

Abstract
De novo heterozygous variants in the brain-specific transcription factor Neuronal Differentiation Factor 2 (NEUROD2) have been recently associated with early-onset epileptic encephalopathy and developmental delay. Here, we report an adolescent with developmental delay without seizures who was found to have a novel de novo heterozygous NEUROD2 missense variant, p.(Leu163Pro). Functional testing using an in vivo assay of neuronal differentiation in Xenopus laevis tadpoles demonstrated that the patient variant of NEUROD2 displays minimal protein activity, strongly suggesting a loss of function effect. In contrast, a second rare NEUROD2 variant, p.(Ala235Thr), identified in an adolescent with developmental delay but lacking parental studies for inheritance, showed normal in vivo NEUROD2 activity. We thus provide clinical, genetic, and functional evidence that NEUROD2 variants can lead to developmental delay without accompanying early-onset seizures, and demonstrate how functional testing can complement genetic data when determining variant pathogenicity.
Keywords: Neurod2, developmental delay
Introduction
Neuronal Differentiation Factor 2 (NEUROD2), encodes for a basic helix-loop-helix (bHLH) transcription factor that is exclusively expressed in the brain and binds to highly conserved enhancer box response elements in the promotor regions of target DNA [Longo and others 2008; Uhlen and others 2015]. The protein is known to play a major role in nervous system development and function, and is essential for neuronal survival and synaptic transmission [Bormuth and others 2013; Chen and others 2016; Guner and others 2017; Olson and others 2001]. NEUROD2 is important for neuronal differentiation, maturation, and circuitry formation [Guzelsoy and others 2019; Hahn and others 2019; Matsushita and others 2017; Pieper and others 2019]. Mice with a mixed C57Bl/6 × 129/Sv background lacking Neurod2 display ataxia, motor deficits, and have a reduced seizure threshold [Olson and others 2001]. They also have failure to thrive with early postnatal death associated with low thyroid hormone levels, though this resolves with backcrossing to pure 129/Sv [Lin and others 2006]. Detailed regional analysis of the brains of null mice has implicated NEUROD2 in development and maturation of thalamocortical projections, survival of cerebellar and hippocampal neurons, formation of inhibitory synapses in the cerebellar cortex, maturation of hippocampal connectivity, and radial migration of cerebral cortical neurons [Guzelsoy and others 2019; Ince-Dunn and others 2006; Olson and others 2001; Pieper and others 2019; Wilke and others 2012].
We previously reported two children with early-onset epileptic encephalopathy and significant global development delay who had de novo heterozygous missense variants in the DNA-binding domain of NEUROD2 (OMIM# 601725) [Sega and others 2019]. Both patients were non-responsive to multiple antiepileptic medications, one becoming seizure-free with a ketogenic diet, the other with a vagal nerve stimulator. We also demonstrated that the patient variants were dysfunctional by using an in vivo model of neuronal induction in tadpoles.
Here we present an additional patient with a de novo missense variant in NEUROD2, predicted to be damaging by in silico tools and confirmed in our in vivo model. Her characteristics are notable primarily for global development delay without the early-onset epileptic encephalopathy of the previously observed phenotype. We also report another patient with developmental delay and a rare missense variant with unknown inheritance in NEUROD2. In this case, however, our in vivo evidence suggests a normally functioning protein, pointing to the importance of functional evidence to support a genetic diagnosis of NEUROD2-related neurologic disease.
Clinical Descriptions
Patient 1 is a 14-year-old female of maternal European/Native American and paternal German/Irish descent with global development delay. She was born full term with bilateral fifth finger clinodactyly and borderline short stature, but without other dysmorphic features. She was diagnosed with failure to thrive in infancy, and since has experienced constipation, childhood hypotonia, respiratory distress, and poor feeding requiring hospital admission. She began crawling at 10–11 months, walking at 18 months, and spoke her first words at 2.5 years. While she does not carry a formal diagnosis of autism, she has pervasive developmental disorder and sensory processing disorder among other learning disabilities. Assessment of developmental milestones at age 12 years showed mild gross motor issues while her handwriting (fine motor) was not markedly impaired. Verbal functioning at that time was at or above average for her age. She had an average performance in language arts but was far below average for visual motor/constructional tasks and math skills. Brain magnetic resonance imaging (MRI) at 9 years of age was normal. Comparative genomic hybridization array and focused genetic testing for Rett Syndrome, cystic fibrosis, and Russell Silver Syndrome were all negative.
Patient 2 is a 12-year-old boy with autism spectrum, attention deficit hyperactivity disorder, sensory processing disorder, and developmental coordination disorder. He was adopted, and therefore family history as well as parental samples were unavailable. Comparative genomic hybridization array, karyotype, fragile X, and targeted Prader Willi testing were all negative.
Identification and Analyses of Patient Variants
Exome sequencing for Patient 1 and her parents was performed at the National Institutes of Health (NIH) Undiagnosed Diseases Network (UDN) sequencing core and analyzed using bioinformatics and variant interpretation pipelines as previously described [Lee and others 2014; Lee and others 2020] [PMID: 31607746, PMID: 25326637]. A de novo variant in NEUROD2 (NM_006160.3), c.488T>C, p.(Leu163Pro), which was not observed in gnomAD was identified (Figure 1). She had a second de novo variant, in NSD2 p.(Gly840Val). NSD2 (OMIM# 602952) lies in a region associated with Wolf-Hirschhorn Syndrome, a well-described syndrome with characteristic dysmorphisms and organ malformations not found in this patient. Exome sequencing for Patient 2 also revealed a novel missense variant in NEUROD2, c.703G>A, p.(Ala235Thr). As parental samples were not available, we were unable to assess whether the variant was de novo, though the variant was not observed in gnomAD, which raised the potential for pathogenicity. He also had rare compound heterozygous variants in a second gene, TMEM76 (OMIM# 610453), which is associated with Sanfilippo syndrome Type C. As neither NSD2 nor TMEM76 appear to be appropriate candidates for these two patients, we focused our attention on NEUROD2.
Figure 1:
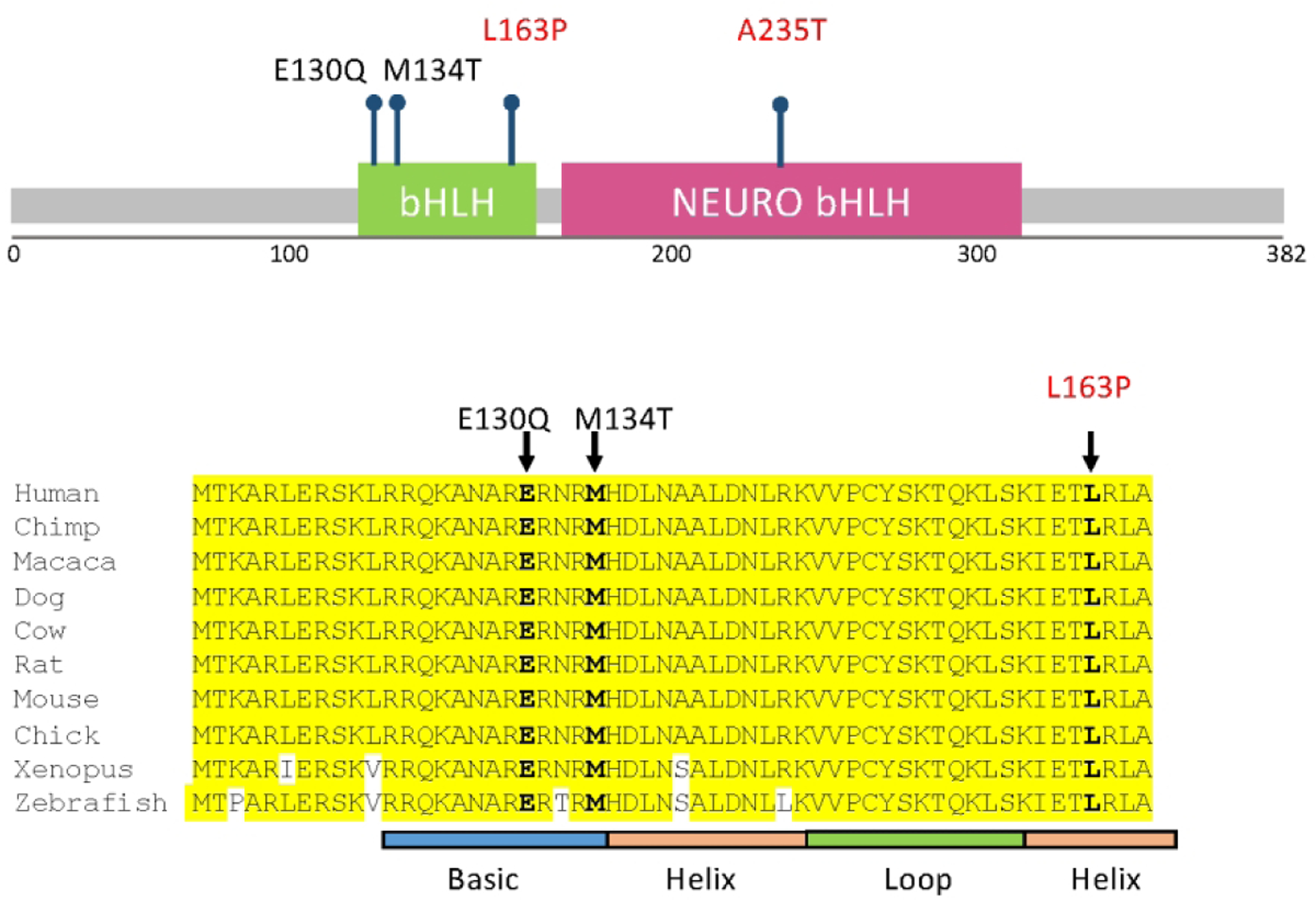
Location of reported variants in NEUROD2. Top panel shows location of all reported variants in relation to bHLH (green box) and NEURO bHLH (magenta box) domains. Bottom panel shows close-up of variants found in the bHLH domain, demonstrating the high degree of amino acid conservation across multiple species. Previously reported variants are in black text, variants reported in this paper are in red text. Adapted from Sega and others, 2019.
In order to determine pathogenicity of the individual variants, we tested them in an in vivo Xenopus laevis model of NEUROD2 function as previously described [Sega and others 2019]. Briefly, we injected wild type or patient variants of NEUROD2 mRNA into only one cell of fertilized X. laevis eggs at the two cell stage. We then compared the induction of ectopic neurons in the resulting tadpoles, with the side developing from the uninjected cell serving as an internal control. Analysis revealed that injection of the p.Leu163Pro mRNA variant resulted in a defective ability to induce ectopic neurons in tadpoles as compared to wild type NEUROD2 mRNA, while the p.Ala235Thr variant functioned similarly to wild type (Figure 2).
Figure 2:
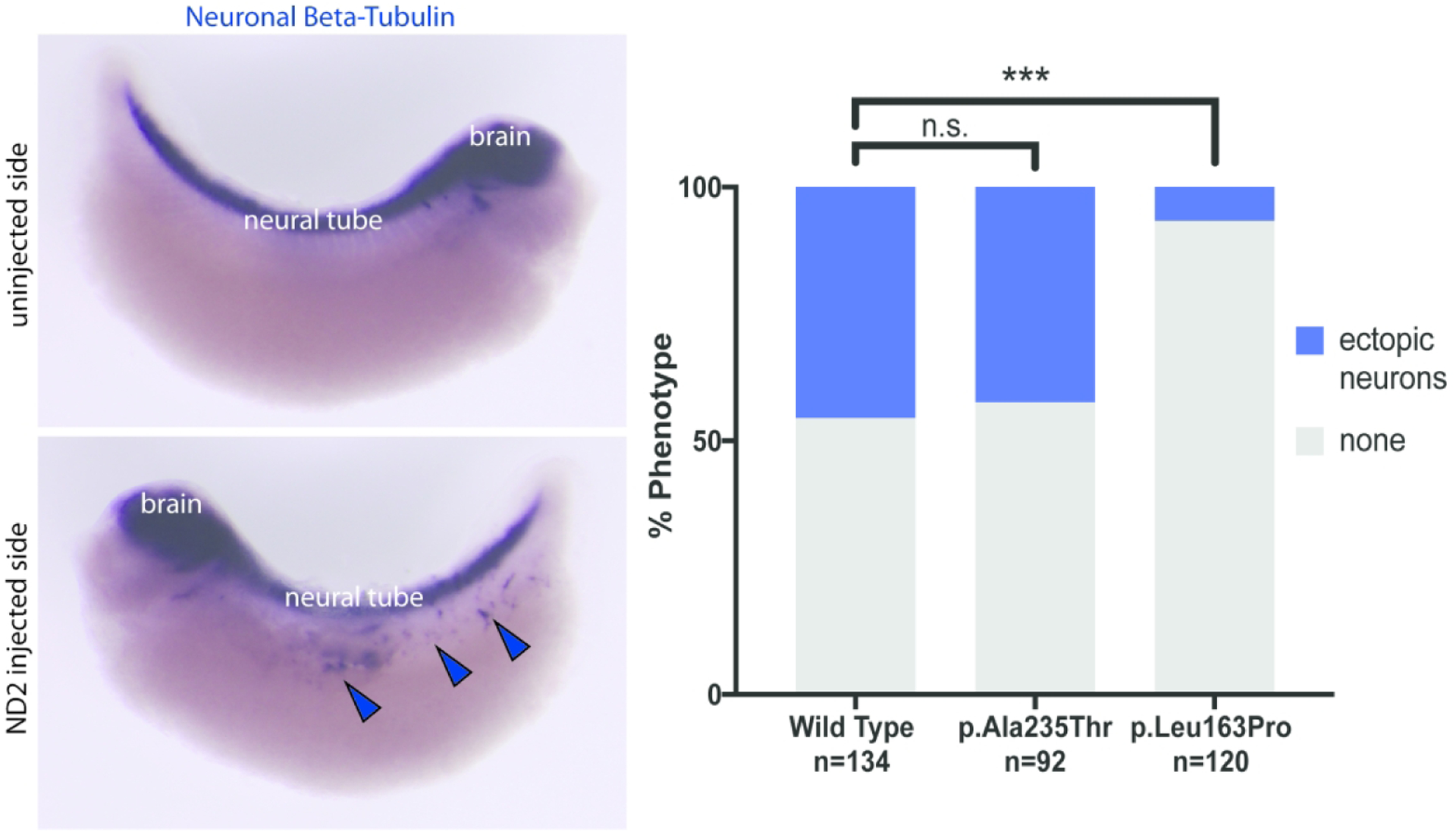
Induction of ectopic neurons in vivo with NEUROD2 overexpression. Left panel with neuronal beta-tubulin staining showing normal neuronal configuration (top) and ectopic neuronal induction (bottom). Chart in right panel shows quantification of ectopic neuronal induction for wild type NEUROD2 and both patient variants tested. “n” denotes number of individual embryos scored for phenotype for the indicated variant. Bars at top denote significance of difference as compared to wild type (n.s. = not significant, *** = significant).
Discussion
We had previously established the association of de novo NEUROD2 variants with early-infantile epileptic encephalopathy in combination with global developmental delay. Here we report another individual with a dysfunctional de novo NEUROD2 variant who presented with global developmental delay but without seizures, thus expanding the phenotype of disorders associated with this gene.
The variant reported here, p.(Leu163Pro), lies in the second helix of the bHLH domain and is highly conserved across multiple species (Figure 1). Based on this location, we suspect that this missense substitution affects the ability of NEUROD2 to bind to target DNA, though direct DNA binding assays are needed to test this hypothesis. It is also possible that the variant could result in an unstable mRNA prone to degredation, although we believe this is unlikely given that NEUROD2 has only a single exon and mRNA decay typically requires at least one intron [Hsu and others 2017; Kurosaki and others 2019]. Notably, however, the ability of the p.(Leu163Pro) mRNA to induce neuronal differentiation was minimal but still detectable. This activity falls between our two previously reported variants p.(Glu130Gln), which had no detectable activity, and p.(Met134Thr), which had roughly 40% of the maximal activity seen with wild type protein [Sega and others 2019]. This suggests that the seizure phenotype seen with some NEUROD2 variants is not exclusively determined by level of protein activity and that other factors, such as epigenetics, modifier genes, or environment, are likely at play. Similarly, although observing ectopic neuron expression is an assay of protein function, our experience with this small number of patients suggests that it cannot be used to predict specific patient phenotypes. It is also unclear whether patients with NEUROD2 variants who do not present with early-onset epileptic encephalopathy remain at risk for developing seizures later in life.
Although Patient 1 is only the third reported patient with dysfunctional NEUROD2 protein, it is notable that all three had variants in the protein’s bHLH domain. Given that this is a key functional domain and necessary for DNA binding, we suspect that variants in this region have a higher likelihood of pathogenicity. The identification of further patients with de novo missense variants in NEUROD2 will help address key questions, such as the nature of genotype-phenotype correlations and whether variants in other regions of the protein can also lead to neurologic phenotypes. Given what is known about the key role of NEUROD2 in cortical neuron development, migration, maintenance, and connectivity, it remains to be seen precisely which functions are disrupted by which variants [Guzelsoy and others 2019; Ince-Dunn and others 2006; Wilke and others 2012].
The second variant that we studied, p.(Ala235Thr), displayed normal function in our assay, leading us to suspect that this p.(Ala235Thr) variant was not the cause of the child’s developmental issues and may have been inherited. It is regrettable that parental data is unavailable to confirm this suspicion, as allele segregation is an important part of classifying variants. Patients who have been adopted or otherwise do not have access to biological relatives exemplify the critical value of functional testing of individual variants. Additionally, while all putatively damaging NEUROD2 variants we have reported thus far have been de novo, it is unclear if recessive or other inherited variants could be problematic. Future research into NEUROD2 roles and interactions could determine other possible means to explore variants functionally, as we have employed only one means of testing specific variants to date. With next generation sequencing increasingly utilized in clinical diagnostics, more rare or novel variants of uncertain significance will be identified. In parallel, functional assays, such as those performed here, will be increasingly important to improve variant classification.
In summary, we provide evidence that heterozygous de novo NEUROD2 variants may result in developmental delay with or without associated seizures, thus expanding this newly-described phenotype. As this is only the third patient reported in the literature with a pathogenic NEUROD2 variant, identification of additional patients will bring further refinements of the clinical presentation, spectrum of pathogenic variants, and course of this condition.
Table 1:
Variants and clinical summaries for patients reported with NEUROD2 variants.
| NEUROD2 de novo Variant | p.(Leu163Pro) | p.(Glu130Gln) | p.(Met134Thr) |
| Variant location notes | Second helix of bHLH | Basic region of first helix of bHLH; Glu130 is strictly conserved across all bHLH transcription factors | Basic region of first helix of bHLH |
| Patient’s current age, sex | 14 years, female | 6 years, female | 5 years, male |
| Seizures | - | History of infantile spasms / hypsarrhythmia, initial control achieved with ketogenic diet. Currently on CBD oil, no seizures × 3 years. | History of infantile spasms / hypsarrhythmia, initial control achieved with vagal nerve stimulator, which remains in place, no seizures × 4 years. |
| Intellectual delay | Pervasive developmental disorder, sensory processing disorder. | Significant global intellectual delay. Nonverbal but interactive. | Significant global intellectual delay. Nonverbal but interactive. |
| Motor delay | Mild gross motor delay | Significant fine and gross motor delay. Limited us of arms and hands, requires turning at night. Able to sit, unable to stand, but has some mobility with specialized walker. | Fine and gross motor delay. Able to feed himself with some help, unable to hold a pencil. Walks and runs without assistance. |
| Brain MRI | Unremarkable | Bilateral increased T2 signal in putamina, parietal periventricular white matter; thin corpus callosum | Mild general cerebral volume loss; absence of pituitary T1 bright spot |
Acknowledgements:
The authors would like to thank the patients and their families for allowing us to share their stories, and Yale New Haven hospital and Sarah and Jeffrey Buell for their support of the Pediatric Genomics Discovery Program.
Funding Sources:
The work was partially supported by awards from the National Institutes of Health (NIH) / National Institute of Child Health and Human Development, 1R01HD102186 (EKM and MKK), the NIH Common Fund, U01HG007703 (SFN, JAMA, and CGSP) and the UCLA California Center for Rare Diseases of the Institute for Precision Health.
Footnotes
Data Availability: Data is available upon reasonable request from authors.
Conflicts: Two authors report part ownership of startup companies unrelated to this work: Qiyas Higher Health (SAL) and Victory Genomics (SAL and MKK). No other authors have any disclosures to report.
This work was approved by Institutional Review Boards for Yale University School of Medicine, University of California San Francisco, and University of California Los Angeles.
References
- Bormuth I, Yan K, Yonemasu T, Gummert M, Zhang M, Wichert S, Grishina O, Pieper A, Zhang W, Goebbels S, Tarabykin V, Nave KA, Schwab MH. 2013. Neuronal basic helix-loop-helix proteins Neurod2/6 regulate cortical commissure formation before midline interactions. J Neurosci 33(2):641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Moran JT, Zhang Y, Ates KM, Yu D, Schrader LA, Das PM, Jones FE, Hall BJ. 2016. The transcription factor NeuroD2 coordinates synaptic innervation and cell intrinsic properties to control excitability of cortical pyramidal neurons. J Physiol 594(13):3729–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guner G, Guzelsoy G, Isleyen FS, Sahin GS, Akkaya C, Bayam E, Kotan EI, Kabakcioglu A, Ince-Dunn G. 2017. NEUROD2 Regulates Stim1 Expression and Store-Operated Calcium Entry in Cortical Neurons. eNeuro 4(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzelsoy G, Akkaya C, Atak D, Dunn CD, Kabakcioglu A, Ozlu N, Ince-Dunn G. 2019. Terminal neuron localization to the upper cortical plate is controlled by the transcription factor NEUROD2. Sci Rep 9(1):19697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MA, Jin SG, Li AX, Liu J, Huang Z, Wu X, Kim BW, Johnson J, Bilbao AV, Tao S, Yim JA, Fong Y, Goebbels S, Schwab MH, Lu Q, Pfeifer GP. 2019. Reprogramming of DNA methylation at NEUROD2-bound sequences during cortical neuron differentiation. Sci Adv 5(10):eaax0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu MK, Lin HY, Chen FC. 2017. NMD Classifier: A reliable and systematic classification tool for nonsense-mediated decay events. PLoS One 12(4):e0174798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince-Dunn G, Hall BJ, Hu SC, Ripley B, Huganir RL, Olson JM, Tapscott SJ, Ghosh A. 2006. Regulation of thalamocortical patterning and synaptic maturation by NeuroD2. Neuron 49(5):683–695. [DOI] [PubMed] [Google Scholar]
- Kurosaki T, Popp MW, Maquat LE. 2019. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Biol 20(7):406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M, Fox M, Fogel BL, Martinez-Agosto JA, Wong DA, Chang VY, Shieh PB, Palmer CG, Dipple KM, Grody WW, Vilain E, Nelson SF. 2014. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312(18):1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Huang AY, Wang LK, Yoon AJ, Renteria G, Eskin A, Signer RH, Dorrani N, Nieves-Rodriguez S, Wan J, Douine ED, Woods JD, Dell’Angelica EC, Fogel BL, Martin MG, Butte MJ, Parker NH, Wang RT, Shieh PB, Wong DA, Gallant N, Singh KE, Tavyev Asher YJ, Sinsheimer JS, Krakow D, Loo SK, Allard P, Papp JC, Undiagnosed Diseases N, Palmer CGS, Martinez-Agosto JA, Nelson SF. 2020. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet Med 22(3):490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tapscott SJ, Olson JM. 2006. Congenital hypothyroidism (cretinism) in neuroD2-deficient mice. Mol Cell Biol 26(11):4311–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo A, Guanga GP, Rose RB. 2008. Crystal structure of E47-NeuroD1/beta2 bHLH domain-DNA complex: heterodimer selectivity and DNA recognition. Biochemistry 47(1):218–229. [DOI] [PubMed] [Google Scholar]
- Matsushita M, Nakatake Y, Arai I, Ibata K, Kohda K, Goparaju SK, Murakami M, Sakota M, Chikazawa-Nohtomi N, Ko SBH, Kanai T, Yuzaki M, Ko MSH. 2017. Neural differentiation of human embryonic stem cells induced by the transgenemediated overexpression of single transcription factors. Biochem Biophys Res Commun 490(2):296–301. [DOI] [PubMed] [Google Scholar]
- Olson JM, Asakura A, Snider L, Hawkes R, Strand A, Stoeck J, Hallahan A, Pritchard J, Tapscott SJ. 2001. NeuroD2 is necessary for development and survival of central nervous system neurons. Developmental biology 234(1):174–187. [DOI] [PubMed] [Google Scholar]
- Pieper A, Rudolph S, Wieser GL, Gotze T, Miessner H, Yonemasu T, Yan K, Tzvetanova I, Castillo BD, Bode U, Bormuth I, Wadiche JI, Schwab MH, Goebbels S. 2019. NeuroD2 controls inhibitory circuit formation in the molecular layer of the cerebellum. Sci Rep 9(1):1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sega AG, Mis EK, Lindstrom K, Mercimek-Andrews S, Ji W, Cho MT, Juusola J, Konstantino M, Jeffries L, Khokha MK, Lakhani SA. 2019. De novo pathogenic variants in neuronal differentiation factor 2 (NEUROD2) cause a form of early infantile epileptic encephalopathy. J Med Genet 56(2):113–122. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. 2015. Proteomics. Tissue-based map of the human proteome. Science (New York, NY) 347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- Wilke SA, Hall BJ, Antonios JK, Denardo LA, Otto S, Yuan B, Chen F, Robbins EM, Tiglio K, Williams ME, Qiu Z, Biederer T, Ghosh A. 2012. NeuroD2 regulates the development of hippocampal mossy fiber synapses. Neural Dev 7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]