

Abstract
We examined the signaling route for fever during localized inflammation in male and female mice, elicited by casein injection into a preformed air pouch. The localized inflammation gave rise to high concentrations of prostaglandins of the E species (PGE2) and cytokines in the air pouch and elevated levels of these inflammatory mediators in plasma. There were also elevated levels of PGE2 in the cerebrospinal fluid, although there was little evidence for PGE2 synthesis in the brain. Global deletion of the PGE2 prostaglandin E receptor 3 (EP3) abolished the febrile response as did deletion of the EP3 receptor in neural cells, whereas its deletion on peripheral nerves had no effect, implying that PGE2 action on this receptor in the CNS elicited the fever. Global deletion of the interleukin-1 receptor type 1 (IL-1R1) also abolished the febrile response, whereas its deletion on neural cells or peripheral nerves had no effect. However, deletion of the IL-1R1 on brain endothelial cells, as well as deletion of the interleukin-6 receptor α on these cells, attenuated the febrile response. In contrast, deletion of the PGE2 synthesizing enzymes cyclooxygenase-2 and microsomal prostaglandin synthase-1 in brain endothelial cells, known to attenuate fever evoked by systemic inflammation, had no effect. We conclude that fever during localized inflammation is not mediated by neural signaling from the inflamed site, as previously suggested, but is dependent on humoral signaling that involves interleukin actions on brain endothelial cells, probably facilitating PGE2 entry into the brain from the circulation and hence representing a mechanism distinct from that at work during systemic inflammation.
Keywords: blood-brain barrier, cytokines, fever, inflammation, mouse, PGE2
Significance Statement
Fever elicited by a localized inflammation has long been suggested to depend on the activation of and signaling by peripheral nerves innervating the inflamed tissue and not on humoral messengers, in contrast to what now is clearly established for systemic inflammation. We here show that fever evoked by the injection of pyrogen into a preformed air pouch, providing an enclosed space lined by a synovial-like membrane, is dependent not on peripheral neural signals but on the activation of CNS PGE2 EP3 receptors, similar to what is the case during systemic inflammation. The mechanisms are nevertheless distinct because fever during localized inflammation, although it depends on cytokine signaling in the brain endothelium, does not require brain PGE2 production.
Introduction
Prostaglandins (PGs) have been known as the critical mediator of fever since the 1970s, when their production by cyclooxygenase (Cox) enzymes was shown to be the target of antipyretic and anti-inflammatory drugs (Vane, 1971; Flower et al., 1972; Flower and Vane, 1972). Prostaglandins of the E species (PGE2), which are synthesized from cyclooxygenase-produced PGH2 by terminal isomerases such as microsomal prostaglandin E synthase-1 (mPGES-1; Jakobsson et al., 1999), were later identified as the pyrogen (Stitt, 1986). The demonstration of the expression of inducible Cox, Cox-2 (Kujubu et al., 1991; Sirois et al., 1992), and mPGES-1 in brain vessels (Cao et al., 1996; Elmquist et al., 1997; Ek et al., 2001; Yamagata et al., 2001) gave rise to the idea that PGE2 was produced in these vessels upon peripheral immune challenge through the action of circulating cytokines and that this centrally produced PGE2 elicited the febrile response. Functional studies have subsequently confirmed this idea by showing that deletion of Cox-2 or mPGES-1 in brain endothelial cells attenuates fever evoked by systemic immune challenge (Wilhelms et al., 2014; Eskilsson et al., 2017).
However, in addition to this blood-borne pathway for the generation of fever, it has been suggested that fever in response to a localized inflammation may be mediated by the activation of peripheral nerves, hence not requiring the activation of central PGE2 synthesis by circulating cytokines. Although the initial observations that surgical transection of the vagus nerve attenuated fever (Watkins et al., 1995; Sehic and Blatteis, 1996; Hansen and Krueger, 1997) later were refuted (Blomqvist and Engblom, 2018), it has been maintained that somatic afferent nerves may mediate the febrile response to a localized inflammation. Dorsal root ganglion (DRG) cells express IL-1 receptors as well as prostaglandin E receptors 3 (EP3; Nakamura et al., 2000; Binshtok et al., 2008), the PGE2 receptor subtype critical for fever (Ushikubi et al., 1998), and nociceptors can be sensitized by IL-1β and PGE2 (Binshtok et al., 2008). The putative role for afferent nerve signaling to the brain in fever has been studied in models of localized inflammation using a subcutaneous air pouch or a subcutaneous Teflon chamber (Miller et al., 1997; Ross et al., 2000; Rummel et al., 2005; Zhang et al., 2008). Although in these models there seems to be leakage of IL-6 into the circulation (Cartmell et al., 2000), no Cox-2 induction in the brain was found, leading the investigators to suggest the presence of a neural rather than humoral route of signaling. Further support for a neuronal signaling pathway for fever comes from studies in which the injection of a local anesthetic at the site of local inflammation or transection of the peripheral nerve innervating the inflamed tissue were reported to attenuate fever (for a discussion of these findings, see Blomqvist and Engblom, 2018).
A conceptual problem with the idea of a neural instead of humoral signaling for fever from the site of a peripheral inflammation is that such a route would either have to feed into the central thermoregulatory system without involving PGE2 release in the preoptic hypothalamus, which seems inconsistent with the antipyresis achieved by the largely centrally acting Cox-inhibitor paracetamol during localized inflammation, such as experimentally induced monoarthritis in animals and otitis media in humans (Kanashiro et al., 2009; van den Anker, 2013), or evoke central prostaglandin release in the hypothalamus by cells other than brain vascular cells. Here, we reexamined the mechanisms of fever during localized inflammation, using injection of the milk protein casein into a preformed air pouch (Zhang et al., 2008). We used a variety of genetically modified mice, with global or tissue-specific gene deletions of inflammatory mediators, to examine the putative pathways by which the localized inflammation gave rise to fever. We show that fever in this model is dependent on humoral signaling that involves interleukin actions on brain endothelial cells, albeit in a way that seems distinct from that at work during systemic inflammation.
Material and Methods
Animals
All experimental procedures were approved by the Animal Ethics Committee in Linköping and followed international guidelines. Adult, age, and sex-matched mice were used. Mice with global gene deletions [EP1 (Stock et al., 2001), EP2 (Tilley et al., 1999), EP3 (Fleming et al., 1998), mPGES-1 (Trebino et al., 2003), or interleukin-1 receptor type 1 (IL-1R1; Glaccum et al., 1997)], as well as mice with conditional deletions [EP3, possessing loxP sites flanking exon 1 of the Ptger3 gene (Lazarus et al., 2007); IL-6 receptor α, possessing loxP sites flanking exons 4–6 of the Il6ra gene (McFarland-Mancini et al., 2010); IL-1R1, possessing loxP sites flanking exon 5 of the Il1r1 gene (Abdulaal et al., 2016); Cox-2, possessing loxP sites flanking exons 4–5 of the Ptgs2 gene (Ishikawa and Herschman, 2006); and mPGES-1, possesing loxP-sites flanking exon 2 in the Ptges1 gene (Wilhelms et al., 2014)] were used. The mice with conditional deletions were crossed with mice expressing Cre recombinase under specific promoters. These included (1) mice expressing Cre recombinase in neuronal and glia cell precursors under the control of the nestin promoter and enhancer, a widely used line for selective deletion in neural cells (Tronche et al., 1999); (2) mice expressing Cre recombinase under the TRPV1 promotor, which, with the exception of some cell groups in the hypothalamus, is expressed specifically by nociceptors in primary sensory ganglia (Cavanaugh et al., 2011). This Cre-line has previously been shown to efficaciously delete genes in these latter structures (Spencer et al., 2018); (3) the human tissue plasminogen activator (HTPA), expressed by neural crest derivates. The HTPA-Cre line is reported to be a pan neural crest cell marker that recombines almost exclusively in neural crest derivates, the exception being a minor component of limb periosteum and tendons. When combined with a reporter line, it produced homogenous staining in spinal dorsal root ganglia, indicating recombination of both glial and neuronal precursors in these structures (Pietri et al., 2003); (4) LysM, expressed by myeloid cells. The LysM-Cre line has demonstrated a deletion efficiency of 83–98% in mature macrophages and nearly 100% in granulocytes, and partial deletion in CD11c splenic dendritic cells but no significant deletion in tail DNA or purified T and B cells (Clausen et al., 1999); and (5) a tamoxifen-inducible CreERT2 from the Slco1c1 promoter, expressed by brain endothelial cells (Ridder et al., 2011). The specificity of this Cre-line has been demonstrated using reporter mice, combined with staining for cell-specific markers (Eskilsson et al., 2014; Fritz et al., 2016, 2018). It preferentially produces recombination in small- to medium-size brain blood vessels (Eskilsson et al., 2017).
The mice were obtained from The Jackson Laboratory or directly from the respective investigator. They were all maintained on a C57BL/6 background and housed under constant ambient temperature (21°C), on a 12 h/12 h light/dark cycle (lights on at 7 A.M.) with food and water ad libitum. Gene deletion in mice with the Slco1c1 CreERT2 construct was induced by intraperitoneal injection of tamoxifen (1 mg tamoxifen diluted in a mixture of 10% ethanol and 90% sunflower seed oil twice a day for 5 d) at least 5 weeks before additional experiments.
Creation of chimeric mice
Mice chimeric for mPGES-1 were created by irradiating mPGES-1 KO and WT mice to an absorbed dose of 9 Gy in two fractions. About 24 h after irradiation, the mice were injected intravenously with 2 million freshly prepared CD45+ bone marrow cells of the same or opposite genotype. They were allowed to survive for ∼5 months before they were used for additional experiments (Engström et al., 2012). This procedure results in extensive replacement of hematopoietically derived cells, both in the periphery and in the brain, as reported previously (Engström et al., 2012).
Surgery
The mice were anesthetized with isoflurane and implanted intraperitoneally with a transponder that records core body temperature (E‐Mitter Telemetry System, STARR Life Sciences) after which 3 ml air sterilized through a PVC filter (Filtropur S plus, 0.2 μm, Sarstedt) was injected subcutaneously at the back of the mouse to create an air pouch. The mice were then transferred to a room in which the ambient temperature was set to 29°C, providing near-thermoneutral conditions (Rudaya et al., 2005). Three d after the first air injection, the pouch was again filled with ∼3 ml sterilized air, followed after another 4 d of injection into the pouch of 0.5–0.8 ml (depending on the weight of the animal) of a casein solution (10% casein, Hammarsten grade; MP Biomedicals) in 50 mm sodium bicarbonate buffer, conducted at ∼7 A.M. The milk protein casein is known to elicit inflammation and fever in animals and humans, although its mode of action is not clear (Kitamura et al., 1986; Moissidis et al., 2005). The air pouch is a closed space covered by a membrane resembling the synovial membrane of a joint that creates a mechanical barrier that retains the inflammatory stimulus and products of the inflammatory response (Edwards et al., 1981; Sedgwick et al., 1983). In the initial experiments, mice were given a brief anesthesia with isoflurane to facilitate injections; however, as this procedure resulted in a drop in body temperature, it was subsequently abandoned. Control animals were injected with buffer solution only. Mice used for real-time PCR analyses, immunoassays, and immunohistochemical stainings were killed 5 h after injection, corresponding to a time point when fever is established and rising in this model (Fig. 1).
Figure 1.

Fever response, and prostaglandin synthesis and PGE2 levels in the air pouch, plasma, and brain elicited by casein injection into the pouch. A, Temperature recordings in WT mice injected at time point 0 with casein or vehicle. The temperature peak seen in both genotypes immediately after injection is because of the handling stress during the injection procedure. Solid lines represent mean, and dotted lines represent SEM. Data are taken from WT mice in the experiment examining the role of brain endothelial IL-1 receptors (Fig. 6E). B–E, qPCR Analysis of Cox-2 and mPGES-1 mRNA in the hypothalamus (B, C) and the air pouch wall (D, E) 5 h after injection of casein or vehicle into the air pouch; n = 6. F–H, Concentration of PGE2 or PGE2 metabolites in the air pouch lavage (F), cerebrospinal fluid (G), and plasma (H) 5 h after casein or vehicle injection; n = 7–10. Error bars represent SEM. I, Microphotograph of the air pouch wall stained for Cox-2 and CD45. Note the strong Cox-2 labeling in the lining of the wall, and the infiltration in the wall of CD45-positive but Cox-2 negative immune cells. Scale bar, 200 μm. J, Cox-2 expressing brain vascular cells (arrowheads) 5 h after casein injection into the air pouch. Scale bar, 200 μm.
Real-time PCR
The mice were killed by asphyxiation with CO2. The wall of the air pouch was dissected, and the brain and in some experiments dorsal root ganglia from the cervical and lumbar segments were collected. The hypothalamus was cut out as described previously (Reyes et al., 2003). Tissue was placed in RNAlater stabilization reagent (Qiagen) and kept at −70°C until further processing. RNA was extracted with RNeasy Plus Universal Kit (Qiagen), and reverse transcription was done with High Capacity cDNA Reverse Transcription Kit (Applied Biosystems); qPCR was performed using Gene Expression Master Mix (Applied Biosystems) on a 96-well plate (7900HT Fast Real-Time PCR System Software; Applied Biosystems). The following TaqMan assays (all from Applied Biosystems) were used: Mm00434228_m1 (IL‐1β), Mm00446190_m1 (IL‐6), Mm00443258_m1 (TNF), Mm00478374_m1 (Cox-2), Mm00452105_m1 (mPGES-1), and Mm99999915_g1 (GAPDH).
Immunoassays
The mice were asphyxiated and blood was drawn from the right atrium and transferred to EDTA-coated tubes (Sarstedt) to which indomethacin (10 μM; Sigma-Aldrich) was added and centrifuged at 7000 × g for 7 min at 4°C. The plasma was immediately frozen on dry ice and kept at −70°C. Air pouch lavage was collected by injecting into the air pouch 1 ml sterile saline, which was subsequently withdrawn. The lavage was centrifuged at 15,000 × g for 10 min at 4°C. The supernatant was collected and frozen on dry ice and kept at −70°C. After blood and lavage had been collected, the carcass was placed in a stereotaxic frame, the atlanto-occipital membrane was exposed, and cerebrospinal fluid was withdrawn from the cisterna magna using a Hamilton syringe mounted on a micromanipulator and immediately frozen. Samples that contained traces of blood were discarded. The whole procedure from when the animals were killed until cerebrospinal fluid was withdrawn and frozen took <10 min.
The concentration of PGE2 in the cerebrospinal fluid (diluted 1:100) and air pouch lavage (diluted 1:1000 after casein injection and 1:10 after vehicle injection) was determined using a High Sensitivity Prostaglandin E2 Enzyme Immunoassay Kit (Assay Designs). The values were calculated using a standard curve ranging from 7.81 to 1000 pg/ml (r2 = 0.998). The kit antiserum showed the following cross reactivity, according to the manufacturer: PGE2 100%, PGE1 70%, PGE3 16.3%, PGF1α 1.4%, PGF2α 0.7%, 6-keto-PGF1α 0.6%, PGA2 0.1%, PGB1 0.1%, and < 0.1% for 13, 14-dihydro-15-ketoPGF2α, 6, 15-keto, 13, 14-dihydro-PGF1α, thromboxane B2, 2-arachidonoylglyerol, anandamide, PGD2 and arachidonic acid. The concentration of PGE2 metabolites in plasma (diluted 1:50) was determined with a Prostaglandin E Metabolite EIA Kit (Cayman Chemical). The values were calculated using a standard curve ranging from 0.2 to 50 pg/ml (r2 = 0.9835-0.9981). The kit antiserum recognizes derivatized 13,14-dihydro-15-ketoPGE1 and 13,14-dihydro-15-ketoPGE2 and bicycloPGE1, but has <0.01% cross reactivity with arachidonic acid, leukotriene B4, tetranor-PGEM, tetranor-PGFM, PGD2, PGE1, 6-keto PGE1, PGE2, PGF1α, 6-keto PGF1α, PGF2α, and thromboxane B2.
Levels of IL-1β were analyzed with a mouse IL-1b/IL-1F2 immunoassay (catalog #MLB00C, R&D Systems) according to the manufacturer's instructions. The lavage from casein-injected mice was diluted 1:2 but left undiluted for mice injected with vehicle and for plasma. The values were calculated using a standard curve ranging from 12.5 to 800 pg/ml (r2 = 1.0). The sensitivity of the assay was 2.31 pg/ml, and according to the manufacturer, it exhibited no cross reactivity with mouse recombinant IL-1; IL-1F10/IL-1HY2; IL-1ra; IL-1 RAPL2/IL-1 R9; IL-1 RI; IL-1 RII; IL-1 Rrp2/IL-1 R6; IL-2–7, 9, 10–13, 15, 20, 23; IL-10ra,; IL-11ra; IL-12p40; IL-23R; or ST2/IL-1 R4.
Levels of TNF and IL-6 were analyzed using a Milliplex Mouse Cytokine/Chemokine Magnetic Panel MCYTOMAG-70K (Merck Millipore), using a dilution of 1:2 for plasma and 1:200 for lavage. According to the manufacturer, assay sensitivity was 1.1 pg/ml for IL-6 and 2.3 pg/ml for TNF, and intra-assay precision was 2.3% and 2.6%, respectively. There was no or negligible cross-reactivity between IL-6 and TNF and other cytokines.
Immunohistochemistry
After asphyxiation with CO2, the animals were perfused transcardially with a phosphate-buffered (0.1 m) paraformaldehyde solution (4%). Tissue of interest was postfixed for 3 h in the same fixative, then cryoprotected with 25% sucrose in PBS and cut at 30 μm on a freezing microtome. The immunohistochemical procedures were performed according to standardized protocols (Engström et al., 2012). The primary antibodies were rabbit anti-Cox-2 (1:500–1:1000; catalog #sc-1747 M-17, Santa Cruz Biotechnology), which was detected with Alexa Fluor 555 donkey anti-rabbit (1:500; Invitrogen) or an avidin-biotin-HRP system (Vector Laboratories) with 3,3′-diaminobenzidine as chromogen, and rat anti-CD45 (1:5000; Serotec), which was detected with Alexa Fluor 488 goat anti-rat (1:500; Invitrogen).
Experimental design and statistical analyses
In all comparisons, littermates were used. Groups were designed to be age and sex matched, but within these limitations, animals were randomly assigned to the respective group. Statistical analyses were done in Prism version 9 (GraphPad). Significant differences were determined using a one-way or two-way ANOVA, both followed by a post hoc test with correction for multiple comparisons controlling the false discovery rate according to the linear step-up multiple-testing procedure (Benjamini et al., 2006) or, for pair-wise comparisons, a nonparametric test (Mann–Whitney). Results were considered statistically significant when p < 0.05. Group size in the temperature recording experiments was considered as follows: With an estimated SD ∼0.5°C, 12 mice in each group are required to permit detection of a difference of 0.6°C with 80% reliability at a significance level of 5%. We strived to obtain that group size; however if there was no or minimal difference in mean temperature between groups after the first rounds of an experiment (encompassing ∼4–8 mice in each group), or if a very clear effect was seen, we did not further pursue that experiment, considering it was not justified from an animal ethics perspective.
Results
Casein injection into a preformed air pouch results in fever and induced PGE2 synthesis
Following casein injection into the air pouch, the mice displayed fever starting ∼3–4 h after the injection and lasting ∼7–8 h (Fig. 1A). Because fever induced by systemic immune challenge has been shown to be elicited by PGE2 (Blomqvist and Engblom, 2018), we first examined if there was increased PGE2 synthesis in this model. We found a significant but weak induction of Cox-2 mRNA but not of mPGES-1 mRNA in the hypothalamus (Fig. 1B,C), and strong induction of Cox-2 as well as significant induction of mPGES-1 mRNA in the air pouch wall (Fig. 1D,E). There was a huge increase in PGE2 levels in the air pouch lavage in the casein-injected mice (Fig. 1F), but increased levels of PGE2 and of PGE2 metabolites were also seen in the cerebrospinal fluid (Fig. 1G) and plasma (Fig. 1H), respectively. Immunohistochemical staining for Cox-2 protein showed strong, induced labeling in the lining toward the lumen of the air pouch (Fig. 1I), previously identified as consisting of fibroblasts and collagen fibers (Sedgwick et al., 1983), as well as induced expression in some brain blood vessels with immunolabeling seen in scattered cells (Fig. 1J). In the air pouch wall, many CD45-positive immune cells were seen, but these were not stained for Cox-2 (Fig. 1I).
The fever response to casein injection is dependent on the synthesis of PGE2 by nonhematopoietic cells
Given the above findings, we examined whether deletion of PGE2 synthesis would inhibit fever induced by casein injection into the air pouch. We found that global deletion of the PGE2 synthesizing enzyme mPGES-1 almost completely abolished the febrile response (Fig. 2A). Similarly, the nonselective Cox inhibitor indomethacin (10 mg/kg body weight; Alpharma) injected either intraperitoneally or into the air pouch, as well as the selective Cox-2 inhibitor parecoxib (10 mg/kg; Dynastat, Pfizer), injected intraperitoneally, also abolished the fever response (Fig. 2B,E). We then examined the role of hematopoietic versus nonhematopoietic cells for the casein-induced fever. We generated mPGES-1 chimeric mice by transplantation of bone marrow of the opposite genotype to mPGES-1 knock-out mice and wild-type mice, and used animals receiving bone marrow of their own genotype as controls (Engström et al., 2012). Wild-type mice transplanted with knock-out bone marrow (KO→WT) displayed fever similar to wild-type mice with wild-type bone marrow (WT→WT). Knock-out mice transplanted with wild-type bone marrow (WT→KO) showed attenuated fever, like knock-out mice with knock-out bone marrow (KO→KO; Fig. 2C). Furthermore, the PGE2 levels in the air pouch lavage were much higher in WT→WT and KO→WT animals than in KO→KO and WT→KO animals (Fig. 2D). These data suggest that nonhematopoietic cells are synthesizing the PGE2 that is critical for the fever response to casein injected into the preformed air pouch. This idea is consistent with our observation that Cox-2 was densely expressed by cells in the lining of the air pouch wall but not by infiltrating immune cells (Fig. 1I).
Figure 2.
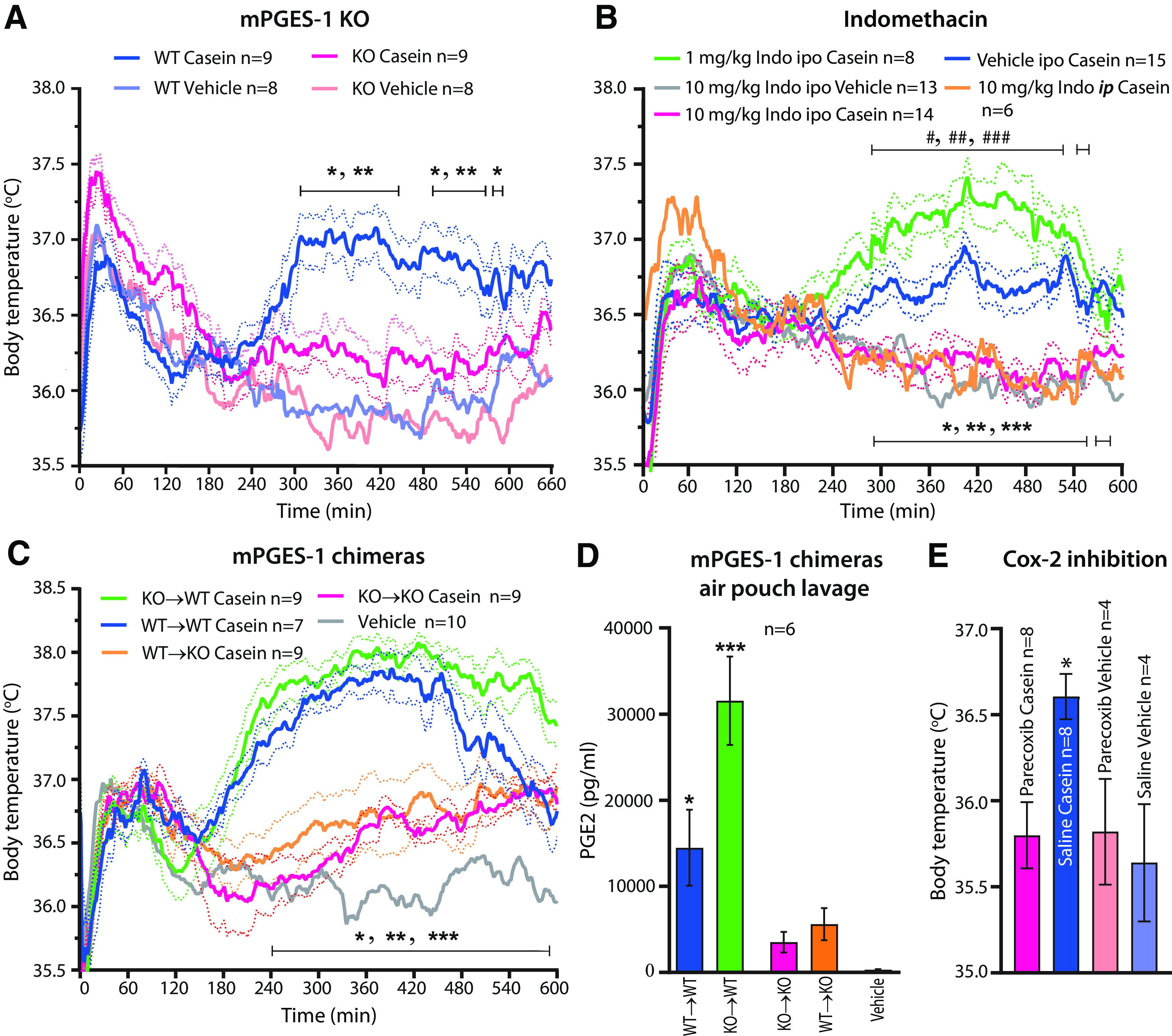
Temperature response and PGE2 production after casein injection into a preformed air pouch in mPGES-1 KO and chimeric mice, and treatment with cyclooxygenase inhibitor. A, Attenuated fever in mPGES-1 KO mice. *p < 0.05 and **p < 0.01 for WT mice versus mPGES-1 KO mice injected with casein (two-way ANOVA for the period of 4–10 h: F(3,30) = 17.71, p < 0.0001). B, Effect of indomethacin (Indo) injected into the air pouch (ipo) or intraperitoneally (ip). Note that both routes of administration of 10 mg/kg indomethacin abolished casein-induced fever. Injection of 1 mg/kg indomethacin into the air pouch resulted in augmented fever to casein. *p < 0.05, **p < 0.01, and ***p < 0.001 for Vehicle ipo Casein versus 10 mg/kg Indo ipo Casein, and #p < 0.05, ##p < 0.01, and ###p < 0.001 for Vehicle ipo Casein versus 1 mg/kg Indo ipo Casein (two-way ANOVA for the period of 4–10 h: F(4,51) = 17.26, p < 0.0001). C, mPGES-1 chimeric mice. WT→WT, WT mice transplanted with WT bone marrow; KO→WT, WT mice transplanted with KO bone marrow; KO→KO, KO mice transplanted with KO bone marrow; and WT→KO, KO mice transplanted with WT bone marrow. *p < 0.05, **p < 0.01, and ***p < 0.001 for KO→WT mice versus WT→KO mice injected with casein (two-way ANOVA for the period of 4–10 h: F(4,39) = 32.02, p < 0.0001). Solid lines represent mean, and dotted lines represent SEM. D, PGE2 concentration in air pouch lavage in mPGES-1 chimeric mice 5 h after casein injection into the pouch. Vehicle, mixed chimeras. Error bars represent SEM. *p = 0.0155 for WT→WT versus KO→KO and *p = 0.0358 for WT→WT versus WT→KO. ***p < 0.0001 for KO→WT versus KO→KO and WT→KO (one-way ANOVA: F(4,25) = 15.66, p < 0.0001). E, Effect of parecoxib on casein-induced fever. Parecoxib, 10 mg/kg, or saline was injected intraperitoneally 3 h after casein or vehicle injection into the air pouch (i.e., immediately before the appearance of the casein-induced fever); the temperature recordings were done 2.5 h later, close to the peak of the casein-induced fever and before the effect of parecoxib disappears (Nilsberth et al., 2009a). *p = 0.0173 for parecoxib casein versus saline casein (one-way ANOVA: F(3,20) = 4.625, p = 0.0129).
EP3 receptors in the CNS are critical for the febrile response to casein injection
We next examined where the PGE2 induced by the casein injection into the air pouch exerted its pyrogenic effect. We first confirmed that deletion of the EP3 receptor, which has been shown to be critical for PGE2-induced fever in other models (Ushikubi et al., 1998; Engblom et al., 2003), abolished the febrile response to casein injection (Fig. 3C). There was no effect of deletion of the EP1 or EP2 receptor (Fig. 3A,B). We then generated mice with deletion of EP3 receptors on peripheral nerves, or on neural cells, by crossing mice with a conditional deletion of the EP3 receptor (Lazarus et al., 2007) with mice expressing Cre recombinase under the HTPA promotor (expressed by neural crest derivates; Pietri et al., 2003) or mice expressing Cre recombinase under the Nestin promotor (expressed by neuronal and glia cell precursors; Tronche et al., 1999). Mice expressing Cre recombinase under the Nestin promotor showed no febrile response to the casein injection (Fig. 3D), whereas mice expressing Cre recombinase under the HTPA promotor displayed the same febrile response as control (wild type) mice (Fig. 3E). We further explored the possibility that nerves innervating the region of the air pouch could be involved by injecting the air pouch with a local anesthetic (8 mg/kg ropivacaine; Narop, Aspen Nordic) 5 min before the injection of casein to block any inflammation-elicited neural signaling from this structure. The dose chosen was the highest that could be administered without risking systemic effects. However, mice injected with the local anesthetic into the air pouch showed the same febrile response to casein as did mice injected with vehicle (Fig. 3G).
Figure 3.

Effect of EP receptor deletion and nerve blockade on casein-induced fever. A, B, No difference in the febrile response between WT and EP1 (A) and EP2 (B) KO mice. C, D, Absence of fever in mice with global deletion of EP3 (C); *p < 0.05 and **p < 0.01 for WT mice versus EP3 KO mice injected with casein (two-way ANOVA for the period of 4–10 h: F(3,28) = 20.29, p < 0.0001) or EP3 deletion in neural cells (D); *p < 0.05 for WT mice versus EP3flox Nestin-Cre mice injected with casein (two-way ANOVA for the period of 4–10 h: F(3,34) = 5.545, p = 0.0033). E, Normal febrile response in mice with deletion of the EP3 receptor in neural crest derivates. Solid lines represent mean, and dotted lines represent SEM. F, qPCR Analysis of the amount of remaining EP3 receptor mRNA in the CNS and DRG after deletion in EP3 floxed mice with Nestin-Cre and HTPA-Cre, respectively. Error bars show SEM. G, No effect of nerve blockade by injection of a local anesthetic (ropivacaine, Narop) into the air pouch (ipo), immediately before the injection of casein into the pouch. To account for any systemic effects of the local anesthetic, the same dose was also injected subcutaneously (sc), on the belly. Solid lines represent mean.
To evaluate the efficacy of the EP3 gene deletion, we compared the EP3 mRNA expression in whole brain and dorsal root ganglia in the EP3 floxed mice expressing Nestin-Cre or HTPA-Cre with that found in littermates not expressing Cre (WT), using qPCR. In offspring expressing Nestin-Cre the amount of EP3 receptor mRNA was ∼10% of that seen in WT littermates both in the CNS and in dorsal root ganglia. In contrast, in offspring expressing HTPA, ∼60% of the EP3 mRNA remained in the CNS but only ∼1% in the dorsal root ganglia (Fig. 3F). Hence, although the absence of fever in the Nestin-Cre EP3 mice could be consistent with EP3 deletion either in the CNS or in afferent peripheral somatic nerves, the presence of fever in the HTPA-Cre EP3 mice cannot be ascribed to the presence of EP3 receptors on peripheral nerves, demonstrating that the EP3 receptors in the CNS are critical.
Casein injection into an air pouch results in high concentrations of cytokines in the pouch but only low levels in the circulation
Although fever in the air pouch model was associated with high levels of PGE2 in the air pouch, as well as elevated PGE2 levels in plasma and cerebrospinal fluid (Fig. 1F–H), and deletion of PGE2 synthesis attenuated fever (Fig. 2A), it was not clear if all PGE2 observed derived from the air pouch or whether PGE2 also was synthesized at other sites and that such synthesis elicited the fever. Because systemic administration of pyrogenic inflammatory mediators, such as lipopolysaccharide, has been shown to result in high levels of circulating proinflammatory cytokines, such as IL-1β, IL-6 and TNF (Elander et al., 2009; Matsuwaki et al., 2017), which are critical for PGE2 production and fever (Leon, 2002; Eskilsson et al., 2014; Matsuwaki et al., 2017), we examined the production of these cytokines in the air pouch and the extent to which they were also found in the circulation. We found that casein injection into the air pouch resulted in high levels in the pouch of IL-1β, but there were also elevated levels in plasma (Fig. 4A), however, at a concentration several magnitudes lower than that seen after immune challenge with LPS (Elander et al., 2009; Matsuwaki et al., 2017). Casein injection resulted in high concentrations in the air pouch also of IL-6 (Fig. 4B) and TNF (Fig. 4C). IL-6 was also elevated in plasma (Fig. 4B), present in a concentration about one order of magnitude lower than that seen after intraperitoneal injection of LPS (Elander et al., 2009; Matsuwaki et al., 2017). TNF levels in plasma were low and did not differ between mice injected with casein and those injected with vehicle (Fig. 4C).
Figure 4.
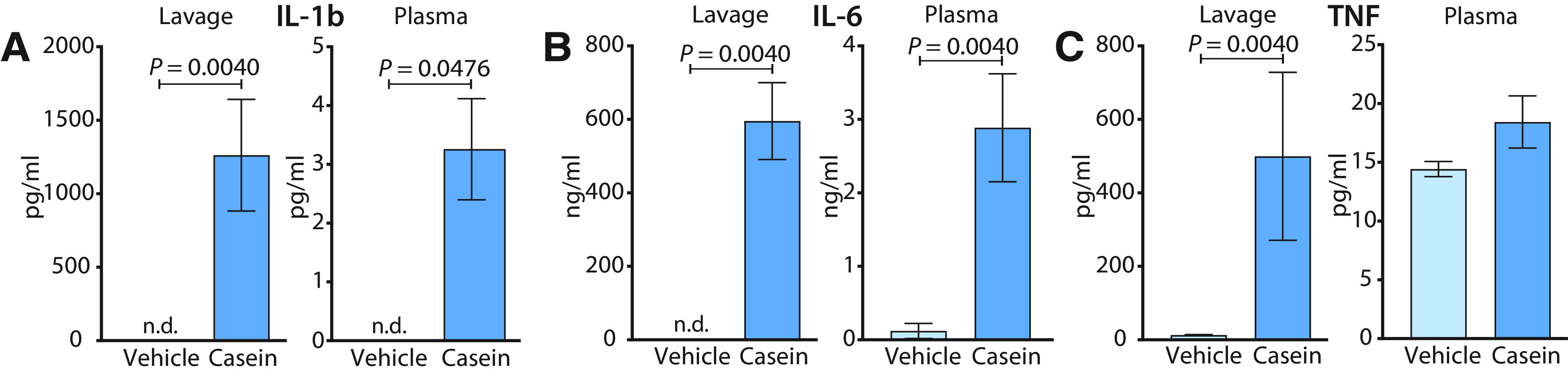
A–C, Levels of IL-1β (A), IL-6 (B), and TNF (C) in air pouch lavage and plasma 5 h after casein or vehicle injection into the air pouch; n = 3–4 in the vehicle groups and n = 6–8 in the casein groups. Error bars represent SEM. n.d., Not detectable.
Prostaglandin synthesis in brain endothelial or neural cells or in hematopoietic/myeloid cells is not critical for fever induced by casein injection
Because circulating pyrogenic cytokines (IL-1β and IL-6) were present in the circulation, although at low levels (Fig. 4A,B), and as there was induction, although weak, of Cox-2 in the brain endothelium (Fig. 1J), we next examined whether deletion of prostaglandin synthesis in brain endothelial cells would affect the casein-induced fever as has been shown for fever induced by peripheral injection of LPS and IL-1β (Wilhelms et al., 2014; Eskilsson et al., 2017). Mice with conditional deletion of Cox-2 (Ishikawa and Herschman, 2006) or mPGES-1 (Wilhelms et al., 2014) were crossed with mice expressing Cre recombinase under the promotor for the thyroxine transporter Slco1c1 (Ridder et al., 2011), which is almost exclusively expressed by brain endothelial cells (Ridder et al., 2011; Eskilsson et al., 2014; Wilhelms et al., 2014; Fritz et al., 2018). However, neither endothelial cell deletion of Cox-2 nor of mPGES-1 affected the fever elicited by casein injection into the air pouch, but mice with these deletions showed temperature responses almost identical to those of wild-type mice (Fig. 5A,B). Neither did deletion of Cox-2 in neural cells using a Nestin-Cre mouse line affect the febrile response (Fig. 5C). We also examined whether deletion of PGE2 synthesis in hematopoietic/myeloid cells would affect casein-induced fever. However, deletion of Cox-2 in cells expressing the LysM domain, by using a LysM-Cre line (Clausen et al., 1999), had no effect on the febrile response to casein injection into the air pouch (Fig. 5D). Although we did not specifically delete any putative PGE2 production by microglial cells, there was little evidence of inflammatory activation of these cells, as shown previously in this model (Zhang et al., 2008), because there was minor or no induced expression in brain tissue of the transcripts for the cytokines IL-1β and IL-6 (Fig. 5E), which are produced by the microglial cells on immune stimulation (Norden et al., 2016). In contrast, both these cytokines are strongly upregulated in models of systemic immune challenge (Nilsberth et al., 2009b).
Figure 5.

A–D, Normal fever to casein injected into a preformed air pouch in mice with tissue-specific deletion of PGE2 synthesizing enzymes. A, Endothelial deletion of Cox-2. B, Endothelial deletion of mPGES-1. C, Cox-2 deletion in neural cells. D, Cox-2 deletion in myeloid cells. Solid lines represent mean, and dotted lines represent SEM. E, Statistically significant but minor induction of IL-1β mRNA but not of IL-6 mRNA in the hypothalamus after casein injection in WT mice. Horizontal bars represent mean.
Hence, although proinflammatory cytokines were present in the circulation, albeit in low concentrations, and a weak induction of Cox-2 occurred in brain vascular cells, there was no evidence that any PGE2 production in the brain or in hematopoietic/myeloid cells was critical for the fever seen in the casein air pouch model.
Cytokine signaling in brain endothelial cells is critical for fever induced by casein injection
As IL-1β has been shown to be critical for the febrile response to localized inflammation, such as a turpentine abscess (Zheng et al., 1995; Horai et al., 1998), and as very high levels of this cytokine were found in the air pouch in the present model, we examined its role for the febrile response. We found that mice with global deletion of IL-1R1 were afebrile following casein injection into the air pouch (Fig. 6A), as reported previously (Zhang et al., 2008). The fever depending on IL-1R1 was not mediated by IL-1R1 on nerves innervating the air pouch because deletion of the IL-1R1 on neural cells or on neural crest derivates or thin sensory peripheral nerves obtained by crossing mice with conditional deletion of the IL-1R1 (Abdulaal et al., 2016) with mice expressing Cre under the Nestin, HTPA, and TRPV1 promoter, respectively, had no effect on the febrile response (Fig. 6B–D). However, when IL-1R1 was deleted in brain endothelial cells, the casein-induced fever was attenuated (Fig. 6E). A similar finding was obtained in mice with deletion of the membrane-bound IL-6Rα. Deletion of IL-6Rα on brain endothelial cells also attenuated the fever induced by casein injection into an air pouch (Fig. 7A), whereas deletion of this receptor on neural cells or sensory peripheral nerves had no effect (Fig. 7B,C).
Figure 6.

Fever response to casein injected into a preformed air pouch in mice with IL-1R1 deletion. A, Absence of fever in mice with global deletion of IL-1R1. *p < 0.05, **p < 0.01, and ***p < 0.001 for WT mice versus IL-1R1 KO mice injected with casein (two-way ANOVA for the time period of 4–10 h: F(3,22) = 27.79, p < 0.0001). B–D, No difference in the febrile response between WT mice and mice with deletion of IL-1R1 in neural cells (B), neural crest derivates (C), or thin afferent nerves (D). E, Attenuated fever in mice with deletion of IL-1R1 in brain endothelial cells. *p < 0.05, and **p < 0.01 for WT mice versus IL-1R1flox Slco1c1-Cre mice injected with casein (two-way ANOVA for the time period of 4–10 h: F(3,39) = 14.72, p < 0.0001). Solid lines represent mean, and dotted lines represent SEM.
Figure 7.

Fever response to casein injected into a preformed air pouch in mice with IL-6Rα deletion. A, Attenuated fever in mice with deletion of IL-6Rα in brain endothelial cells. *p < 0.05 for WT mice versus IL-6Rαflox Slco1c1-Cre mice injected with casein (two-way ANOVA for the time-period of 4–10 h: F(3,33) = 20.39, p < 0.0001). B, C, No difference in the febrile response between WT mice and mice with deletion of IL-6Rα in neural cells (B) or thin afferent nerves (C). Solid lines represent mean, and dotted lines represent SEM.
Discussion
Several previous studies of localized inflammation in which pyrogen was injected into a preformed air pouch or into an artificial subcutaneously implanted Teflon chamber have suggested that the febrile response seen in such models is elicited by neural signaling from the site of inflammation (Miller et al., 1997; Ross et al., 2003; Rummel et al., 2005; Zhang et al., 2008; Quan, 2014) and not by a humoral pathway as is the case for systemic inflammation (Blomqvist and Engblom, 2018). This idea has been further fostered by the observations that dorsal root ganglia express IL-1R1 and EP3 receptors and that peripheral nerves respond to IL-1β and PGE2 (Nakamura et al., 2000; Binshtok et al., 2008), present at a high concentration at the site of inflammation (Miller et al., 1997; Rummel et al., 2005; Zhang et al., 2008; present study). Furthermore, in several studies of localized inflammation, previous investigators have failed to detect any induction in the brain of Cox-2 (Rummel et al., 2005; Zhang et al., 2008; but see Rummel et al., 2006), considered to be an obligatory step in the synthesis of pyrogenic PGE2 (Ivanov and Romanovsky, 2004). Nevertheless, in the models of localized inflammation, there is almost invariably elevated levels of IL-6 in plasma (Cartmell et al., 2000; Ross et al., 2003; Rummel et al., 2005, 2006; Zhang et al., 2008), and in studies in which an IL-6 antiserum was given systemically, the febrile response was abolished (Cartmell et al., 2000; Rummel et al., 2006), suggesting the presence of a humoral route for eliciting fever also in these models (Rummel et al., 2011).
Here, we examined the signaling pathway for the febrile response to a localized inflammation, casein injection into a preformed air pouch, by using genetically modified mice. We confirmed previous data that the localized inflammation gives rise to high concentrations of PGE2 and cytokines at the site of inflammation and moderate levels of IL-6 in the circulation (Cartmell et al., 2000; Ross et al., 2003; Rummel et al., 2005; Zhang et al., 2008), but we also detected trace levels of IL-1β and TNF in plasma. We found that the PGE2 in the air pouch was generated by nonhematopoietic cells, most likely those lining the inner surface of the pouch that displayed Cox-2 immunoreactivity, but that elevated PGE2 levels were present also in plasma and cerebrospinal fluid. Whereas global deletion of the PGE2 EP3 receptor abolished fever, deletion of this receptor on neural crest derivates (and hence on peripheral nerves) had no effect. Similarly, whereas global deletion of the IL-1R1 abolished fever, its deletion in neural crest derivates as well as specifically on thin afferent nerves was without effect. These data refute the idea that binding of PGE2 or IL-1β onto receptors on peripheral nerves is responsible for the fever seen during localized inflammation. Furthermore, blocking neural signaling from the inflamed site by the injection of a local anesthetic into the air pouch did not affect the febrile response. Hence, our findings provide no evidence that neural signaling from the site of inflammation is critically involved.
Instead, the present data show that fever evoked by a localized inflammation, at least in the present experimental paradigm, is elicited by humoral signaling but by a mechanism that appears distinct from that eliciting fever during systemic inflammation, although they have some characteristics in common (Fig. 8). It is now well established that following peripheral immune challenge with, for example, intraperitoneal or intravenous injection of LPS or IL-1β, fever is elicited by the synthesis of PGE2 by brain endothelial cells in the preoptic region of the hypothalamus and that this synthesis in turn is driven by binding of IL-1β and IL-6 to their receptors on the endothelial cells and the subsequent activation of the NfκB and STAT3 signaling pathways, respectively (Ek et al., 2001; Yamagata et al., 2001; Ridder et al., 2011; Engström et al., 2012; Eskilsson et al., 2014, 2017; Wilhelms et al., 2014). Here, we demonstrate that although endothelial deletion of receptors for IL-1 and IL-6 attenuated the fever, similar to what has been shown for systemic inflammation (Eskilsson et al., 2014; Matsuwaki et al., 2017), endothelial deletion of the PGE2 synthesizing enzymes Cox-2 and mPGES-1 had no effect, which contrasts with the strong attenuation of fever seen after such deletions in models of systemic inflammation (Wilhelms et al., 2014; Eskilsson et al., 2017). Furthermore, whereas systemic immune challenge results in the release of high concentrations of proinflammatory cytokines into the circulation, blood cytokine levels were low in the present model, and although the localized inflammation elicited an induction of Cox-2 in the brain, it was feeble (and therefore likely overlooked in several previous studies). Also, in contrast to the generalized inflammatory activation of brain cytokines seen after systemic immune challenge (Nilsberth et al., 2009b), there was only a weak induced transcription of IL-1β and none of IL-6 in the brain parenchyma.
Figure 8.

Suggested pathways across the blood-brain barrier for the generation of fever during localized (left) versus systemic (right) inflammation. During localized inflammation, high concentrations of PGE2 and cytokines are generated at the inflamed site. The PGE2, and to some extent the cytokines (in particular, IL-6), leak into the circulation to reach the brain. Through a yet unknown mechanism, partly dependent on the presence of receptors for IL-1 and IL-6 on brain endothelial cells (IL1R, IL6R), PGE2 may enter the brain to reach PGE2 EP3 receptor-expressing (EP3R) thermoregulatory neurons in the median preoptic nucleus of the hypothalamus (MnPO). In contrast, systemic inflammation yields high concentrations of cytokines in the circulation. The binding of the cytokines to their receptors on brain endothelial cells results, via the STAT3 and TAK1 pathways, respectively, in induced expression of Cox-2 and mPGES-1 and the subsequent production of PGE2 in these cells and its transport into the brain parenchyma.
However, similar to what has been found for fever elicited by systemic immune challenge, fever elicited by casein injection into an air pouch depended on the formation of PGE2 and its binding to EP3 receptors in the CNS (Lazarus et al., 2007). Consistent with this finding, PGE2 levels were elevated in the cerebrospinal fluid. This PGE2 could originate from the brain and/or the circulation. Although there was only sparse induction of Cox-2 protein in the brain vessels and only minor upregulation of Cox-2 mRNA (and none of mPGES-1 mRNA) in the hypothalamus, and although endothelial deletion of Cox-2 or mPGES-1 did not affect the fever, it cannot be excluded that some cytokine-driven PGE2 production occurred in cells other than brain endothelial cells. However, this idea is inconsistent with the lack of effect by deletion of prostaglandin synthesis in neural cells and myeloid cells and the feeble microglial activation. Alternatively, the brain endothelial cells in which recombination does not occur (∼15% in the hypothalamus; Wilhelms et al., 2014) could be responsible, but this idea is difficult to reconcile with the strong attenuation of fever seen in other models of peripheral immune challenge after deletion of brain endothelial Cox-2 or mPGES-1, using the same genetically modified mouse lines (Wilhelms et al., 2014) as used here. It is also difficult to reconcile with the attenuation of the febrile response observed here when receptors for IL-1 and IL-6 were deleted in endothelial cells using the same Cre lines as used for deleting the PGE2 synthesizing enzymes. These findings lead to the second possibility that the elevated PGE2 concentrations in the brain, and the central PGE2 that elicited fever, derive from PGE2 in the circulation, which also was elevated in this model. The PGE2 in the circulation in turn is likely to be a result of leakage from the air pouch, which contained very high PGE2 levels, similar to what has been shown for IL-6 (Cartmell et al., 2000).
The role of circulating PGE2 for the generation of fever has been a matter of controversy. Circulating PGE2 is rapidly metabolized, with >90% being cleared by a single passage through the lungs (Hamberg and Samuelsson, 1971), suggesting that it would be unlikely to reach the brain in any significant amount. Nevertheless, intravenous injection of PGE2 has been shown to elicit fever (Steiner et al., 2006; Ootsuka et al., 2008). Some of the same investigators also demonstrated that lung macrophages rapidly produced PGE2 after immune challenge and that peripheral administration of neutralizing antibodies attenuated the first phase of fever (seen during the first 60 min after intravenous injection of LPS; Romanovsky et al., 1998; Rudaya et al., 2005), suggesting that this febrile phase was elicited by circulating PGE2 (Steiner et al., 2006). However, this idea has been challenged: It was shown that mice with deletion of the prostaglandin synthesizing enzyme mPGES-1 in hematopoietic cells displayed a normal first phase of fever, whereas mice that expressed mPGES-1 only in hematopoietic cells showed no fever. The latter mice nevertheless showed significantly elevated levels of PGE2 metabolites in plasma but not of PGE2 in the cerebrospinal fluid (Engström et al., 2012). In the same vein, in a rat tumor model, high levels of PGE2 were recorded in plasma, yet there was no fever and no elevated PGE2 levels in the cerebrospinal fluid (Ruud et al., 2013). However, the situation could be different when elevated PGE2 levels occur during sustained inflammatory conditions that could change the permeability of the blood-brain barrier. Cytokines such as IL-6 have been shown to increase endothelial and blood-brain barrier permeability (Paul et al., 2003; Alsaffar et al., 2018), which could include the transport of PGE2. Such a mechanism would be consistent with the attenuated fever in the present study in mice with endothelial deletion of IL-1R1 and IL-6Rα. Alternatively, the genetic interventions might have altered the impact of endogenous cryogens such as α melanocyte-stimulating hormone, nitrous oxide, vasopressin, or glucocorticoids (Kozak et al., 2000), for example, by facilitating the release or action of any of these substances. Both these putative mechanisms need further examination.
Footnotes
This work was supported by the Swedish Research Council (Grants 2020-00881 and 2018-02929), the Swedish Brain Foundation (Grant FO2019-0033), the Swedish Cancer Foundation (Grant 190304 Pj) and the Knut and Alice Wallenberg Foundation (Grant WAF 2012). We thank Dr. Ari Waisman, Dr. Werner Müller, and Dr. Emmanuel Pinteaux for providing the floxed IL-1R1 mouse, Dr. Harvey Herschman for providing the floxed Cox-2 mouse, Dr. Sylvie Dufour for providing the HTPA-Cre line, and Dr. Markus Schwaninger for providing the Slco1c1-CreERT2 line.
The authors declare no competing financial interests.
References
- Abdulaal WH, Walker CR, Costello R, Redondo-Castro E, Mufazalov IA, Papaemmanouil A, Rothwell NJ, Allan SM, Waisman A, Pinteaux E, Müller W (2016) Characterization of a conditional interleukin-1 receptor 1 mouse mutant using the Cre/LoxP system. Eur J Immunol 46:912–918. 10.1002/eji.201546075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsaffar H, Martino N, Garrett JP, Adam AP (2018) Interleukin-6 promotes a sustained loss of endothelial barrier function via Janus kinase-mediated STAT3 phosphorylation and de novo protein synthesis. Am J Physiol Cell Physiol 314:C589–C602. 10.1152/ajpcell.00235.2017 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Krieger AM, Yekutieli D (2006) Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93:491–507. 10.1093/biomet/93.3.491 [DOI] [Google Scholar]
- Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji R-R, Bean BP, Woolf CJ, Samad TA (2008) Nociceptors are interleukin-1beta sensors. J Neurosci 28:14062–14073. 10.1523/JNEUROSCI.3795-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomqvist A, Engblom D (2018) Neural mechanisms of inflammation-induced fever. Neuroscientist 24:381–399. 10.1177/1073858418760481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Matsumura K, Yamagata K, Watanabe Y (1996) Endothelial cells of the rat brain vasculature express cyclooxygenase-2 mRNA in response to systemic interleukin-1 beta: a possible site of prostaglandin synthesis responsible for fever. Brain Res 733:263–272. 10.1016/0006-8993(96)00575-6 [DOI] [PubMed] [Google Scholar]
- Cartmell T, Poole S, Turnbull AV, Rothwell NJ, Luheshi GN (2000) Circulating interleukin-6 mediates the febrile response to localised inflammation in rats. J Physiol 526:653–661. 10.1111/j.1469-7793.2000.00653.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R, O'Donnell D, Nicoll RA, Shah NM, Julius D, Basbaum AI (2011) Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J Neurosci 31:5067–5077. 10.1523/JNEUROSCI.6451-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8:265–277. 10.1023/a:1008942828960 [DOI] [PubMed] [Google Scholar]
- Edwards JC, Sedgwick AD, Willoughby DA (1981) The formation of a structure with the features of synovial lining by subcutaneous injection of air: an in vivo tissue culture system. J Pathol 134:147–156. 10.1002/path.1711340205 [DOI] [PubMed] [Google Scholar]
- Ek M, Engblom D, Saha S, Blomqvist A, Jakobsson PJ, Ericsson-Dahlstrand A (2001) Inflammatory response: pathway across the blood-brain barrier. Nature 410:430–431. 10.1038/35068632 [DOI] [PubMed] [Google Scholar]
- Elander L, Engström L, Ruud J, Mackerlova L, Jakobsson PJ, Engblom D, Nilsberth C, Blomqvist A (2009) Inducible prostaglandin E2 synthesis interacts in a temporally supplementary sequence with constitutive prostaglandin-synthesizing enzymes in creating the hypothalamic-pituitary-adrenal axis response to immune challenge. J Neurosci 29:1404–1413. 10.1523/JNEUROSCI.5247-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmquist JK, Breder CD, Sherin JE, Scammell TE, Hickey WF, Dewitt D, Saper CB (1997) Intravenous lipopolysaccharide induces cyclooxygenase 2-like immunoreactivity in rat brain perivascular microglia and meningeal macrophages. J Comp Neurol 381:119–129. [DOI] [PubMed] [Google Scholar]
- Engblom D, Saha S, Engström L, Westman M, Audoly LP, Jakobsson PJ, Blomqvist A (2003) Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat Neurosci 6:1137–1138. 10.1038/nn1137 [DOI] [PubMed] [Google Scholar]
- Engström L, Ruud J, Eskilsson A, Larsson A, Mackerlova L, Kugelberg U, Qian H, Vasilache AM, Larsson P, Engblom D, Sigvardsson M, Jönsson J-I, Blomqvist A (2012) Lipopolysaccharide-induced fever depends on prostaglandin E2 production specifically in brain endothelial cells. Endocrinology 153:4849–4861. 10.1210/en.2012-1375 [DOI] [PubMed] [Google Scholar]
- Eskilsson A, Mirrasekhian E, Dufour S, Schwaninger M, Engblom D, Blomqvist A (2014) Immune-induced fever is mediated by IL-6 receptors on brain endothelial cells coupled to STAT3-dependent induction of brain endothelial prostaglandin synthesis. J Neurosci 34:15957–15961. 10.1523/JNEUROSCI.3520-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskilsson A, Matsuwaki T, Shionoya K, Mirrasekhian E, Zajdel J, Schwaninger M, Engblom D, Blomqvist A (2017) Immune-induced fever is dependent on local but not generalized prostaglandin E2 synthesis in the brain. J Neurosci 37:5035–5044. 10.1523/JNEUROSCI.3846-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming EF, Athirakul K, Oliverio MI, Key M, Goulet J, Koller BH, Coffman TM (1998) Urinary concentrating function in mice lacking EP3 receptors for prostaglandin E2. Am J Physiol 275:F955–F961. 10.1152/ajprenal.1998.275.6.F955 [DOI] [PubMed] [Google Scholar]
- Flower RJ, Vane JR (1972) Inhibition of prostaglandin synthetase in brain explains the anti- pyretic activity of paracetamol (4-acetamidophenol). Nature 240:410–411. 10.1038/240410a0 [DOI] [PubMed] [Google Scholar]
- Flower RJ, Gryglewski R, Herbaczyńska-Cedro K, Vane JR (1972) Effects of anti-inflammatory drugs on prostaglandin biosynthesis. Nat New Biol 238:104–106. 10.1038/newbio238104a0 [DOI] [PubMed] [Google Scholar]
- Fritz M, Klawonn AM, Nilsson A, Singh AK, Zajdel J, Wilhelms DB, Lazarus M, Löfberg A, Jaarola M, Kugelberg UÖ, Billiar TR, Hackam DJ, Sodhi CP, Breyer MD, Jakobsson J, Schwaninger M, Schütz G, Parkitna JR, Saper CB, Blomqvist A, et al. (2016) Prostaglandin-dependent modulation of dopaminergic neurotransmission elicits inflammation-induced aversion in mice. J Clin Invest 126:695–705. 10.1172/JCI83844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz M, Klawonn AM, Jaarola M, Engblom D (2018) Interferon-ɣ mediated signaling in the brain endothelium is critical for inflammation-induced aversion. Brain Behav Immun 67:54–58. 10.1016/j.bbi.2017.08.020 [DOI] [PubMed] [Google Scholar]
- Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, Livingston DJ, Peschon JJ, Morrissey PJ (1997) Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol 159:3364–3371. [PubMed] [Google Scholar]
- Hamberg M, Samuelsson B (1971) On the metabolism of prostaglandins E 1 and E 2 in man. J Biol Chem 246:6713–6721. 10.1016/S0021-9258(19)45905-X [DOI] [PubMed] [Google Scholar]
- Hansen MK, Krueger JM (1997) Subdiaphragmatic vagotomy blocks the sleep- and fever-promoting effects of interleukin-1beta. Am J Physiol 273:R1246–R1253. 10.1152/ajpregu.1997.273.4.R1246 [DOI] [PubMed] [Google Scholar]
- Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, Takahashi M, Iwakura Y (1998) Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med 187:1463–1475. 10.1084/jem.187.9.1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T-O, Herschman HR (2006) Conditional knockout mouse for tissue-specific disruption of the cyclooxygenase-2 (Cox-2) gene. Genesis 44:143–149. 10.1002/gene.20192 [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Romanovsky AA (2004) Prostaglandin E2 as a mediator of fever: synthesis and catabolism. Front Biosci 9:1977–1993. 10.2741/1383 [DOI] [PubMed] [Google Scholar]
- Jakobsson PJ, Thorén S, Morgenstern R, Samuelsson B (1999) Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A 96:7220–7225. 10.1073/pnas.96.13.7220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanashiro A, Pessini AC, Machado RR, Malvar DC, Aguiar FA, Soares DM, do Vale ML, de Souza GE (2009) Characterization and pharmacological evaluation of febrile response on zymosan-induced arthritis in rats. Am J Physiol Regul Integr Comp Physiol 296:R1631–R1640. 10.1152/ajpregu.90527.2008 [DOI] [PubMed] [Google Scholar]
- Kitamura M, Goto F, Ohkawara S, Yoshinaga M (1986) Production of pyrogen by polymorphonuclear leukocytes during the course of casein-induced peritonitis in rabbits. Acta Pathol Jpn 36:791–803. 10.1111/j.1440-1827.1986.tb03114.x [DOI] [PubMed] [Google Scholar]
- Kozak W, Kluger MJ, Tesfaigzi J, Kozak A, Mayfield KP, Wachulec M, Dokladny K (2000) Molecular mechanisms of fever and endogenous antipyresis. Ann N Y Acad Sci 917:121–134. 10.1111/j.1749-6632.2000.tb05376.x [DOI] [PubMed] [Google Scholar]
- Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR (1991) TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem 266:12866–12872. [PubMed] [Google Scholar]
- Lazarus M, Yoshida K, Coppari R, Bass CE, Mochizuki T, Lowell BB, Saper CB (2007) EP3 prostaglandin receptors in the median preoptic nucleus are critical for fever responses. Nat Neurosci 10:1131–1133. 10.1038/nn1949 [DOI] [PubMed] [Google Scholar]
- Leon LR (2002) Invited review: cytokine regulation of fever: studies using gene knockout mice. J Appl Physiol 92:2648–2655. 10.1152/japplphysiol.01005.2001 [DOI] [PubMed] [Google Scholar]
- Matsuwaki T, Shionoya K, Ihnatko R, Eskilsson A, Kakuta S, Dufour S, Schwaninger M, Waisman A, Müller W, Pinteaux E, Engblom D, Blomqvist A (2017) Involvement of interleukin-1 type 1 receptors in lipopolysaccharide-induced sickness responses. Brain Behav Immun 66:165–176. 10.1016/j.bbi.2017.06.013 [DOI] [PubMed] [Google Scholar]
- McFarland-Mancini MM, Funk HM, Paluch AM, Zhou M, Giridhar PV, Mercer CA, Kozma SC, Drew AF (2010) Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J Immunol 184:7219–7228. 10.4049/jimmunol.0901929 [DOI] [PubMed] [Google Scholar]
- Miller AJ, Luheshi GN, Rothwell NJ, Hopkins SJ (1997) Local cytokine induction by LPS in the rat air pouch and its relationship to the febrile response. Am J Physiol 272:R857–R861. 10.1152/ajpregu.1997.272.3.R857 [DOI] [PubMed] [Google Scholar]
- Moissidis I, Chaidaroon D, Vichyanond P, Bahna SL (2005) Milk-induced pulmonary disease in infants (Heiner syndrome). Pediatr Allergy Immunol 16:545–552. 10.1111/j.1399-3038.2005.00291.x [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kaneko T, Yamashita Y, Hasegawa H, Katoh H, Negishi M (2000) Immunohistochemical localization of prostaglandin EP3 receptor in the rat nervous system. J Comp Neurol 421:543–569. [DOI] [PubMed] [Google Scholar]
- Nilsberth C, Elander L, Hamzic N, Norell M, Lönn J, Engström L, Blomqvist A (2009a) The role of interleukin-6 in lipopolysaccharide-induced fever by mechanisms independent of prostaglandin E2. Endocrinology 150:1850–1860. 10.1210/en.2008-0806 [DOI] [PubMed] [Google Scholar]
- Nilsberth C, Hamzic N, Norell M, Blomqvist A (2009b) Peripheral lipopolysaccharide administration induces cytokine mRNA expression in the viscera and brain of fever-refractory mice lacking microsomal prostaglandin E synthase-1. J Neuroendocrinol 21:715–721. 10.1111/j.1365-2826.2009.01888.x [DOI] [PubMed] [Google Scholar]
- Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP (2016) Sequential activation of microglia and astrocyte cytokine expression precedes increased iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 64:300–316. 10.1002/glia.22930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ootsuka Y, Blessing WW, Steiner AA, Romanovsky AA (2008) Fever response to intravenous prostaglandin E2 is mediated by the brain but does not require afferent vagal signaling. Am J Physiol Regul Integr Comp Physiol 294:R1294–R1303. 10.1152/ajpregu.00709.2007 [DOI] [PubMed] [Google Scholar]
- Paul R, Koedel U, Winkler F, Kieseier BC, Fontana A, Kopf M, Hartung HP, Pfister HW (2003) Lack of IL‐6 augments inflammatory response but decreases vascular permeability in bacterial meningitis. Brain 126:1873–1882. 10.1093/brain/awg171 [DOI] [PubMed] [Google Scholar]
- Pietri T, Eder O, Blanche M, Thiery JP, Dufour S (2003) The human tissue plasminogen activator-Cre mouse: a new tool for targeting specifically neural crest cells and their derivatives in vivo. Dev Biol 259:176–187. 10.1016/s0012-1606(03)00175-1 [DOI] [PubMed] [Google Scholar]
- Quan N (2014) In-depth conversation: spectrum and kinetics of neuroimmune afferent pathways. Brain Behav Immun 40:1–8. 10.1016/j.bbi.2014.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes TM, Walker JR, DeCino C, Hogenesch JB, Sawchenko PE (2003) Categorically distinct acute stressors elicit dissimilar transcriptional profiles in the paraventricular nucleus of the hypothalamus. J Neurosci 23:5607–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridder DA, Lang M-F, Salinin S, Röderer J-P, Struss M, Maser-Gluth C, Schwaninger M (2011) TAK1 in brain endothelial cells mediates fever and lethargy. J Exp Med 208:2615–2623. 10.1084/jem.20110398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanovsky AA, Kulchitsky VA, Simons CT, Sugimoto N (1998) Methodology of fever research: why are polyphasic fevers often thought to be biphasic? Am J Physiol 275:R332–R338. 10.1152/ajpregu.1998.275.1.R332 [DOI] [PubMed] [Google Scholar]
- Ross G, Roth J, Störr B, Voigt K, Zeisberger E (2000) Afferent nerves are involved in the febrile response to injection of LPS into artificial subcutaneous chambers in guinea pigs. Physiol Behav 71:305–313. 10.1016/s0031-9384(00)00358-9 [DOI] [PubMed] [Google Scholar]
- Ross G, Hübschle T, Pehl U, Braun HA, Voigt K, Gerstberger R, Roth J (2003) Fever induction by localized subcutaneous inflammation in guinea pigs: the role of cytokines and prostaglandins. J Appl Physiol 94:1395–1402. 10.1152/japplphysiol.00485.2002 [DOI] [PubMed] [Google Scholar]
- Rudaya AY, Steiner AA, Robbins JR, Dragic AS, Romanovsky AA (2005) Thermoregulatory responses to lipopolysaccharide in the mouse: dependence on the dose and ambient temperature. Am J Physiol Regul Integr Comp Physiol 289:R1244–R1252. 10.1152/ajpregu.00370.2005 [DOI] [PubMed] [Google Scholar]
- Rummel C, Barth SW, Voss T, Korte S, Gerstberger R, Hübschle T, Roth J (2005) Localized vs. systemic inflammation in guinea pigs: a role for prostaglandins at distinct points of the fever induction pathways? Am J Physiol Regul Integr Comp Physiol 289:R340–R347. 10.1152/ajpregu.00104.2005 [DOI] [PubMed] [Google Scholar]
- Rummel C, Sachot C, Poole S, Luheshi GN (2006) Circulating interleukin-6 induces fever through a STAT3-linked activation of COX-2 in the brain. Am J Physiol Regul Integr Comp Physiol 291:R1316–R1326. 10.1152/ajpregu.00301.2006 [DOI] [PubMed] [Google Scholar]
- Rummel C, Matsumura K, Luheshi GN (2011) Circulating IL-6 contributes to peripheral LPS-induced mPGES-1 expression in the rat brain. Brain Res Bull 86:319–325. 10.1016/j.brainresbull.2011.09.006 [DOI] [PubMed] [Google Scholar]
- Ruud J, Nilsson A, Engström Ruud L, Wang W, Nilsberth C, Iresjö BM, Lundholm K, Engblom D, Blomqvist A (2013) Cancer-induced anorexia in tumor-bearing mice is dependent on cyclooxygenase-1. Brain Behav Immun 29:124–135. 10.1016/j.bbi.2012.12.020 [DOI] [PubMed] [Google Scholar]
- Sedgwick AD, Sin YM, Edwards JC, Willoughby DA (1983) Increased inflammatory reactivity in newly formed lining tissue. J Pathol 141:483–495. 10.1002/path.1711410406 [DOI] [PubMed] [Google Scholar]
- Sehic E, Blatteis CM (1996) Blockade of lipopolysaccharide-induced fever by subdiaphragmatic vagotomy in guinea pigs. Brain Res 726:160–166. [PubMed] [Google Scholar]
- Sirois J, Simmons DL, Richards JS (1992) Hormonal regulation of messenger ribonucleic acid encoding a novel isoform of prostaglandin endoperoxide H synthase in rat preovulatory follicles. Induction in vivo and in vitro. J Biol Chem 267:11586–11592. [PubMed] [Google Scholar]
- Spencer NJ, Magnúsdóttir EI, Jakobsson JET, Kestell G, Chen BN, Morris D, Brookes SJ, Lagerström MC (2018) CGRPα within the Trpv1-Cre population contributes to visceral nociception. Am J Physiol Gastrointest Liver Physiol 314:G188–G200. 10.1152/ajpgi.00188.2017 [DOI] [PubMed] [Google Scholar]
- Steiner AA, Ivanov AI, Serrats J, Hosokawa H, Phayre AN, Robbins JR, Roberts JL, Kobayashi S, Matsumura K, Sawchenko PE, Romanovsky AA (2006) Cellular and molecular bases of the initiation of fever. PLoS Biol 4:e284. 10.1371/journal.pbio.0040284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt JT (1986) Prostaglandin E as the neural mediator of the febrile response. Yale J Biol Med 59:137–149. [PMC free article] [PubMed] [Google Scholar]
- Stock JL, Shinjo K, Burkhardt J, Roach M, Taniguchi K, Ishikawa T, Kim H-S, Flannery PJ, Coffman TM, McNeish JD, Audoly LP (2001) The prostaglandin E2 EP1 receptor mediates pain perception and regulates blood pressure. J Clin Invest 107:325–331. 10.1172/JCI6749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley SL, Audoly LP, Hicks EH, Kim H-S, Flannery PJ, Coffman TM, Koller BH (1999) Reproductive failure and reduced blood pressure in mice lacking the EP2 prostaglandin E2 receptor. J Clin Invest 103:1539–1545. 10.1172/JCI6579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP (2003) Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci U S A 100:9044–9049. 10.1073/pnas.1332766100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23:99–103. 10.1038/12703 [DOI] [PubMed] [Google Scholar]
- Ushikubi F, Segi E, Sugimoto Y, Murata T, Matsuoka T, Kobayashi T, Hizaki H, Tuboi K, Katsuyama M, Ichikawa A, Tanaka T, Yoshida N, Narumiya S (1998) Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature 395:281–284. 10.1038/26233 [DOI] [PubMed] [Google Scholar]
- van den Anker JN (2013) Optimising the management of fever and pain in children. Int J Clin Pract Suppl 67:26–32. 10.1111/ijcp.12056 [DOI] [PubMed] [Google Scholar]
- Vane JR (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231:232–235. 10.1038/newbio231232a0 [DOI] [PubMed] [Google Scholar]
- Watkins LR, Goehler LE, Relton JK, Tartaglia N, Silbert L, Martin D, Maier SF (1995) Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: evidence for vagal mediation of immune-brain communication. Neurosci Lett 183:27–31. 10.1016/0304-3940(94)11105-r [DOI] [PubMed] [Google Scholar]
- Wilhelms DB, Kirilov M, Mirrasekhian E, Eskilsson A, Kugelberg UÖ, Klar C, Ridder D, Herschman HR, Schwaninger M, Blomqvist A, Engblom D (2014) Deletion of prostaglandin E2 synthesizing enzymes in brain endothelial cells attenuates inflammatory fever. J Neurosci 34:11684–11690. 10.1523/JNEUROSCI.1838-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Matsumura K, Inoue W, Shiraki T, Suzuki K, Yasuda S, Sugiura H, Cao C, Watanabe Y, Kobayashi S (2001) Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin- induced fever. J Neurosci 21:2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ching S, Chen Q, Li Q, An Y, Quan N (2008) Localized inflammation in peripheral tissue signals the CNS for sickness response in the absence of interleukin-1 and cyclooxygenase-2 in the blood and brain. Neuroscience 157:895–907. 10.1016/j.neuroscience.2008.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann KJ, Conn CA, Soszynski D, Grabiec C, Trumbauer ME, Shaw A, Kostura MJ, Stevens K, Rosen H, North RJ, Chen HY, Tocci MJ, Kluger MJ, Van der Ploeg LHT (1995) Resistance to fever induction and impaired acute-phase response in interleukin-1 beta-deficient mice. Immunity 3:9–19. 10.1016/1074-7613(95)90154-x [DOI] [PubMed] [Google Scholar]