

Abstract
Mitochondrial fission is sustained through contact with several organelles, including the endoplasmic reticulum, lysosomes, and the actin cytoskeleton. Nagashima et al. (2020) now demonstrate that PI(4)P-containing Golgi-derived vesicles also modulate mitochondrial fission, driven by Arf1 and PI(4)KIIIβ activity, identifying a new organelle contact involved in maintaining mitochondrial homeostasis.
Mitochondrial fission and fusion are crucial events that maintain the structure and function of the mitochondrial network. Mitochondrial fission is guided by the endoplasmic reticulum (ER) (Friedman et al., 2011). Sites of mitochondrial-ER contacts (MERCs) initiate fission and mark the site of constriction. This process is followed by recruitment of the GTPase Dynamin-related-protein 1 (Drp1) to the outer mitochondrial membrane. Drp1 oligomerizes to encircle the mitochondria and drives the constriction of the mitochondrial membrane through GTP hydrolysis (Chang and Blackstone, 2010). Other factors such as phospholipids, calcium, and lysosomes have been implicated at different stages of mitochondrial fission, suggesting the contribution of additional organelles is needed for efficient fragmentation of the mitochondrial network.
ADP-ribosylation factor 1 (Arf1) is a small GTPase of the Ras superfamily that is recruited to the Golgi apparatus and activated by a Golgi-localized exchange factor to promote the generation of COP1-coated vesicles (Sztul et al., 2019). Arf1 also activates phosphatidylinositol 4-kinase-III-β (PI(4)KIIIβ), which mediates the phosphorylation of phosphatidylinositol to generate phosphatidylinositol 4-phosphate (PI(4)P). PI(4)P interacts with proteins on the ER and Golgi to remodel these membranes and regulate ER-Golgi contact sites (Chung et al., 2015). A previous report demonstrated loss of Arf1 activity in Caenorhabditis elegans resulted in mitochondrial hyperfusion (Ackema et al., 2014); however, the exact mechanism was not clear. Given the involvement of Arf1 and PI(4)KIIIβ in membrane remodeling, Nagashima and collaborators set out to address the mechanisms by which PI(4)P pools contribute to mitochondrial homeostasis (Nagashima et al., 2020).
To test specifically whether Arf1 deficiency leads to mitochondrial remodeling in mammalian cells, the authors knocked down Arf1 in HeLa cells. Arf1 silencing led to a hyperfused and branched morphology of the mitochondrial network, confirming the phenotype shown in C. elegans is conserved in human cells. Transmission electron microscopy revealed excess mitochondrial branching and abnormal sites of hyperconstriction in both Arf1- and PI(4)KIIIβ-deficient cells. Hyperconstricted sites were characterized by the appearance of an exceptionally long, narrow neck with the inner and outer membranes running in parallel. Further, knockdown of other PI(4)K family members or ceramide transfer protein (CERT), another Arf1 effector, did not cause mitochondrial hyperfusion, indicating the role of Arf1 in mitochondrial fission is specific to its regulation of PI(4)KIIIβ. These observations raise the possibility that Arf1 or PI(4)KIIIβ silencing has an effect on the maintenance of cristae morphology/dynamics; characterizing the cristae in this model could identify potential impairment of electron transport chain machinery and metabolic output. The authors showed that the ER was found in close proximity to hyperconstriction sites, suggesting the role of Arf1 is downstream of MERC formation at sites of fission. Further, there were no detected changes in expression levels of profission or pro-fusion regulators, and no excessive accumulation of the mitochondrial fusion machinery proteins. In fact, silencing of Mfn1 and Mfn2, two GTPases crucial for mitochondrial outer membrane fusion, did not rescue the hyperfusion defects seen in PI(4)KIIIβ-silenced cells. This intriguing observation implies the hyperfused phenotype in PI(4)KIIIβ-deficient cells is modulated through lipid-mediated remodeling of the mitochondrial network rather than overactive fusion.
Though both Arf1 and PI(4)KIIIβ primarily localize to the Golgi, Arf1 and PI(4)KIIIβ foci were observed at MERC sites. To uncover the order of events in fission, the authors used live-cell imaging to show that GFP-tagged Arf1 is recruited to the majority of fission events following Drp1 recruitment. The Arf1-GFP foci did not fully co-localize with ER markers and displayed distinct recruitment from lysosomes, suggesting Arf1 is recruited to MERCs from a different cellular compartment. Indeed, the authors found that Arf1-GFP foci are recruited to sites of mitochondrial constriction via trans-Golgi network (TGN)-derived vesicles positive for TGN46 and PI(4)P. PI(4)P foci were enriched at MERC-induced constriction sites, and accumulation of these PI(4)P foci depends upon expression of Arf1 and PI(4)KIIIβ. The authors speculate that the PI(4)P pools may aid in the final steps of mitochondrial division by recruiting adaptor proteins necessary for actin polymerization (Dong et al., 2016). However, the exact mechanism by which PI(4)P contributes to mitochondrial fission remains unclear. The lack of PI(4)P pools at the mitochondria in Arf1-silenced cells presumably prevents membrane remodeling, suggesting that the hyperfused phenotype is due to lack of curvature-inducing lipids capable of dividing membranes at fission sites.
While mitochondria were first described as dynamic organelles over 100 years ago (Lewis and Lewis, 1914), the development of new imaging technologies has made it easier to track mitochondria in live cells. The remarkable ability of mitochondria to continually divide and fuse is crucial for normal physiology. This study by Nagashima et al. provides new mechanistic insight into the complex process of mitochondrial fission, in which contacts between ER, lysosome, mitochondria, and now Golgi membranes drive the majority of fission events (Figure 1). Like all important discoveries, this report also raises fundamental new questions. Why have cells evolved such complex organelle communication to drive a single mitochondrial fission event? Contacts between different organelles may provide ways of communication, adaptability, and integration of distinct molecular events that might impact overall cell physiology. Due to these organelle contact sites, alterations or signals at one organelle can have repercussions and generate responses in distant, interconnected locations. How are inter-organelle contacts coupled to the excision of damaged mitochondria and/or the activity of the mitophagy machinery? Examining whether Golgi-derived vesicles also carry proteins involved in cell death or mitophagy to the mitochondria could reveal novel aspects of this crosstalk. It is also unclear if Golgi-derived vesicles are targeted to the ER first, where they can be brought to mitochondria through MERCs, or if they are recruited directly to the mitochondrial membrane. Interestingly, mitochondrial membrane lipid content is regulated in part by interactions with the ER membrane, leading to fine-tuning of metabolic processes and coordination of cell death (Chipuk et al., 2012). Whether Golgi-derived vesicles also influence the lipid composition of the mitochondrial membrane could be a fascinating area to investigate. Importantly, de novo Arf1 mutations affecting its GDP/GTP-binding domain have been reported to cause brain malformations and impaired neuronal migration (Ge et al., 2016) by undetermined mechanisms. It is plausible that dysfunctional mitochondrial network phenotypes in neuronal cells could explain the aberrant neurodevelopmental processes. Overall, the exciting findings described by Nagashima and colleagues provide the first clear indication that the trans-Golgi network is also required for mitochondrial fission. Understanding the molecular mechanisms that guide these highly coordinated organelle contacts will pave the way toward elucidating how mitochondrial health and quality control underlie human development and disease.
Figure 1. A Simplified Look at the Process of Mitochondrial Fission.
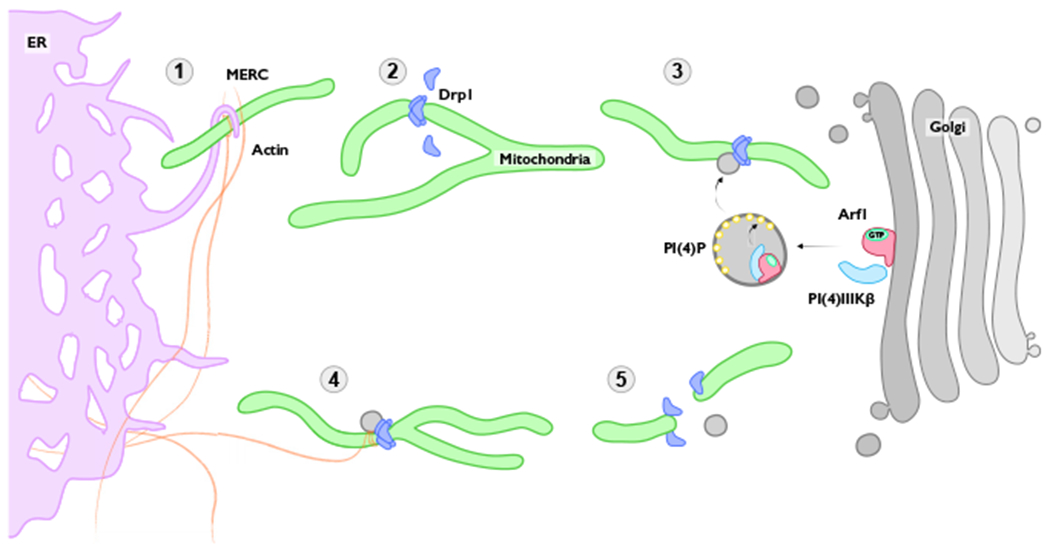
(1) ER-mitochondria contacts (MERCs) are formed to mark future sites of fission. F-actin polymerization concurrently aids in pre-constriction of the mitochondria. (2) Drp1 is recruited to its receptor proteins on the outer mitochondrial membrane through post-translational modifications. Drp1 matures and oligomerizes to initiate further constriction. (3) PI(4)P-containing Golgi-derived vesicles accumulate at Drp1 foci. (4) Further constriction of the outer mitochondrial membrane is promoted by F-actin filaments and Drp1 GTP hydrolysis. (5) Dynamic activity of all components results in complete fission of the mitochondria, with Golgi-derived vesicles dissociating prior to Drp1.
ACKNOWLEDGMENTS
We thank our colleague Dr. Kristopher Burkewitz for expert advice. Funding was provided by an American Heart Association pre-doctoral fellowship (19PRE34380515 to M.L.R.); National Institutes of Health (NIH)/National Institute of General Medical Sciences (NIGMS) Cellular, Biochemical, and Molecular Sciences (CBMS) T32 (to G.L.R.); NIH/NIGMS 1R35GM128915-01 (to V.G.); and NIH/National Cancer Institute 1R21 CA227483-01A1 (to V.G.).
REFERENCES
- Ackema KB, Hench J, Böckler S, Wang SC, Sauder U, Mergentaler H, Westermann B, Bard F, Frank S, and Spang A (2014). The small GTPase Arf1 modulates mitochondrial morphology and function. EMBO J. 33, 2659–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-R, and Blackstone C (2010). Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N Y Acad. Sci 1201, 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, and Green DR (2012). Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, and De Camilli P (2015). Intracellular transport. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 349, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R, Saheki Y, Swarup S, Lucast L, Harper JW, and De Camilli P (2016). Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell 166, 408–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, and Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Gong H, Dumas K, Litwin J, Phillips JJ, Waisfisz Q, Weiss MM, Hendriks Y, Stuurman KE, Nelson SF, et al. (2016). Missense-depleted regions in population exomes implicate ras superfamily nucleotide-binding protein alteration in patients with brain malformation. NPJ Genom. Med. 1, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MR, and Lewis WH (1914). Mitochondria in tissue culture. Science 39, 330–333. [DOI] [PubMed] [Google Scholar]
- Nagashima S, Tábara L-C, Tilokani L, Paupe V, Anand H, Pogson JH, Zunino R, McBride HM, and Prudent J (2020). Golgi-derived PI(4)P-containing vesicles drive late steps of mitochondrial division. Science 367, 1366–1371. [DOI] [PubMed] [Google Scholar]
- Sztul E, Chen P-W, Casanova JE, Cherfils J, Dacks JB, Lambright DG, Lee FS, Randazzo PA, Santy LC, Schürmann A, et al. (2019). ARF GTPases and their GEFs and GAPs: concepts and challenges. Mol. Biol. Cell 30, 1249–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]