victim (k = 4) the association between ELS and IL-6 was non-significant, z = −.05, SE = .06, p = .49, 95%CI = −.16 – .07, while when the reporter was the caregiver (k = 2) or a third party (k = 1) the association was positive, z = .39, SE = .07, p < .0001, 95%CI = .25 – .51 and z = .86, SE = .20, p < .0001, 95%CI = .46 – 1.26, respectively.
3.2. Early life adversity and inflammation using stimulated cytokine production
We identified 8 studies that have investigated the association between early life adversity and inflammatory processes using stimulated cytokine production in vitro with mixed results (Ayaydin et al., 2016; Azad et al., 2012; Chen et al., 2003, 2017; Ehrlich et al., 2016; Miller and Chen, 2010; Ramratnam et al., 2017; Wright et al., 2004). These studies could not be subjected to meta-analysis due to the significant heterogeneity across studies in the immune cells stimulated, stimulants used, and cytokines measured. Table 3 summarizes each of these studies, their methods, and results. Of the 8 studies published in this area, 6 observed a positive association between early adversity and at least one measure of in vitro production of inflammatory cytokines (Azad et al., 2012; Chen et al., 2017, 2003; Ehrlich et al., 2016; Miller and Chen, 2010; Wright et al., 2004), while three studies observed negative associations between early adversity and in vitro production of pro-inflammatory cytokines (Ayaydin et al., 2016; Ramratnam et al., 2017; Wright et al., 2004). The methodological heterogeneity across these studies may have contributed to some of these mixed results, and few conclusions can be drawn from comparisons across these studies at present.
Table 3.
Childhood adversity and inflammation using stimulated cytokine production.
| Study | Sample size | Age | Health status | Early life stress | Cells stimulated | Pathogen | Inflammatory outcomes | Main result |
|---|---|---|---|---|---|---|---|---|
| Wright et al. (2004) | 114 | 2 | at risk for asthma or allergy | Caregiver stress | PBMC | dust mite antigen; cockroach extract; phytohemagglutinin | IFN-y; TNF-α; IL-10; IL-13 |
|
|
||||||||
| Ramratnam et al. (2017) | 419 | 3 | at risk for respiratory disorder | Maternal stress; maternal depression; composite of stress and depression; measured prenatally, child age 1, 2, and 3 | PBMC | rhinovirus; respiratory syncytial virus; polyclonal stimulus; antigenic stimulus; cockroach extract, dust mite extract; CD3/aCD28 monoclonal antibody; phytohemagglutinin;tetanus toxoid | IFN-y; IL-4; IL-5; IL-13; examined separately |
|
|
||||||||
| Azad et al. (2012) | 267 | 8 to 10 | asthma | Family SES | PBMC | LPS | IL-6 |
|
| Chen et al. (2017) | 150 | 9 to 17 | diagnosed asthma | Childhood SES; family relationship stress | PBMC | PMA/INO; Poly I:C | Th1 composite (IFN-y; IL-10); Th2 composite (IL-2, IL-4, IL-5, IL-13); inflammatory composite (IL-1beta, IL-6, TNF-α) |
|
|
||||||||
|
||||||||
|
||||||||
| Chen et al. (2003) | 30 | 13–18 | Adolescents with persistent asthma | Low neighborhood SES and cumulative life stress | PBMC | phorbol myristate acetate (PMA 25 ng/ml) ionomycin (INO 1 M/ml) | IL-4, IL-5, and IFN-γ |
|
| Ayaydin et al. (2016) | 43 | 13 to 18 | PTSD | Sexual abuse | PBMC | PMA/INO | IFN-γ; TNF-α |
|
|
||||||||
| Ehrlich et al. (2016) | 147 | 15 to 19 | at risk for depression | Composite based on whether participants were born to a teenage mother, experienced parent death or parental separation, had parents with an affective illness of parents or came from low family SES backgrounds | monocytes | LPS | IL-6; GC sensitivity |
|
| Miller and Chen (2010) | 135 | 17–19 | Women at risk for affective disorders | Harsh family climate and episodic recent stressors | White blood cells | LPS | IL-6 |
|
|
Note: PMA = phorbol myristate acetate, INO = ionomycin, PBMC = peripheral blood mononuclear cells, LPS = Lipopolysaccharide.
4. Discussion
To date, this is the first meta-analysis focused specifically on early life adversity and inflammation in pediatric samples. Results from the meta-analysis illustrate the current state of the field and provide preliminary evidence for understanding the association between adversity and inflammatory processes in youth. Of note, only circulating levels of CRP and IL-6 were measured consistently enough across studies to qualify for quantitative analysis. The effect sizes for the association between early life adversity and both CRP and IL-6 were small, and only significant for CRP. This association appeared to be driven by studies conducted in infancy and adolescence. From our descriptive analysis of stimulated cytokine production in vitro, early life adversity also appears to be related to greater production of cytokines by immune cells following in vitro stimulation, although these studies were also quite mixed in both methods and results. These results should be interpreted with caution because the studies that have been published in this research area are limited in number and quite heterogeneous in study design, population, and inflammatory measures.
Contrary to our hypotheses, the associations between early life adversity exposure on circulating IL-6 and CRP were very small in magnitude. The effect sizes for IL-6 and CRP were < .20 and only significant for CRP. This finding contrasts somewhat with the similarly small, but uniformly significant, effect sizes observed in a meta-analysis of similar studies in adult samples, specifically z = 0.10 for CRP, z = 0.08 for IL-6, and z = 0.23 for TNF-α (Baumeister et al., 2016). Importantly, our confidence intervals were wider, and there were too few studies that measured TNF-α (the largest effect in adults) in pediatric populations to subject to meta-analysis. Taken together, there are two possible explanations for the pattern of associations observed in these pediatric studies, one in which dysregulated immune function measured via circulating inflammatory markers does not emerge until later in human development, and one that suggests sensitive periods of immune development that warrant further investigation in the context of early adversity.
In the first scenario, early life adversity may exert proinflammatory effects on immune cells throughout development that are only reliably observed when immune cells are stimulated in vitro, but not necessarily when measured in circulation. This may happen because increasing stress during childhood is associated with parallel increases in activity of the HPA-axis and its end-product, cortisol (Kuhlman et al., 2016, 2018a). When cortisol binds to glucocorticoid receptors in healthy immune cells, the production of inflammatory cytokines is inhibited (Barnes, 1998; Waage et al., 1990). Thus, upregulations in the HPA-axis may be masking the pro-inflammatory phenotype in children as measured via circulating markers. Indeed, the HPA-axis is socially regulated by caregivers during much of childhood such that youth exhibit attenuated HPA-axis activity around supportive caregivers (Gunnar and Donzella, 2002) while youth exposed to maltreatment and neglect exhibit elevated concentrations of cortisol throughout the day (Dozier et al., 2006; Tarullo and Gunnar, 2006). Thus, children exposed to high early life adversity may exhibit lower circulating markers of inflammation as a function of the inhibitory effect elevated circulating glucocorticoids have on the system. This may also explain why a more consistent pattern of exaggerated cytokine production is observed in studies using in vitro stimulation of immune cells, where the influence of glucocorticoids on production of cytokines is either missing or tightly controlled and evaluated. Adolescence is an important turning point for many of these processes in part because the HPA-axis is substantially altered during pubertal development (Gunnar and Quevedo, 2007; Tarullo and Gunnar, 2006), and the influence of caregivers on regulation of the axis is reduced; for example, adolescents exposed to trauma and maltreatment do not as consistently exhibit elevated diurnal cortisol throughout the day (Kuhlman et al., 2015). Consistent with this idea, adolescents exposed to harsh family environments also appear to have immune cells that are less sensitive to the inhibitory effects of glucocorticoids over time (Miller and Chen, 2010). As these glucocorticoid resistant cells accumulate over time, circulating measures of inflammatory proteins are more likely to be observed. These observations underscore the importance of measuring glucocorticoid concentrations in addition to inflammatory markers whenever possible, yet, only one third of the published studies to our knowledge have included an index of HPA-axis functioning in addition to their inflammatory index. The field would greatly benefit if future studies in children simultaneously measured cortisol when assessing inflammatory markers in order to account for whether null associations between early life adversity and inflammation are accounted for by elevated cortisol in exposed youth. Adolescence is also a likely time for proinflammatory phenotypes to emerge in circulating markers of immune function due to increases in risky behaviors, such as smoking, lack of exercise, and unhealthy eating habits that contribute to weight gain (Raposa et al., 2014) as well as impulsive behaviors that increase stress exposure (Hanson et al., 2015; Lovallo, 2013; Rudolph et al., 2000; Rudolph and Hammen, 2003). These risky behaviors may be particularly likely to occur during adolescence given the increase in psychosocial stress exposure that occurs during this phase of development (Rudolph and Hammen, 2003) and decreasing supervision of behavior by adults. Thus, as children develop into young adults there are multiple biological and behavioral inputs into the immune system that contribute to the production and regulation of inflammation that lead to more reliable elevations in circulating inflammatory markers among at risk populations. This is an important possibility because it introduces a great deal of opportunity for prevention in youth populations.
Alternatively, while age was not a statistically significant moderator, positive associations between early life adversity and inflammation were observed most robustly within infancy and late childhood/adolescent samples, but not in early childhood samples (e.g., Fig. 4). This observation underscores the importance of taking development into account in future studies of inflammatory processes in children, and further supports theoretical frameworks that see infancy and adolescence as sensitive periods for immune development (Kuhlman et al., 2017). That being said, a very small number of studies have been conducted in infancy or early childhood, which leaves us with very little data speaking to whether the association between early life adversity and elevated markers of inflammation is linear or non-linear across development.
From the limited number of studies looking at in vitro stimulation of pro-inflammatory cytokines, there is more consistent evidence for a positive association between early adversity exposure and stimulated cytokine production, although the methodological approaches and subsequent findings have been quite mixed. As a result of this heterogeneity, we were unable to summarize these studies quantitatively, but rather provide a qualitative synthesis of their findings. Together these studies portray a complex story about the potential effects of early adversity on stimulated production of inflammatory cytokines. There may be several reasons for these mixed findings, including the different stimulants used to evoke an inflammatory response, specific cytokines assessed and their role in pathogen defense, the type of adversity exposure, and the age of the sample. For instance, across studies, stimulants used included LPS, PMA/INO, rhinovirus, dust mite extract, cockroach extract, and tetanus toxoid. Similarly, a range of cytokines have been assessed, and while some studies focused on a single cytokine (Azad et al., 2012; Ehrlich et al., 2016), other studies assessed multiple cytokines, with some of these studies aggregating across cytokines (Chen et al., 2017) and others examining each cytokine separately (Ramratnam et al., 2017). It should also be noted that all of these studies focused on youth with or at risk for a physical or psychiatric disorder and findings may differ for otherwise healthy samples of youth. Lastly, studies varied in the types of adversities assessed as well as the age of the participants at the time of the blood collection and in vitro stimulation assay, ranging from toddlerhood to adolescence (i.e., 2–19 years). Although assessing various stressors and focusing on different developmental stages can provide a better understanding of differential stressor and timing effects (see Kuhlman et al., 2017 for review), currently there are too few studies to make any definitive conclusions.
Interest in the role of early life adversity in inflammatory processes has increased since the first study in this area was published thirty years ago (See Dantzer and Kelley, 2007 for a history of this field). The studies reviewed here represent a relatively large number of youth from a wide range of backgrounds and have utilized a wide range of methodological approaches. More attention to the role of different types of adversity in the development of the immune system will advance the translational value of this research over time. Of greatest importance will be to better understand the immune implications of chronic, compared with acute, forms of childhood adversity. For example, the stimulated cytokine studies varied in their operational definitions of early adversity, including sexual abuse (Ayaydin et al., 2016), maternal stress or mental health (Ramratnam et al., 2017; Wright et al., 2004), low childhood socioeconomic status (Azad et al., 2012; Chen et al., 2017), to mixed adverse childhood events (Ehrlich et al., 2016). See Kuhlman et al. (2017) for more on how different types of adversity may influence psychoneuroimmune development. There is also a need for more consistency in inflammatory measures across studies to garner a clear understanding of the emergence of exaggerated inflammatory processes in adversity exposed youth. For example, studies examining in vitro production of pro-inflammatory cytokines tend to look at multiple outcomes, including the production of Th1 or Th2 cytokines. Th1 cytokines including IFN-γ defend against intracellular parasites such as bacteria and viruses, whereas Th2 cytokines including IL-4, IL-5, IL-13, protect against helminths and other extracellular parasites (Romagnani, 1994). Yet, no conclusions can be drawn about whether early life adversity is linked to alterations in any specific patterns of cytokine responses. Further, there was a great deal of inconsistency across studies in the adoption of best practices for research on inflammatory biomarkers, particularly with respect to what factors should be controlled for in analyses (O’Connor et al., 2009) which may differ in adult and pediatric samples. Finally, five of the studies included in our meta-analysis used salivary measures of IL-6 or CRP (Cicchetti et al., 2015; David et al., 2017; Measelle and Ablow, 2018; Riis et al., 2016; Tyrka et al., 2015) which have not yet been validated to the extent that blood-based markers have been and may not be comparable. However, given the small number of studies in the field and the importance of non-invasive measures in studies of children, we thought it important to include their results. There was no evidence that the effect of early life adversity on IL-6 or CRP was moderated by collection of these markers in blood or saliva but these analyses need to be replicated as more studies are published.
The association between early life adversity, as measured by CRP and IL-6, appears to be small across youth samples, was only observable in CRP, and may be more consistent in studies of stimulated cytokine production in vitro. Overall, alterations to lifelong inflammatory processes may not already be instantiated in youth exposed to early life adversity. However, future investigations of this relationship are needed that break down the role of different types of adversity exposure, their timing, and their chronicity. Further, the largest effect size of childhood adversity on inflammation in adults was for TNF-α, which has only been examined in two pediatric samples to our knowledge. The considerable associations between early life adversity and inflammatory markers in adults (Baumeister et al., 2016) may also be compensated for by interdependent systems (i.e., HPA axis), perpetuated by chronic stress exposure, or behaviorally mediated and therefore largely preventable. These associations may be most effectively interrogated in studies using in vitro stimulation of immune cells and experimental models that provoke the immune system in pediatric populations, such as using psychological stress (Steptoe et al., 2007), or vaccines (Kuhlman et al., 2018c). These paradigms may be particularly useful in pediatric populations given the generally low circulating markers of inflammation seen in young people, despite exposure to severe adversity, and therefore more sensitively inform the nuanced development of immune perturbations that follow adverse experiences.
Acknowledgements
The composition of this manuscript was made possible by the National Institute of Mental Health via a Mentored Clinical Scientist Research Career Development Award awarded to Dr. Kuhlman (K08MH112773). We would like to thank the following research assistants for their tireless help reviewing abstracts and extracting pertinent methodological data from articles: Priya Kainth, Christina Keyheyan, Emily Koos, Julia Nakamura, Afrida Sara, So Yen “Jamie” Shin, and Katherine Zhuo.
Footnotes
The RE model was also run with only one of the analyses (SES and CRP) from David et al., 2017. The effect size was did not change meaningfully.
References
- Ayaydin H, Abali O, Akdeniz NO, Kok BE, Gunes A, Yildirim A, Deniz G, 2016. Immune system changes after sexual abuse in adolescents. Pediatr. Int. Off. J. Jpn. Pediatr. Soc 58, 105–112. 10.1111/ped.12767. [DOI] [PubMed] [Google Scholar]
- Azad MB, Lissitsyn Y, Miller GE, Becker AB, HayGlass KT, Kozyrskyj AL, 2012. Influence of socioeconomic status trajectories on innate immune responsiveness in children. PloS One 7, e38669. 10.1371/journal.pone.0038669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin JR, Arseneault L, Caspi A, Fisher HL, Moffitt TE, Odgers CL, Pariante C, Ambler A, Dove R, Kepa A, Matthews T, Menard A, Sugden K, Williams B, Danese A, 2018. Childhood victimization and inflammation in young adulthood: a genetically sensitive cohort study. Brain. Behav. Immun 67, 211–217. 10.1016/j.bbi.2017.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ, 1998. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin. Sci. Lond. Engl 1979 (94), 557. [DOI] [PubMed] [Google Scholar]
- Baumeister D, Akhtar R, Ciufolini S, Pariante CM, Mondelli V, 2016. Childhood trauma and adulthood inflammation: a meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-α Mol. Psychiatry 21, 642. 10.1038/mp.2015.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick J, Nguyen V, Leng L, Piecychna M, Crowley MJ, Bucala R, Mayes LC, Grigorenko EL, 2015. Preliminary associations between childhood neglect, MIF, and cortisol: potential pathways to long-term disease risk. Dev. Psychobiol 57, 131–139. 10.1002/dev.21265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bücker J, Fries GR, Kapczinski F, Post RM, Yatham LN, Vianna P, Bogo Chies JA, Gama CS, Magalhães PV, Aguiar BW, Pfaffenseller B, Kauer-Sant’Anna M, 2015. Brain-derived neurotrophic factor and inflammatory markers in school-aged children with early trauma. Acta Psychiatr. Scand 131, 360–368. 10.1111/acps.12358. [DOI] [PubMed] [Google Scholar]
- Canadian Paediatric Society, 2003. Age limits and adolescents. Paediatr. Child Health 8, 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E, Fisher EB, Bacharier LB, Strunk RC, 2003. Socioeconomic status, stress, and immune markers in adolescents with asthma. Psychosom. Med 65, 984–992. [DOI] [PubMed] [Google Scholar]
- Chen E, Lee WK, Cavey L, Ho A, 2013. Role models and the psychological characteristics that buffer low-socioeconomic-status youth from cardiovascular risk. Child Dev. 84, 1241–1252. 10.1111/cdev.12037. [DOI] [PubMed] [Google Scholar]
- Chen E, Shalowitz MU, Story RE, Ehrlich KB, Manczak EM, Ham PJ, Le V, Miller GE, 2017. Parents’ childhood socioeconomic circumstances are associated with their children’s asthma outcomes. J. Allergy Clin. Immunol 140, 828–835.e2. 10.1016/j.jaci.2016.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E, Turiano NA, Mroczek DK, Miller GE, 2016. Association of reports of childhood abuse and all-cause mortality rates in women. JAMA Psychiatry 73, 920–927. 10.1001/jamapsychiatry.2016.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti D, Handley ED, Rogosch FA, 2015. Child maltreatment, inflammation, and internalizing symptoms: investigating the roles of C-reactive protein, gene variation, and neuroendocrine regulation. Dev. Psychopathol 27, 553–566. 10.1017/S0954579415000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese A, Moffitt TE, Harrington H, et al. , 2009. Adverse childhood experiences and adult risk factors for age-related disease: depression, inflammation, and clustering of metabolic risk markers. Arch. Pediatr. Adolesc. Med 163, 1135–1143. 10.1001/archpediatrics.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese A, Pariante CM, Caspi A, Taylor A, Poulton R, 2007. Childhood maltreatment predicts adult inflammation in a life-course study. Proc. Natl. Acad. Sci 104, 1319–1324. 10.1073/pnas.0610362104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Kelley KW, 2007. Twenty years of research on cytokine-induced sickness behavior. Brain. Behav. Immun 21, 153–160. 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW, 2008. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci 9, 46–56. 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David J, Measelle J, Ostlund B, Ablow J, 2017. Association between early life adversity and inflammation during infancy. Dev. Psychobiol 59, 696–702. 10.1002/dev.21538. [DOI] [PubMed] [Google Scholar]
- Dozier M, Manni M, Gordon MK, Peloso E, Gunnar MR, Stovall-McClough KC, Eldreth D, Levine S, 2006. Foster children’s diurnal production of cortisol: an exploratory study. Child Maltreat. 11, 189–197. [DOI] [PubMed] [Google Scholar]
- Egger M, Smith GD, Phillips AN, 1997. Meta-analysis: principles and procedures. BMJ 315, 1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich KB, Ross KM, Chen E, Miller GE, 2016. Testing the biological embedding hypothesis: is early life adversity associated with a later proinflammatory phenotype? Dev. Psychopathol 28, 1273–1283. 10.1017/S0954579416000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR, 2010. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol. Psychiatry 68, 748–754. 10.1016/j.biopsych.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcone T, Janigro D, Lovell R, Simon B, Brown CA, Herrera M, Myint AM, Anand A, 2015. S100B blood levels and childhood trauma in adolescent inpatients. J. Psychiatr. Res 62, 14–22. 10.1016/j.jpsychires.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felitti VJ, Anda RF, Nordenberg D, Williamson DF, Spitz AM, Edwards V, Koss MP, Marks JS, 1998. Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults: the Adverse Childhood Experiences (ACE) Study. Am. J. Prev. Med 14, 245–258. 10.1016/S0749-3797(98)00017-8. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Campisi J, 2014. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A 69, S4–S9. 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- Gimeno D, Kivimäki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A, Kumari M, Lowe GDO, Rumley A, Marmot MG, Ferrie JE, 2009. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol. Med 39, 413–423. 10.1017/S0033291708003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goosby BJ, Malone S, Richardson EA, Cheadle JE, Williams DT, 2015. Perceived discrimination and markers of cardiovascular risk among low-income African American youth. Am. J. Hum. Biol. Off. J. Hum. Biol. Council 27, 546–552. 10.1002/ajhb.22683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnar M, Quevedo K, 2007. The neurobiology of stress and development. Annu. Rev. Psychol 58, 145–173. 10.1146/annurev.psych.58.110405.085605. [DOI] [PubMed] [Google Scholar]
- Gunnar MR, Donzella B, 2002. Social regulation of the cortisol levels in early human development. Psychoneuroendocrinology 27, 199–220. 10.1016/S0306-4530(01)00045-2. [DOI] [PubMed] [Google Scholar]
- Hadley C, Decaro JA, 2014. Testing hypothesized predictors of immune activation in Tanzanian infants and children: community, household, caretaker, and child effects. Am. J. Hum. Biol. Off. J. Hum. Biol. Council 26, 523–529. 10.1002/ajhb.22558. [DOI] [PubMed] [Google Scholar]
- Hagel I, Lynch NR, Di Prisco MC, Sanchez J, Pérez M, 1995. Nutritional status and the IgE response against Ascaris lumbricoides in children from a tropical slum. Trans. R. Soc. Trop. Med. Hyg 89, 562–565. [DOI] [PubMed] [Google Scholar]
- Hanson JL, Nacewicz BM, Sutterer MJ, Cayo AA, Schaefer SM, Rudolph KD, Shirtcliff EA, Pollak SD, Davidson RJ, 2015. Behavioral problems after early life stress: contributions of the hippocampus and amygdala. Biol. Psychiatry 77, 314–323. 10.1016/j.biopsych.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RE, 2007. Cyclooxygenase-2 (COX-2) and the inflammogenesis of Cancer. In: Inflammation in the Pathogenesis of Chronic Diseases, Subcellular Biochemistry. Springer, Dordrecht, pp. 93–126. 10.1007/1-4020-5688-5_4. [DOI] [PubMed] [Google Scholar]
- Harris RE, Casto BC, Harris ZM, 2014. Cyclooxygenase-2 and the inflammogenesis of breast cancer. World J. Clin. Oncol 5, 677–692. 10.5306/wjco.v5.i4.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges LV, Vevea JL, 1998. Fixed-and random-effects models in meta-analysis. Psychol. Meth 3, 486. [Google Scholar]
- Higgins JPT, Thompson SG, 2002. Quantifying heterogeneity in a meta-analysis. Stat. Med 21, 1539–1558. 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- Horn S, Chiang J, Bower J, Kuhlman K, 2017. Methodological approaches to the study of neuroendocrine and inflammatory biomarkers of childhood trauma: a systematic review. Biol. Psychiatry 81, S395–S396. 10.1016/j.biopsych.2017.02.704. [DOI] [Google Scholar]
- Kalmakis KA, Chandler GE, 2015. Health consequences of adverse childhood experiences: a systematic review. J. Am. Assoc. Nurse Pract 27, 457–465. 10.1002/2327-6924.12215. [DOI] [PubMed] [Google Scholar]
- Kelly-Irving M, Lepage B, Dedieu D, Bartley M, Blane D, Grosclaude P, Lang T, Delpierre C, 2013. Adverse childhood experiences and premature all-cause mortality. Eur. J. Epidemiol 28, 721–734. 10.1007/s10654-013-9832-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, McLaughlin KA, Green JG, Gruber MJ, Sampson NA, Zaslavsky AM, Aguilar-Gaxiola S, Alhamzawi AO, Alonso J, Angermeyer M, Benjet C, Bromet E, Chatterji S, de Girolamo G, Demyttenaere K, Fayyad J, Florescu S, Gal G, Gureje O, Haro JM, Hu C, Karam EG, Kawakami N, Lee S, Lépine J-P, Ormel J, Posada-Villa J, Sagar R, Tsang A, Üstün TB, Vassilev S, Viana MC, Williams DR, 2010. Childhood adversities and adult psychopathology in the WHO World Mental Health Surveys. Br. J. Psychiatry 197, 378–385. 10.1192/bjp.bp.110.080499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kılıç S, Ozlem K, Murat T, 2017. Inflammatory parameters in sexually abused children. Saudi Med. J 38, 1213–1218. 10.15537/smj.2017.12.21463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig W, Sund M, Fröhlich M, Fischer H-G, Löwel H, Döring A, Hutchinson WL, Pepys MB, 1999. C-Reactive Protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation 99, 237–242. 10.1161/01.CIR.99.2.237. [DOI] [PubMed] [Google Scholar]
- Korkeila J, Vahtera J, Korkeila K, Kivimäki M, Sumanen M, Koskenvuo K, Koskenvuo M, 2010. Childhood adversities as predictors of incident coronary heart disease and cerebrovascular disease. Heart 96, 298–303. 10.1136/hrt.2009.188250. [DOI] [PubMed] [Google Scholar]
- Kuhlman KR, Chiang JJ, Horn S, Bower JE, 2017. Developmental psychoneuroendocrine and psychoneuroimmune pathways from childhood adversity to disease. Neurosci. Biobehav. Rev 80, 166–184. 10.1016/j.neubiorev.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman KR, Geiss EG, Vargas I, Lopez-Duran NL, 2015. Differential associations between childhood trauma subtypes and adolescent HPA-axis functioning. Psychoneuroendocrinology 54, 103–114. 10.1016/j.psyneuen.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman KR, Repetti RL, Reynolds BM, Robles TF, 2018a. Interparental conflict and child HPA-axis responses to acute stress: insights using intensive repeated measures. J. Fam. Psychol 10.1037/fam0000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman KR, Repetti RL, Reynolds BM, Robles TF, 2016. Change in parent-child conflict and the HPA-axis: Where should we be looking and for how long? Psychoneuroendocrinology 68, 74–81. 10.1016/j.psyneuen.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman KR, Robles TF, Bower JE, Carroll JE, 2018b. Screening for childhood adversity: the what and when of identifying individuals at risk for lifespan health disparities. J. Behav. Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman KR, Robles TF, Dooley LN, Boyle CC, Haydon MD, Bower JE, 2018c. Within-subject associations between inflammation and features of depression: using the flu vaccine as a mild inflammatory stimulus. Brain. Behav. Immun 69, 540–547. 10.1016/j.bbi.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovallo WR, 2013. Early life adversity reduces stress reactivity and enhances impulsive behavior: implications for health behaviors. Int. J. Psychophysiol. Off. J. Int. Organ. Psychophysiol 90, 8–16. 10.1016/j.ijpsycho.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews KA, Chang Y-F, Thurston RC, Bromberger JT, 2014. Child abuse is related to inflammation in mid-life women: role of obesity. Brain. Behav. Immun 36, 29–34. 10.1016/j.bbi.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Measelle J, Ablow J, 2018. Contributions of early adversity to pro-inflammatory phenotype in infancy: the buffer provided by attachment security. Attach. Hum. Dev 20, 1–23. 10.1080/14616734.2017.1362657. [DOI] [PubMed] [Google Scholar]
- Miller AH, Raison CL, 2016. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat. Rev. Immunol 16, 22–34. 10.1038/nri.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, 2010. Harsh family climate in early life presages the emergence of a proinflammatory phenotype in adolescence. Psychol. Sci 21, 848–856. 10.1177/0956797610370161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Parker KJ, 2011. Psychological stress in childhood and susceptibility to the chronic diseases of aging: moving toward a model of behavioral and biological mechanisms. Psychol. Bull 137, 959–997. 10.1037/a0024768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Cole SW, 2012. Clustering of depression and inflammation in adolescents previously exposed to childhood adversity. Biol. Psychiatry Endocrinol. Epigen. Extinction Early Life Traumatization 72, 34–40. 10.1016/j.biopsych.2012.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH, 2009. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev 22, 240–273. 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moher D, Liberati A, Tetzlaff J, Altman DG, Group TP, 2009. Preferred Reporting Items for Systematic Reviews and Meta-Analyses : The PRISMA Statement 6. 10.1371/journal.pmed.1000097. [DOI] [PMC free article] [PubMed]
- O’Connor M-F, Bower JE, Cho HJ, Creswell JD, Dimitrov S, Hamby ME, Hoyt MA, Martin JL, Robles TF, Sloan EK, Thomas KS, Irwin MR, 2009. To assess, to control, to exclude: effects of biobehavioral factors on circulating inflammatory markers. Brain. Behav. Immun 23, 887–897. 10.1016/j.bbi.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RS, Enwonwu CO, Okolo S, Hassan A, 2004. Metabolic effects of acute measles in chronically malnourished Nigerian children. J. Nutr. Biochem 15, 281–288. 10.1016/j.jnutbio.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Ramratnam SK, Visness CM, Jaffee KF, Bloomberg GR, Kattan M, Sandel MT, Wood RA, Gern JE, Wright RJ, 2017. Relationships among maternal stress and depression, type 2 responses, and recurrent wheezing at age 3 years in low-income urban families. Am. J. Respir. Crit. Care Med 195, 674–681. 10.1164/rccm.201602-0272OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposa EB, Bower JE, Hammen CL, Najman JM, Brennan PA, 2014. A developmental pathway from early life stress to inflammation: the role of negative health behaviors. Psychol. Sci 25, 1268–1274. 10.1177/0956797614530570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, 2003. Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation 107, 363–369. 10.1161/01.CIR.0000053730.47739.3C. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH, 1997. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med 336, 973–979. 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- Riis JL, Granger DA, Minkovitz CS, Bandeen-Roche K, DiPietro JA, Johnson SB, 2016. Maternal distress and child neuroendocrine and immune regulation. Soc. Sci. Med 1982 (151), 206–214. 10.1016/j.socscimed.2015.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnani S, 1994. Lymphokine production by human T cells in disease states. Annu. Rev. Immunol 12, 227. [DOI] [PubMed] [Google Scholar]
- Rudolph KD, Hammen C, 2003. Age and gender as determinants of stress exposure, generation, and reactions in youngsters: a transactional perspective. Child Dev. 70, 660–677. 10.1111/1467-8624.00048. [DOI] [PubMed] [Google Scholar]
- Rudolph KD, Hammen C, Burge D, Lindberg N, Herzberg D, Daley SE, 2000. Toward an interpersonal life-stress model of depression: the developmental context of stress generation. Dev. Psychopathol 12, 215–234. [DOI] [PubMed] [Google Scholar]
- Slopen N, Koenen KC, Kubzansky LD, 2012. Childhood adversity and immune and inflammatory biomarkers associated with cardiovascular risk in youth: a systematic review. Brain. Behav. Immun 26, 239. [DOI] [PubMed] [Google Scholar]
- Steptoe A, Hamer M, Chida Y, 2007. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain. Behav. Immun 21, 901–912. 10.1016/j.bbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Sterne JAC, Egger M, 2001. Funnel plots for detecting bias in meta-analysis: guidelines on choice of axis. J. Clin. Epidemiol 54, 1046–1055. 10.1016/S0895-4356(01)00377-8. [DOI] [PubMed] [Google Scholar]
- Tarullo AR, Gunnar MR, 2006. Child maltreatment and the developing HPA axis. Horm. Behav 50, 632–639. 10.1016/j.yhbeh.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Tyrka AR, Parade SH, Valentine TR, Eslinger NM, Seifer R, 2015. Adversity in preschool-aged children: effects on salivary interleukin-1β Dev. Psychopathol. 27, 567–576. 10.1017/S0954579415000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer-Yaniv A, Djalovski A, Yirmiya K, Halevi G, Zagoory-Sharon O, Feldman R, 2018. Maternal immune and affiliative biomarkers and sensitive parenting mediate the effects of chronic early trauma on child anxiety. Psychol. Med 48, 1020–1033. 10.1017/S0033291717002550. [DOI] [PubMed] [Google Scholar]
- Waage A, Slupphaug G, Shalaby R, 1990. Glucocorticoids inhibit the production of IL 6 from monocytes, endothelial cells and fibroblasts. Eur. J. Immunol 20, 2439–2443. 10.1002/eji.1830201112. [DOI] [PubMed] [Google Scholar]
- Walsh K, Basu A, Werner E, Lee S, Feng T, Osborne LM, Rainford A, Gilchrist M, Monk C, 2016. Associations among child abuse, depression, and interleukin-6 in pregnant adolescents: paradoxical findings. Psychosom. Med 78, 920–930. 10.1097/PSY.0000000000000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthman CM, Panter-Brick C, 2008. Homeless street children in Nepal: use of allostatic load to assess the burden of childhood adversity. Dev. Psychopathol 20, 233–255. 10.1017/S0954579408000114. [DOI] [PubMed] [Google Scholar]
- Wright RJ, Finn P, Contreras JP, Cohen S, Wright RO, Staudenmayer J, Wand M, Perkins D, Weiss ST, Gold DR, 2004. Chronic caregiver stress and IgE expression, allergen-induced proliferation, and cytokine profiles in a birth cohort pre-disposed to atopy. J. Allergy Clin. Immunol 113, 1051. 10.1016/j.jaci.2004.03.032. [DOI] [PubMed] [Google Scholar]