

Abstract
[FeFe] hydrogenases are highly active catalysts for the interconversion of molecular hydrogen with protons and electrons. Here, we use a combination of isotopic labeling, 57Fe nuclear resonance vibrational spectroscopy (NRVS), and density functional theory (DFT) calculations to observe and characterize the vibrational modes involving motion of the 2-azapropane-1,3-dithiolate (ADT) ligand bridging the two iron sites in the [2Fe]H subcluster. A –13C2H2– ADT labeling in the synthetic diiron precursor of [2Fe]H produced isotope effects observed throughout the NRVS spectrum. The two precursor isotopologues were then used to reconstitute the H-cluster of [FeFe] hydrogenase from Chlamydomonas reinhardtii (CrHydA1), and NRVS was measured on samples poised in the catalytically crucial Hhyd state containing a terminal hydride at the distal Fe site. The 13C2H isotope effects were observed also in the Hhyd spectrum. DFT simulations of the spectra allowed identification of the 57Fe normal modes coupled to the ADT ligand motions. Particularly, a variety of normal modes involve shortening of the distance between the distal Fe–H hydride and ADT N–H bridgehead hydrogen, which may be relevant to the formation of a transition state on the way to H2 formation.
Molecular hydrogen is viewed as an ideal carbon-free energy carrier that could be part of a transition to a sustainable economy without CO2 emissions.1,2 At the moment, the majority of industrial hydrogen is produced by high-temperature steam reforming of natural gas which leads to the release of at least one molecule of CO2 for every 4 H2 produced.3 Ideally, electrochemical energy from solar, wind, or other carbon-free sources could be used to drive the water-splitting or “hydrogen evolution reaction” (HER) without CO2 release.1,4 Highly efficient catalysts with low overpotentials are essential for electrochemical conversions of hydrogen, and the high prices and scarcity of the current Pt or other noble metal HER catalysts have led to the search for systems that use earth-abundant materials.5–7 One source of inspiration driving this search is Nature, which uses plentiful transition metals Fe or Fe with Ni in the active sites of hydrogenases.8,9
Hydrogenases are enzymes that catalyze the reversible interconversion of molecular hydrogen with protons and electrons: H2 ⇌ 2H+ + 2e−. [FeFe] hydrogenases contain an active site “H-cluster” consisting of a [4Fe–4S]H cluster linked via a cysteine residue to a unique [2Fe]H subcluster (Figure 1a).11 This subcluster carries a 2-azapropane-1,3-dithiolate (ADT) ligand bridging a pair of CO and CN− ligated Fe ions. The ADT bridgehead nitrogen has been implicated as part of a proton transfer relay extending through a neighboring cysteine.12–16 In the Chlamydomonas reinhardtii [FeFe] hydrogenase (CrHydA1), the conserved relay consists of C169, a water molecule, and oxygens from E141, S189, and E144 residues.
Figure 1.
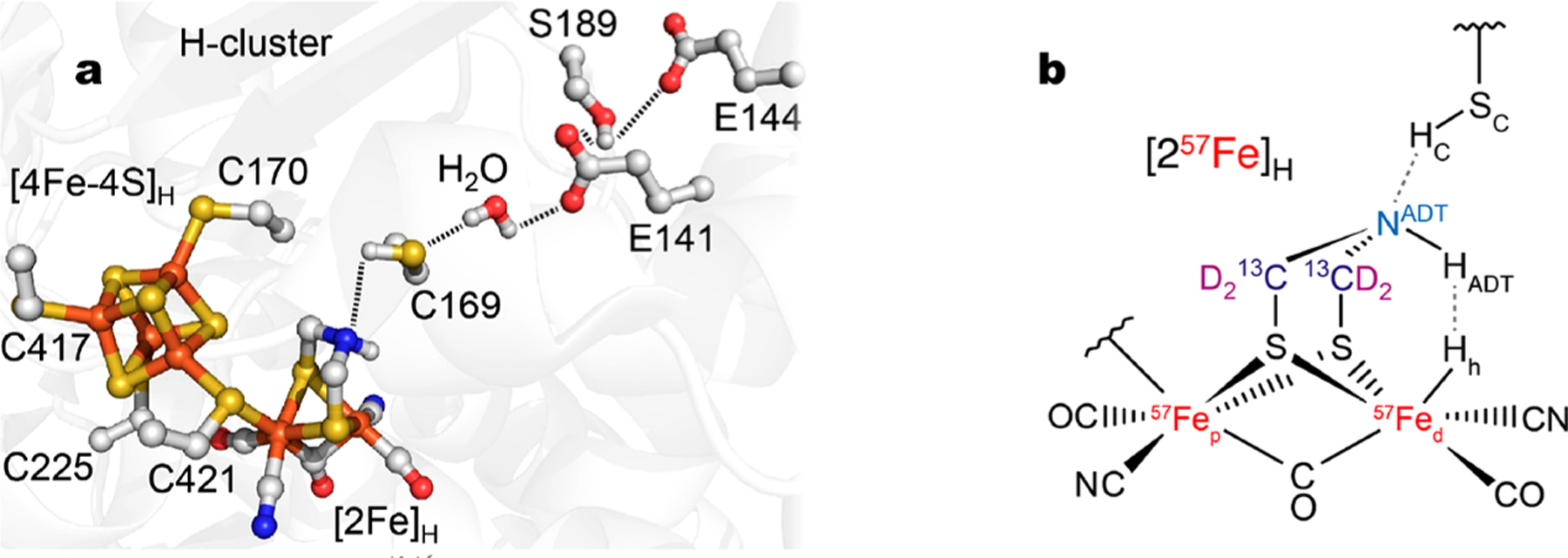
(a) [FeFe] hydrogenase active site including key amino acids in the proton transfer pathway (based on the PDB 4XDC10 structure of the CpI enzyme from Clostridium pasteurianum, but using CrHydA1 sequence numbering). (b) Schematic structure of the [2Fe]H subcluster in the Hhyd state, showing the isotopically labeled nuclei 57Fe, 13C, and D (i.e. 2H). The important hydrogens, Hh (catalytic hydride at the distal Fed iron), HADT (at the ADT NADT nitrogen), and HC (at the SC C169 sulfur), are shown.
An iron hydride form of [FeFe] hydrogenase, Hhyd, is a key intermediate of the catalytic cycle, and it has been studied by multiple spectroscopic and molecular modeling techniques.17–24 The Hhyd species contains a terminal Fed–Hh (hydride) at the [2Fe]H iron site distal to [4Fe–4S]H (Figure 1b), with a [4Fe–4S]H+ –Fep(II)Fed(II) redox state for the H-cluster, along with the –NHADT– amine form of the ADT bridgehead.18–21
Nuclear resonance vibrational spectroscopy (NRVS) has become a popular technique for elucidating the element-selective normal modes of appropriate Mössbauer isotopes.25–31 In previous work on the CrHydA1 and DdHydAB (from Desulfovibrio desulfuricans) enzymes,20–22 we have shown that 57Fed–Hh bending modes can be observed using 57Fe-NRVS for the Hhyd species and that these modes exhibit peak positions that are characteristic of the local environment. To better identify additional normal modes of Hhyd, we proceeded to label the [2Fe]H subcluster not only with 57Fe but also with 13C and D in the methylene groups of the ADT ligand. We accomplished this by preparing a [2Fe]H precursor, the 57Fe-labeled salt (Et4N)2 [57Fe2[(SCH2)2NH]-(CN)2(CO)4] (1) as well as its variant also labeled with 13C and D on the two methylene groups of the ADT ligand (13CD-1).32 We then used these samples to reconstitute an apo form of CrHydA1 containing the [4Fe–4S]H cluster but lacking the [2Fe]H subsite.33
We first examine NRVS spectra for the precursor isotopologues 1 vs 13CD-1 in Figure 2a. Close inspection reveals a number of subtle changes to band positions and intensities in the broad ~100–700 cm−1 range, most of them well reproduced by the DFT simulation shown in Figure 2b. We note that this is the first demonstration of NRVS isotope shifts from labeling in the second and third coordination spheres of 57Fe, although such shifts have been seen before in resonance Raman spectra.34,35 In the following, when referring to the bands observed (or vibrational frequencies calculated) for the two isotopologues, we use a nomenclature x → y (cm−1) where x and y represent 1 and 13CD-1, respectively.
Figure 2.
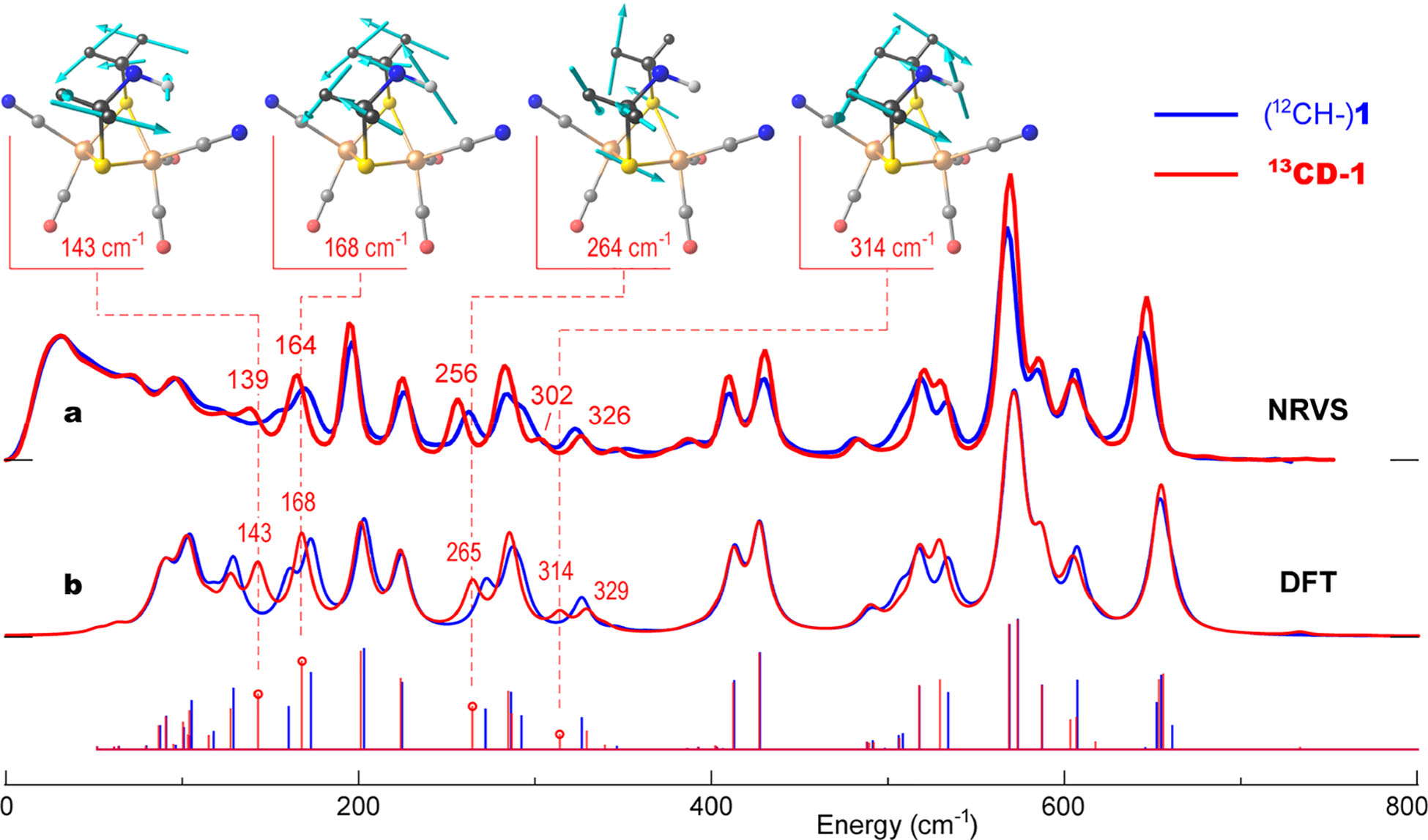
57Fe-PVDOS for the [2Fe]H precursor isotopologues 1 (blue) vs 13CD-1 (red) from (a) NRVS experiments and (b) DFT calculations. Sticks correspond to individual DFT normal mode energies and intensities before lineshape convolution. For 13CD-1, important band positions are labeled, and atomic motions in selected normal modes are shown.
Since the bands from 400 to 660 cm−1 are dominated by Fe–CN and Fe–CO motions, we focus instead on differences in the region from 100 to 350 cm−1, which contains delocalized bending and torsional modes as well as Fe–S stretching. In the 13CD-1 spectra, several bands exhibit clear downshifts from the (12CH–)1 data, for example, at 150 → 139, 168 → 164, and 260 → 256 cm−1 (Figure 2a). This pattern is echoed in the 13CD-1 DFT simulations, with downshifted bands at 161 → 143, 173 → 168, and 273 → 265 cm−1 (Figure 2b). The normal-mode analysis also reveals an isotope-dependent redistribution of the intensities underlying the DFT bands at 326 → 329/314 cm−1, mapping onto the NRVS features at 322 → 326/302 cm−1.
Having identified the most significant isotope shifts in the precursor spectra, we now illustrate the atomic motions deduced from the DFT calculations. As displayed in Figure 2 for 13CD-1, the normal mode calculated at 143 cm−1 is mostly out-of-phase rotation of the two ADT –μS13CD2– groups around their S–C axes, combined with some motion of the μS pivot points due to Fe–S–Fe bending (see animated representations of the calculated vibrational modes as part of the Supporting Information and their characterization in Table S2). The large amount of methylene motion explains the significant isotope shift. In contrast, the 168 cm−1 mode involves rocking of the entire –D213C–NH–13 CD2– assembly in one direction while the underlying Fe2S2 cluster (and associated ligands) rotates in the opposite direction. At higher frequencies, the 264 cm−1 mode involves out-of-phase displacements of the –μS13CD2– fragments with substantial Fe–S stretching character, while the 314 cm−1 mode is an in-phase –13CD2– methylene group motion, accompanied by wagging of the –NH– bridgehead in the opposite direction.
Our key observations from these precursor studies are (i) that the 13CD substitution in the Fe-bridging ADT ligand induces measurable isotope shifts in the 57Fe NRVS spectra, on the order of the 8 cm−1 instrumental resolution; (ii) that the DFT calculations are sufficiently accurate to reproduce these shifts, allowing confidence in the motions assigned to these modes; and (iii) that the calculations predict a variety of ADT flexing modes with significant motion of the –NH– bridgehead.
Precursors 1 and 13CD-1 were used for maturation of the apo CrHydA1 containing natural-abundance Fe in the [4Fe–4S]H cluster. This yielded holo CrHydA1 labeled with 57Fe in the [2Fe]H subcluster, and with either a natural-abundance ADT ligand (1-CrHydA1) or –13CD2– in the methylene portions of ADT (13CD-1-CrHydA1). These samples were poised in the Hhyd state by reduction with 100 mM sodium dithionite at pH 6. As shown by infrared (IR) spectra in Figure S1, both samples exhibited the standard Hhyd IR signature, with minimal contributions from other redox states.
NRVS data for 1-CrHydA1 and 13CD-1-CrHydA1 are shown in Figure 3a, with the corresponding DFT simulations in Figure 3b. The calculated spectra were generated using a DFT model of Hhyd including the entire H-cluster and its immediate protein environment;21,22,36 see DFT methods and model coordinates in the Supporting Information for further details. Again, we focus first on differences in the low-energy region, where we see the most obvious isotope effects. These include NRVS downshifts at 281 → 274 and 313 → 305 cm−1, with the DFT simulations yielding corresponding modifications at 296 → 290 and 314 → 308 cm−1. The 150–200 cm−1 isotope-dependent NRVS region (~174 → 166 cm−1) of Hhyd essentially repeats in the DFT spectra (~171 → 163 cm−1), indicating overlapping contributions from different modes. A complementary DFT simulation for the [4Fe–4S]H2+ –Fep(II)Fed(I) redox state of the H-cluster, Hox, reveals comparable 12CH → 13CD spectral shifts in the broader ~150–330 cm−1 region (Figure S2); this indicates that the ADT labeling effects observed in NRVS are stable against potential impurities from additional redox states of the H-cluster.
Figure 3.
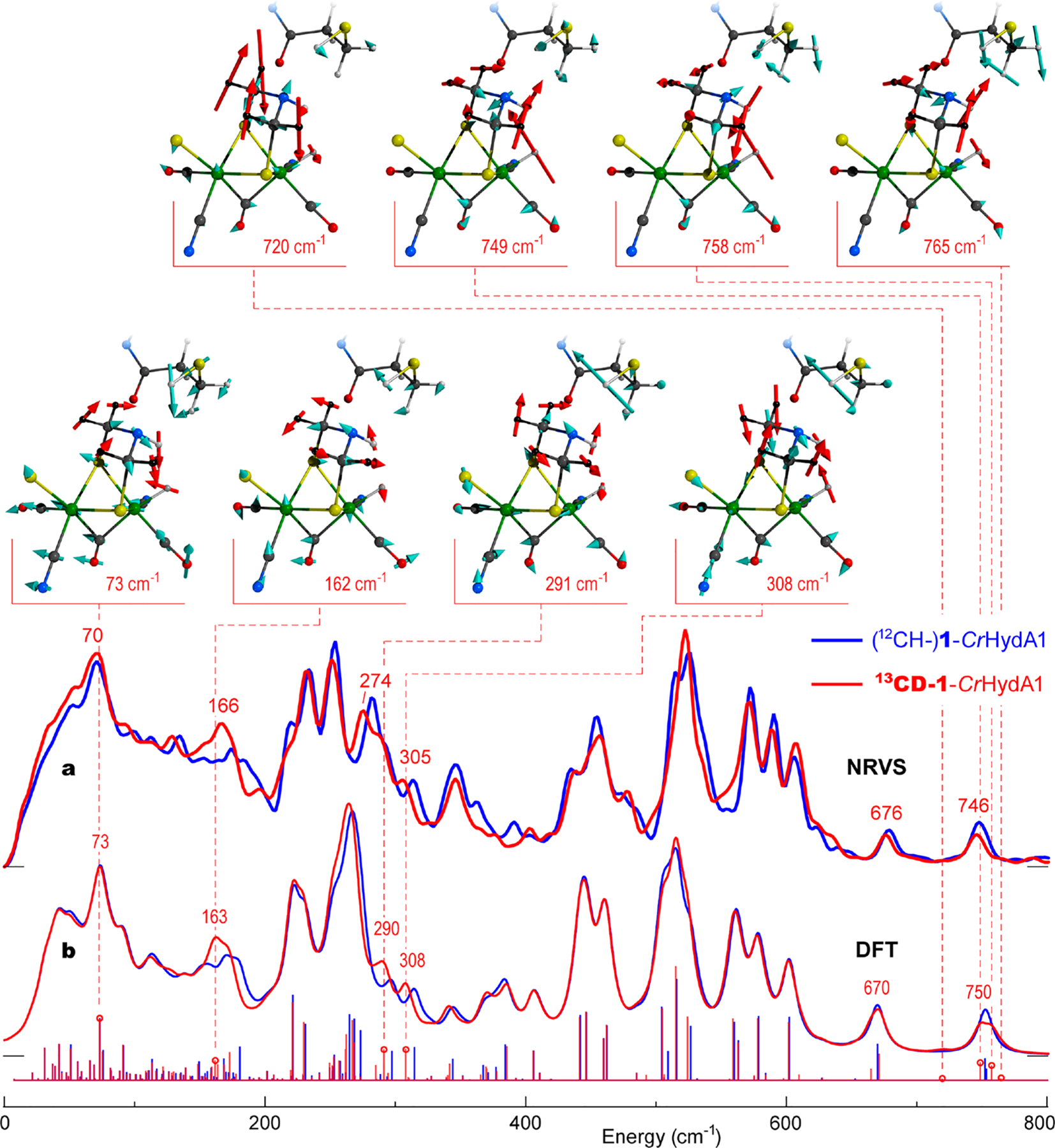
57Fe-PVDOS for the Hhyd state isotopologues 1-CrHydA1 (blue) vs 13CD-1-CrHydA1 (red) from (a) an NRVS experiment and (b) DFT calculations. Sticks correspond to individual DFT normal mode energies and intensities before broadening. For 13CD-1-CrHydA1, important band positions are labeled, and atomic motions in selected normal modes are shown. Only the [2Fe]H and C169 fragments of the DFT model are shown with the methylene, Hh, and HADT hydrogen nuclei displacements indicated by red arrows.
The atomic motions deduced from the DFT calculations on Hhyd are displayed in Figure 3 and animated as part of the Supporting Information. The 162 cm−1 band of 13CD-1-CrHydA1 contains [2Fe]H modes heavily mixed with the protein environment, but an important feature here is rocking of the –NHADT– bridgehead toward the distal iron hydride Fed–Hh, along with out-of-phase rotation of the ADT –13CD2– groups; this character matches the 13CD-1 precursor mode at 143 cm−1. At higher energies, the 291 cm−1 mode exhibits a breathing motion of the Fe2S2 moiety, which leads to changing the distance between Fep and the [4Fe–4S]H cluster; in this case, there is an in-phase motion of the ADT methylene groups in the opposite direction of the amine bridgehead, similar to the 13CD-1 mode at 314 cm−1 described above. The 308 cm−1 mode exhibits an entire ADT fragment wagging/rotation relative to Fep and Fed, equivalent to the 13CD-1 mode at 287 cm−1. We also illustrate the 73 cm−1 mode, which is highly delocalized with torsional motions of the entire H-cluster.
We now turn to the higher-energy side of the Hhyd spectra, which contains two distinct Fed–Hh bending mode peaks observed at 679/676 and 748/746 cm−1, and calculated at 670/670 and 753/750 cm−1 (Figure 3). These were the focus of previous studies because they characterize the terminal iron hydride bonding and its interactions with the surroundings.20–22 The two main features arise from the relatively pure Hh hydride bending motion perpendicular to and parallel to the plane defined by the Fep–Fed axis and the Fed–Hh bond, respectively. Although the isotope-dependent shifts in these bands are small and nearly unmeasurable, the fine structure of the underlying normal modes displays a difference. In the current DFT analysis there are 1-/13CD-1-CrHydA1 “perpendicular” modes at 670/665,671 cm−1, and “parallel” modes at 752,754/749,758 cm−1 respectively. The 13CD-labeling introduces NADT–HADT bending admixtures to the Fed–Hh modes, where the HADT and Hh nuclei displace either in- or out-of-phase; e.g., the 758 cm−1 “parallel” mode (Figure 3) brings Hh and HADT closer during half of each excursion cycle. The calculations suggest an increased involvement of the heavier 13CD-ADT fragment in the Fed–Hh bends, with rotations of the two –13CD2– methylene groups contributing at least 16% to the vibrational kinetic energy. Similar modes are calculated in the ADT-labeled 13CD-1 precursor in the ~670–770 cm−1 region, while the unlabeled (12CH–)1 variant produces their counterparts at frequencies only above 800 cm−1 (Table S2).
The DFT analysis therefore indicates that some modes in the Fed–Hh bending region involve mixing with motions inherent to the 13CD-labeled ADT ligand. A search for such “satellite” modes is what initially prompted our isotopic labeling investigation. The experimental data might show weak “satellite” features on either side of the main Fed–Hh bending peaks (Figure 3). However, despite prolonged data collection in this region to improve the signal-to-noise (S/N) ratio, firm assignment of the small differences to “satellites” is not yet possible. The exact calculated energies of the “satellites” should also be taken with caution, because they are governed by motion of a very light Hh nucleus that mediates interaction between 57Fed and the ADT bridgehead. Further experimental insight into these modes will require a significantly higher NRVS photon flux, which may be available in the next generation of synchrotron sources, e.g., PETRA-IV.37
The accuracy of the DFT calculations at reproducing the experimental NRVS spectra of the unlabeled and isotopically labeled precursor and [2Fe]H, here and in our previous work,20–22,36 gives us confidence that it is valuable to consider the predicted “satellite” modes in Hhyd, whether or not they can be conclusively detected by NRVS. Illustrations of these “satellite” modes at 720 and 765 cm−1 are included in Figure 3. The latter two modes involve “parallel” Fed–Hh bending, similar to the 749 and 758 cm−1 modes. Interestingly, some of these modes involve motion of the nearby cysteine at the end of the proton transfer channel leading to the ADT ligand. These vibrational modes appear to represent a pathway for coupled proton transfer from (C169)SC–HC to NADT and from NADT–HADT to Fed–Hh.
Are any other modes relevant to H2 production catalysis? We inspected the DFT calculations for changes in HADT···Hh and NADT···HC distances that occur during normal mode displacements (see Figure S3). The results for the modes with the greatest distance changes are summarized in Table S1. The equilibrium 2.06 Å HADT···Hh distance is already firmly in the 1.7–2.2 Å range for a “dihydrogen bond”,38 and it is similar to the 2.02 Å value seen as the shortest H···H distance in solid BH3NH3.39 We found that a few modes contribute a disproportionate amount of motion involving the HADT···Hh distance as well as the NADT···HC distance. In particular, the “parallel” Fed–Hh bending modes at 752/758 cm−1 yield the record ~0.14/0.15 Å contractions in the HADT···Hh distance across the entire vibrational spectra. For the NADT···HC distance, the largest vibrational contraction of ~0.11 Å is achieved in the SC–HC stretching mode calculated at 2449/2449 cm−1.
From time-resolved photochemical IR studies, Sanchez et al. have shown that the decay of Hhyd is kinetically competent as a near-final step in the [FeFe] hydrogenase catalytic cycle.40 However, since the pKa for a neutral secondary amine such as the ADT bridgehead nitrogen is extremely high, an intervening protonated ADT –NH2+– intermediate, HhydH+, has often been included in the catalytic cycle.17,24,41–44 Our results, which document the role of ADT flexibility in normal modes that bring HADT and Hh closer together, offer the possibility of a mechanism update.
In this speculative scenario, high-frequency modes such as at 752/758 and 2449/2449 cm−1, combined with low-frequency modes such as at 73 cm−1, would involve coordinated motion of HADT toward Hh, while HC moves toward NADT. This might precipitate a “deep tunneling” transfer of HADT to Hh, while SC–HC transfer replenishes the NADT–HADT, and with the SC–HC proton reloaded from the H2O in the proton transfer chain. Champion and co-workers have shown that deep tunneling can allow high pKa residues to participate in proton transfer chains, as invoked for a serine residue in the green fluorescent protein.45 If the transfer reaction for Hhyd were facilitated by electron transfer from the [4Fe–4S]H+ to the [FeIIFeII]H subsite, the overall PCET reaction would yield an Hox electronic state with bound H2. This scenario agrees with calculations on the reverse reaction of H2 activation by Greco et al.46
In summary, we have investigated vibrations of the [FeFe] hydrogenase active site in the Hhyd state through 57Fe, 13C, and D isotopic labeling, combined with 57Fe NRVS measurements and DFT calculations. This represents the first observation of second and third coordination sphere isotope effects using NRVS. We identified normal modes involving the flexing of the bridging ADT ligand that point to its unique properties as an active site ligand. The combined motions of the Fed–Hh, NADT–HADT, and (C169)SC–HC protons are presumably coupled to the remainder of the proton transfer chain as well as electron transfer. These effects may be important for catalysis and will be investigated in future studies.
Supplementary Material
ACKNOWLEDGMENTS
V.P. acknowledges funding by the Deutsche Forschungsge-meinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy, EXC 2008-390540038, UniSysCat. J.A.B. acknowledges funding from the DFG SPP 1927 “Iron-Sulfur for Life” project (Project BI 2198/1-1). E.J.R., J.A.B., and W.L. would like to thank the Max Planck Society for continuous financial support. The contributions of T.B.R. and C.P.R. were funded by the U.S. National Institutes of Health through GM61153. S.P.C. was funded by NIH GM65440. Some computational work was performed under the XSIM project on the CORI computing system at NERSC, a U.S. Department of Energy Office of Science User Facility operated under Contract DE-AC02-05CH11231. The authors gratefully acknowledge the assistance by Giorgio Caserta in acquisition of the NRVS data.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c02323.
Experimental and computational procedures and additional data and figures including FTIR spectra, 57Fe-PVDOS results, and relative displacements (PDF) Animated vibrational modes of the Hhyd DFT model as GIF files (ZIP)
Animated vibrational modes of the precursor DFT model as GIF files (ZIP)
Coordinates of the precursor, Hhyd, and Hox DFT models as XYZ files (ZIP)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c02323
The authors declare no competing financial interest.
Contributor Information
Vladimir Pelmenschikov, Institut für Chemie, Technische Universität Berlin, 10623 Berlin, Germany;.
James A. Birrell, Max Planck Institute for Chemical Energy Conversion, 45470 Mülheim an der Ruhr, Germany;.
Leland B. Gee, Department of Chemistry, Stanford University, Stanford, California 94305, United States;.
Casseday P. Richers, School of Chemical Sciences, University of Illinois, Urbana, Illinois 61801, United States;.
Edward J. Reijerse, Max Planck Institute for Chemical Energy Conversion, 45470 Mülheim an der Ruhr, Germany;.
Hongxin Wang, SETI Institute, Mountain View, California 94043, United States;.
Simon Arragain, IFP Energies nouvelles, 92852 Rueil-Malmaison, France; Department of Chemistry, University of California, Davis, California 95616, United States;.
Nakul Mishra, Department of Chemistry, University of California, Davis, California 95616, United States;.
Yoshitaka Yoda, Precision Spectroscopy Division, SPring-8/JASRI, Sayo, Hyogo 679-5198, Japan;.
Hiroaki Matsuura, Life Science Research Infrastructure Group, Advanced Photon Technology Division, RIKEN/SPring-8 Center, Sayo, Hyogo 679-5148, Japan;.
Lei Li, Hyogo Science and Technology Association, Synchrotron Radiation Research Center, Tatsuno-shi, Hyogo 679-5165, Japan;.
Kenji Tamasaku, Research and Utilization Division, SPring-8/JASRI, Sayo, Hyogo 679-5198, Japan;.
Thomas B. Rauchfuss, School of Chemical Sciences, University of Illinois, Urbana, Illinois 61801, United States;.
Wolfgang Lubitz, Max Planck Institute for Chemical Energy Conversion, 45470 Mülheim an der Ruhr, Germany;.
Stephen P. Cramer, SETI Institute, Mountain View, California 94043, United States;.
REFERENCES
- (1).Glenk G; Reichelstein S Economics of converting renewable power to hydrogen. Nat. Energy 2019, 4 (3), 216–222. [Google Scholar]
- (2).Armaroli N; Balzani V The Hydrogen Issue. ChemSusChem 2011, 4 (1), 21–36. [DOI] [PubMed] [Google Scholar]
- (3).Ávila Neto C; Dantas SC; Silva FA; Franco TV; Romanielo L; Hori C; Assis AJ Hydrogen production from methane reforming: Thermodynamic assessment and autothermal reactor design. J. Nat. Gas Sci. Eng 2009, 1, 205–215. [Google Scholar]
- (4).Hosseini SE; Wahid MA Hydrogen from solar energy, a clean energy carrier from a sustainable source of energy. Int. J. Energy Res 2020, 44 (6), 4110–4131. [Google Scholar]
- (5).Ren XF; Lv QY; Liu LF; Liu BH; Wang YR; Liu AM; Wu G Current progress of Pt and Pt-based electrocatalysts used for fuel cells. Sust. En. Fuels 2020, 4 (1), 15–30. [Google Scholar]
- (6).Bullock RM; Helm ML Molecular Electrocatalysts for Oxidation of Hydrogen Using Earth-Abundant Metals: Shoving Protons Around with Proton Relays. Acc. Chem. Res 2015, 48 (7), 2017–2026. [DOI] [PubMed] [Google Scholar]
- (7).Fukuzumi S; Lee YM; Nam W Thermal and photocatalytic production of hydrogen with earth-abundant metal complexes. Coord. Chem. Rev 2018, 355, 54–73. [Google Scholar]
- (8).Lubitz W; Ogata H; Rüdiger O; Reijerse E Hydrogenases. Chem. Rev 2014, 114 (8), 4081–4148. [DOI] [PubMed] [Google Scholar]
- (9).Peters JW; Schut GJ; Boyd ES; Mulder DW; Shepard EM; Broderick JB; King PW; Adams MWW [FeFe]-and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta, Mol. Cell Res 2015, 1853 (6), 1350–1369. [DOI] [PubMed] [Google Scholar]
- (10).Esselborn J; Muraki N; Klein K; Engelbrecht V; Metzler-Nolte N; Apfel UP; Hofmann E; Kurisu G; Happe T A structural view of synthetic cofactor integration into [FeFe]-hydrogenases. Chem. Sci 2016, 7 (2), 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wittkamp F; Senger M; Stripp ST; Apfel UP FeFe-Hydrogenases: recent developments and future perspectives. Chem. Commun 2018, 54 (47), 5934–5942. [DOI] [PubMed] [Google Scholar]
- (12).Cornish AJ; Gartner K; Yang H; Peters JW; Hegg EL Mechanism of Proton Transfer in FeFe-Hydrogenase from Clostridium pasteurianum. J. Biol. Chem 2011, 286 (44), 38341–38347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hong G; Cornish AJ; Hegg EL; Pachter R On understanding proton transfer to the biocatalytic [Fe-Fe]H sub-cluster in [Fe-Fe] H2 ases: QM/MM MD simulations. Biochim. Biophys. Acta, Bioenerg 2011, 1807 (5), 510–517. [DOI] [PubMed] [Google Scholar]
- (14).Knörzer P; Silakov A; Foster CE; Armstrong FA; Lubitz W; Happe T Importance of the Protein Framework for Catalytic Activity of FeFe-Hydrogenases. J. Biol. Chem 2012, 287 (2), 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Long H; King PW; Chang CH Proton Transport in Clostridium pasteurianum FeFe Hydrogenase I: A Computational Study. J. Phys. Chem. B 2014, 118 (4), 890–900. [DOI] [PubMed] [Google Scholar]
- (16).Ginovska-Pangovska B; Ho MH; Linehan JC; Cheng YH; Dupuis M; Raugei S; Shaw WJ Molecular dynamics study of the proposed proton transport pathways in FeFe-hydrogenase. Biochim. Biophys. Acta, Bioenerg 2014, 1837 (1), 131–138. [DOI] [PubMed] [Google Scholar]
- (17).Mulder DW; Ratzloff MW; Bruschi M; Greco C; Koonce E; Peters JW; King PW Investigations on the Role of Proton-Coupled Electron Transfer in Hydrogen Activation by FeFe-Hydrogenase. J. Am. Chem. Soc 2014, 136 (43), 15394–15402. [DOI] [PubMed] [Google Scholar]
- (18).Mulder DW; Guo Y; Ratzloff MW; King PW Identification of a Catalytic Iron-Hydride at the H-Cluster of [FeFe]-Hydrogenase. J. Am. Chem. Soc 2017, 139, 83–86. [DOI] [PubMed] [Google Scholar]
- (19).Silakov A; Wenk B; Reijerse E; Lubitz W 14N HYSCORE investigation of the H-cluster of FeFe hydrogenase: evidence for a nitrogen in the dithiol bridge. Phys. Chem. Chem. Phys 2009, 11 (31), 6592–6599. [DOI] [PubMed] [Google Scholar]
- (20).Reijerse EJ; Pham CC; Pelmenschikov V; Gilbert-Wilson R; Adamska-Venkatesh A; Siebel JF; Gee LB; Yoda Y; Tamasaku K; Lubitz W; Rauchfuss TB; Cramer SP Direct observation of an iron bound terminal hydride intermediate in [FeFe] hydrogenase. J. Am. Chem. Soc 2017, 139 (12), 4306–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pelmenschikov V; Birrell JA; Pham CC; Mishra N; Wang HX; Sommer C; Reijerse E; Richers CP; Tamasaku K; Yoda Y; Rauchfuss TB; Lubitz W; Cramer SP Reaction Coordinate Leading to H2 Production in [FeFe] Hydrogenase Identified by Nuclear Resonance Vibrational Spectroscopy and Density Functional Theory. J. Am. Chem. Soc 2017, 139 (46), 16894–16902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pham CC; Mulder DW; Pelmenschikov V; King PW; Ratzloff MW; Wang H; Mishra N; Alp EE; Zhao J; Hu MY; Tamasaku K; Yoda Y; Cramer SP Terminal Hydride Species in [FeFe]-Hydrogenases are Vibrationally Coupled to the Active Site Environment. Angew. Chem., Int. Ed 2018, 57, 10605–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rumpel S; Sommer C; Reijerse E; Fares C; Lubitz W Direct Detection of the Terminal Hydride Intermediate in FeFe Hydrogenase by NMR Spectroscopy. J. Am. Chem. Soc 2018, 140 (11), 3863–3866. [DOI] [PubMed] [Google Scholar]
- (24).Lorent C; Katz S; Duan J; Kulka CJ; Caserta G; Teutloff C; Yadav S; Apfel U-P; Winkler M; Happe T; Horch M; Zebger I Shedding light on proton and electron dynamics in [FeFe] hydrogenases. J. Am. Chem. Soc 2020, 142, 5493–5497. [DOI] [PubMed] [Google Scholar]
- (25).Seto M; Yoda Y; Kikuta S; Zhang XW; Ando M Observation of Nuclear Resonant Scattering Accompanied by Phonon Excitation Using Synchrotron Radiation. Phys. Rev. Lett 1995, 74, 3828–3831. [DOI] [PubMed] [Google Scholar]
- (26).Chumakov A; Rüffer R Nuclear inelastic scattering. Hyperfine Interact. 1998, 113 (1), 59–79. [Google Scholar]
- (27).Scheidt WR; Li JF; Sage JT What Can Be Learned from Nuclear Resonance Vibrational Spectroscopy: Vibrational Dynamics and Hemes. Chem. Rev 2017, 117 (19), 12532–12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hu MY Some notes on data analysis for nuclear resonant inelastic x-ray scattering. Hyperfine Interact. 2016, 237, 64. [Google Scholar]
- (29).Sage JT; Paxson C; Wyllie GRA; Sturhahn W; Durbin SM; Champion PM; Alp EE; Scheidt WR Nuclear resonance vibrational spectroscopy of a protein active-site mimic. J. Phys.: Condens. Matter 2001, 13, 7707–7722. [Google Scholar]
- (30).Leu BM; Zgierski MZ; Wyllie GRA; Scheidt WR; Sturhahn W; Alp EE; Durbin SM; Sage JT Quantitative Vibrational Dynamics of Iron in Nitrosyl Porphyrins. J. Am. Chem. Soc 2004, 126 (13), 4211–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cramer SP Nuclear Resonance Vibrational Spectroscopy. In X-Ray Spectroscopy with Synchrotron Radiation: Fundamentals and Applications; Springer International Publishing: Cham, 2020; pp 257–278. [Google Scholar]
- (32).Reijerse EJ; Pelmenschikov V; Birrell JA; Richers CP; Kaupp M; Rauchfuss TB; Cramer SP; Lubitz W Asymmetry in the Ligand Coordination Sphere of the [FeFe] Hydrogenase Active Site Is Reflected in the Magnetic Spin Interactions of the Azapropanedithiolate Ligand. J. Phys. Chem. Lett 2019, 10 (21), 6794–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Berggren G; Adamska A; Lambertz C; Simmons TR; Esselborn J; Atta M; Gambarelli S; Mouesca JM; Reijerse E; Lubitz W; Happe T; Artero V; Fontecave M Biomimetic assembly and activation of FeFe-hydrogenases. Nature 2013, 499 (7456), 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Han S; Czernuszewicz RS; Kimura T; Adams MWW; Spiro TG Fe2S2 Protein Resonance Raman Revisited: Structural Variations among Adrenodoxin, Ferredoxin, and Red Paramagnetic Protein. J. Am. Chem. Soc 1989, 111 (10), 3505–3511. [Google Scholar]
- (35).Fu W; Drozdzewski PM; Davies MD; Sligar SG; Johnson MK Resonance Raman and Magnetic Circular Dichroism Studies of Reduced [2Fe-2S] Proteins. J. Biol. Chem 1992, 267 (22), 15502–15510. [PubMed] [Google Scholar]
- (36).Birrell JA; Pelmenschikov V; Mishra N; Wang H; Yoda Y; Tamasaku K; Rauchfuss TB; Cramer SP; Lubitz W; DeBeer S Spectroscopic and Computational Evidence that [FeFe] Hydrogenases Operate Exclusively with CO-Bridged Intermediates. J. Am. Chem. Soc 2020, 142 (1), 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Schroer CG; Agapov I; Brefeld W; Brinkmann R; Chae YC; Chao HC; Eriksson M; Keil J; Nuel Gavalda X; Rohlsberger R; Seeck OH; Sprung M; Tischer M; Wanzenberg R; Weckert E PETRA IV: the ultralow-emittance source project at DESY. J. Synchrotron Radiat 2018, 25 (5), 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Custelcean R; Jackson JE Dihydrogen Bonding: Structures, Energetics, and Dynamics. Chem. Rev 2001, 101 (7), 1963–1980. [DOI] [PubMed] [Google Scholar]
- (39).Klooster WT; Koetzle TF; Siegbahn PEM; Richardson TB; Crabtree RH Study of the N—H. H—B dihydrogen bond including the crystal structure of BH3NH3 by neutron diffraction. J. Am. Chem. Soc 1999, 121 (27), 6337–6343. [Google Scholar]
- (40).Sanchez MLK; Sommer C; Reijerse E; Birrell JA; Lubitz W; Dyer RB Investigating the Kinetic Competency of CrHydA1 FeFe Hydrogenase Intermediate States via Time-Resolved Infrared Spectroscopy. J. Am. Chem. Soc 2019, 141 (40), 16064–16070. [DOI] [PubMed] [Google Scholar]
- (41).Sommer C; Adamska-Venkatesh A; Pawlak K; Birrell JA; Rudiger O; Reijerse EJ; Lubitz W Proton Coupled Electronic Rearrangement within the H-Cluster as an Essential Step in the Catalytic Cycle of FeFe Hydrogenases. J. Am. Chem. Soc 2017, 139 (4), 1440–1443. [DOI] [PubMed] [Google Scholar]
- (42).Duan JF; Senger M; Esselborn J; Engelbrecht V; Wittkamp F; Apfel UP; Hofmann E; Stripp ST; Happe T; Winkler M Crystallographic and spectroscopic assignment of the proton transfer pathway in FeFe-hydrogenases. Nat. Commun 2018, 9, 4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ratzloff MW; Artz JH; Mulder DW; Collins RT; Furtak TE; King PW CO-Bridged H-Cluster Intermediates in the Catalytic Mechanism of FeFe-Hydrogenase CaI. J. Am. Chem. Soc 2018, 140 (24), 7623–7628. [DOI] [PubMed] [Google Scholar]
- (44).Arrigoni F; Bertini L; Bruschi M; Greco C; De Gioia L; Zampella G H2 Activation in [FeFe]-Hydrogenase Cofactor Versus Diiron Dithiolate Models: Factors Underlying the Catalytic Success of Nature and Implications for an Improved Biomimicry. Chem. - Eur. J 2019, 25 (5), 1227–1241. [DOI] [PubMed] [Google Scholar]
- (45).Salna B; Benabbas A; Sage JT; van Thor J; Champion PM Wide-dynamic-range kinetic investigations of deep proton tunnelling in proteins. Nat. Chem 2016, 8 (9), 874–880. [DOI] [PubMed] [Google Scholar]
- (46).Greco C; Bruschi M; Fantucci P; Ryde U; De Gioia L Mechanistic and physiological implications of the interplay among iron-sulfur clusters in [FeFe]-hydrogenases. A QM/MM perspective. J. Am. Chem. Soc 2011, 133 (46), 18742–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.