

Abstract
Background
This is an updated version of the original Cochrane Review published in 2008 and updated in 2013.
Epilepsy is a common neurological condition which affects up to 1% of the population. Approximately 30% of people with epilepsy do not respond to treatment with currently available drugs. The majority of these people have focal epilepsy. Vigabatrin is an antiepileptic drug licensed for use in drug‐resistant epilepsy.
Objectives
To assess the efficacy and tolerability of vigabatrin as an add‐on therapy for people with drug‐resistant focal epilepsy.
Search methods
For the latest update of this review, we searched the following databases on 1 November 2018: Cochrane Register of Studies (CRS Web), MEDLINE (Ovid 1946 to 31 October 2018), ClinicalTrials.gov and the World Health Organization International Clinical Trials Registry Platform. The Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL) are both included in the Cochrane Register of Studies (CRS Web). We checked reference lists of retrieved studies for additional reports of relevant studies and contacted Hoechst Marion Roussel (manufacturers of vigabatrin) in 2000.
Selection criteria
We included randomised, double‐blind, placebo‐controlled, fully published trials of vigabatrin in people of any age with drug‐resistant focal epilepsy.
Data collection and analysis
Two review authors assessed trials for inclusion and extracted data using the standard methodological procedures expected by Cochrane. Primary analysis was by intention‐to‐treat (ITT). We evaluated: 50% or greater reduction in seizure frequency, treatment withdrawal, adverse effects, dose‐response analysis, cognitive outcomes and quality of life. We presented results as risk ratios (RR) with 95% or 99% confidence intervals (CI).
Main results
We identified 11 trials that included 756 participants (age range: 10 to 64 years). The trials tested vigabatrin doses between 1 g/day and 6 g/day. All 11 trials displayed a risk of bias across at least three risk of bias domains. Predominantly, the risk of bias was associated with: allocation concealment (selection bias), blinding of outcome assessment (detection bias) and incomplete outcome data (attrition bias).
Participants treated with vigabatrin may be two to three times more likely to obtain a 50% or greater reduction in seizure frequency compared with those treated with placebo (RR 2.60, 95% CI 1.87 to 3.63; 4 studies; low‐certainty evidence). Those treated with vigabatrin may also be three times more likely to have treatment withdrawn although we are uncertain (RR 2.86, 95% CI 1.25 to 6.55; 4 studies; very low‐certainty evidence).
Compared to placebo, participants given vigabatrin were more likely to experience adverse effects: dizziness/light‐headedness (RR 1.74, 95% CI 1.05 to 2.87; 9 studies; low‐certainty evidence), fatigue (RR 1.65, 95% CI 1.08 to 2.51; 9 studies; low‐certainty evidence), drowsiness (RR 1.70, 95% CI 1.18 to 2.44; 8 studies) and depression (RR 3.28, 95% CI 1.30 to 8.27; 6 studies).
Although the incidence rates were higher among participants receiving vigabatrin compared to those receiving placebo, the effect was not significant for the following adverse effects: ataxia (RR 2.76, 95% CI 0.96 to 7.94; 7 studies; very low‐certainty evidence), nausea (RR 3.57, 95% CI 0.63 to 20.30; 4 studies), abnormal vision (RR 1.64, 95% CI 0.67 to 4.02; 5 studies; very low‐certainty evidence), headache (RR 1.23, 95% CI 0.79 to 1.92; 9 studies), diplopia (RR 1.76, 99% CI 0.94 to 3.30) and nystagmus (RR 1.53, 99% CI 0.62 to 3.76; 2 studies; low‐certainty evidence).
Vigabatrin had little to no effect on cognitive outcomes or quality of life.
Authors' conclusions
Vigabatrin may significantly reduce seizure frequency in people with drug‐resistant focal epilepsy. The results largely apply to adults and should not be extrapolated to children under 10 years old. Short‐term follow‐up of participants showed that some adverse effects were associated with its use. Analysis of longer‐term observational studies elsewhere, however, has demonstrated that vigabatrin use can lead to the development of visual field defects.
Plain language summary
Vigabatrin add‐on for drug‐resistant focal epilepsy
Background
Epilepsy is a common neurological disorder affecting approximately 1 in 100 people. Around 30% of these people cannot control their epilepsy with currently available antiepileptic medication and are said to have drug‐resistant epilepsy. Most often, drug‐resistant epilepsy is focal in nature, meaning that the seizures start in one specific area of the brain. Vigabatrin is an antiepileptic medication that can be used as an add‐on treatment for drug‐resistant focal epilepsy, meaning that it is taken in addition to other antiepileptic medications.
Main results
We found 11 clinical trials, involving 756 people, which investigated vigabatrin as an add‐on for people with drug‐resistant focal epilepsy. The people in the trials were aged between 10 to 64 years and were given doses of 1 g/day to 6 g/day vigabatrin.
We found that people given vigabatrin may be two to three times more likely to experience a 50% or greater reduction in seizure frequency than people given placebo (a non‐active treatment). We also suggest that people given vigabatrin may be up to three times more likely to stop treatment than people given placebo. People given vigabatrin were more likely to experience side effects: dizziness/light‐headedness, fatigue, drowsiness and depression, than people given placebo. However, the evidence suggested that they should not be more likely to experience: ataxia (disorders that affect co‐ordination, balance and speech), feeling sick (nausea), abnormal vision, headache, seeing double (diplopia), or involuntary movement of the eyes (nystagmus) than people given placebo.
Reliability of the evidence
We judged that all studies had significant risk of bias. The studies did not explain how people were allocated to treatment groups and did not say whether investigators knew which treatment people were receiving. Alongside other reasons, this reduced our confidence (certainty) in the results. Overall, we are uncertain to very uncertain whether the results we have reported are reliable. The true effect of vigabatrin could be significantly different to that reported here.
Conclusions
Vigabatrin may significantly reduce seizure frequency for people with drug‐resistant focal epilepsy but we are uncertain. Most of the evidence was gathered from adults, therefore, the effects of vigabatrin could be different in children. All of the included trials were of short duration; therefore, we cannot report any long‐term effects of vigabatrin. Importantly, reviews of long‐term studies have reported that long‐term vigabatrin use can lead to the development of visual problems.
Summary of findings
Summary of findings 1. Vigabatrin compared to placebo for drug‐resistant focal epilepsy.
| Vigabatrin compared to placebo for drug‐resistant focal epilepsy | ||||||
|
Patient or population: people (aged 10–64 years) with drug‐resistant focal epilepsy Setting: outpatients Intervention: vigabatrin (doses used: 2 g/day, 3 g/day, 1–4 g/day, 6 g/day) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI, 99% CI for adverse effect outcomes) | Relative effect (95% CI, 99% CI for adverse effect outcomes) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with vigabatrin | |||||
| ≥ 50% reduction in seizure frequency (ITT analysis) Duration of treatment (range): 16–36 weeks |
Study population | RR 2.60 (1.87 to 3.63) | 513 (4 RCTs) | ⊕⊕⊝⊝ Lowa,b | The other 7 studies did report the outcome; however, the data were not usable. The studies compared seizure frequency with vigabatrin to the placebo treatment period rather than the baseline period, as per our definition. | |
| 180 per 1000 | 468 per 1000 (337 to 654) | |||||
|
Treatment withdrawal for any reason Duration of treatment (range): 12 (3 months) to 36 weeks |
Study population | RR 2.86 (1.25 to 6.55) | 370 (4 RCTs) | ⊕⊝⊝⊝ Very lowa,c | — | |
| 30 per 1000 | 87 per 1000 (38 to 199) | |||||
|
Adverse effects: ataxia Duration of treatment (range): 7–36 weeks |
Study population | RR 2.76 (0.96 to 7.94) | 481 (7 RCTs) | ⊕⊝⊝⊝ Very lowa,c | — | |
| 14 per 1000 | 38 per 1000 (13 to 108) | |||||
|
Adverse effects: dizziness/light‐headedness Duration of treatment (range): 7–36 weeks |
Study population | RR 1.74 (1.05 to 2.87) | 709 (9 RCTs) | ⊕⊕⊝⊝ Lowa,b | — | |
| 84 per 1000 | 145 per 1000 (88 to 240) | |||||
|
Adverse effects: fatigue Duration of treatment (range): 8–36 weeks |
Study population | RR 1.65 (1.08 to 2.51) | 709 (9 RCTs) | ⊕⊕⊝⊝ Lowa,b | — | |
| 113 per 1000 | 186 per 1000 (122 to 284) | |||||
|
Adverse effects: nystagmus Duration of treatment (range): 16–18 weeks |
Study population | RR 1.53 (0.62 to 3.76) | 357 (2 RCTs) | ⊕⊕⊝⊝ Lowa,b | — | |
| 89 per 1000 | 136 per 1000 (55 to 334) | |||||
|
Adverse effects: abnormal vision Duration of treatment (range): 8–36 weeks |
Study population | RR 1.64 (0.67 to 4.02) | 430 (5 RCTs) | ⊕⊝⊝⊝ Very lowa,c | — | |
| 45 per 1000 | 74 per 1000 (30 to 182) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; ITT: intention‐to‐treat; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for risk of bias. We judged all included studies at unclear risk of bias for allocation concealment and blinding of outcome assessment. Furthermore, we assessed most studies at unclear risk of selection bias and attrition bias with three studies displaying high risk of selection bias. bDowngraded one level for imprecision: the number of events (fewer than 400) did not satisfy the optimal information size. cDowngraded two levels for imprecision: the number of events (fewer than 100) did not satisfy the optimal information size.
Background
Description of the condition
Epilepsy is a common neurological condition with an estimated incidence of 61.4 per 100,000 person‐years and a global lifetime‐prevalence of 7.6 per 1000 (Fiest 2017). While the majority of people who are diagnosed with epilepsy will go into remission, up to 30% will develop drug‐resistant epilepsy despite treatment with combinations of antiepileptic drugs (AEDs) (Cockerell 1995). The majority of people who are resistant to drug treatment have localised related seizures, commonly known as focal seizures. These are divided into the three types: focal aware, focal impaired‐awareness and secondarily generalised tonic‐clonic seizures (Fisher 2017).
Description of the intervention
Since 1993, numerous second‐generation AEDs have been licensed for use as add‐on therapy in drug‐resistant epilepsy (Chung 2008). One of the earliest second‐generation drugs to be approved for use was vigabatrin (vinyl‐GABA or VGB). Vigabatrin was initially licensed in 1989 as add‐on therapy for adults with drug‐resistant focal epilepsy and was considered an efficacious treatment (Marson 1996). In 1997, concerns were first raised about visual field constrictions observed in people taking vigabatrin (Eke 1997). Since these initial concerns were raised, numerous observational studies have investigated the prevalence of visual field constrictions in people taking vigabatrin. In one systematic review of 1678 people taking vigabatrin, 44% had visual field loss compared to just 7% of the 406 people taking a control (Maguire 2010). One large longitudinal study of 563 people, of whom 432 were tested, found 31% of those aged eight to 12 years and 42% of those older than 12 years had a visual field defect; risks increased with exposure duration and increased mean dose (Wild 2007).
How the intervention might work
Vigabatrin is a structural analogue of gamma‐aminobutyric acid (GABA), which irreversibly inhibits GABA‐transaminase (Grant 1991), and increases brain levels of the inhibitory transmitter GABA (Schechter 1984).
Why it is important to do this review
Vigabatrin is also used in the treatment of infantile spasms (West syndrome), particularly when associated with tuberous sclerosis (Hancock 2008; Wheless 2005). However, as this review focuses on the use of vigabatrin in drug‐resistant focal epilepsy, neither its efficacy nor tolerability is investigated here for the treatment of infantile spasms, tuberous sclerosis or West syndrome.
Objectives
To assess the efficacy and tolerability of vigabatrin as an add‐on therapy for people with drug‐resistant focal epilepsy.
Methods
Criteria for considering studies for this review
Types of studies
Studies had to meet all of the following criteria:
randomised controlled trial (RCT);
double‐blinded;
placebo‐controlled;
parallel‐group or cross‐over design.
Types of participants
People of any age with drug‐resistant focal epilepsy (that is, experiencing focal aware, focal impaired‐awareness or secondary generalised tonic‐clonic seizures). We did not consider the treatment of people with infantile spasms, tuberous sclerosis or West syndrome in this review.
Types of interventions
Active treatment group: vigabatrin in addition to an existing AED regimen being taken at the time of randomisation.
Control group: matched placebo in addition to an existing AED regimen being taken at the time of randomisation.
Types of outcome measures
Primary outcome
50% or greater reduction in seizure frequency: we chose the proportion of participants with a 50% or greater reduction in seizure frequency as the measure of treatment efficacy. The seizure frequency during the treatment period was compared to the prerandomisation baseline period. This outcome was chosen as it is commonly reported in this type of study.
Secondary outcomes
Treatment withdrawal for any reason: we chose the proportion of participants having treatment withdrawn during the course of the treatment period as a measure of global effectiveness. Treatment may be withdrawn due to adverse effects, lack of efficacy, a combination of both, or sometimes due to other reasons unrelated to treatment. However, in studies of relatively short duration, such as the studies included here, adverse effects are likely to be the main reason for treatment withdrawal.
-
Adverse effects:
-
the proportion of participants experiencing any of the following five adverse effects that are considered by the review authors to be common and important adverse effects of AEDs:
ataxia (co‐ordination problems);
dizziness or light headedness;
fatigue or drowsiness;
nausea;
depression;
-
vigabatrin has a known association with visual field defects. In this review of short‐term RCTs, it was not possible to identify associated visual field constrictions as they are often asymptomatic. The association with long‐term visual field constrictions is the subject of another systematic review (Maguire 2010). Therefore, we instead assessed the proportion of participants experiencing the following visual adverse effects which could be detected within the time course of the studies:
abnormal vision;
diplopia (double vision);
nystagmus (involuntary eye movement).
-
Dose‐response analysis: we evaluated the proportion of participants experiencing a 50% or greater reduction in seizure frequency by dose of the drug.
Cognitive outcomes and quality of life: there is no consensus on which instruments should be used to assess the effects of AEDs on cognition and quality of life outcomes (Cochrane 1998). Due to this, we provided a simple descriptive summary of findings of the effects of vigabatrin on cognitive and quality of life outcomes. We did not synthesise results in a meta‐analysis.
Search methods for identification of studies
Electronic searches
This search was run for the original review in March 2008 and subsequent searches on 25 October 2010, 12 October 2012 and 4 February 2014. For the most recent update of this review, we searched the following databases on 1 November 2018:
Cochrane Register of Studies (CRS Web) using the search strategy set out in Appendix 1;
MEDLINE (Ovid, 1946 to 31 October 2018) using the search strategy set out in Appendix 2;
ClinicalTrials.gov (clinicaltrials.gov/) using the search strategy set out in Appendix 3;
World Health Organization (WHO) International Clinical Trials Registry Platform (apps.who.int/trialsearch/) using the search strategy set out in Appendix 4.
The Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL) are included in CRS Web.
The search strategies used for the earlier version of this review are set out in Appendix 5.
Searching other resources
We checked reference lists of retrieved studies for additional reports of relevant studies.
We contacted the manufacturers of vigabatrin, Hoechst Marion Roussel, in 2000 requesting information about any unpublished or ongoing studies. The company has since rebranded as Sanofi. We contacted Sanofi, again requesting information regarding any unpublished or ongoing studies, during the most recent literature search in 2018.
We included only fully published and peer‐reviewed studies. We imposed no language restrictions.
Data collection and analysis
Selection of studies
Two review authors (RB and MG) independently assessed trials for inclusion and extracted information from the trials. Any disagreements were resolved by discussion.
Data extraction and management
We extracted the following information from the included trials.
-
Methodological and trial design
Method of randomisation concealment (e.g. allocation of sequentially sealed packages of medication, sealed opaque envelopes, telephone randomisation).
Method of blinding.
Whether any participants had been excluded from the reported analyses.
Duration of baseline period.
Duration of treatment period.
Duration of any washout period.
Dose(s) of vigabatrin tested.
-
Participant, demographic information
Total number of participants allocated to each treatment group.
Age range and number of each sex.
Types of epilepsy or seizure types.
Number of concomitant AEDs.
-
Outcomes
We recorded the number of participants experiencing each outcome (see Types of outcome measures) per randomised group.
Assessment of risk of bias in included studies
Two review authors (RB and MG) independently assessed risk of bias for each trial using the Cochrane 'Risk of bias' tool as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We discussed and resolved any disagreements. We judged the studies at high, low or unclear risk of bias across six chosen domains: random sequence generation, allocation concealment, blinding of participants and study personnel, blinding of outcome assessment, incomplete outcome data, selective outcome reporting and other sources of bias.
Measures of treatment effect
We synthesised data using the risk ratio (RR). For the outcomes 50% reduction in seizure frequency and treatment withdrawal, we have quoted 95% confidence intervals (CIs). For individual adverse effects, we quoted 99% CIs to make an allowance for multiple testing. We did not use RR for cognitive or quality of life outcomes because we instead narratively synthesised the evidence for these outcomes. We had intended to evaluate dose‐response relationships using a regression model; however, only two trials provided data that could be used to evaluate this outcome. Therefore, we found it necessary to partake a narrative synthesis for this outcome.
Unit of analysis issues
For the cross‐over trials, we used data from both periods and assumed that there was no carry‐over effect. Ideally, we would have only included data from the first treatment period to replicate a parallel‐study design, however, the data were largely unavailable with only one trial providing fully paired data. The other cross‐over trials provided no information on paired responses, and only the total number of responders under either treatment or placebo was available. In such cases, it is possible to calculate the RR estimate but not possible to calculate the exact estimate for the variance of the RR. To avoid excluding such trials, we calculated an approximate estimate of the variance of the RR, assuming independence between outcomes under placebo and vigabatrin. This approximate estimate is an overestimate and, therefore, may be regarded as a more conservative estimate. For the one trial which did provide fully paired information, the approximate estimate was similar to the exact estimate of variance in the paired analysis. In essence, the extra information of knowing that the sample receiving the placebo was the same sample as those receiving the treatment would improve our analysis. However, due to the way that the trials reported results, this information was not available.
Participants randomised to cross‐over trials, therefore, contributed two observations. The first observation consisted of their responder status under placebo and the second observation consisted of their responder status under the intervention drug, vigabatrin. The terms 'participants' and 'observations' have been used to distinguish these differences.
Dealing with missing data
All analyses included all participants in the treatment groups to which they had been allocated. For the efficacy outcome (50% or greater reduction in seizure frequency), we undertook the following analyses (two of which were sensitivity analyses).
-
Primary (ITT) analysis: participants from both treatment groups (vigabatrin or placebo) not completing follow‐up or with inadequate seizure data were assumed to be non‐responders.
Worst‐case analysis (sensitivity analysis): participants not completing follow‐up or with inadequate seizure data were assumed to be non‐responders in the intervention (vigabatrin) group, but were assumed to be responders in the placebo group.
Best‐case analysis (sensitivity analysis): participants not completing follow‐up or with inadequate seizure data were assumed to be responders in the intervention (vigabatrin) group, but were assumed to be non‐responders in the placebo group.
Assessment of heterogeneity
We assessed clinical heterogeneity by comparing the distribution of important participant factors between trials (age, seizure type, duration of epilepsy, number of AEDs taken at time of randomisation) and trial factors (randomisation concealment, blinding, losses to follow‐up).
We assessed statistical heterogeneity by using the I2 statistic, with an I2 greater than 50% indicating significant heterogeneity. We found statistical heterogeneity to be present for several outcomes. Where results differed between choice of model, we presented results based on both fixed‐effect and random‐effects models.
Assessment of reporting biases
To assess any possible small‐study effect bias, we had planned to create a funnel plot by plotting the RR against the standard error of the RR. Unfortunately, due to the limited number of studies that reported each outcome (fewer than 10 studies), we were unable to construct a funnel plot, in accordance with Cochrane guidelines (Sterne 2011).
Data synthesis
We undertook analyses using Review Manager 5, the Cochrane software package (Review Manager 2014). Our preferred effect estimator for calculating the RR was the Mantel‐Haenszel method. In the case that no clinical or statistical heterogeneity (I2 of 50% or less) was detected, we used a fixed‐effect model. In contrast, when there was either clinical or statistical heterogeneity (I2 greater than 50%), we utilised a random‐effects model for the meta‐analysis.
Due to the reporting methods of the original trial articles, we had to make some approximations when calculating the effect estimates. In all cases, any approximation made was conservative. That is, it was more in favour of a greater efficacy of the placebo than vigabatrin. We have detailed these approximations in the Unit of analysis issues section.
Subgroup analysis and investigation of heterogeneity
We completed subgroup analysis according to the participant factor or trial factor identified as, or suspected of, producing any clinical heterogeneity detected during meta‐analysis.
Sensitivity analysis
We employed a sensitivity analysis for the efficacy outcome, 50% or greater reduction in seizure frequency, whereby we completed a best‐ and worst‐case ITT analysis (described in Dealing with missing data). We also carried out a sensitivity analysis to determine whether the differences between a random‐effects model and fixed‐effect model influenced the conclusions drawn.
Summary of findings and assessment of the certainty of the evidence
We used the GRADE approach, as outlined in the Handbook for Grading the Quality of Evidence and the Strength of Recommendations using the GRADE approach (Schünemann 2013), to assess the certainty of evidence and to interpret the review findings. We used GRADEpro GDT software (GRADEpro GDT 2015), which imports data from Review Manager 5 (Review Manager 2014), to create a 'Summary of findings' table for the following outcomes: 50% or greater reduction in seizure frequency, treatment withdrawal for any reason, ataxia, dizziness or light‐headedness, fatigue or drowsiness, abnormal vision and nystagmus.
Results
Description of studies
Results of the search
The results for all searches, collated, are available in Figure 1.
1.
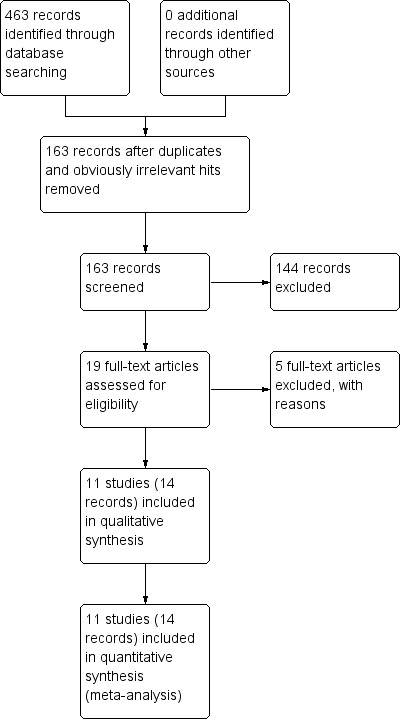
Study flow diagram.
The searches conducted in March 2008 and October 2010 identified 58 records through MEDLINE, 202 records through CENTRAL and 122 records through the Cochrane Epilepsy Group Specialised Register. Removing obvious duplicates and irrelevant records from the identified records resulted screening of 149 records. Detailed checks of the abstracts of these 149 records resulted in the exclusion of eight records which referred to single‐blinded studies, 62 records which were conference proceedings, 21 records which related to the treatment of infantile spasms or other syndromes, five records which were head‐to‐head trials, and 17 records which studied visual field defects. A further six records had no efficacy data available, six records were not placebo‐controlled and five records were long‐term follow‐up studies.
We consequently screened 19 records at the full‐text stage. We determined that 14 records, related to 11 individual studies were eligible for inclusion in this review (Beran 1996; Bruni 2000; Dean 1999; French 1996; Gram 1985; Grunewald 1994; Loiseau 1986; McKee 1993; Rimmer 1984; Tartara 1986; Tassinari 1987). The other five records were excluded at the full‐text stage for reasons described in Excluded studies (Chiron 1996; Provinciali 1996; Reynolds 1991; Ring 1990; Zahner 1999).
We updated our searches in October 2012 and identified 12 records from MEDLINE and two records from CENTRAL. After removing duplicates and irrelevant records, we found no new studies for inclusion in this review.
Similarly, we updated our searches in February 2014 and identified a further 21 records. Following the removal of duplicates and obviously irrelevant records, we found no new studies for inclusion in this review.
In November 2018, we updated our searches and we identified 46 records. After removing duplicates and obviously irrelevant records, we screened 14 records at the abstract stage. We found no new studies that were eligible for inclusion in this review update. Of the 14 records that were excluded at the abstract stage: eight records were either literature reviews, meta‐analyses or involved pooled data; five records referred to studies that were not RCTs and one record referred to an active‐controlled trial.
Included studies
We found 11 randomised, placebo‐controlled, double‐blind studies of vigabatrin as an add‐on treatment in drug‐resistant focal epilepsy, which reported on the primary efficacy outcome and met the inclusion criteria (see Characteristics of included studies table). Four of these trials were parallel‐group studies (Bruni 2000; Dean 1999; French 1996; Grunewald 1994), and seven were cross‐over trials (Beran 1996; Gram 1985; Loiseau 1986; McKee 1993; Rimmer 1984; Tartara 1986; Tassinari 1987). The 11 trials randomised 756 participants, with the vast majority of participants having focal epilepsy. For some of the outcomes, it was possible to exclude participants with seizures other than focal types but for some outcomes, due to the way results were reported in the original reports, this was not possible. For this reason, the number of participants contributing to the various outcomes differed. All participants had treatment‐resistant epilepsy and were taking between one and four concomitant AEDs.
Beran 1996 was an Australian multicentre, cross‐over study in 1996 on 97 participants under double‐blind conditions. Inclusion criteria were defined as participants aged 16 to 65 years with uncontrolled focal seizures. Treatment arms included 2 g/day or 3 g/day of vigabatrin and two placebo arms (2 g/day and 3 g/day placebo). After a run‐in period of eight weeks, participants were randomised to one of the four treatment arms for a treatment period of eight weeks. Participants then entered a washout period, where doses of vigabatrin were titrated down over a period of four weeks. At the end of the four‐week washout period, participants were crossed over to the matching treatment arm, which was followed by a further four‐week washout period. 80 participants were included in the reported analysis.
Bruni 2000 was a randomised, double‐blind, placebo‐controlled, parallel‐group Canadian multicentre study in 2000 with 111 participants, aged between 18 and 50 years. After a baseline assessment period of 12 weeks, participants were randomised to one of two treatment arms and entered a 32‐week titration period, followed by a maintenance period lasting a further four weeks. Final daily doses of vigabatrin were between 1 g and 4 g. Of the 111 participants, 11 were excluded from the primary efficacy analysis due to major protocol violations.
Dean 1999 reported a randomised, double‐blind, placebo‐controlled US multicentre, parallel‐group study with 174 participants aged between 18 and 63 years. There were four treatment arms: placebo and 1 g/day, 3 g/day and 6 g/day of vigabatrin. After a baseline assessment period of 12 weeks, participants were randomised to one of four treatment arms, and entered a six‐week titration period, followed by a further maintenance period lasting 12 weeks.
French 1996 reported a randomised, double‐blind, placebo‐controlled, parallel‐group study in 1996 with 183 participants. Participants were randomised to one of two treatment arms consisting of a placebo or 3 g/day of vigabatrin. Randomised participants were aged 18 to 60 years with drug‐resistant focal impaired‐awareness seizures with or without secondary generalisation. After a 12‐week baseline period, participants entered a four‐week titration period followed by a 12‐week maintenance period. Follow‐up during the maintenance period was at two, four, eight and 12 weeks.
Gram 1985 reported a randomised, double‐blind, placebo‐controlled, cross‐over trial with 21 participants aged between 17 and 63 years, all with focal epilepsy. Participants were randomised to one of two treatment arms, placebo or 3 g/day of vigabatrin, for 12 weeks and then crossed over to the other treatment arm for a further 12 weeks without a washout period. A final single‐blind placebo period was incorporated to test for any carry‐over effect. Only the last eight weeks of the treatment period were included in the analysis to incorporate a four‐week washout period. Participants were followed up every fourth week during the treatment phases.
Grunewald 1994 reported a randomised, double‐blind, placebo‐controlled, parallel‐group study based in the UK. A total of 45 participants, aged between 15 and 61 years, were randomised to one of two treatment arms, either placebo or 3 g/day of vigabatrin. Participants initially entered an eight‐week baseline period followed by a two‐week titration period and then an 18‐week maintenance period.
Loiseau 1986 reported a randomised, double‐blind, placebo‐controlled, cross‐over study with 23 participants randomised to either placebo or 3 g/day of vigabatrin. Participants were aged between 10 and 58 years. Of the 23 randomised participants, four had seizure types other than focal epilepsy and were excluded from all primary outcome results in this review. After a five‐week baseline period, participants were randomised to one of the two treatment arms for 10 weeks. Participants were then crossed over to the other treatment arm for a further 10 weeks without undergoing a washout period. The study was followed by a final five‐week, single‐blind placebo arm to evaluate any carry‐over effect, and one week from each treatment period was excluded from the analysis to incorporate a one‐week washout period. Participants were followed up every fifth week during the treatment phases. Four participants dropped out of the trial, two of whom had focal epilepsy.
McKee 1993 reported a randomised, double‐blind, placebo‐controlled, cross‐over study with 24 participants, aged between 17 and 53 years, who were randomised to either placebo or 3 g/day of vigabatrin. After a baseline period of four weeks, participants were randomised to one of the two treatment arms for 12 weeks followed by a four‐week washout period after which participants were crossed over to the other treatment arm for a further 12 weeks. Of the 24 participants, two had generalised tonic‐clonic epilepsy and, due to the way in which results were reported, had to be included in the analysis within this review. Participants were followed up at two, six and 12 weeks of treatment. Two participants had incomplete seizure data.
Rimmer 1984 reported a randomised, double‐blind, placebo‐controlled, cross‐over study with 24 participants, aged between 16 and 61 years, who were randomised to either placebo or 3 g/day of vigabatrin. After a baseline period of four weeks, participants were randomised to one of the two treatment arms for nine weeks. After the first treatment, participants were crossed over to the other treatment arm for a further nine weeks without a washout period. The 24 participants were reported to have 'mainly' focal impaired‐awareness epilepsy, and so it was possible that a small proportion of the study population may not have had focal epilepsy.
Tartara 1986 reported a randomised, double‐blind, placebo‐controlled, cross‐over Italian study with 23 participants, aged between 17 and 50 years, who were randomised to either placebo or 2 g/day or 3 g/day of vigabatrin (dose dependent on bodyweight). Participants were randomised to one of the two treatment arms for seven weeks. After the first treatment, participants did not undergo a washout period but were immediately crossed over to the other treatment arm for a further seven weeks. The study included 23 participants, three of whom had seizure types other than focal epilepsy and who were excluded from all primary outcome results. Participants were followed up at two, four and seven weeks of treatment.
Tassinari 1987 reported a randomised, double‐blind, placebo‐controlled, cross‐over Italian study with 31 participants, aged between 10 and 58 years, who were randomised to either placebo or 2 g/day or 3 g/day dose of vigabatrin (dose dependent on bodyweight). After a baseline period of two months, participants were randomised to one of the two treatment arms for three months. Participants were then crossed over to the other treatment arm for a further three months without a washout period. The study included 31 participants, one of whom had progressive myoclonic epilepsy and was excluded from all primary outcome results. Participants were followed up every six weeks during the treatment phases.
Clinical heterogeneity
All 11 trials were similar in both the age range and types of epilepsies included. Five trials included participants treated with two or fewer concomitant AEDs (Bruni 2000; Dean 1999; French 1996; McKee 1993; Tartara 1986); five trials included participants treated with three or fewer concomitant AEDs (Beran 1996; Gram 1985; Grunewald 1994; Loiseau 1986; Rimmer 1984); and one trial included participants treated with up to four concomitant AEDs (Tassinari 1987). A sensitivity analysis (not shown) revealed no significant departures in primary efficacy outcomes between trials of participants with one or two concomitant AEDs compared to trials of participants with three or more concomitant AEDs. Where information on duration of epilepsy was provided, there were no significant differences in the ranges of duration of epilepsy between the trials (Bruni 2000; Dean 1999; Gram 1985; Loiseau 1986; McKee 1993; Rimmer 1984; Tartara 1986).
Excluded studies
We excluded five studies at the full‐text screening stage (Chiron 1996; Provinciali 1996; Reynolds 1991; Ring 1990; Zahner 1999; see Characteristics of excluded studies table). Specifically, we excluded four trials that used a response‐conditional design (Chiron 1996; Reynolds 1991; Ring 1990; Zahner 1999). In these trials, participants were randomised to either vigabatrin or placebo under the condition that they had demonstrated a response to vigabatrin in an earlier, baseline phase of the trial. We subsequently identified one study as being single‐blinded, although this had not been stated in the article abstract or title, and so we excluded it (Provinciali 1996).
Risk of bias in included studies
The risk of bias judgements for each study are presented in the 'Risk of bias' table located in the Characteristics of included studies table. Figure 2 provides an illustrative summary of the risk of bias judgements across all of the included studies, while Figure 3 is a graph displaying the proportion of studies awarded a high, unclear or low risk of bias judgement for each risk of bias domain.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Nine of the 11 included studies specifically reported that they were RCTs. However, only two studies described valid methods for random sequence generation (Beran 1996; Grunewald 1994). Specifically, Beran 1996 used a computer‐generated randomisation schedule and Grunewald 1994 used a random number generated code. Therefore, we both studies were at low risk of selection bias for random sequence generation. We rated six studies at unclear risk of bias for random sequence generation because they did not provide details of how randomisation was achieved (Bruni 2000; Dean 1999; French 1996; Loiseau 1986; McKee 1993; Rimmer 1984). In contrast, Tartara 1986 did describe a method of randomisation, however, we deemed the method of random sequence generation to be inadequate. Tartara 1986 randomised participants to treatment dependent on their order of enrolment on the study. This would produce a predictive randomisation pattern which personnel could then use to predict treatment allocation. Therefore, we judged that this study at high risk of selection bias due to random sequence generation.
Two studies did not specify in the publication text whether they were randomised trials (Gram 1985; Tassinari 1987). However, we assumed that the two studies were randomised because both studies were described as being double‐blind. This would suggest that the personnel were not aware of treatment allocation, thus implying that randomisation had been conducted. For this reason, we continued to include the two studies in the review but awarded them high risk of bias for random sequence generation to convey our uncertainty.
All 11 studies failed to provide any methods for allocation concealment (Beran 1996; Bruni 2000; Dean 1999; French 1996; Gram 1985; Grunewald 1994; Loiseau 1986; McKee 1993; Rimmer 1984; Tartara 1986; Tassinari 1987). Consequently, we judged that all of the included studies were at unclear risk of selection bias for allocation concealment.
Blinding
All 11 studies stated that placebo was either 'matched' or 'identical' in appearance (Beran 1996; Bruni 2000; Dean 1999; French 1996; Gram 1985; Grunewald 1994; Loiseau 1986; McKee 1993; Rimmer 1984; Tartara 1986; Tassinari 1987), with two trials masking for taste (Gram 1985; Loiseau 1986). Therefore, we judged all 11 studies at low risk of performance bias.
In contrast, none of the included studies reported whether, or how, outcome assessment was blinded. As a result, we deemed all 11 studies at unclear risk of detection bias.
Incomplete outcome data
Three studies reported attrition and conducted an ITT analysis, although, on occasion, this was not specifically stated (Bruni 2000; Rimmer 1984; Tassinari 1987). Hence, these three studies were at low risk of attrition bias. Dean 1999 also conducted an ITT analysis and did report the overall attrition rate for the study, but did not specify the attrition rate per treatment group. Consequently, this study was at unclear risk of attrition bias. We also judged that Grunewald 1994 was at unclear risk of attrition bias. Attrition was reported; however, it was not clear whether ITT analysis had been used.
We judged that six other studies were at unclear risk of attrition bias (Beran 1996; French 1996; Gram 1985Loiseau 1986; McKee 1993; Tartara 1986). The six studies fully reported attrition; however, did not conduct an ITT analysis.
Selective reporting
Nine studies reported all outcomes that they had specified in the methods section of their full‐text publications in their respective results section (Beran 1996; Bruni 2000; French 1996; Grunewald 1994; Loiseau 1986; McKee 1993; Rimmer 1984; Tartara 1986; Tassinari 1987). Therefore, these nine studies were at low risk of reporting bias.
We judged that Dean 1999 was at unclear risk of reporting bias after the outcome treatment withdrawal was only partially reported. As alluded to earlier, Dean 1999 reported the total number of participants who withdrew from the study but did not specify the number of participants who withdrew per treatment group. Consequently, we were unable to use the include data from the study for the outcome, treatment withdrawal for any reason, and were also unable to complete an ITT analysis for the other outcomes, including 50% or greater reduction in seizure frequency. For this reason, the study was at unclear risk of reporting bias.
Gram 1985 specified no outcomes in the methods section of the publication text. As a result, we were unable to determine whether all intended outcomes had been reported. Therefore, we judged that Gram 1985 was at unclear risk of reporting bias.
Other potential sources of bias
Four trials were cross‐over designs without washout periods (Loiseau 1986; Rimmer 1984; Tartara 1986; Tassinari 1987). As a consequence, there could have been a carryover effect between the two treatments that could have biased the results. Hence, we judged the four cross‐over studies at unclear risk of other bias. Loiseau 1986 did exclude data collected during the first week of each treatment arm to compensate for not including a washout period. However, we considered that this time period was of inadequate length to avoid any carryover effect and, therefore, this study was at unclear risk of other bias. Another cross‐over study did not include a baseline period (Gram 1985). Without a baseline period to assess participants' basal seizure frequency, it was not possible to determine whether the severity of epilepsy was balanced between treatment groups. Consequently, this study was at unclear risk of other bias.
We did not detect any other potential sources of bias for the other six included studies (Beran 1996; Bruni 2000; Dean 1999; French 1996; Grunewald 1994; McKee 1993). Therefore, these studies were at low risk of other bias.
Effects of interventions
See: Table 1
The main results of the review can be found in Table 1.
50% or greater reduction in seizure frequency
Four studies reported 50% or greater reduction in seizure frequency in accordance with the definition in the Types of outcome measures subsection (Bruni 2000; Dean 1999; French 1996; Grunewald 1994). Specifically, we defined the outcome as the proportion of participants who experienced a 50% or greater reduction in seizure frequency during the treatment period when compared to the prerandomisation baseline period. Unfortunately, although the other seven included studies did report the outcome, the definition that the studies used did not adhere to our definition (Beran 1996; Gram 1985; Loiseau 1986; McKee 1993; Rimmer 1984; Tartara 1986; Tassinari 1987). In these studies, percentage seizure reduction was considered as the difference in seizure frequency during the vigabatrin treatment arm compared to the placebo treatment arm, rather than being compared against the seizure frequency during the baseline period. For this reason, we were unable to incorporate data from these seven studies into the meta‐analysis.
The four studies that reported the outcome were all parallel‐group studies and included 513 participants (Bruni 2000; Dean 1999; French 1996; Grunewald 1994). The meta‐analysis pooling across doses (vigabatrin 1 g/day to 6 g/day) showed some evidence of heterogeneity (I2 = 34% in the ITT analysis); however, according to our methods, this was not statistically significant. Thus, we utilised a fixed‐effect model. Participants allocated to vigabatrin were more likely to achieve a 50% or greater reduction in seizure frequency (RR 2.60, 95% CI 1.87 to 3.63; P < 0.001; low‐certainty evidence; Analysis 1.1).
1.1. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 1: 50% or greater reduction in seizure frequency (ITT analysis)
For the best‐case scenario, there was no significant heterogeneity, therefore, we continued to utilise a fixed‐effect model for the analysis. The best‐case analysis indicated that participants allocated to vigabatrin were nearly three times as likely to achieve a 50% or greater reduction in seizure frequency than those allocated to placebo (RR 2.78, 95% CI 2.02 to 3.82; I2 = 0%; Analysis 1.2). In contrast, there was significant heterogeneity within the worst‐case analysis data set so we employed a random‐effects model (I2 = 61%). The worst‐case scenario demonstrated that participants allocated to vigabatrin remained more likely to achieve a 50% or greater reduction in seizure frequency than those allocated to placebo (RR 1.65, 95% CI 0.98 to 2.76; Analysis 1.3); however, the effect size calculated was not statistically significant (P = 0.06). We then conducted a sensitivity analysis to investigate whether the use of a random‐effects model over a fixed‐effect model influenced the findings. The sensitivity analysis revealed little difference in the overall RR calculated (RR 1.61, 95% CI 1.20 to 2.16), but did result in the effect size calculated becoming statistically significant (P = 0.001).
1.2. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 2: 50% or greater reduction in seizure frequency (best‐case analysis)
1.3. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 3: 50% or greater reduction in seizure frequency (worst‐case analysis)
We were unable to construct a funnel plot for this outcome as too few studies reported the outcome according to our definition of 50% or greater seizure reduction. Therefore, we were unable to comment on whether there was reporting bias towards smaller studies.
Treatment withdrawal for any reason
Four trials reported sufficient information to calculate the number of participants withdrawing from each treatment arm (Bruni 2000; French 1996; Grunewald 1994; Tassinari 1987). The four studies consisted of 370 individual participants. Notably, Tassinari 1987 was a cross‐over study so data from the 31 included participants was included in both treatment groups for the purposes of our current meta‐analysis. All other included studies reported total numbers of participants withdrawing from treatment but did not provide sufficient information for us to calculate which treatment arm these participants had belonged to at the time of treatment withdrawal (Beran 1996; Dean 1999; Gram 1985; Loiseau 1986; McKee 1993; Rimmer 1984; Tartara 1986).
An analysis pooling across doses showed evidence of no heterogeneity between the four studies for which data on withdrawal by treatment group were presented (I2 = 0%). Therefore, we used a fixed‐effect model. Participants allocated to vigabatrin were nearly three times more likely to have treatment withdrawn for any reason compared to those taking placebo (RR 2.86, 95% CI 1.25 to 6.55; P = 0.01; very low‐certainty evidence; Analysis 1.4).
1.4. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 4: Treatment withdrawal due to any reason
Adverse effects
All 11 included studies (756 participants) reported adverse effects data. The data included more participants with non‐focal epilepsy than did the analysis of the primary outcome above. This was because, although seizure reduction data were reported by the individual or by seizure type, adverse effect data were not. Although the vast majority of participants had focal epilepsy, several studies included some participants with other seizure types (Loiseau 1986: four; McKee 1993: two; Rimmer 1984: number with non‐focal type unclear; Tartara 1986: three; Tassinari 1987: one). In addition, due to the way in which the data were reported, the denominators for the adverse effect data sometimes differed slightly to the number in each treatment arm (Dean 1999; Tartara 1986), although these discrepancies were small (one in both cases).
An analysis pooling data from all trials showed no heterogeneity for any of the 10 adverse effects considered (I2 statistics ranged from 0% to 50%). The data set for one adverse event, nystagmus, did display some heterogeneity (I2 = 49%); however, according to our stated methods, this level of heterogeneity did not reach the threshold at which we would consider using a random‐effects model (i.e. I2 > 50%). Therefore, we employed a fixed‐effect model for all 10 adverse effects outcomes assessed.
The analyses pooling data across doses indicated that the following adverse effects were significantly associated with vigabatrin: dizziness/light‐headedness (RR 1.74, 99% CI 1.05 to 2.87; 9 studies; 709 participants; low‐certainty evidence; Analysis 1.6), fatigue (RR 1.65, 99% CI 1.08 to 2.51; 9 studies; 709 participants; low‐certainty evidence; Analysis 1.7), drowsiness (RR 1.70, 99% CI 1.18 to 2.44; 8 studies; 666 participants; Analysis 1.8), and depression (RR 3.28, 99% CI 1.30 to 8.27; 6 studies; 546 participants; Analysis 1.10). Although we observed increased incidence rates in the vigabatrin group compared to placebo for: ataxia (RR 2.76, 99% CI 0.96 to 7.94; 7 studies; 481 participants; very low‐certainty evidence; Analysis 1.5), nausea (RR 3.57, 99% CI 0.63 to 20.30; 4 studies; 251 participants; Analysis 1.9), headache (RR 1.23, 99% CI 0.79 to 1.92; 9 studies; 709 participants; Analysis 1.11), abnormal vision (RR 1.64, 99% CI 0.67 to 4.02; 5 studies; 430 participants; very low‐certainty evidence; Analysis 1.12), diplopia (RR 1.76, 99% CI 0.94 to 3.30; 7 studies, 655 participants; Analysis 1.13), and nystagmus (RR 1.53, 99% CI 0.62 to 3.76; 2 studies; 357 participants; low‐certainty evidence; Analysis 1.14); these overall effect sizes were not statistically significant. Notably, fewer than half of the studies recorded any occurrences of abnormal vision, diplopia or nystagmus.
1.6. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 6: Dizziness/light‐headedness
1.7. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 7: Fatigue
1.8. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 8: Drowsiness
1.10. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 10: Depression
1.5. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 5: Ataxia
1.9. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 9: Nausea
1.11. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 11: Headache
1.12. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 12: Abnormal vision
1.13. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 13: Diplopia
1.14. Analysis.

Comparison 1: Vigabatrin versus placebo, Outcome 14: Nystagmus
Dose‐response analysis
Six trials randomised participants to placebo or vigabatrin 3 g/day (French 1996; Gram 1985; Grunewald 1994; Loiseau 1986; McKee 1993; Rimmer 1984), two trials randomised participants to either 2 g/day or 3 g/day depending on bodyweight (Tartara 1986; Tassinari 1987), and Bruni 2000 titrated to maximum doses of 4 g/day.
Two trials examined response rates over different doses of vigabatrin. Beran 1996 compared percentage reduction in mean back‐transformed seizure rate between vigabatrin 2 g/day and 3 g/day, relative to placebo. The study demonstrated a 36% reduction in mean back‐transformed seizure rate with 2 g/day versus 31% reduction with 3 g/day. There were no significant differences in seizure reduction between doses. Dean 1999 compared response rates between placebo, and vigabatrin 1 g/day, 3 g/day and 6 g/day. The proportion of participants achieving a 50% or greater reduction in seizure frequency were: placebo: 7%; 1 g/day: 24%; 3 g/day: 51% and 6 g/day: 53%. The difference between 6 g/day and 3 g/day was not significant.
Cognitive outcomes and quality of life
Two trials evaluated cognitive and quality of life outcomes (Dean 1999; French 1996), while one study reported only cognitive outcomes (McKee 1993). Dean 1999 (174 participants) and French 1996 (183 participants) were both large multicentre trials, whereas McKee 1993 was a small cross‐over study with 24 participants.
The two larger trials included the same eight cognitive tests of abilities (Lafayette Grooved Pegboard, Stroop Test, Benton Visual Retention Test, Controlled Oral Word Association Test, Symbol Digit Modalities Test, Rey Auditory Verbal Learning Test, Wonderlic Personnel Test and the Digit Cancellation Test), and the same three mood tests (Profile of Mood States, Washington Psychosocial Seizure Inventory and Mood Rating Scale) (Dean 1999; French 1996). There were no differences between the placebo and vigabatrin groups at the end of the study for any of the eight tested outcomes for French 1996, whereas Dean 1999 reported outcomes from the Digit Cancellation Test decreased with increasing doses of the drug. In summary, vigabatrin had little impact on tests of cognitive abilities or quality of life.
McKee 1993 employed an assortment of three psychomotor test, four memory tests (Forward Digit Span Test, Backward Digit Span Test, Paired Associate Learning Test and Rivermead Behavioural Memory Test) and three self‐reported rating scales to assess cognition. Similarly, this study found no significant differences in test or scale scores between participants who received vigabatrin or placebo at any time point.
Discussion
Summary of main results
This systematic review and meta‐analysis of short‐term RCTs of vigabatrin suggests that vigabatrin may be effective in reducing seizure frequency in drug‐resistant focal epilepsy, at least in the short term. Specifically, people with drug‐resistant focal epilepsy who are given vigabatrin may be two to three times more likely to achieve a 50% or greater reduction in seizure frequency compared to those given placebo.
In addition reducing seizure frequency, an AED needs to have good tolerability, with limited adverse effects. Overall, tolerability can be measured by the proportion of participants withdrawing from treatment and incidence rates of adverse effects reported while taking the drug. In this review, we demonstrated that several of the investigated adverse effects may be significantly associated with vigabatrin, such as dizziness/light‐headedness, fatigue, drowsiness and depression. Furthermore, the evidence also suggested that participants may be up to three times more likely to withdraw from treatment with vigabatrin compared to placebo. Interestingly, none of the three visual adverse effects were associated with vigabatrin compared to placebo. However, this is not surprising given the short duration of the included studies.
Overall completeness and applicability of evidence
This review only synthesised evidence from trials comparing vigabatrin to placebo and did not compare vigabatrin to other AEDs. Furthermore, and perhaps more importantly, it does not inform us of how frequently constriction of the visual field occurs. Although vigabatrin may have proven efficacy in reducing seizures, associated adverse effects (visual field constrictions) are common and can be severe (Nicolson 2002). From assessing three visual adverse effects that we knew could be detected within a short‐term follow‐up period, we found that none were significantly associated with vigabatrin treatment compared to placebo. It is important to consider that such visual defects may not have been detected due to the short treatment durations and follow‐ups of the included studies. Long‐term observational studies may provide a more accurate representation of the adverse effect profile of vigabatrin.
Moreover, this review cannot inform readers about the efficacy or tolerability of vigabatrin in children. Two studies enrolled populations aged 10 to 58 years. However, the majority of the other studies enrolled people aged 17 or 18 and older. From this perspective, the findings of this review may not be applicable to children. Separate studies should be completed in order to confirm this.
Certainty of the evidence
We evaluated that all of the included studies demonstrated unclear risk of bias across at least three risk of bias domains. For this reason, we downgraded the evidence for all GRADE‐assessed outcomes once for risk of bias (see Table 1). We also noted imprecision in the results due to the low number of events recorded. For three outcomes (50% or greater reduction in seizure frequency, dizziness/light‐headedness, fatigue, and nystagmus), there were between 100 and 400 events across the treatment groups. The low number of events did not satisfy the suggested optimal information size, therefore, we downgraded the evidence for these outcomes once. This resulted in a GRADE assessment of low‐certainty evidence for the three outcomes. Moreover, there were fewer than 100 events across treatment groups for the remaining three outcomes (treatment withdrawal due to any reason, ataxia, and abnormal vision). Due to the very limited number of events, we downgraded the evidence for these outcomes twice. Consequently, we assessed the evidence for the remaining four outcomes as very low‐certainty.
Potential biases in the review process
Two of the seven cross‐over trials incorporated formal washout periods within the trial design (Beran 1996; McKee 1993), and three trials added single‐blind placebo periods to the end of the study to test for carry‐over effects (Gram 1985; Loiseau 1986; Tassinari 1987), which were not significant in two trials (Loiseau 1986; Tassinari 1987). Two trials excluded part of the treatment period from the analysis to incorporate a washout period (Loiseau 1986 excluded one treatment week and Gram 1985 excluded four weeks). Two of the seven cross‐over trials had no washout period (Rimmer 1984; Tartara 1986). Analysis here assumed that there was no carry‐over effect of vigabatrin when participants crossed from vigabatrin to placebo. There is some evidence to support this assumption (Loiseau 1986; Tassinari 1987). A sensitivity analysis excluding trials first with no washout period and then excluding all cross‐over trials revealed no systematic departures in overall conclusions.
The review might also present slightly skewed results regarding the adverse effect profile associated with short‐term vigabatrin due to the inclusion of data from participants with seizure types other than focal seizures. However, the vast majority of data were collected from participants with drug‐resistant focal epilepsy, so any impact from the inclusion of participants with any other seizure type should be minimal.
Agreements and disagreements with other studies or reviews
An analysis of short‐term RCTs, such as the analysis presented here, cannot detect adverse effects that take longer (compared to the length of an RCT) to develop or present, or adverse effects that are very rare. Longer‐term observational studies of vigabatrin have identified that visual field defects are a common adverse effect associated with the prolonged use of vigabatrin (Nicolson 2002; Wild 2007). This important adverse effect has been observed in both adults and children, and the incidence rate increases with increased dose and with longer duration of use (Wild 2007). The associated visual field defects are, however, often asymptomatic. As a result, this adverse effect would most likely not be detected by RCTs.
In this systematic review, we included three visual adverse effects, any abnormality of vision, diplopia and nystagmus. It cannot be assumed that summary statistics produced by a systematic review of RCTs will provide an adequate representation of associated visual field defects; in fact, they are expected to underestimate true risks. An analysis of the extent of visual field defects associated with vigabatrin use is the subject of another systematic review which has shown that an estimated 44% of participants exposed to vigabatrin develop visual field loss (Maguire 2010).
Authors' conclusions
Implications for practice.
When used as an add‐on for people with drug‐resistant focal epilepsy, vigabatrin may significantly reduce seizure frequency. Add‐on vigabatrin may also cause more people to withdraw from treatment. Although not reported here, work by others has shown that long‐term vigabatrin is associated with visual field constrictions which occur in a significant number of people taking vigabatrin. We suggest that we were unable to detect these visual adverse effects because of their asymptomatic presentation and due to the short duration of RCTs. Given the seriousness of such visual adverse effects, the implications of long‐term vigabatrin use should still be considered before commencing vigabatrin add‐on therapy.
Implications for research.
Further research is required to identify the risk of visual field defects and those who are susceptible to it. Further randomised controlled trials are also necessary to examine the efficacy and tolerability of add‐on vigabatrin in children with drug‐resistant focal epilepsy, as well as comparing the efficacy of add‐on vigabatrin to using other antiepileptic medications as add‐on therapies.
What's new
| Date | Event | Description |
|---|---|---|
| 1 November 2018 | New citation required but conclusions have not changed | Conclusions unchanged. |
| 1 November 2018 | New search has been performed | Searches updated 1 November 2018; no new studies found. |
History
Protocol first published: Issue 1, 2000 Review first published: Issue 3, 2008
| Date | Event | Description |
|---|---|---|
| 12 October 2012 | New citation required but conclusions have not changed | Conclusions remain the same. |
| 12 October 2012 | New search has been performed | Searches updated 12 October 2012; no new studies found. |
Acknowledgements
We would like to acknowledge the contribution of the previous authors to this review: Dr Azmi Rasid, Karla Hemming and Jane Hutton.
Appendices
Appendix 1. CRS Web search strategy
1. MESH DESCRIPTOR Vigabatrin EXPLODE ALL AND CENTRAL:TARGET
2. (vigabatrin* OR sabril):AB,KW,MC,MH,TI AND CENTRAL:TARGET
3. #1 OR #2
4. MESH DESCRIPTOR Epilepsy EXPLODE ALL AND CENTRAL:TARGET
5. MESH DESCRIPTOR Seizures EXPLODE ALL AND CENTRAL:TARGET
6. (epilep* OR seizure* OR convuls*):AB,KW,MC,MH,TI AND CENTRAL:TARGET
7. #4 OR #5 OR #6 AND CENTRAL:TARGET
8. #3 AND #7
9. #8 AND >04/02/2014:CRSCREATED
Appendix 2. MEDLINE search strategy
This strategy is based on the Cochrane Highly Sensitive Search Strategy for identifying randomised trials (published in Lefebvre 2011).
1. exp VIGABATRIN/
2. (vigabatrin$ or sabril).tw.
3. 1 or 2
4. exp Epilepsy/
5. exp Seizures/
6. (epilep$ or seizure$ or convuls$).tw.
7. 4 or 5 or 6
8. exp *Pre‐Eclampsia/ or exp *Eclampsia/
9. 7 not 8
10. (randomized controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
11. clinical trials as topic.sh.
12. trial.ti.
13. 10 or 11 or 12
14. exp animals/ not humans.sh.
15. 13 not 14
16. 3 and 9 and 15
17. limit 16 to ed=20140204‐20181101
18. 16 not (1$ or 2$).ed.
19. 18 and (2014$ or 2015$ or 2016$ or 2017$ or 2018$).dt.
20. 17 or 19
21. remove duplicates from 20
Appendix 3. ClinicalTrials.gov search strategy
Interventional Studies | Epilepsies, Partial | Vigabatrin OR sabril | First posted from 02/04/2014 to 11/01/2018
Appendix 4. ICTRP search strategy
Condition: Partial epilepsy OR Focal epilepsy
Intervention: vigabatrin OR sabril
Recruitment status: All
Date of registration between 04/02/2014 and 01/11/2018
Appendix 5. Original search strategies
We conducted searches of the following databases:
a) Cochrane Epilepsy Group Specialised Register, 3 April 2008, using the search terms 'vigabatrin' and 'Sabril';
b) Cochrane Central Register of Controlled Trials (CENTRAL) (the Cochrane Library, Issue 1, 2008) using the search terms 'vigabatrin' and 'Sabril';
c) MEDLINE (Ovid) from 1950 to March week 4, 2008, using the search strategy set out below. These search terms were combined with phases 1 and 2 of the Cochrane highly sensitive search strategy for MEDLINE as set out in Appendix 5b of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2005):
1. exp Vigabatrin/ 2. (vigabatrin or Sabril).tw. 3. 1 OR 2 4. exp Epilepsy/ OR epilep$.tw. 5. exp Seizures/ OR seizure$.tw. 6. convulsion$.tw. 7. 4 OR 5 OR 6 8. 3 AND 7
Data and analyses
Comparison 1. Vigabatrin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 50% or greater reduction in seizure frequency (ITT analysis) | 4 | 513 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.60 [1.87, 3.63] |
| 1.2 50% or greater reduction in seizure frequency (best‐case analysis) | 3 | 339 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.78 [2.02, 3.82] |
| 1.3 50% or greater reduction in seizure frequency (worst‐case analysis) | 3 | 339 | Risk Ratio (M‐H, Random, 95% CI) | 1.65 [0.98, 2.76] |
| 1.4 Treatment withdrawal due to any reason | 4 | 401 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.86 [1.25, 6.55] |
| 1.5 Ataxia | 7 | 677 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.76 [0.96, 7.94] |
| 1.6 Dizziness/light‐headedness | 9 | 905 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.74 [1.05, 2.87] |
| 1.7 Fatigue | 9 | 905 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.65 [1.08, 2.51] |
| 1.8 Drowsiness | 8 | 864 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.70 [1.18, 2.44] |
| 1.9 Nausea | 4 | 328 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.57 [0.63, 20.30] |
| 1.10 Depression | 6 | 690 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.28 [1.30, 8.27] |
| 1.11 Headache | 9 | 905 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.23 [0.79, 1.92] |
| 1.12 Abnormal vision | 5 | 575 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.64 [0.67, 4.02] |
| 1.13 Diplopia | 7 | 797 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.76 [0.94, 3.30] |
| 1.14 Nystagmus | 2 | 357 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.53 [0.62, 3.76] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Beran 1996.
| Study characteristics | ||
| Methods |
Study design: randomised, double‐blind, PBO‐controlled, multicentre, cross‐over study Country: Australia Number of treatment arms: 3 Study duration: Phase 1: baseline period (8 weeks) Phase 2: first treatment period (8 weeks) Phase 3: washout period (4 weeks) Phase 4: second treatment period (8 weeks) Phase 5: washout period (4 weeks) |
|
| Participants |
Total number of participants randomised: 97 Number of participants included in efficacy analysis: 80 (all randomised participants were included in the analysis of adverse event data). Gender: 45 males; 52 females Age: range 17–64 years Number of concomitant AEDs: ≤ 3 Epilepsy type: drug‐resistant focal seizures |
|
| Interventions |
Treatment arm 1: VGB 2 g/day Treatment arm 2: PBO 2 g/day Treatment arm 3: VGB 3 g/day Treatment arm 4: PBO 3 g/day |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Source of funding unknown. Correspondence regarding the study was to be addressed to Dr B Cooper of Hoechst Marion Roussel, the manufacturers of VGB. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "All medications were administered under double‐blind conditions according to a computer‐generated randomization schedule." |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Each dose of active vigabatrin or matching inactive placebo was packed in a foil sachet containing three tablets which comprised drug and/or placebo (i.e. each sachet contained 0, 2 or 3 placebo tablets and 0, 2 or 3 vigabatrin tablets." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: attrition reported but intention‐to‐treat analysis not conducted. |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes listed in the methods section of the publication were reported in the results section. |
| Other bias | Low risk | Comment: none detected. |
Bruni 2000.
| Study characteristics | ||
| Methods |
Study design: randomised, double‐blind, PBO‐controlled, multicentre, parallel‐group study Country: Canada Number of treatment arms: 2 Study duration: Phase 1: baseline period (12 weeks) Phase 2: titration period (32 weeks) Phase 3: maintenance period (4 weeks) |
|
| Participants |
Total number of participants randomised: 111 (VGB: 58; PBO: 53) Number of participants included in efficacy analysis: 111 (VGB: 58; PBO: 53) Gender: 61 males; 50 females Duration of epilepsy: range 3–43 years; mean 20 (SEM 0.9) years Age: range 18–50 years; mean 34 (SEM 0.8) years Number of concomitant AEDs: ≤ 2 Epilepsy type: focal impaired‐awareness seizures with or without secondary generalisation |
|
| Interventions |
Treatment arm 1: VGB 1–4 g/day Treatment arm 2: PBO |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Source of funding unknown. 2 study authors were either current employees of, or previous employees, of Hoechst Marion Roussel, the manufacturers of VGB, at the time of publication. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no details regarding random sequence generation provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study medication was supplied in double‐blind condition as 500 mg tablets of active medication or matching placebo." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: attrition reported and intention‐to‐treat analysis conducted. |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes listed in the methods section of the publication were reported in the results section. |
| Other bias | Low risk | Comment: none detected. |
Dean 1999.
| Study characteristics | ||
| Methods |
Study design: randomised, double‐blind, PBO‐controlled, multicentre, parallel‐group study Country: USA Number of treatment arms: 4 Study duration: Phase 1: baseline period (12 weeks) Phase 2: titration period (6 weeks) Phase 3: maintenance period (12 weeks) |
|
| Participants |
Total number of participants randomised: 174 (VGB 1 g/day: 45; VGB 3 g/day: 43; VGB 6 g/day: 41; PBO: 45) Gender: 83 males; 91 females Duration of epilepsy: mean 22 (SD 10) years Age: range 18–63 years; mean 35 (SD 10) years Number of concomitant AEDs: ≤ 2 Epilepsy type: focal impaired‐awareness seizures or focal seizure with secondary generalisation |
|
| Interventions |
Treatment arm 1: VGB 1 g/day Treatment arm 2: VGB 3 g/day Treatment arm 3: VGB 6 g/day Treatment arm 4: PBO |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Study funded through a grant from Hoechst Marion Roussel, the manufacturers of VGB. 1 study author was affiliated with Hoechst Marion Roussel. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no details regarding random sequence generation provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Matching placebo tablets were provided, and double‐blind conditions were maintained throughout the study." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: attrition reported and intention‐to‐treat analysis conducted. However, attrition per treatment group not specified. |
| Selective reporting (reporting bias) | Unclear risk | Comment: all outcomes listed in methods section of the publication were reported in results section. However, treatment withdrawal per treatment arm was not specified. Therefore, impossible to calculate withdrawal rates by treatment arm, or best‐case or worst‐case scenarios. |
| Other bias | Low risk | Comment: none detected. |
French 1996.
| Study characteristics | ||
| Methods |
Study design: randomised, double‐blind, PBO‐controlled, multicentre, parallel‐group study Country: USA Number of treatment arms: 2 Study duration: Phase 1: baseline period (12 weeks) Phase 2: titration period (4 weeks) Phase 3: maintenance period (12 weeks) |
|
| Participants |
Total number of participants randomised: 183 (VGB: 93; PCB: 90; 1 participant withdrew from the VGB treatment group prior to receiving any study drug and therefore the authors did not provide any characteristics data for this participant) Gender: 80 males; 102 females Age: range 18–60 years; mean 34 years Number of concomitant AEDs: ≤ 2 Epilepsy type: focal impaired‐awareness seizures |
|
| Interventions |
Treatment arm 1: VGB 3 g/day Treatment arm 2: PBO |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Source of funding unknown. Some of the study authors were affiliated with Hoechst Marion Roussel, the manufacturer of VGB. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no details regarding random sequence generation provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Vigabatrin was supplied as 0.5‐g tablets. Matching inactive placebo was provided." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: attrition reported; however, intention‐to‐treat analysis not performed. Also, 20 participants were randomised but then withdrew prior to taking any study medication and the study did not report which group they were originally randomised to. Therefore, we were unable to conduct a full intention‐to‐treat analysis. |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes listed in the methods section of the publication were reported in the results section. |
| Other bias | Low risk | Comment: none detected. |
Gram 1985.
| Study characteristics | ||
| Methods |
Study design: double‐blind, PBO‐controlled, single‐centre, cross‐over study Country: Denmark Number of treatment arms: 2 Study duration: Phase 1: first treatment period (12 weeks) Phase 2: single‐blind PBO period (4 weeks) Phase 3: second treatment period (12 weeks) Phase 4: single‐blind PBO period (4 weeks) (Note: only data collected during the latter 8 weeks of each treatment period was included in the analysis as an attempt to avoid a carry‐over effect.) |
|
| Participants |
Total number of participants randomised: 21 Gender: 11 males, 10 females Duration of epilepsy: range 8–47 years; median 26 years Age: range 17–63 years Number of concomitant AEDs: ≤ 3 Epilepsy type: drug‐resistant focal impaired‐awareness epilepsy |
|
| Interventions |
Treatment arm 1: VGB 3 g/day Treatment arm 2: PBO |
|
| Outcomes |
Primary outcome
Secondary outcome
|
|
| Notes | Source of funding unknown. VGB was supplied by Centre de Recherche Merrell International, France. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Comment: no details regarding random sequence generation provided. Publication did not explicitly state whether the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "identical placebo packets contained quinine as a taste corrigent." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: attrition reported but intention‐to‐treat analysis not conducted. |
| Selective reporting (reporting bias) | Unclear risk | Comment: no outcomes were described in the methods section of the publication, therefore, we were unable to determine whether all intended outcomes had been fully reported. |
| Other bias | Unclear risk | Comment: study design did not include a baseline period. Percent change in the total number of seizures therefore reflected the difference in seizure frequency from when participants were receiving PBO to when they were receiving VGB, rather than baseline to treatment period. |
Grunewald 1994.
| Study characteristics | ||
| Methods |
Study design: randomised, double‐blind, PBO‐controlled, parallel‐group study Country: UK Number of treatment arms: 2 Study duration: Phase 1: baseline period (8 weeks) Phase 2: titration period (2 weeks) Phase 3: maintenance period (18 weeks) |
|
| Participants |
Total number of participants randomised: 45 (VGB: 22; PBO 23) Gender: 24 males; 21 females Age: range 15–61 years Number of concomitant AEDs: ≤ 3 Epilepsy type: focal seizures |
|
| Interventions |
Treatment arm 1: VGB 3 g/day Treatment arm 2: PBO |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Marion Merrell Dow, Inc., supported this study and provided the VGB and matching PBO. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "using a random number generated code…" |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "matching placebo." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: attrition reported. However, unclear whether intention‐to‐treat analysis used. |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes listed in the methods section of the publication were reported in the results section. |
| Other bias | Low risk | Comment: study did not include a washout period; however, the first 4 weeks data for each treatment arm were discounted and only latter 8 weeks contributed to the analysis, thus avoiding carry‐over effect. |
Loiseau 1986.
| Study characteristics | ||
| Methods |
Study design: randomised, double‐blind, PBO‐controlled, multicentre, cross‐over study Country: France Number of treatment arms: 2 Study duration: Phase 1: baseline period (5 weeks) Phase 2: first treatment period (10 weeks) Phase 3: second treatment period (10 weeks) Phase 4: single blind PBO period (5 weeks) (Note: data collected during the first week of each treatment period were excluded from the analysis as an attempt to incorporate a 1‐week washout period.) |
|
| Participants |
Total number of participants randomised: 23 Number of participants included in efficacy analysis: 19 Gender: 7 males; 16 females Age: range 10–58 years; mean 28.9 (SD 14.9) years Duration of epilepsy: range 2–40 years; mean 15.1 (SD 10.6) years Number of concomitant AEDs: ≤ 2 Epilepsy type: 19 participants had focal impaired awareness seizures (10 with secondary generalised seizures), 4 participants had primary generalised epilepsy and were therefore excluded from our analysis. |
|
| Interventions |
Treatment arm 1: VGB 3 g/day Treatment arm 2: PBO |
|
| Outcomes |
Primary outcomes
Secondary outcome
|
|
| Notes | Source of funding unknown. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no details regarding random sequence generation provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Identical placebo sachets contained 1.5 g of lactose and I mg of quinine sulfate for taste‐masking." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: attrition reported but intention‐to‐treat analysis not conducted. |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes listed in the methods section of the publication were reported in the results section. |
| Other bias | Unclear risk | Quote: "Only the final 9 weeks of the two double‐blind treatment periods and the final 4 weeks of the single‐blind placebo period (Pll,) were included, thus eliminating the I‐week crossover periods, during which only half the dose was used." Comment: study did not include a washout period. Discarding only 1 week of data at the entry point of each treatment period is unlikely to avoid carryover. |
McKee 1993.
| Study characteristics | ||
| Methods |
Study design: randomised, double‐blind, PBO‐controlled, single centre, cross‐over study Country: Scotland, UK Number of treatment arms: 2 Study duration: Phase 1: baseline period (4 weeks) Phase 2: first treatment period (12 weeks) Phase 3: washout period (4 weeks) Phase 4: second treatment period (12 weeks) |
|
| Participants |
Total number of participants randomised: 24 Gender: 8 males; 16 females Age: range 17–53 years; mean 32.9 (SD 9.9) years Duration of epilepsy: range 4–43 years; mean 19.8 (SD 11.8) years Number of concomitant AEDs: ≤ 2 Epilepsy type: 22 participants with focal impaired‐awareness seizure, of which 14 had secondary generalisation. 2 participants had generalised tonic‐clonic seizures. |
|
| Interventions |
Treatment arm 1: VGB 2 g/day for 6 weeks, then VGB 3 g/day for a further 6 weeks Treatment arm 2: PBO |
|
| Outcomes |
|
|
| Notes | Study funded by Marion Merrell Dow, Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no details regarding random sequence generation provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "matched placebo…" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: attrition reported but intention‐to‐treat analysis not conducted. |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes listed in the methods section of the publication were reported in the results section. |
| Other bias | Low risk | Comment: none detected. |
Rimmer 1984.
| Study characteristics | ||
| Methods |
Study design: randomised, double‐blind, PBO‐controlled, single centre, cross‐over trial Country: Wales, UK Number of treatment arms: 2 Study duration: Phase 1: baseline period (4 weeks) Phase 2: first treatment period (9 weeks) Phase 3: second treatment period (9 weeks) |
|
| Participants |
Total number of participants randomised: 24 Gender: 9 males; 15 females Age: range 16–61 years; mean 33 years Duration of epilepsy: range 6–45 years; mean 21 years Number of concomitant AEDs: ≤ 3 Epilepsy type: mainly focal impaired‐awareness seizures with or without secondary generalisation |
|
| Interventions |
Treatment arm 1: VGB 3 g/day Treatment arm 2: PBO |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Funded by a Wellcome Foundation Research Grant. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no details regarding random sequence generation provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "…matching placebo." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: attrition reported and, although not stated in text, an intention‐to‐treat analysis was conducted. |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes listed in the methods section of the publication were reported in the results section. |
| Other bias | Unclear risk | Comment: study did not include a washout period between treatment arms. |
Tartara 1986.
| Study characteristics | ||
| Methods |
Study design: randomised, double‐blind, PBO‐controlled, multicentre, cross‐over study Country: France and Italy Number of treatment arms: 2 Study duration: Phase 1: first treatment period (7 weeks) Phase 2: second treatment period (7 weeks) |
|
| Participants |
Total number of participants randomised: 23 Gender: 13 males; 10 females Age: range 17–50 years; mean 30.5 (SD 9.7) years Duration of epilepsy: range 2–42 years; mean 17.9 (SD 10.5) years Number of concomitant AEDs: ≤ 2 Epilepsy type: drug‐resistant focal epilepsy in all except 3 participants who were excluded from the results |
|
| Interventions |
Treatment arm 1: VGB 2 g/day or 3 g/day (dependent on bodyweight) Treatment arm 2: PBO |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Source of funding unknown. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Each dose group was individually randomised for sequence of drug/placebo administration, depending on order of entrance into the study." |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "vigabatrin and placebo were supplied as identical sachets, each containing one‐half the daily dosage in the form of a powder which was dissolved in water prior to ingestion." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: attrition reported but intention‐to‐treat analysis not conducted. |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes listed in the methods section of the publication were reported in the results section. |
| Other bias | Unclear risk | Comment: study did not include a washout period and did not exclude any data from the initial crossover. Consequently, carryover was not avoided. The study also did not comprise a baseline period. |
Tassinari 1987.
| Study characteristics | ||
| Methods |
Study design: double‐blind, PBO‐controlled, cross‐over study Country: Italy Number of treatment arms: 2 Study duration: Phase 1: baseline period (2 months) Phase 2: first treatment period (3 months) Phase 3: second treatment period (3 months) Phase 4: single‐blind PBO period (1 month) |
|
| Participants |
Total number of participants randomised: 31 Gender: 16 males; 15 females Age: range 10–58 years; mean 28.9 (SD 11.5) years Number of concomitant AEDs: ≤ 4 Epilepsy type: drug‐resistant focal epilepsy, except for 1 participant who had progressive myoclonic epilepsy and was excluded from the results |
|
| Interventions |
Treatment arm 1: VGB 2 g/day or 3 g/day (dependent on bodyweight) Treatment arm 2: PBO |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Source of funding unknown. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Comment: no details regarding random sequence generation provided. Publication did not explicitly state whether the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details regarding allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Vigabatrin and placebo were supplied in identical individual packets containing 1.0 g or 1.5 g of ingredient to be dissolved in water before ingestion." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no details regarding the blinding of outcome assessment provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: attrition reported and, although not stated in the text, an intention‐to‐treat analysis was conducted. |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes listed in the methods section of the publication were reported in the results section. |
| Other bias | Unclear risk | Comment: study did not include a washout period and did not exclude any data from the initial crossover. Consequently, carryover was not avoided. |
AED: antiepileptic drug; PBO: placebo; SD: standard deviation; SEM: standard error of the mean; VGB: vigabatrin.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Chiron 1996 | Irrelevant study population as it included children with infantile spasms and Lennox‐Gastaut syndrome. Additionally, the study design appeared to be response conditional. Participants who only partially responded to VGB in the first 3 months of treatment were randomised but not included in study. |
| Provinciali 1996 | Single‐blind. |
| Reynolds 1991 | Design was response conditional in that only those participants responding during the initial open phase were entered into the double‐blind phase. |
| Ring 1990 | Design was response conditional in that only those participants responding during the initial open phase were entered into the double‐blind phase. |
| Zahner 1999 | Study was alternate dose‐controlled rather than placebo‐controlled. |
VGB: vigabatrin.
Differences between protocol and review
The methods have evolved since the review was last published; therefore, the methods have been updated such that they still adhere to the standard methodological guidelines expected by Cochrane.
Contributions of authors
RB was responsible for the reporting of the results and final presentation and wording of the review update.
MG acted as second assessor for the screening of search results.
MJM contributed to the previous versions of this review.
CTS contributed to the previous versions of this review.
AGM acted as third author and arbitrated any disagreements.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK
This review update was funded by the National Institute for Health Research (NIHR) [Clinically effective treatments for central nervous system disorders in the NHS, with a focus on Epilepsy and Movement Disorders (SRPG project 16/114/26)]. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Declarations of interest
RB: none known.
MG: none known.
MJM: none known.
CTS: none known.
AGM: UCB Phama funds the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool. Professor Tony Marson is part funded by the National Institute for Health Research Applied Research Collaboration North West Coast (NIHR ARC NWC).
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Beran 1996 {published data only}
- Beran RG, Berkovic SF, Buchanan N, Danta G, Mackenzie R, Schapel G, et al. A double-blind, placebo-controlled crossover study of vigabatrin 2 g/day and 3 g/day in uncontrolled partial seizures. Seizure 1996;5:259-65. [DOI] [PubMed] [Google Scholar]
Bruni 2000 {published data only}
- Bruni J, Guberman A, Vachon L, Desforges C. Vigabatrin as add-on therapy for adult complex partial seizures: a double-blind, placebo-controlled multicentre study. The Canadian Vigabatrin Study Group. Seizure 2000;9:224-32. [DOI] [PubMed] [Google Scholar]
Dean 1999 {published data only}
- Dean C, Mosier M, Penry K. Dose-response study of vigabatrin as add-on therapy in patients with uncontrolled complex partial seizures. Epilepsia 1999;40:74-82. [DOI] [PubMed] [Google Scholar]
- Dodrill CB, Arnett JL, Sommerville KW, Sussman NW. Effects of differing dosages of vigabatrin (Sabril) on cognitive abilities and quality of life in epilepsy. Epilepsia 1995;36(2):164-73. [DOI] [PubMed] [Google Scholar]
French 1996 {published data only}
- Dodrill CB, Arnett JL, Sommerville KW, Sussman NM. Evaluation of the effects of vigabatrin on cognitive abilities and quality of life in epilepsy. Neurology 1993;43:2501-7. [DOI] [PubMed] [Google Scholar]
- French JA, Mosier M, Walker S, Sommerville K, Sussman N. A double-blind, placebo-controlled study of vigabatrin three g/day in patients with uncontrolled complex partial seizures. Neurology 1996;46:54-61. [DOI] [PubMed] [Google Scholar]
Gram 1985 {published data only}
- Gram L, Klosterskov P, Dam M. Gamma-vinyl GABA: a double-blind placebo-controlled trial in partial epilepsy. Annals of Neurology 1985;17:262-6. [DOI] [PubMed] [Google Scholar]
Grunewald 1994 {published data only}
- Grunewald RA, Thompson PJ, Corcoran R, Corden Z, Jackson GD, Duncan JS. Effects of vigabatrin on partial seizures and cognitive function. Journal of Neurology, Neurosurgery, and Psychiatry 1994;57:1057-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Loiseau 1986 {published data only}
- Loiseau P, Hardenberg JP, Pestre M, Guyot M, Schechter PJ, Tell GP. Double blind, placebo controlled study of vigabatrin (gamma vinyl GABA) in drug resistant epilepsy. Epilepsia 1986;27:115-20. [DOI] [PubMed] [Google Scholar]
McKee 1993 {published data only}
- Gilham RA, Blacklaw J, McKee PJ, Brodie MJ. Effect of vigabatrin on sedation and cognitive function in patients with refractory epilepsy. Journal of Neurology 1993;56:1271-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee PJ, Blacklaw J, Friel E, Thompson GG, Gillham RA, Brodie MJ. Adjuvant vigabatrin in refractory epilepsy: a ceiling to effective dosage in individual patients? Epilepsia 1993;34(5):937-43. [DOI] [PubMed] [Google Scholar]
Rimmer 1984 {published data only}
- Rimmer EM, Richens A. Double-blind study of gamma-vinyl GABA in patients with refractory epilepsy. Lancet 1984;1(8370):189-90. [DOI] [PubMed] [Google Scholar]
Tartara 1986 {published data only}
- Tartara A, Manni R, Galimberti CA, Hardenberg J, Orwin J, Perucca E. Vigabatrin in the treatment of epilepsy: a double blind, placebo controlled study. Epilepsia 1986;27:717-23. [DOI] [PubMed] [Google Scholar]
Tassinari 1987 {published data only}
- Tassinari CA, Michelucci R, Ambrosetto G, Salvi F. Double blind study of vigabatrin in the treatment of drug resistant epilepsy. Archives of Neurology 1987;44:907-10. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Chiron 1996 {published data only}
- Chiron C, Dulac O, Mumford J. Vigabatrin withdrawal randomised study in children. Epilepsy Research 1996;25:209-15. [DOI] [PubMed] [Google Scholar]
Provinciali 1996 {published data only}
- Provinciali L, Bartolini M, Mari F, Del Pesce M, Ceravolo MG. Influence of vigabatrin on cognitive performances and behaviour in patients with drug-resistant epilepsy. Acta Neurologica Scandinavica 1996;94:12-8. [DOI] [PubMed] [Google Scholar]
Reynolds 1991 {published data only}
- Reynolds EH, Ring HA, Farr IN, Heller AJ, Elwes RD. Open, double blind and long term study of vigabatrin in chronic epilepsy. Epilepsia 1991;32:530-8. [DOI] [PubMed] [Google Scholar]
Ring 1990 {published data only}
- Ring HA, Farr IN, Reynolds EH. Vigabatrin: rational treatment for chronic epilepsy. Journal of Neurology, Neurosurgery, and Psychiatry 1990;53:1051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zahner 1999 {published data only}
- Zahner B, Stefan H, Blankenhorn V, Kramer G, Richens A, Thumler R, et al. Once-daily versus twice daily vigabatrin: is there a difference? The results of a double blind pilot study. Epilepsia 1999;40:311-5. [DOI] [PubMed] [Google Scholar]
Additional references
Chung 2008
- Chung AM, Eiland LS. Use of second-generation antiepileptic drugs in the pediatric population. Paediatric drugs 2008;10(4):217-54. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cochrane 1998
- Cochrane HC, Marson AG, Baker GA, Chadwick DW. Neuropsychological outcomes in randomised controlled trial of antiepileptic drugs. A systematic review of methodology and reporting standards. Epilepsia 1998;39(10):1088-97. [DOI] [PubMed] [Google Scholar]
Cockerell 1995
- Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the national general practice study of epilepsy. Lancet 1995;346:140-4. [DOI] [PubMed] [Google Scholar]
Eke 1997
- Eke T, Talbot JF, Lawden MC. Severe persistent visual field constriction associated with vigabatrin. British Medical Journal 1997;314(7075):180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fiest 2017
- Fiest Kirsten M, Sauro Khara M, Wiebe Samuel, Patten Scott B, Kwon Churl-Su, Dykeman Jonathan, et al. Prevalence and incidence of epilepsy: A systematic review and meta-analysis of international studies. Neurology 2017;88(3):296-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fisher 2017
- Fisher Robert S, Cross J Helen, French Jacqueline A, Higurashi Norimichi, Hirsch Edouard, Jansen Floor E, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):522-30. [DOI: 10.111/epi.13670] [DOI] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Version accessed 6 January 2020. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015. Available at gradepro.org.
Grant 1991
- Grant SM, Heel RC. Vigabatrin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in epilepsy and disorders of motor control. Drugs 1991;41(6):889-926. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hancock 2008
- Hancock EC, Osborne JP, Edwards SW. Treatment of infantile spasms. Cochrane Database of Systematic Reviews 2008, Issue 4. Art. No: CD001770. [DOI: 10.1002/14651858.CD001770.pub2] [DOI] [PubMed] [Google Scholar]
Higgins 2005
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Chichester (UK): John Wiley & Sons, 2005. [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Maguire 2010
- Maguire MJ, Hemming K, Wild JM, Hutton JL, Marson AG. Prevalence of visual field loss following exposure to vigabatrin therapy: a systematic review. Epilepsia 2010;51(12):2423-31. [DOI: 10.1111/j.1528-1167.2010.02772] [DOI] [PubMed] [Google Scholar]
Marson 1996
- Marson AG, Kadir ZA, Chadwick DW. New antiepileptic drugs: a systematic review of their efficacy and tolerability. British Medical Journal 1996;313(7066):1169-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nicolson 2002
- Nicolson A, Leach JP, Chadwick CW, Smith DF. The legacy of vigabatrin in a regional epilepsy clinic. Journal of Neurology, Neurosurgery, and Psychiatry 2002;73:327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Schechter 1984
- Schechter PJ, Hanke NF, Grove J, Huebert N, Sjoerdsma A. Biochemical and clinical effects of gamma-vinyl GABA in patients with epilepsy. Neurology 1984;34(2):182-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s) GRADE Working Group. Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach (updated October 2013). Available from guidelinedevelopment.org/handbook.
Sterne 2011
- Sterne JA, Egger M, Moher D. Chapter 10: Addressing reporting biases. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Wheless 2005
- Wheless JW, Clarke DF, Carpenter D. Treatment of pediatric epilepsy: expert opinion. Journal of Child Neurology 2005;20 Suppl:1-56. [DOI] [PubMed] [Google Scholar]
Wild 2007
- Wild JM, Ahn HS, Baulac M, Bursztryn J, Chrion C, Gandolfo E, et al. Vigabatrin and epilepsy: lessons learned. Epilepsia 2007;48:1318-27. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Hemming 2008
- Hemming K, Maguire MJ, Hutton JL, Marson AG. Vigabatrin for refractory partial epilepsy. Cochrane Database of Systematic Reviews 2008, Issue 3. Art. No: CD007302. [DOI: 10.1002/14651858.CD007302] [DOI] [PubMed] [Google Scholar]
Hemming 2013
- Hemming K, Maguire MJ, Hutton JL, Marson AG. Vigabatrin for refractory partial epilepsy. Cochrane Database of Systematic Reviews 2013, Issue 1. Art. No: CD007302. [DOI: 10.1002/14651858.CD007302.pub2] [DOI] [PubMed] [Google Scholar]