

Abstract
COVID‐19 is caused by the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), which infects host cells by binding its viral spike protein receptor‐binding domain (RBD) to the angiotensin converting enzyme 2 (ACE2) on host cells. Blocking the SARS‐CoV‐2‐RBD/ACE2 interaction is, therefore, a potential strategy to inhibit viral infections. Using a novel biopanning strategy, a small anti‐ACE2 peptide is discovered, which shows high affinity and specificity to human ACE2. It blocks not only the SARS‐CoV‐2‐RBD/ACE2 interaction but also the SARS‐CoV‐1‐RBD/ACE2 interaction. Moreover, it inhibits SARS‐CoV‐2 infection in Vero‐E6 cells. The peptide shows negligible cytotoxicity in Vero‐E6 cells and Huh7 cells. In vivo short‐term lung toxicity study also demonstrates a good safety of the peptide after intratracheal administration. The anti‐ACE2 peptide can be potentially used as a prophylactic or therapeutic agent for SARS‐CoV‐2 or other ACE2‐mediated viruses. The strategy used in this study also provides a fast‐track platform to discover other antiviral peptides, which will prepare the world for future pandemics.
Keywords: ACE2, peptide inhibitor, phage biopanning, RBD, SARS‐CoV‐2
Peptide therapeutics is gaining increased interest in recent years due to its high selectivity, efficacy, and tolerability. Small anti‐ACE2 peptides that can effectively block the SARS‐CoV‐2‐RBD/ACE2 interaction are discovered using peptide phage biopanning. These peptides show high affinity and specificity toward human ACE2 and are capable of inhibiting the SARS‐CoV‐2 infection. This strategy can be used as a fast‐track platform to discover other antiviral peptides.
1. Introduction
Since the outbreak of the coronavirus disease 2019 (COVID‐19) in December 2019, the disease has quickly spread to 219 countries and caused more than 131 million infections. The COVID‐19 pandemic has not only claimed more than 2.8 million lives in the world but also greatly shaken the global economy.[ 1 ] COVID‐19 is caused by the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), which shares nearly 80% genome sequence identity to the severe acute respiratory syndrome coronavirus (SARS‐CoV‐1).[ 2 ] SARS‐CoV‐2, SARS‐CoV‐1, and the Middle East respiratory syndrome coronavirus (MERS‐CoV) belong to the Betacoronavirus genus, a type of positive‐sense, single‐stranded RNA (+ssRNA) viruses. Similar to SARS‐CoV‐1 and MERS, SARS‐CoV‐2 infects the lower respiratory tract of human patients and causes pneumonia.[ 2 ]
Both SARS‐CoV‐2 and SARS‐CoV‐1 infect host cells by binding their viral spike (S) proteins to the angiotensin converting enzyme 2 (ACE2) on host cells.[ 3 , 4 ] The S glycoprotein contains two subunits: the N‐terminal S1 subunit for binding to host receptors and the C‐terminal S2 subunit for the fusion of viral and cellular membranes. The S protein binds to the extracellular peptidase domain (PD) of ACE2 through the receptor‐binding domain (RBD) in the S1 subunit. Upon binding, the S protein is cleaved at the S1/S2 site by the transmembrane serine protease 2 (TMPRSS2), and the S2 subunit facilitates the fusion of the virus with host cells.[ 3 , 5 ] Although the interaction between SARS‐CoV‐2‐RBD and ACE2 is similar to that between SARS‐CoV‐1‐RBD and ACE2, the binding affinity of SARS‐CoV‐2‐RBD to ACE2 is much higher because of several sequence variations in the binding interface of its RBD.[ 3 , 4 ]
ACE2 was first discovered as a key enzyme in the renin‐angiotensin system (RAS) and plays important roles in maintaining heart function, regulation of blood pressure, and diabetes.[ 6 , 7 ] ACE2 is abundantly expressed in many organs, such as the lung, heart, kidney, small intestine, stomach, and oral mucosa.[ 8 ] Particularly, the lung is the most vulnerable target organ of SARS‐CoV‐2 because of its very large surface area, leading to high susceptibility to inhaled viruses.[ 9 ] Moreover, the majority of ACE2‐expressing lung cells are alveolar epithelial type II cells, which are more vulnerable to viral infection because of their high expressions of genes related to viral replication and transmission.[ 10 ]
Until now, there is no specific antiviral therapy for COVID‐19. In December 2020, The U.S. Food and Drug Administration (FDA) issued an emergency use authorization for the first COVID‐19 vaccine. Since then, the FDA has approved three COVID‐19 vaccines for emergency use. A general opinion about COVID‐19 is that a vaccine will bring the pandemic to a decisive end. However, more than 35% of Americans would choose not to get vaccinated against SARS‐CoV‐2 even if a vaccine is FDA‐approved and free.[ 11 ] The situation could be more complicated if a vaccine is only partially effective. In addition, the number of mutated SARS‐CoV‐2 is growing, which may compromise the efficiency of the approved vaccines. Therefore, there remains an urgent need to develop antiviral drugs for SARS‐CoV‐2 even after vaccines approved for emergency use.
Binding of SARS‐CoV‐2‐RBD to ACE2 is the first and essential step in viral infections. As a result, blocking the interaction of SARS‐CoV‐2‐RBD to ACE2 is a potential strategy to inhibit viral infections. For example, monoclonal antibodies, nanobody, and cellular nanosponges have been developed to bind SARS‐CoV‐2 and subsequently prevent the interaction of its RBD to ACE2.[ 9 , 12 , 13 ] Development of anti‐ACE2 inhibitors is another strategy to block the interaction between SARS‐CoV‐2‐RBD and ACE2. However, ACE2 plays important functions in the body, and anti‐ACE2 inhibitors should not affect its physiological functions. A small and highly specific inhibitor is, therefore, an ideal candidate to block the SARS‐CoV‐2 infection.
Peptide therapeutics is gaining popularity in recent years with 15 new peptide drugs being approved by the FDA in the last five years. Scenesse (a 13‐mer linear peptide for the treatment of skin damage) and Vyleesi (a 7‐mer cyclic peptide for premenopausal women with hypoactive sexual desire disorder) are examples of the peptide drugs approved by the FDA in 2019.[ 14 ] In general, peptides are highly selective, effective, and safe. Compared to small molecules that often trigger side effects by toxic metabolites or nonspecific accumulation in the body, peptides can be metabolized to amino acids in the body and have a rare incidence of side effects.[ 15 , 16 ] Phage display technology is an attractive tool for the discovery of novel peptides or proteins. The 2018 Nobel Prize in Chemistry was awarded to George Smith and Sir Gregory Winter for their contribution in the phage display of peptides and antibodies. Phage display biopanning is a high‐throughput screening of a phage library that displays billions of different peptides or proteins. Phage display has been successfully adopted to identify protein or peptide ligands against a wide variety of molecular targets including animal tissues, cells, and proteins.[ 17 , 18 , 19 ] These peptide ligands have been widely used as targeting ligands for drug delivery or imaging. Moreover, they have been explored as therapeutic agents, such as vaccines,[ 20 ] immunotherapy agents,[ 21 ] and inhibitors of a target protein.[ 22 , 23 , 24 , 25 ] For example, the world's best‐selling drug, HUMIRA, was discovered using the phage display technology. We recently developed a novel biopanning strategy to discover small peptides that can specifically bind to programmed death‐ligand 1 (PD‐L1) and block its interaction with the programmed cell death 1 (PD‐1).[ 19 ]
In the present study, we report the discovery of small anti‐ACE2 peptides using a peptide phage display library. The peptides bind to ACE2 with high affinity and high specificity. Binding of the peptides to ACE2 specifically blocks the interaction between the SARS‐CoV‐2‐RBD and ACE2.
2. Results and Discussions
The FDA has recently approved three mRNA‐based vaccines for COVID‐19, but no therapeutics have been proven effective for the treatment of COVID‐19.[ 26 ] A variety of therapeutic approaches like inhibition of RNA‐dependent RNA polymerase, inhibition of protease enzyme, inhibition of virus‐cell membrane fusion, modulating the immune system, and neutralizing inflammatory response are undergoing clinical trials.[ 27 ] Several antiviral agents including remdesivir, favipiravir, and lopinavir‐ritonavir have been used for off‐label treatment of COVID‐19. Although Remdesivir initially attracted the most attention as a potential anti‐COVID‐19 drug, a randomized and double‐blind multicenter trial found that remdesivir use was not associated with statistically significant clinical benefits.[ 28 ] In a case study, favipiravir relieves the symptoms of COVID‐19 patients with severe to critical conditions.[ 29 ] A recent clinical trial found no benefit of the lopinavir‐ritonavir therapy in severe COVID‐19 patients beyond standard care.[ 26 ] Similar result was reported in a systematic benefit‐risk assessment of the lopinavir‐ritonavir treatment.[ 30 ] Antimalarial drugs like chloroquine and hydroxychloroquine have also been used as potential therapeutics for COVID‐19. However, in a randomized clinical trial including more than 4700 patients hospitalized with COVID‐19, hydroxychloroquine did not reduce the incidence of death compared to usual care.[ 31 ]
The SARS‐CoV‐2‐RBD/ACE2 interaction can be blocked either by antivirus (e.g., anti‐RBD) inhibitors or by anti‐ACE2 inhibitors. Antivirus inhibitors may potentially block the epitopes on the virus surface, which will compromise the host immune responses against the virus. Also, targeting viral spike RBD may be less effective for mutated viruses. By contrast, targeting ACE2 on host cells may avoid the potential problem associated with virus mutation. Therefore, we hypothesize that anti‐ACE2 inhibitors are more efficient than anti‐RBD inhibitors in blocking the SARS‐CoV‐2‐RBD/ACE2 interaction. Furthermore, anti‐ACE2 inhibitors can be used as prophylactic or therapeutic agents for SARS‐CoV‐2.
Although anti‐ACE2 antibodies can block the RBD/ACE2 interaction, their clinical applications are mainly hampered by their expensive and time‐consuming manufacturing processes in eukaryotic systems. In addition to a long time in developing monoclonal antibodies, large‐scale production of monoclonal antibodies takes at least 3–6 months, making it difficult to combat a pandemic, such as COVID‐19, in a timely manner.[ 13 ] By contrast, the discovery of small peptides using phage display is a rapid process (approximately 3 weeks for biopanning), and peptides can be easily synthesized on a large scale, making peptides better candidates to combat a pandemic like COVID‐19. A typical Current Good Manufacturing Practices (cGMP) peptide synthesizer can chemically synthesize small peptides (up to 50 aa) on a multikilogram scale. Small peptides have other advantages, such as low immunogenicity and lack of Fc‐mediated side effects, such as antibody‐dependent enhancement (ADE).[ 32 ] Moreover, a small peptide can interact with residues in ACE2 that are not accessible by a large antibody. In summary, peptide‐based anti‐ACE2 inhibitors are considered attractive candidates to combat the COVID‐19 pandemic.
2.1. Discovery of Anti‐ACE2 Peptides Using Phage Biopanning
In this study, we aim to discover an anti‐ACE2 peptide that can block the SARS‐CoV‐2‐RBD/ACE2 interaction, thereby blocking the virus infection (Figure 1a). We previously developed a novel biopanning strategy, which can discover small peptides that only bind to specific residues of a protein.[ 19 ] Using the same biopanning strategy, we screened a 12‐mer peptide phage library against human ACE2 protein. After five rounds of biopanning, we observed a significant enrichment of eluted phages (Figure 1b). Eighty‐six phage clones were randomly selected, and 26 unique peptide sequences were discovered (Figure 1c).
Figure 1.
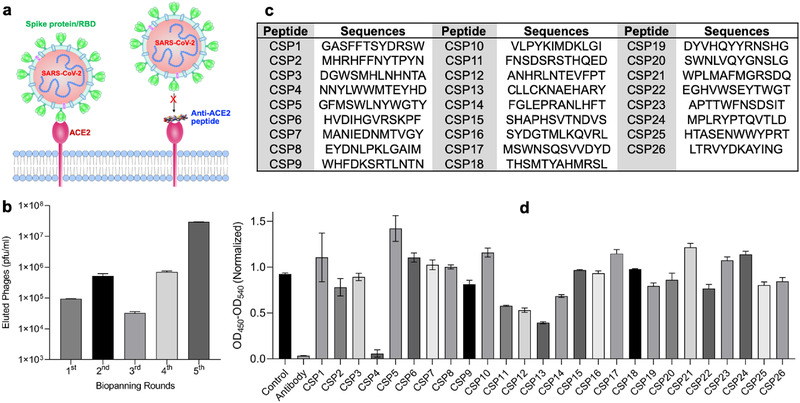
Discovery of anti‐ACE2 peptides using phage display biopanning. a) Schematic of blocking SARS‐CoV‐2 infections by anti‐ACE2 peptides. b) The number of eluted phages after each round of biopanning. c) Sequences of identified anti‐ACE2 peptides. d) Blocking effect of the anti‐ACE2 peptides (10 × 10−6 m) and an anti‐ACE2 antibody (200 × 10−9 m) on the SARS‐CoV‐2‐RBD/ACE2 interaction. All results are presented as the mean ± SD (n = 3).
2.2. Anti‐ACE2 Peptides Block the SARS‐CoV‐2‐RBD/ACE2 Interaction
We first screened the blocking effect of these peptides (10 × 10−6 m) on the interaction of SARS‐CoV‐2‐RBD and human ACE2 protein. As shown in Figure 1d, the CSP4 peptide exhibited the highest blocking effect, followed by CSP11, CSP12, and CSP13 peptides. An anti‐ACE2 antibody (Cat# AF933, R&D Systems, Minneapolis, MN) was used as a positive control in this assay.
Next, we determined the half‐maximal inhibitory concentration (IC50) of the anti‐ACE2 antibody, peptide CSP4, and peptide CSP13 (Figure 2 ). The anti‐ACE2 antibody blocked the SARS‐CoV‐2‐RBD/ACE2 interaction with an IC50 of 43 × 10−9 m. The IC50 values of CSP4 and CSP13 peptides are 635 × 10−9 and 709 × 10−9 m, respectively. However, the maximal blocking efficiency of the CSP13 peptide is only about 30%, while the anti‐ACE2 antibody and the CSP4 peptide completely blocked the interaction at high concentrations. The low blocking efficiency of the CSP13 peptide at high concentrations is possibly because its binding site on ACE2 is not fully overlapped with the SARS‐CoV‐2‐RBD/ACE2 interaction interface.
Figure 2.
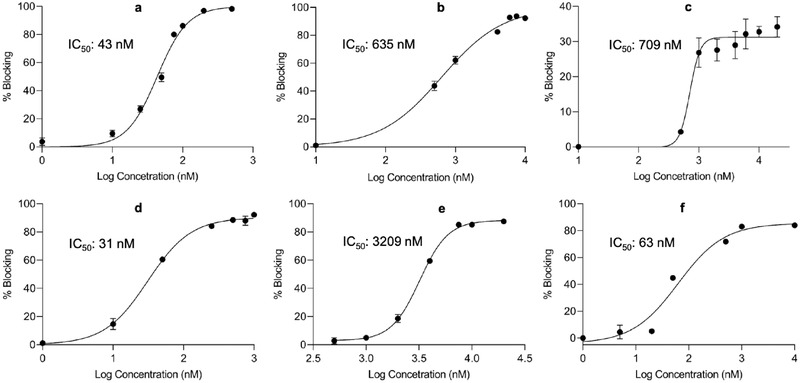
Blockade assay of the SARS‐CoV‐2‐RBD/ACE2 and SARS‐CoV‐1‐RBD/ACE2 interactions by anti‐ACE2 peptides. Blocking efficiency and IC50 of a) the anti‐ACE2 antibody, b) CPS4 peptide, c) CPS13 peptide, and d) the dimer of CPS4 peptide against the SARS‐CoV‐2‐RBD/ACE2 interaction. Blocking efficiency and IC50 of e) the CPS4 peptide and f) CPS4 dimer against the SARS‐CoV‐1‐RBD/ACE2 interaction. All results are presented as the mean ± SD (n = 3).
It is known that the multimerization of a peptide ligand can increase its affinity.[ 18 , 19 ] We, therefore, synthesized a dimer of the CSP4 peptide by linking two monomers to the ─NH2 groups of a Lysine. As illustrated in Figure 2d, dimerization of the CSP4 peptide dramatically decreased its IC50 by 20‐fold to 31 × 10−9 m. Very recently, the high‐resolution Cryo‐EM structure of full‐length human ACE2 revealed the dimerization of ACE2 on cell membrane.[ 3 ] This explains why the dimerization of the CSP4 peptide dramatically increases binding to ACE2.
Both SARS‐CoV‐1 and SARS‐CoV‐2 use the homotrimer spike protein to bind human ACE2 for viral infections. Although the binding affinity of SARS‐CoV‐1‐RBD/ACE2 is weaker than that of SARS‐CoV‐2‐RBD/ACE2, the overall structure of SARS‐CoV‐1‐RBD is similar to that of SARS‐CoV‐2‐RBD. Additionally, the overall binding mode of the SARS‐CoV‐1‐RBD to ACE2 is nearly the same as that of the SARS‐CoV‐2‐RBD to ACE2.[ 4 , 33 ] We, therefore, evaluated whether the CSP4 peptide and its dimer can also block the SARS‐CoV‐1‐RBD/ACE2 interaction (Figure 2e,f). Both the CSP4 peptide and its dimer showed a notable blocking effect with IC50 values of 3209 × 10−9 and 63 × 10−9 m, respectively. Considering the fact that the CSP4 peptide was screened against the SARS‐CoV‐2‐RBD/ACE2 interaction, it is not surprising that the peptides exhibited slightly lower blocking effect on the SARS‐CoV‐1‐RBD/ACE2 interaction. Nevertheless, these results suggest that the newly discovered anti‐ACE2 peptide can be potentially used to block the infectivity of both SARS‐CoV‐2 and SARS‐CoV‐1.
2.3. Evaluation of Binding Affinity and Blocking Efficiency Using Competition Surface Plasmon Resonance (SPR)
SPR is a classic technique to study the interaction of two different proteins. However, direct assay of the interaction between small molecules, such as small peptides, and a protein by SPR is always troublesome due to the low responses from small molecules.[ 34 ] We, therefore, adopted competition SPR to evaluate the binding of the peptide CSP4 and its dimer to the ACE2 protein using a five‐channel BI‐4500 SPR. Approximately 750 RU (RU—response unit) of biotinylated SARS‐CoV‐2‐RBD (Acro Biosystems) was immobilized on a CM Dextran Chip. PBS (pH 7.4) was used as the running buffer, and 10 × 10−3 m NaOH was used for regeneration. The flow rate was set at 60 µL min−1. Competition SPR was used to assess the ability of the anti‐ACE2 peptides to block the interaction between human ACE2 and SARS‐CoV‐2‐RBD. As illustrated in Figure 3a, peptides with different concentrations were incubated with 5 × 10−9 m human ACE2 protein at 37 °C for 30 min before injecting into the chip coated with SARS‐CoV‐2‐RBD. Free human ACE2 solution (5 × 10−9 m) was injected as a control. Increasing the concentration of the peptide CSP4 or CSP4 dimer decreases the binding signal of the ACE2 protein to SARS‐CoV‐2‐RBD coated on the chip, indicating a concentration‐dependent binding of the peptides to the ACE2 protein in the mobile phase (Figure 3b–g). In accordance with the blocking data in Figure 2, the dimer of the CSP4 peptide exhibited higher blocking efficiency to the ACE2 protein (Figure 3e–g).
Figure 3.
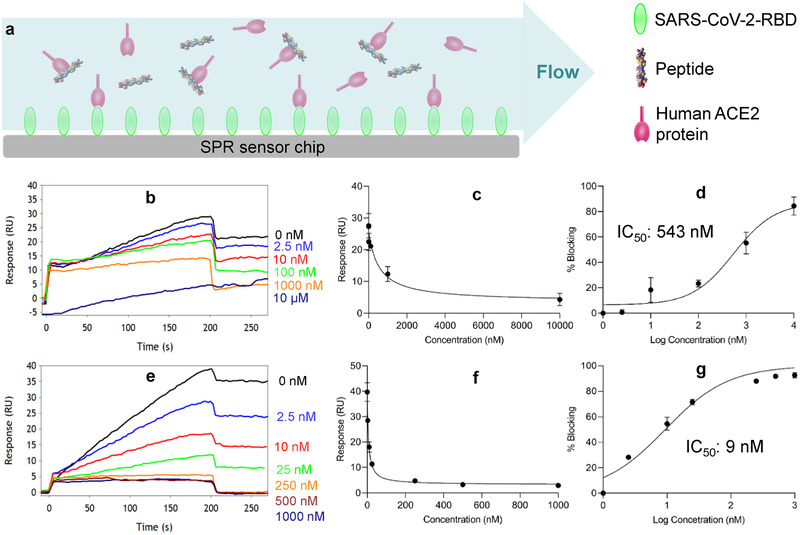
Evaluation of binding affinity and blocking efficiency of the peptides to ACE2 using competition SPR. a) Schematic of the competition of SPR. b) SPR sensorgram, c) response curve, and d) blocking curve of the CSP4 peptide. e) SPR sensorgram, f) response curve, and g) blocking curve of the CSP4 peptide dimer. All results are presented as the mean ± SD (n = 3).
2.4. Binding Specificity of the Peptides to ACE2
Having shown a high binding affinity of the anti‐ACE2 peptides to recombinant ACE2 protein, we next examined whether these peptides also specifically bind to the ACE2 protein expressed on cells. A549 cells are human lung epithelial carcinoma cells with a low expression of endogenous ACE2.[ 35 ] We stably expressed human ACE2 protein in A549 cells and compared the binding affinity of Cy5‐labeled anti‐ACE2 peptides to A549 and A549/ACE2 cells. As shown in Figure 4a, binding of the peptide CSP4 to A549/ACE2 cells is significantly higher than binding to A549 cells, indicating a good specificity of the peptide to the ACE2 protein expressed on A549 cells. Similar specificity was observed for the peptide CSP13 and the dimer of CSP4 (Figure 4b,c). It is notable to mention that 10% FBS was included in the medium when these peptides were incubated with suspended cells, suggesting that the binding affinity and specificity of these peptides to ACE2 may not be compromised by serum proteins in the body.
Figure 4.
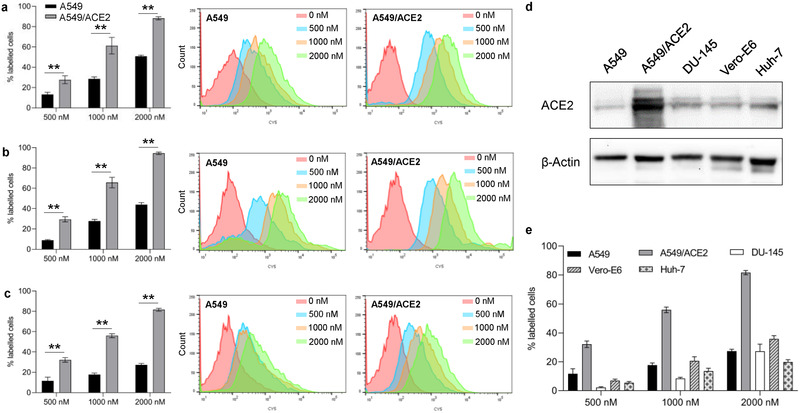
Binding specificity of anti‐ACE2 peptides to ACE2. Percent of A549 and A549/ACE2 cells that bind to Cy5‐labeled peptides a) CSP4, b) CSP13, and c) CSP4 dimer at different concentrations and their corresponding histogram plots. Statistical significance was determined by two‐tailed Student's t‐test (**P < 0.01). d) Representative western blot images of ACE2 expression levels in A549, A549/ACE2, DU‐145, Vero‐E6, and Huh‐7 cells. e) Binding of CSP4 dimer at different concentrations (500 × 10−9 , 1000 × 10−9 , and 2000 × 10−9 m) to A549, A549/ACE2, DU‐145, Vero‐E6, and Huh‐7 cells. All results are presented as the mean ± SD (n = 3).
To further confirm the specificity of CSP4 dimer toward ACE2, we carried out the binding study of CSP4 dimer in different cell lines with varying expression levels of ACE2. As illustrated in Figure 4d, the A549/ACE2 stable cell line shows the highest expression of ACE2, while A549, DU‐145, Vero‐E6 and Huh‐7 show low expression of ACE2. In accordance with the ACE2 expression, CSP4 dimer exhibited the highest binding to A549/ACE2 stable cell line but low binding to other cell lines (Figure 4e). This result further confirms the specificity of CSP4 dimer toward ACE2.
2.5. Cell Toxicity and Stability Studies
Cell cytotoxicity of the anti‐ACE2 peptides was evaluated in Vero‐E6 monkey kidney cells and Huh7 human liver cells, both of which endogenously express ACE2. As shown in Figure 5a,b, the peptides CSP4, CSP13, and CSP4 dimer did not induce toxicity in Vero‐E6 and Huh7 cells at 50 × 10−6 m after 24 h incubation, demonstrating an excellent safety of these peptides.
Figure 5.
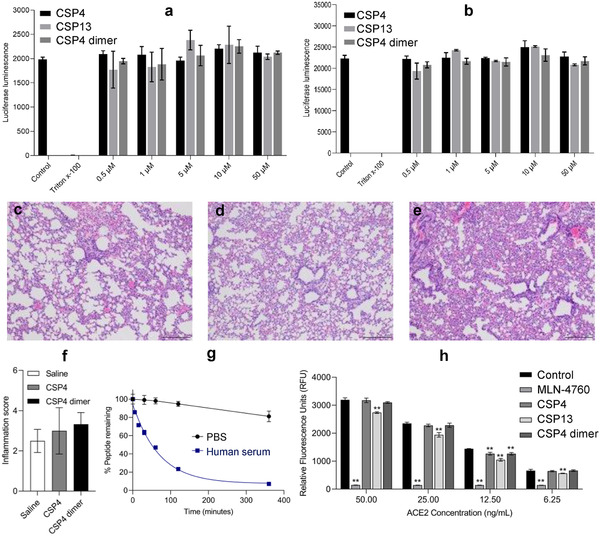
Safety and stability evaluation of the anti‐ACE2 peptides. a) Cytotoxicity of CSP4, CSP13, and CSP4 dimer peptides in Vero‐E6 cells at different concentrations. Triton X‐100 was used as a positive control. b) Cytotoxicity of CSP4, CSP13, and CSP4 dimer peptides in Huh‐7 cells at different concentrations. Triton X‐100 was used as a positive control. Representative H&E staining of lung specimens from the mice (3–4 mice per group) 72 h after intratracheal administration of c) saline, d) CSP4, and e) CSP4 dimer peptides (Scale bar: 100 µm). The peptides were administrated at a dose of 2 mg kg−1. f) Inflammation scores (0–5 scale) of the lung specimens. g) Stability of CSP4 dimer peptide in PBS and human serum. h) Effect of anti‐ACE2 peptides on ACE2 enzyme activity. The ACE2 inhibitor MLN‐4760 was used as a positive control. Cytotoxicity and ACE2 enzyme activity results are presented as the mean ± SD (n = 3). Statistical significance was determined by one‐way ANOVA (a, b, f, and h) with Tukey's multiple comparison. ** p < 0.01.
Because SARS‐CoV‐2 most often affects the airway and lungs of patients, inhalation is the most appropriate route to deliver the anti‐ACE2 peptides. Moreover, inhalation allows for rapid onset of action and high dose to the lung while minimizing systemic exposure.[ 36 , 37 ] We, therefore, conducted an in vivo short‐term lung toxicity study in mice to evaluate the safety of the anti‐ACE2 peptides after intratracheal administration at a dose of 2 mg kg−1. After 72 h, the lungs were harvested for histological analysis. As illustrated in Figure 5c–e, all lungs showed somewhat alveolar septal lymphohistiocytic inflammation and infiltrates, which are caused by intratracheal administration of liquids. All specimens show mild to moderate inflammation. However, no pulmonary vascular changes were observed. Moreover, inflammation of the lungs was quantified using a scale of 0–5 (Figure 5f). No statistical significance was found among the saline, CSP4, and CSP4 dimer groups, suggesting that peptide itself does not induce toxicities in the lung.
We next evaluated the stability of the CSP4 dimer in PBS and human serum (Figure 5g). The CSP4 dimer exhibited good stability in PBS with over 80% of the peptides remaining intact after incubation for 6 h. In human serum, the half‐life of the CSP4 dimer was found to be 48.19 min, which is typical for a short natural peptide. Because the lung is the most vulnerable target organ of SARS‐CoV‐2, the best way to administer the anti‐ACE2 peptides is through pulmonary route. Considering the fact that the lungs contain less enzymes, the relatively short half‐life of the peptides in the serum may not be a hurdle for their application as aerosols. Moreover, the stability of the peptides can be improved by terminal modification, incorporation of non‐natural amino acids, or cyclization.[ 38 ]
2.6. Assessment of the Enzymatic Activity of ACE2 in the Presence of Anti‐ACE2 Peptides
ACE2 is a key enzyme in the RAS and plays important roles in maintaing heart function, regulation of blood pressure, and diabetes.[ 6 , 7 ] ACE2 is abundantly expressed in many organs, such as the lung, heart, kidney, small intestine, stomach, and oral mucosa.[ 8 ] It is, therefore, critical that anti‐ACE2 agents should not interfere with ACE2's physiological activities. We performed an ACE2 enzymatic activity to evaluate whether the anti‐ACE2 peptides interfere with the biological activity of recombinant human ACE2 at different concentrations. CPS4, CPS13, and CPS4 dimer at a concentration of 10 × 10−6 m were incubated with four different concentrations of ACE2 (50, 25, 12.5, and 6.25 ng mL−1) to assess the ACE2 activity. As illustrated in Figure 5h, the ACE2 inhibitor (MLN‐4760) dramatically inhibited the activity of ACE2, and CPS13 slightly reduced the enzymatic activity of ACE2. By contrast, CPS4 and CPS4 dimer did not affect the enzymatic activity of ACE2, except for a slight reduction of ACE2 activity at 12.5 ng mL−1. These findings suggest that anti‐ACE2 peptides CPS4 and CPS4 dimer are effective in blocking the SARS‐CoV‐2 binding without affecting the physiological activity of ACE2 protein.
2.7. Plaque Reduction Neutralization Assay Using SARS‐CoV‐2 Virus
Vero‐E6 cell line has been widely used as a model for SARS‐CoV‐2 infection assays.[ 9 , 39 ] We, therefore, used Vero‐E6 cells for the plaque reduction neutralization assay in a BSL‐3 laboratory. As illustrated in Figure 6 , the CSP4 dimer exhibited a dose‐dependent neutralization effect, suggesting that the peptide can inhibit the infectivity of SARS‐CoV‐2 in Vero‐E6 cells. By contrast, the CSP4 peptide only showed moderate neutralization effect at higher concentration, which is consistent with its relatively poor blocking effect in Figures 2 and 3.
Figure 6.
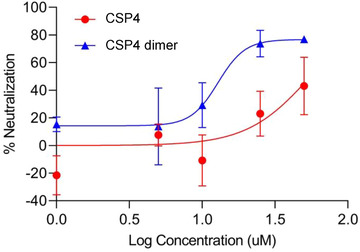
The CSP4 dimer inhibits the infectivity of SARS‐CoV‐2 in Vero‐E6 cells. Vero‐E6 cells were incubated with the anti‐ACE2 peptides at 37 ℃ for 1 h, followed by incubation with 100 pfu of SARS‐CoV‐2 virus at 37 ℃ for 1 h. After removing the virus, overlay media were added, and the plate were incubated at 37 ℃ for 4 d. The cells were then stained, and plaques were counted to determine the inhibitory effect of the peptides. The results are presented as the mean ± SD (n = 3).
3. Conclusion
In conclusion, we discovered an anti‐ACE2 peptide, which shows high affinity and specificity to human ACE2. Dimerization of the peptide dramatically increases its blocking efficiency. It blocks not only the SARS‐CoV‐2‐RBD/ACE2 interaction but also the SARS‐CoV‐1‐RBD/ACE2 interaction. Moreover, it inhibits SARS‐CoV‐2 infection in Vero‐E6 cells. The anti‐ACE2 peptide can be potentially used as a prophylactic or therapeutic agent for SARS‐CoV‐2 or other ACE2‐mediated viruses. The strategy used in this study may also establish a fast‐track platform to discover other antiviral peptides, which will prepare us for future pandemics.
4. Experimental Section
Cells
Huh‐7 cell line (JCRB0403) was purchased from Sekisui XenoTech, LLC (Kansas City, KS). Vero‐E6 (CRL‐1586), DU‐145 (HTB‐81) and A549 (CCL‐185) cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA). A549 cells stably expressing ACE2 (A549‐ACE2) were generated by transducing a human ACE2‐expressing lentivirus and cultured in the presence of blasticidin hydrochloride at 10 µg mL−1 for five passages. These cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (FBS) (10%), streptomycin (100 µg mL−1), and penicillin (100 units mL−1).
Phage Biopanning Procedure
Biopanning was conducted in 96‐well plates as was reported before.[ 19 ] Two wells in a plate were coated with recombinant human ACE2 protein (200 ng) (catalog# 933‐ZN‐010, R&D Systems, Minneapolis, MN) overnight at 4 ℃ with gentle shaking. The first well was blocked with BSA (2%) for 2 h, followed by incubation with 2019‐nCov Spike Protein RBD (500 ng) (catalog# 40592‐V05H, Sino Biological, Wayne, PA) for 2 h. The Ph.D.‐12 Phage Display Peptide Library (catalog # E81102, New England BioLabs, Ipswich, MA) was added to the first well and incubated at room temperature for 1 h. Unbound phages from the first well were then transferred to the second well and incubated for 1 h. The bound phages were eluted and amplified for the next round of biopanning. After five rounds of biopanning, randomly selected colonies of phages were sequenced, and encoded peptides were synthesized using a PurePep Chorus peptide synthesizer (Gyros Protein Technologies, Tucson, AZ) and then purified with HPLC.
Blockade of the Spike Protein‐ACE2 Interaction
A 96‐well plate was coated with SARS‐CoV‐2 S protein RBD (50 ng) (catalog# SPD‐C52H3, Acro Biosystems, Newark, DE) per well overnight at 4 ℃ and then blocked with BSA (2%) at room temperature for 1.5 h. Biotinylated human ACE2 protein (4 ng/100 µL) (catalog# AC2‐H82E6, Acro Biosystems, Newark, DE) was preincubated with anti‐ACE2 peptides or anti‐ACE2 antibody (catalog# AF933, R&D Systems, Minneapolis, MN) at room temperature for 1 h and then added to the plate. After incubation for 1 h, Streptavidin‐HPR and substrate were added to each well to measure absorbance (at 450 nm).
Evaluation of Binding Affinity by Competition Surface Plasmon Resonance
The binding affinity of the anti‐ACE2 peptides to ACE2 was evaluated using a five‐channel BI‐4500 SPR (Biosensing Instrument Inc., Tempe, AZ). ≈750 RU of biotinylated SARS‐CoV‐2 S protein RBD (catalog# SPD‐C82E9, Acro Biosystems, Newark, DE) was immobilized on a CM Dextran Chip pre‐functionalized with streptavidin. PBS (pH 7.4) was used as the running buffer, and NaOH (10 × 10−3 m) was used for regeneration following each binding interaction. The flow rate was set at 60 µL min−1. Competition SPR was used to assess the ability of the anti‐ACE2 peptides to block the interaction between human ACE2 (catalog# AC2‐H52H8, Acro Biosystems, Newark, DE) and SARS‐CoV‐2 S protein RBD. Peptides with different concentrations were incubated with 5 × 10−9 m human ACE2 protein at 37 °C for 30 min before injection into the chip coated with SARS‐CoV‐2 S protein RBD. Free human ACE2 solution (5 × 10−9 m) was injected as a control. The data were analyzed with SPR Data Analysis Version 3.8.4 (Biosensing Instrument Inc., Tempe, AZ).
Flow Cytometry‐Based Binding Assay
Binding specificity of the anti‐ACE2 peptides to A549 cells, A549‐ACE2 stable cells, Vero‐E6 cells, Huh‐7 cells, and DU‐145 cells was evaluated as described before with modifications.[ 18 , 19 ] The cells were detached from flasks using nonenzymatic cell dissociation solution (Catalog#13151‐014, Gibco, Waltham, MA) and re‐suspended to a density of 1 × 106 cells mL−1 in DMEM containing FBS (10%). Cy5‐labeled peptides with various concentrations were incubated with the suspended cells (0.5 mL) at 37 °C for 2 h. After washing with PBS, the cells were subjected to fluorescence analysis using a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ).
Western Blot
ACE2 expression in A549, A549/ACE2 stable cells, Vero‐E6 cells, Huh‐7 cells, and DU‐145 cells was compared using western blot. These cells were lysed with RIPA Lysis and Extraction Buffer (Catalog#89 901, Thermo Fisher Scientific, IL), and total proteins were quantified using the bicinchoninic acid (BCA) assay. Twenty micrograms of total cell lysate protein from each cell line were separated on a 12% SDS‐PAGE gel, transferred to a polyvinylidene difluoride (PVDF) membrane, and blotted with the ACE2 antibody (Catalog#AF933, R&D Systems, Minneapolis, MN). The membrane was also blotted with a β‐actin antibody (Catalog#MAB8929, R&D Systems, Minneapolis, MN) as internal control.
Cell Viability
Cell viability was determined using the CellTiter‐Glo Luminescent Cell Viability Assay (Catalog#G7572, Promega, Madison, WI) as per the protocol. Vero‐E6 and Huh7 cells were seeded in a 96‐well plate at a density of 5000 cells per well. After 24 h of incubation, the medium was replaced with fresh medium containing various concentrations of ACE‐2 peptides (0.25, 0.5, 1, 5,10, 50 × 10−6 m). After incubation for another 24 h, the medium was removed, and CellTiter‐Glo Reagent (80 µL) was added into each well. The plate was mixed in a shaker for 2 min and incubated at room temperature for 10 min before luminescence was measured.
Measurement of ACE2 Activity in the Presence of Anti‐ACE2 Peptides
ACE2 activity was measured as previously described.[ 40 ] The experiment was carried out in a 96‐well black clear bottom plate. Briefly, 10 × 10−6 m of CPS4, CPS13, CPS4 dimer, or MLN‐4760 (ACE2 activity inhibitor, Catalog # 530 616, Sigma‐Aldrich, St. Louis, MO) were incubated with recombinant human ACE2 protein (Catalog# 10108‐H08B, Sino Biological, Wayne, PA) and ACE2 fluorescent substrate (Catalog# AS‐60757, AnaSpec, Fremont, CA) at room temperature for 16 h. ACE2 activity was then determined by measuring the intramolecularly quenched ACE2 fluorogenic substrate on a SpectraMax M5e (Molecular Devices, Sunnyvale, CA) microplate reader with excitation wavelength of 320 × 10−9 m and emission wavelength of 405 × 10−9 m.
Serum Stability
The anti‐ACE2 peptides were incubated with PBS or 50% human serum (Catalog# BP2525‐100, Fisher Scientific, NJ) at a final concentration of 20 × 10−6 m. After 30, 60, 120, or 360 min, an aliquot was collected and incubated with acetonitrile containing 0.1% trifluoroacetic acid on ice for 15 min. It was then centrifuged at 12 000 g for 10 min at 4˚C, and the supernatant was analyzed using an AB/SCIEX API 4000 QTrap mass spectrometer (Foster City, CA).
In Vivo Short‐Term Lung Toxicity
The animal protocol (protocol number: 2050) was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Missouri‐Kansas City. To evaluate the short‐term toxicity, the anti‐ACE2 peptides were intratracheally administered to Balb/c mice (3–4 mice per group) at a specific dose (2 mg kg−1), and the lungs were harvested after 72 h for histological analysis. The whole section of each mouse's lung specimen was evaluated for inflammation using a score scale of 0–5: scale 0—no inflammation (no inflammatory cells noted in peri‐alveolar/interstitial space); scale 1—mild inflammation (focal/patchy areas of chronic inflammatory cells in peri‐alveolar/interstitial space); scale 2—mild inflammation (involvement of most but not all peri‐alveolar/interstitial spaces by mild chronic inflammation); scale 3—moderate inflammation (most peri‐alveolar/interstitial spaces involved by moderate chronic inflammation); scale 4—severe inflammation (diffuse/severe chronic inflammation in near all peri‐alveolar/interstitial space); scale 5—severe inflammation (diffuse/severe chronic inflammation in all peri‐alveolar/interstitial space).
SARS‐CoV‐2 and Propagation
All experimental procedures with SARS‐CoV‐2 were approved by the Institutional Biosafety Committee (IBC) of the University of Kansas Medical Center. Work with SARS‐CoV‐2 was conducted in the BSL3 Lab (Hemenway 4037) of the University of Kansas Medical Center. The SARS‐CoV‐2 virus, isolate USA‐WA1/2020 (NR‐52281), was obtained from Biodefense and Emerging Infections Research Resources Repository (BEI Resources), NIAID, NIH. The virus was propagated in Vero‐E6 cells once, titrated by plaque assay,[ 41 ] aliquoted, and stored at −80 °C. The virus titer of the wild‐type SARS‐CoV‐2 was 1.0 × 107 pfu mL−1, which equals a physical titer of 8.2 × 109 viral genome copies (vgc) mL−1 determined by reverse transcription‐quantitative PCR (RT‐qPCR).[ 41 ]
Plaque Reduction Neutralization Assay Using SARS‐CoV‐2
Vero‐E6 cells were seeded in a 24‐well plate at a density of 0.5 × 106 cells and grown to confluence the second day. The anti‐ACE2 peptides with different concentrations were added into each well in DMEM with 2% FBS (200 µL) and were incubated for 1 h at 37 °C. 100 pfu of SARS‐CoV‐2 were then added to each well and incubated for another hour at 37 °C on a rocking rotator. The medium containing viruses was removed, and ≈0.5 mL of overlay media (1% methylcellulose in DMEM containing 5% FBS) were added to each well. The plates were incubated at 37 °C under 5% CO2 for 4 d. After removing the methylcellulose overlays, cells were fixed using 10% formaldehyde solution for 30 min, stained with 1% crystal violet solution, and washed twice with distilled water. Plaques in each well were manually counted to determine the inhibitory efficacy of the peptide on SARS‐CoV‐2 infection.
Statistical Analysis
Data were presented as the mean ± standard deviation (SD). An independent Student's t‐test was used to compare differences if there were two groups or one‐way ANOVA followed by Tukey's multiple comparison test to compare differences when there were more than two groups of data. All tests were two‐sided, and p values below the 5% level were regarded as significant. All the data were analyzed and graphed using GraphPad Prism (version 8).
Acknowledgements
P.A. and S.K. contributed equally to this work. This work was supported in part by the National Institutes of Health (R01AA021510, R01CA231099, and R01GM121798).
Data Availability Statement
Research data are not shared.
References
- 1. COVID‐19 Coronavirus Pandemic (Reported Cases and Deaths by Country, Territory, or Conveyance), 2020. https://www.worldometers.info/coronavirus/.
- 2. Zhou P., Yang X. L., Wang X. G., Hu B., Zhang L., Zhang W., Si H. R., Zhu Y., Li B., Huang C. L., Chen H. D., Chen J., Luo Y., Guo H., Jiang R. D., Liu M. Q., Chen Y., Shen X. R., Wang X., Zheng X. S., Zhao K., Chen Q. J., Deng F., Liu L. L., Yan B., Zhan F. X., Wang Y. Y., Xiao G. F., Shi Z. L., Nature 2020, 579, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q., Science 2020, 367, 1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Othman H., Bouslama Z., Brandenburg J. T., da Rocha J., Hamdi Y., Ghedira K., Srairi‐Abid N., Hazelhurst S., Biochem. Biophys. Res. Commun. 2020, 527, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walls A. C., Park Y. J., Tortorici M. A., Wall A., McGuire A. T., Veesler D., Cell 2020, 181, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Somasundaram R., Choraria A., Antonysamy M., Int. Immunopharmacol. 2020, 85, 106654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turner A. J., Hiscox J. A., Hooper N. M., Trends Pharmacol. Sci. 2004, 25, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hamming I., Timens W., Bulthuis M. L., Lely A. T., Navis G., van Goor H., J. Pathol. 2004, 203, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Q., Honko A., Zhou J., Gong H., Downs S. N., Vasquez J. H., Fang R. H., Gao W., Griffiths A., Zhang L., Nano Lett. 2020, 20, 5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao Y., Zhao Z., Wang Y., Zhou Y., Ma Y., Zuo W., Am J. Respir. Crit. Care Med. 2020, 202, 756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O'Keefe S. M., One in Three Americans Would Not Get COVID‐19 Vaccine , 2020. https://news.gallup.com/poll/317018/one-three-americans-not-covid-vaccine.aspx.
- 12. Ejemel M., Li Q., Hou S., Schiller Z. A., Tree J. A., Wallace A., Amcheslavsky A., Yilmaz N. K., Buttigieg K. R., Elmore M. J., Godwin K., Coombes N., Toomey J. R., Schneider R., Ramchetty A. S., Close B. J., Chen D. Y., Conway H. L., Saeed M., Ganesa C., Carroll M. W., Cavacini L. A., Klempner M. S., Schiffer C. A., Wang Y., Nat. Commun. 2020, 11, 4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu Y., Li C., Xia S., Tian X., Kong Y., Wang Z., Gu C., Zhang R., Tu C., Xie Y., Yang Z., Lu L., Jiang S., Ying T., Cell Host Microbe 2020, 27, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de la Torre B. G., Albericio F., Molecules 2020, 25, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahrens V. M., Bellmann‐Sickert K., Beck‐Sickinger A. G., Future Med. Chem. 2012, 4, 1567. [DOI] [PubMed] [Google Scholar]
- 16. Smith M. C., Gestwicki J. E., Expert Rev. Mol. Med. 2012, 14, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith G. P., Display P., Angew. Chem., Int. Ed. Engl. 2019, 58, 14428. [DOI] [PubMed] [Google Scholar]
- 18. Chen Z., Jin W., Liu H., Zhao Z., Cheng K., Mol. Pharmaceutics 2015, 12, 2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu H., Zhao Z., Zhang L., Li Y., Jain A., Barve A., Jin W., Liu Y., Fetse J., Cheng K., J. ImmunoTher. Cancer 2019, 7, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frenzel A., Schirrmann T., Hust M., mAbs 2016, 8, 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luzar J., Strukelj B., Lunder M., Allergy 2016, 71, 1526. [DOI] [PubMed] [Google Scholar]
- 22. Titus J. K., Kay M. K., Glaser C. J. J., J. Venom Res. 2017, 8, 19. [PMC free article] [PubMed] [Google Scholar]
- 23. Marcozzi A., Masini T., Zhu D., Pesce D., Illarionov B., Fischer M., Herrmann A., Hirsch A. K. H., ChemBioChem 2018, 19, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Villard S., Lacroix‐Desmazes S., Kieber‐Emmons T., Piquer D., Grailly S., Benhida A., Kaveri S. V., Saint‐Remy J. M., Granier C., Blood 2003, 102, 949. [DOI] [PubMed] [Google Scholar]
- 25. Warren J. G., Kasun G. W., Leonard T., Kirkpatrick B. C., Mol. Plant Pathol. 2016, 17, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., Li X., Xia J., Chen N., Xiang J., Yu T., Bai T., Xie X., Zhang L., Li C., Yuan Y., Chen H., Li H., Huang H., Tu S., Gong F., Liu Y., Wei Y., Dong C., Zhou F., Gu X., N. Engl. J. Med. 2020, 382, 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tu Y. F., Chien C. S., Yarmishyn A. A., Lin Y. Y., Luo Y. H., Lin Y. T., Lai W. Y., Yang D. M., Chou S. J., Yang Y. P., Wang M. L., Chiou S. H., Int. J. Mol. Sci. 2020, 21, 2657. [Google Scholar]
- 28. Wang Y., Zhang D., Du G., Du R., Zhao J., Jin Y., Fu S., Gao L., Cheng Z., Lu Q., Hu Y., Luo G., Wang K., Lu Y., Li H., Wang S., Ruan S., Yang C., Mei C., Wang Y., Ding D., Wu F., Tang X., Ye X., Ye Y., Liu B., Yang J., Yin W., Wang A., Fan G., Lancet 2020, 395, 1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takahashi H., Iwasaki Y., Watanabe T., Ichinose N., Okada Y., Oiwa A., Kobayashi T., Moriya M., Oda T., Int. J. Infect. Dis. 2020, 100, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Osborne V., Davies M., Lane S., Evans A., Denyer J., Dhanda S., Roy D., Shakir S., Drug Saf. 2020, 43, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Group R. C., Horby P., Mafham M., Linsell L., Bell J. L., Staplin N., Emberson J. R., Wiselka M., Ustianowski A., Elmahi E., Prudon B., Whitehouse T., Felton T., Williams J., Faccenda J., Underwood J., Baillie J. K., Chappell L. C., Faust S. N., Jaki T., Jeffery K., Lim W. S., Montgomery A., Rowan K., Tarning J., Watson J. A., White N. J., Juszczak E., Haynes R., Landray M. J., N. Engl. J. Med. 2020, 383, 2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vlieghe P., Lisowski V., Martinez J., Khrestchatisky M., Drug Discovery Today 2010, 15, 40. [DOI] [PubMed] [Google Scholar]
- 33. Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X., Nature 2020, 581, 215. [DOI] [PubMed] [Google Scholar]
- 34. de Mol N. J., Methods Mol. Biol. 2010, 627, 101. [DOI] [PubMed] [Google Scholar]
- 35. Ma D., Chen C. B., Jhanji V., Xu C., Yuan X. L., Liang J. J., Huang Y., Cen L. P., Ng T. K., Eye 2020, 34, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kipshidze N., Iversen P., Porter T. R., Kipshidze N., Siddiqui F., Dangas G., Fareed J., Clin. Appl. Thromb./Hemostasis 2020, 26, 107602962095491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Boer A. H., Hagedoorn P., Hoppentocht M., Buttini F., Grasmeijer F., Frijlink H. W., Expert Opin. Drug Delivery 2017, 14, 499. [DOI] [PubMed] [Google Scholar]
- 38. Werle M., Bernkop‐Schnurch A., Amino Acids 2006, 30, 351. [DOI] [PubMed] [Google Scholar]
- 39. Sun Z., Chen C., Li W., Martinez D. R., Drelich A., Baek D. S., Liu X., Mellors J. W., Tseng C. T., Baric R. S., Dimitrov D. S., mAbs 2020, 12, 1778435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao F., Burns K. D., Methods Mol. Biol. 2017, 1527, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hao S., Ning K., Kuz C. A., Vorhies K., Yan Z., Qiu J., mBio 2020, 11, e02852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Research data are not shared.