

There is a Blood Commentary on this article in this issue.
Key Points
RiBi impairment leads to p53-mediated apoptosis and MYC-driven lymphoma regression independent of protein synthesis lesions.
The impaired ribosome biogenesis checkpoint triggers the selective degradation of the antiapoptotic form of MCL-1 by the proteasome.
Abstract
MYC-driven B-cell lymphomas are addicted to increased levels of ribosome biogenesis (RiBi), offering the potential for therapeutic intervention. However, it is unclear whether inhibition of RiBi suppresses lymphomagenesis by decreasing translational capacity and/or by p53 activation mediated by the impaired RiBi checkpoint (IRBC). Here we generated Eμ-Myc lymphoma cells expressing inducible short hairpin RNAs to either ribosomal protein L7a (RPL7a) or RPL11, the latter an essential component of the IRBC. The loss of either protein reduced RiBi, protein synthesis, and cell proliferation to similar extents. However, only RPL7a depletion induced p53-mediated apoptosis through the selective proteasomal degradation of antiapoptotic MCL-1, indicating the critical role of the IRBC in this mechanism. Strikingly, low concentrations of the US Food and Drug Administration–approved anticancer RNA polymerase I inhibitor Actinomycin D (ActD) dramatically prolonged the survival of mice harboring Trp53+/+;Eμ-Myc but not Trp53–/–;Eμ-Myc lymphomas, which provides a rationale for treating MYC-driven B-cell lymphomas with ActD. Importantly, the molecular effects of ActD on Eμ-Myc cells were recapitulated in human B-cell lymphoma cell lines, highlighting the potential for ActD as a therapeutic avenue for p53 wild-type lymphoma.
Visual Abstract

Introduction
The transcription factor MYC regulates numerous cellular processes, including cell growth, cell cycle progression, and protein synthesis, but it predisposes cells to apoptosis under stress conditions.1 In response to mitogens, MYC critically controls ribosome biogenesis (RiBi), a highly complex and energy-consuming multistep process2 required for cell growth and proliferation.2,3 MYC directly controls this process at numerous levels, including the transcription of 47S precursor ribosomal RNA (pre-rRNA), ∼80 distinct ribosomal proteins (RPs), messenger RNAs (mRNAs), and 5S rRNA, through upregulation or activation of RNA polymerase I (Pol I), Pol II, and Pol III, respectively.2,3 This corresponds to an ancient MYC function, because the RP genes are highly conserved in eukaryotes and seem to have evolved as the first primordial MYC regulon.4
The importance of MYC is underscored in human cancer, in which its expression is deregulated in up to 70% of patients.5 Deregulation of MYC triggers an intrinsic tumor suppressor program that leads to p53 activation and either cell senescence or apoptosis, depending on cell context.6 Critical for MYC-driven lymphoma progression is the evasion of apoptotic cell death,7 facilitated by the disruption of the ARF-MDM2-p53 tumor suppressor pathway,8,9 by the loss of proapoptotic BCL-2 family members,10,11 or by the overexpression of antiapoptotic BCL-2 family members.12 High-resolution analyses of somatic copy-number alterations in a large spectrum of cancers consistently revealed that genes for antiapoptotic BCL-2 family members MCL1 and BCLXL were most often amplified.13 Of note, in more than 60% of patients, the MYC gene was coamplified with MCL1 and BCLXL.13
MYC is translocated or overexpressed in the most aggressive lymphomas, including Burkitt lymphoma (BL) and diffuse large B-cell lymphoma (DLBCL).14 In the Eμ-Myc transgenic mouse model, which phenocopies many of the clinical pathologic features of human BL,15 MYC is overexpressed under the control of the immunoglobulin heavy chain enhancer (Eμ).16 Recent studies showed that loss of a single allele of Mcl1 was sufficient to substantially reduce the incidence and the sustained expansion of Eμ-Myc lymphomas in mice,17 whereas loss of Bclxl had only a modest effect on tumor expansion.5 These findings and others have provided the rationale for the development of small-molecule BH3 mimetics for cancer therapy,18 most recently mimicking MCL-1.19
Earlier studies demonstrated that Eμ-Myc lymphomas depend on RiBi for growth and proliferation,3 and that RiBi is an Achilles heel of MYC-driven tumors.20,21 Indeed, when Eμ-Myc mice were crossed with Belly Spot and Tail (Bst) mice, which are hypomorphic for the Rpl24 gene (or eL24), global translation rates in the B cells of these mice were largely brought to normal, extending their lymphoma-free survival.22 Survival benefit was attributed to decreased protein synthetic capacity; however, others showed that depletion of RPL24 leads to p53-mediated cell cycle arrest.23 We previously demonstrated that as a consequence of impaired RiBi, a precursor complex made up of RPL5 (or uL18), RPL11 (or uL5), and the 5S rRNA,24 which we termed the impaired RiBi checkpoint (IRBC) complex,2,25 is redirected to the binding and inhibition of MDM2, leading to p53 activation. Critically, Eμ-Myc mice harboring a point mutation in MDM2, which prevents its binding to the IRBC complex while retaining normal regulation of p53 by DNA damage or p19ARF, succumb to lymphomagenesis much more rapidly than control Eμ-Myc mice.26 This finding is compatible with a recent study from our laboratory indicating that the IRBC acts as an intrinsic tumor suppressor in deregulated MYC-expressing cells27 and also in studies showing that inhibition of Pol I-dependent rDNA transcription leads to p53-mediated apoptosis in Eμ-Myc lymphomas.20 Although the mechanism that causes cell death remains uncertain, these data suggest that targeting RiBi alleviates MYC-driven tumor development.
Here we set out to define the mechanism by which RiBi inhibition suppresses MYC-driven tumorigenesis. To address the contribution of decreased global translational capacity vs IRBC activation, we partially depleted either the 60S RPL7a (or eL8) or RPL11 in the Eμ-Myc lymphoma model. Both proteins are required at the same stage of RiBi,28 but only RPL11 is required for IRBC complex formation.24,29 Our results show that approximately equivalent depletion of either RP in Eμ-Myc lymphoma cells reduced RiBi, nascent protein synthesis, and proliferation to similar levels. However, depletion of RPL7a, but not RPL11, induced p53-dependent apoptotic cell death, implicating the IRBC in this response. Further analyses in RPL7a-depleted cells showed that upstream of caspase activation, the expression of antiapoptotic MCL-1 was substantially reduced because of the IRBC and p53 activation. These observations were recapitulated by low doses of Actinomycin D (ActD), the first US Food and Drug Administration (FDA)-approved antibiotic for cancer therapy, which selectively inhibits Pol I–mediated rDNA transcription30 and was extended to human BL and DLBCL cell lines that harbor wild-type (wt) p53. Strikingly, low-dose ActD treatment greatly delayed the in vivo growth of Trp53+/+;Eμ-Myc lymphomas but had no impact on the expansion of Trp53–/–;Eμ-Myc lymphomas. The dependency of the human B-cell lymphoma cell lines on p53 and the in vivo mouse data provide evidence that supports the application of ActD as a therapeutic strategy for MYC-driven B-cell lymphomas stratified for wt p53.
Methods
All reagents and drug treatments and additional methods not mentioned below are detailed in the supplemental Data (available on the Blood Web site).
Cell lines
The murine Eμ-Myc lymphoma Trp53+/+ (clones 4242, 107, and 226), Trp53–/– (clones 3239, KA540, and 3391) cell lines, the short hairpin Ren (shRen)-, shRPL7a-, and shRPL11-expressing Eμ-Myc cell lines (supplemental Table 1), and the stable 3xFLAG-tagged wt MCL-1 (MCL-1wt) and KR-mutant MCL-1 (MCL-1KR) Trp53+/+;Eμ-Myc cell lines were cultured in Anne Kelso media and maintained in 10% CO2 at 37 ºC as described previously.20 Mcl1wt and Mcl1–/– (clones 1 and 2) mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco’s modified Eagle medium (DMEM) (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U/mL penicillin, and 100 mg/mL streptomycin and maintained at 5% CO2 at 37 ºC. The BL-derived cell lines BL2, Raji, and Ramos and the DLBCL-derived cell lines SUDHL5, SUDHL6, DOOH2, and SUDHL4 were cultured in RPMI-1640 (Sigma-Aldrich) media supplemented with 10% or 20% FBS and 2 mM GlutaMAX (Gibco) at 5% CO2, as described.31,32 The Oci-LY3 DLBCL cell line was cultured in Iscove modified Dulbecco medium (IMDM) (Sigma-Aldrich) media supplemented with 20% FBS, 2 mM GlutaMAX, and 50 μM 2-mercaptoethanol and maintained at 5% CO2. All cell lines were routinely tested for mycoplasma contamination by polymerase chain reaction and found to be negative.
Mice
Four-week-old male C57BL/6J and congenic C57BL/6J-Ly5.1 (B6.SJL-PtprcaPepcb/BoyJ) mice were obtained from the Garvan Institute of Medical Research (Sydney, NSW, Australia) or from Charles River Laboratories (Calco, Italy). The Eμ-Myc lymphomas were generated by injecting 200 000 Eμ-Myc lymphoma cells into the lateral tail veins of 6- to 8-week-old mice, as previously described.20 Trp53+/+;Eμ-Myc lymphoma cells (clone 4242-GFP) were transplanted into C57BL/6J mice, whereas Trp53–/–;Eμ-Myc lymphoma cells (clone 3239) were transplanted into C57BL/6J-Ly5.1 mice to distinguish the Trp53–/–;Eμ-Myc lymphoma cells that express the CD45.2 antigen from the host cells that express the CD45.1 antigen. Then, 0.1 mg/kg ActD (BioVision) in 1:1 polyethylene glycol (PEG)400/phosphate-buffered saline (PBS) or vehicle (1:1 PEG400/PBS) was administrated intraperitoneally when 20% to 30% Eμ-Myc lymphoma cells were detected in the peripheral blood, starting 9 to 10 days after transplantation. In each treatment group, 3 to 5 mice were used to analyze on-target effects after a single administration, 5 to 7 mice were used to assess the effects of ActD on immune cells populations in hematopoietic tissues, and another 9 mice were kept to record survival. Experiments comparing Trp53+/+;Eμ-Myc and Trp53–/–;Eμ-Myc lymphoma-bearing mice were performed side by side and conducted with the approval of the Animal Experimentation Ethics Committee at Peter MacCallum Cancer Centre (Melbourne, VIC, Australia). Analyses of the on-target effects of 1 single administration of ActD and of the effects of ActD on lymphoid and myeloid populations in hematopoietic and lymphoid tissues in control or Trp53+/+;Eμ-Myc lymphoma-transplanted C57BL/6J mice were conducted following the guidelines and with the approval of the Bellvitge Biomedical Research Institute Animal Care and Use Committee (Barcelona, Spain).
Survival analysis
For Kaplan-Meier survival analyses at 9 to 10 days posttransplantation, mice were treated with either vehicle or 0.1 mg/kg per day ActD (n = 9 mice per treatment group). Treatment was administered intraperitoneally in the first cycle of 5 days on/6 days off and in 2 additional cycles of 4 days on/4 days off resuming at day 32 posttransplantation. Lymphoma progression was followed up by flow cytometry analysis of peripheral blood extracted by tail-vein bleeding before and 3 days after treatment initiation. Mice were weighed once per day, monitored, and euthanized at an ethical end point (hunched posture, ruffled fur, enlarged lymph nodes, labored breathing, weight loss greater than 20% of the original weight, limited mobility, or paralysis).
Statistical analysis
Statistical analyses were performed using GraphPad Prism v.7.0a. Comparisons between 2 groups with normally distributed data (by Lilliefors test) were performed by using a Student t test. Experiments with more than 2 groups were compared by 1-way analysis of variance with post hoc Tukey’s test. Data sets with 2 independent factors were compared by 2-way analysis of variance post hoc Tukey’s test. Survival curves were analyzed using log-rank Mantel-Cox test and Gehan-Breslow-Wilcoxon test. The statistical test used, number of replicates (n), and measures of distribution and deviation are indicated in the figure legends. P values <.05 were considered statistically significant.
Results
Impaired RiBi leads to IRBC induction and p53 activation in Eμ-Myc lymphoma cells
To understand how inhibiting RiBi suppresses MYC-driven tumorigenesis, we used the tetracycline (Tet)–regulated microRNA 30 (miR-30)-shRNA system33 to deplete either 60S RPL7a or RPL11 proteins. We generated stable Trp53+/+;Eμ-Myc cell lines in which the mRNA levels of RPL7a or RPL11 were reduced to ∼50% (Figure 1A), similar to the level predicted for the hypomorphic Rpl24 mutation in the Bst mouse,22,23 leading to undetectable levels of nascent RPL7a or RPL11 protein, respectively, in lysates cleared of mature ribosomes by ultracentrifugation (Figure 1B).24 This led to a selective decrease in native 60S ribosomes (supplemental Figure 1A) and nascent 28S rRNA (Figure 1C; supplemental Figure 1B). The total amount of ribosomal particles and the rate of protein synthesis were consistently reduced (supplemental Figure 1C; Figure 1D). However, only depletion of RPL7a, but not loss of RPL11, induced p53, which was accompanied by increased p21 expression and both caspase-3 and poly (ADP-ribose) polymerase 1 (PARP1) cleavage (Figure 1E) (markers of p53-induced apoptosis). Similar results were obtained with low concentrations of ActD (supplemental Figure 1D), which decreased 47S pre-rRNA expression (supplemental Figure 1E). To determine the role of the IRBC complex in mediating p53 activation, RPL5 was immunoprecipitated from the Eμ-Myc lymphoma ribosome–free lysates.24 In parallel to p53 activation, the amounts of MDM2 and RPL11 associated with RPL5 were increased in lysates from RPL7a-depleted cells vs control cells but were completely absent in lysates from the RPL11-depleted cells (Figure 1F). Likewise, low concentrations of ActD induced the formation of the IRBC (supplemental Figure 1F). These findings showed that despite a similar effect on global translation in Eμ-Myc lymphoma cells, depletion of RPL7a, but not loss of RPL11, leads to p53 activation, an effect mediated by the IRBC.
Figure 1.
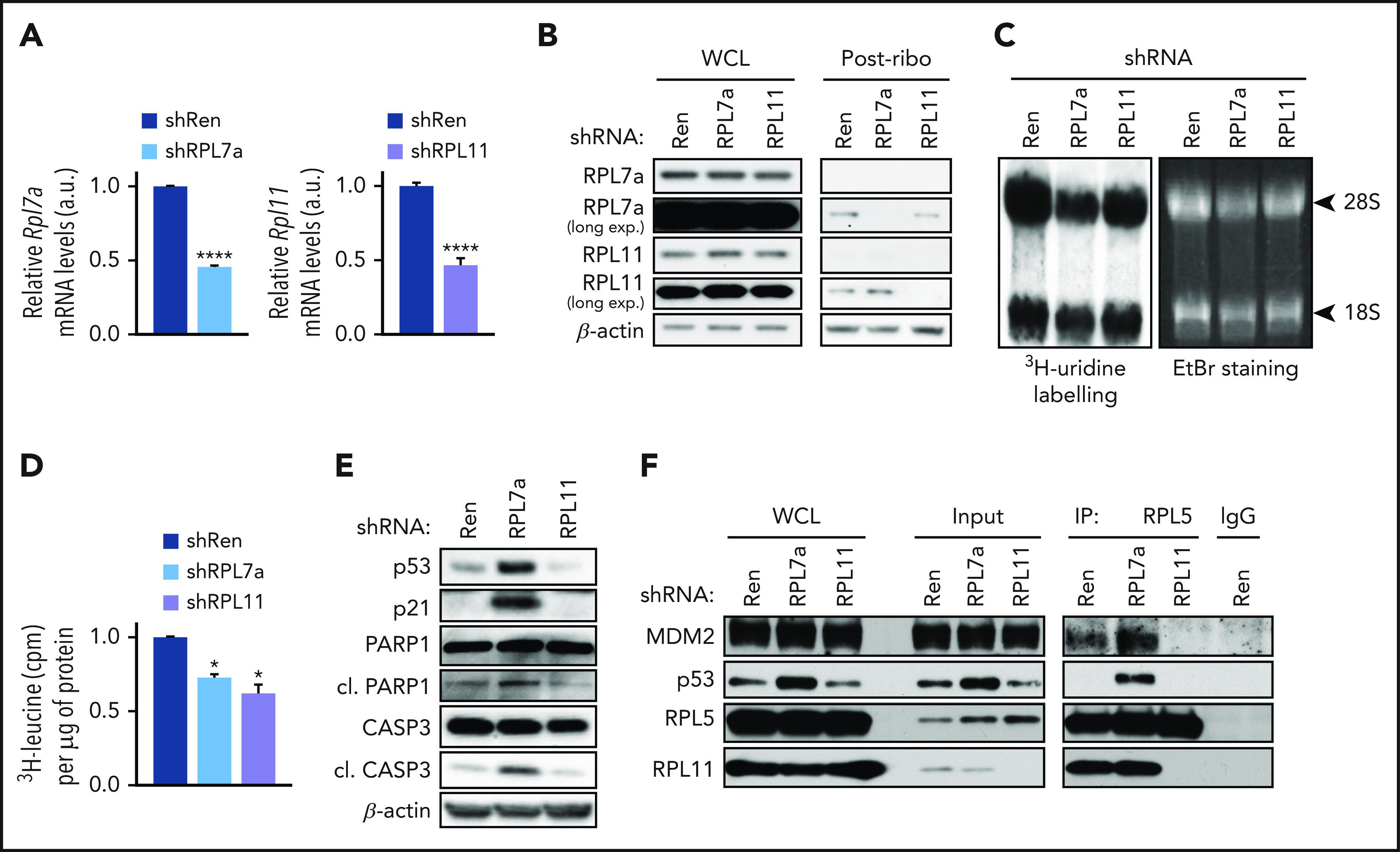
Effects of RP depletion on nascent protein synthesis and activation of the IRBC. RPL7a and RPL11 shRNA vector-containing Eμ-Myc cell lines were treated for 22 hours with 1 μg/mL or 10 ng/mL doxycycline, respectively, to achieve ∼50% depletion of their corresponding RP mRNA. Renilla (Ren) shRNA-expressing cells, used as a control, were treated for 22 hours with 1 μg/mL doxycycline. (A) Rpl7a and Rpl11 transcript levels in correspondingly RP-depleted cells relative to control cells (shRen), determined by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) and normalized to β-actin (n = 4). ****P < .0001 (unpaired Student t test). (B) Representative western blot analysis of RPL7a and RPL11 levels in whole cell lysates (WCLs) and in postribosomal supernatants (Post-ribo) containing 20 and 60 μg protein, respectively, from control (shRen) and from RPL7a- and RPL11-depleted cells (n = 2). (C) 3H-uridine autoradiogram (left panel) and ethidium bromide (EtBr)-stained agarose gel (right panel) of 28S and 18S shRNA from control (shRen) and from RPL7a- and RPL11-depleted cells pulsed with 3H-uridine for 2 hours. (D) Nascent protein synthesis rate, quantified by incorporation of 3H-leucine and normalized to total protein concentration (n = 3). *P < .05 (1-way analysis of variance (ANOVA) test). (E-F) Representative western blot analyses (E) WCLs (n > 4), post-ribosomal supernatants (Input), and coimmunoprecipitates of MDM2, p53, RPL11, and RPL5 in control (shRen) and RPL7a- and RPL11-depleted cells (n = 4) (F). A nonspecific rabbit immunoglobulin G (IgG) antibody was used as control for the immunoprecipitation (IP). a.u., arbitrary units; CASP3, caspase-3; cl., cleaved; cpm, counts per minute; long exp., long exposure. Data are presented as mean ± standard error of the mean (SEM).
Activation of the IRBC leads to caspase-dependent cell death mediated by p53
In line with p53 activation and caspase-3 cleavage (Figure 1E), depletion of RPL7a for 48 hours caused a stronger reduction in cell number than depletion of RPL11 (Figure 2A). To determine the contribution of p53 in this response, we generated Trp53–/–;Eμ-Myc lymphoma cell lines expressing Tet-inducible shRNA against Rpl7a or Rpl11. Despite a similar 50% depletion of RP mRNA levels in Trp53+/+;Eμ-Myc cells (Figure 1A) and Trp53–/–;Eμ-Myc cells (supplemental Figure 2A), which led to an equivalent reduction in protein synthesis (compare Figure 1D and supplemental Figure 2B), the effect of RPL7a depletion on the number of viable cells in Trp53–/–;Eμ-Myc cells was not as severe as that in Trp53+/+;Eμ-Myc cells (Figure 2A). Concomitantly, acute treatment with low-dose ActD for 12 hours dramatically reduced the number of Trp53+/+;Eμ-Myc lymphoma viable cells but had less impact on Trp53–/–;Eμ-Myc lymphoma cells (Figure 2B), whereas the general apoptotic inducers staurosporine or ionomycin had similar effects on Trp53+/+;Eμ-Myc and Trp53–/–;Eμ-Myc lymphoma cell viability (supplemental Figure 2C-D).
Figure 2.
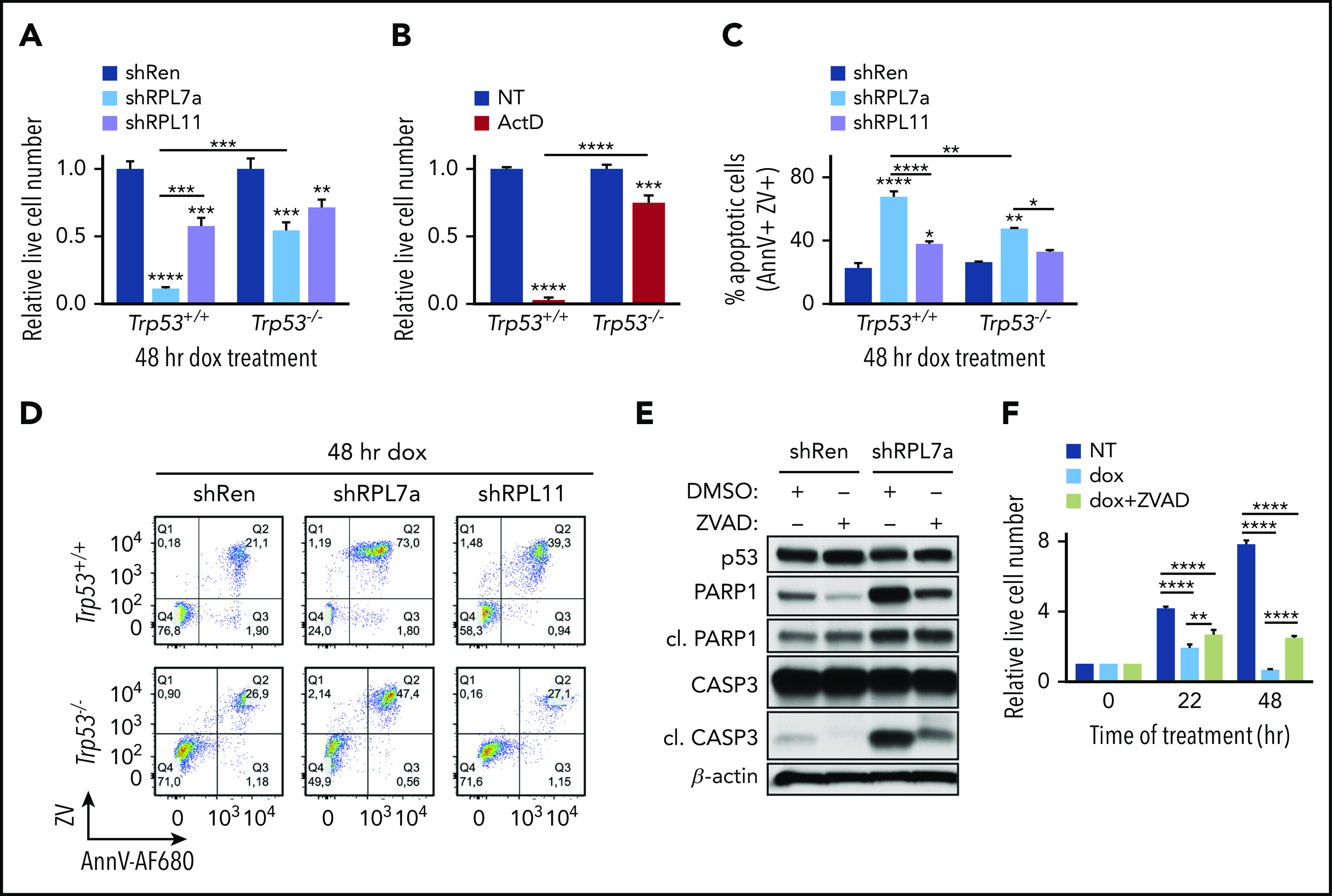
Induction of the IRBC stabilizes p53 and leads to caspase-dependent cell death in Trp53+/+;Eμ-Myc, but not in Trp53–/–;Eμ-Myc lymphoma cells. (A-B) Proliferation assays of control (shRen) and RPL7a and RPL11-depleted Trp53+/+;Eμ-Myc or Trp53–/–;Eμ-Myc lymphoma cells treated for 48 hours with doxycycline (dox) as in Figure 1 and of (A) Trp53+/+;Eμ-Myc or Trp53–/–;Eμ-Myc lymphoma cells treated for 12 hours with 5 nM ActD or dimethyl sulfoxide (DMSO)-vehicle (NT) (B). Data are shown relative to their respective Trp53+/+;Eμ-Myc or Trp53–/–;Eμ-Myc lymphoma cell controls (n = 3-5). (C) Proportion of apoptotic shRen, shRPL7a, and shRPL11 Trp53+/+;Eμ-Myc or Trp53–/–;Eμ-Myc lymphoma cells from panel A determined by annexin V (AnnV) and Zombie Violet (ZV) staining and flow cytometry (n = 2-3). (D) Representative dot plots from panel C. shRen-expressing control and RPL7a- and RPL11-depleted Trp53+/+;Eμ-Myc (top panels) or Trp53–/–;Eμ-Myc (bottom panels) cells are shown. (E) Representative western blot analysis of lysates from shRen and shRPL7a Trp53+/+;Eμ-Myc lymphoma cells treated for 22 hours with doxycycline with or without the pan-caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone) (Z-VAD-FMK; ZVAD) at 20 μM (n = 3). (F) Proliferation assay of RPL7a-depleted Trp53–/–;Eμ-Myc lymphoma cells in NT, treated under doxycycline or doxycycline and ZVAD conditions at 22 and 48 hours (n = 3). AnnV-AF680, Alexa Fluor 680-conjugated annexin V. Data are presented as mean ± SEM. *P < .05; **P < .01; ***P < .001; ****P < .0001 (2-way ANOVA test).
Consistent with the induction of the IRBC leading to p53-dependent cell death, flow cytometric analyses of RPL7a-depleted Eμ-Myc cell lines showed a dramatic increase in the proportion of early and late apoptotic cells, which was dependent on the presence of p53 (Figure 2C-D; supplemental Figure 2E-F). Moreover, this apoptotic increase was significantly lower in RPL11-depleted Eμ-Myc cells (Figure 2C-D; supplemental Figure 2E-F), suggesting that the p53-independent effects of RPL11 depletion or the effects of RPL7a depletion on Trp53–/–;Eμ-Myc live cell numbers (Figure 2A) were ascribed, at least in part, to a decrease in protein synthesis and cell proliferation. The generation of Trp53+/+;Eμ-Myc clones expressing another independent sequence of shRPL7a or shRPL11 (supplemental Figure 2G) recapitulated our previous findings, because RPL7a depletion specifically induced p53 (supplemental Figure 2H) and led to a more severe reduction in cell number and viability than RPL11 depletion (supplemental Figure 2I-J). Critically, treatment of RPL7a-depleted Trp53+/+;Eμ-Myc lymphoma cells with the pan-caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone) (Z-VAD-FMK) significantly reduced both caspase-3 and PARP1 cleavage (Figure 2E) and partially rescued cell survival, a response that increased with time (Figure 2F). The incomplete response may be explained by a partial inhibition of apoptosis or by the contribution of nonapoptotic cell death. These data support the notion that while decreased rates of protein synthesis slow down proliferation to a certain extent, impaired RiBi rapidly activates the IRBC, p53, and apoptosis.
Activation of the IRBC induces the selective loss of the antiapoptotic MCL-1
Mediators of p53-induced cell death include proapoptotic BH3-only proteins, particularly, PUMA and NOXA,34 which activate the apoptotic effectors BAX and BAK either by direct binding or indirectly through inhibition of antiapoptotic BCL-2 family members.35 Analysis of the level of 3 main antiapoptotic BCL-2 family members (BCL-XL, BCL-2, and MCL-1) showed that RPL7a depletion or ActD treatment had little effect on antiapoptotic BCL-XL and BCL-2 (Figure 3A; supplemental Figure 3A). In contrast, the upper band of MCL-1, reported to be the antiapoptotic form,36 was selectively reduced in RPL7a-depleted cells (Figure 3A; supplemental Figure 3B) and more dramatically after treatment with ActD (supplemental Figure 3A). We confirmed that this band represents MCL-1 by comparing MCL-1–deleted MEFs with wt MEFs (supplemental Figure 3C). Moreover, the reduction in MCL-1 seems to be dependent on the IRBC and p53, because MCL-1 levels were not affected by RPL11 depletion (Figure 3A; supplemental Figure 3B) or by the depletion of RPL7a in Trp53–/–;Eμ-Myc cells (Figure 3B). A time course analysis after ActD treatment in Trp53+/+;Eμ-Myc cells showed that the rapid p53 induction was followed by loss of the MCL-1 upper band and induction of classical hallmarks of apoptosis (PARP1 and caspase-3 cleavage), whereas none of these responses were observed in Trp53–/–;Eμ-Myc lymphoma cells (Figure 3C). We verified that the effects of ActD are conserved in distinct Trp53+/+;Eμ-Myc and Trp53−/−;Eμ-Myc cell lines (supplemental Figure 3D) and that the use of the Pol I inhibitors CX-546137 and BMH-2138 (supplemental Figure 3E) (both with distinct mechanisms of action) led to the rapid reduction in number and viability of Trp53+/+;Eμ-Myc cells (supplemental Figure 3F-G). Importantly, both inhibitors induced p53, the emergence of the apoptotic markers, and the reduction of antiapoptotic MCL-1 levels selectively in Trp53+/+;Eμ-Myc lymphoma cells (Figure 3C).
Figure 3.
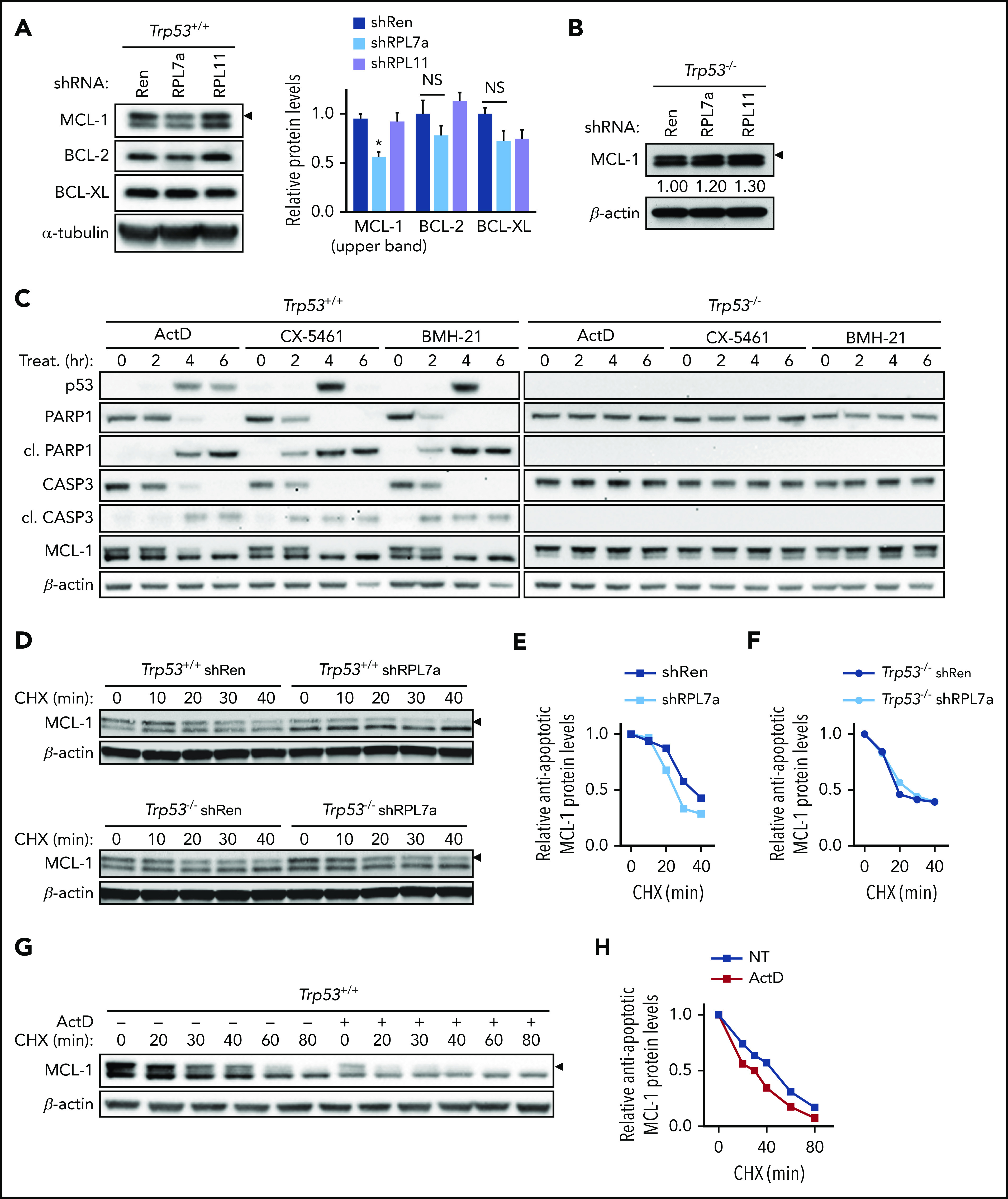
Antiapoptotic MCL-1 protein expression and stability are decreased in RPL7a-depleted and ActD-treated Trp53+/+;Eμ-Myc lymphoma cells. (A) Representative western blot analyses of antiapoptotic BCL-2 proteins in control (shRen) and RPL7a- and RPL11-depleted Eμ-Myc lymphoma cells treated as described in Figure 1. Antiapoptotic MCL-1, upper band, is indicated (arrowhead). MCL-1, BCL-2, and BCL-XL protein expression was normalized to α-tubulin and shown as relative to shRen cells (n = 3). (B) Representative western blot analyses of MCL-1 in control (shRen) and RPL7a- and RPL11-depleted Trp53–/–;Eμ-Myc lymphoma cells (n = 3). Levels of antiapoptotic MCL-1 form (arrowhead), normalized to β-actin, are indicated below the corresponding blot as relative to shRen cells. (C) Time-course western blot analyses in Trp53+/+;Eμ-Myc and Trp53–/–;Eμ-Myc lymphoma cells treated with the Pol I inhibitors ActD (5 nM), CX-5461 (50 nM), and BMH-21 (1 μM) for the indicated times (n = 2). (D) Representative western blots showing MCL-1 stability in control (shRen) and RPL7a-depleted Trp53+/+;Eμ-Myc (top panel) or Trp53–/–;Eμ-Myc (bottom panel) lymphoma cells pretreated with doxycycline for 22 hours and harvested after treatment with 100 μg/mL cycloheximide (CHX) at the indicated time points (n = 2-3). Band corresponding to antiapoptotic MCL-1 is indicated (arrowhead). (E-F) Line graphs showing MCL-1 stability over time in control (shRen) and RPL7a-depleted Trp53+/+;Eμ-Myc (E) and Trp53–/–;Eμ-Myc (F) lymphoma cells determined from MCL-1 immunoblots (D) and shown as relative to MCL-1 level at the time of CHX addition. (G-H) Representative western blot and line graph showing MCL-1 stability in Trp53+/+;Eμ-Myc lymphoma cells treated with or without 5 nM ActD for 4 hours (n = 2), determined by CHX chase at the indicated time points, as described in panel E. Band corresponding to antiapoptotic MCL-1 is indicated (arrowhead). NS, not significant. Data are presented as mean ± SEM. *P < .05 (2-way ANOVA test).
The reduction of MCL-1 in RPL7a-depleted Trp53+/+;Eμ-Myc cells did not seem to be mediated by a decrease of its transcription, because there was no significant difference in the amount of Mcl1 mRNA in RPL7a-depleted vs control cells (supplemental Figure 3H). Likewise, although selective translational control of Mcl1 mRNA has been implicated in regulating its protein levels,39 there was no apparent redistribution of Mcl1 mRNA to inactively translating polysomes in cell extracts from RPL7a-depleted Trp53+/+;Eμ-Myc cells (supplemental Figure 3I). However, MCL-1 protein is highly unstable,40,41 and western blot analyses in the presence of the protein synthesis inhibitor cycloheximide (CHX) showed that the half-life of the MCL-1 upper band decreased from ∼35 minutes to ∼25 minutes in RPL7a-depleted Trp53+/+;Eμ-Myc cells (Figure 3D, top panel; Figure 3E). In contrast, there was no difference in the half-life of the MCL-1 upper band in RPL7a-depleted Trp53–/–;Eμ-Myc lymphoma cells (Figure 3D, bottom panel; Figure 3F). Likewise, ActD treatment reduced the half-life of the MCL-1 upper band in Trp53+/+;Eμ-Myc cells (Figure 3G-H). These findings demonstrate that IRBC-dependent activation of p53 causes increased degradation of antiapoptotic MCL-1.
Loss of MCL-1 is associated with enhanced levels of ubiquitination and increased interaction with NOXA
Caspase-3–mediated proteolysis42,43 and ubiquitin-dependent and -independent proteasomal degradations have been implicated in MCL-1 degradation.41,44 After ActD treatment, caspase activation and loss of MCL-1 closely parallel each another (Figure 3C). However, despite treatment with the pan-caspase inhibitor quinoline-Val-Asp-difluorophenoxymethylketone (QVD-OPh), which prevents caspase-3 cleavage induced either by ActD treatment or by RPL7a depletion, reduction in MCL-1 was not prevented (Figure 4A; supplemental Figure 4A). In parallel to MCL-1 loss, ActD treatment or RPL7a depletion induced expression of the Noxa mRNA (supplemental Figure 4B-C). NOXA has been reported to bind and inhibit MCL-1, dissociating MCL-1–BAK complexes45,46 that contribute to p53-induced apoptosis.34 Induction of Noxa mRNA led to an increase in its protein and its association with MCL-1 in immunoprecipitates from ActD-treated Trp53+/+;Eμ-Myc cells (Figure 4B). Under these conditions, MCL-1 interaction with BAK was reduced (Figure 4B), suggesting that binding of NOXA to MCL-1 may displace BAK from MCL-1 inhibition, thus enhancing apoptotic cell death. In contrast, although the activation of p53 by ActD treatment also resulted in increased PUMA mRNA and protein levels (Figure 4B; supplemental Figure 4D), PUMA association with MCL-1 was decreased (Figure 4B), which is consistent with NOXA having the more important role in this setting. In addition to competing with BAK for MCL-1 binding, NOXA has been reported to trigger MCL-1 ubiquitination and degradation through recruitment of the E3-ubiquitin ligase ARF-BP1/MULE47-49 or by directly targeting MCL-1 to the proteasome in an ubiquitin-independent manner.50 In MCL-1 immunoprecipitates from ActD-treated or RPL7a-depleted Trp53+/+;Eμ-Myc cells, we observed a dramatic increase in the amount of ubiquitinated proteins that was not observed in Trp53–/–;Eμ-Myc lymphoma cells (Figure 4C; supplemental Figure 4E). These findings fit a model in which IRBC activation leads to the p53-dependent ubiquitination of MCL-1 or an associated protein, the proteasomal degradation of MCL-1, and the subsequent release of BAK and induction of apoptosis.
Figure 4.
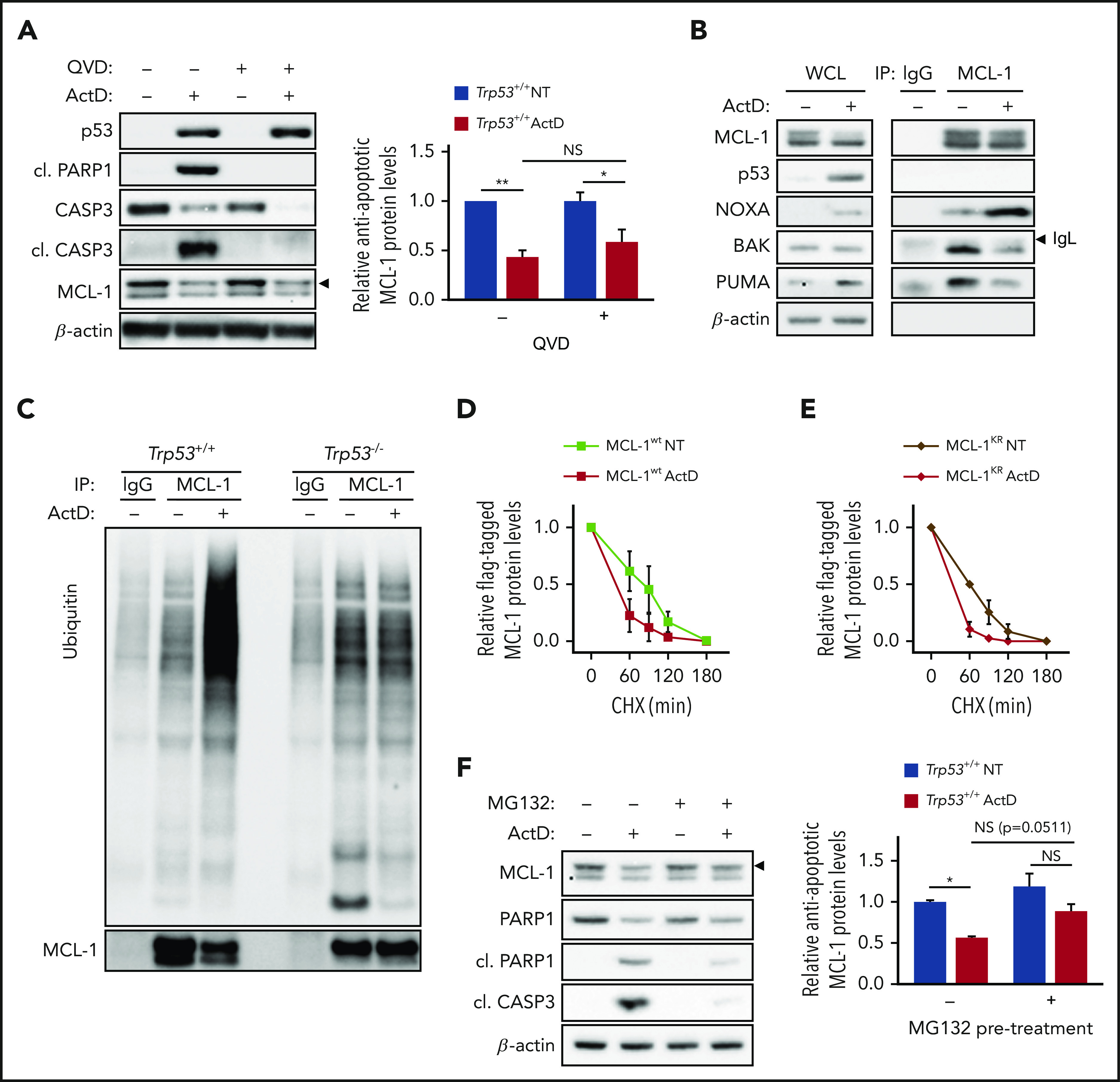
ActD induces ubiquitin-independent but proteasome-dependent degradation of MCL-1. (A) Representative western blot analysis of Trp53+/+;Eμ-Myc lymphoma cells treated for 4 hours with 5 nM ActD, with or without 20 μM quinoline-Val-Asp-difluorophenoxymethylketone (QVD-OPh; QVD). Expression of antiapoptotic MCL-1 (arrowhead), determined as in Figure 3A, is shown as relative to MCL-1 levels in NT cells in the absence of QVD (n = 3). (B) Co-IP of NOXA, PUMA, and BAK with MCL-1 in Trp53+/+;Eμ-Myc lymphoma cells treated for 4 hours with 5 nM ActD compared with NT cells (n = 3). Indicated band (arrowhead) corresponds to the immunoglobulin light chain (IgL) of the antibodies used for IP. (C) Ubiquitination assay in MCL-1 immunoprecipitates from Trp53+/+;Eμ-Myc and Trp53–/–;Eμ-Myc lymphoma cells treated for 6 hours with 5 nM ActD compared with NT cells (n = 2). (D-E) FLAG-tagged MCL1 stability of either FLAG-tagged MCL-1wt- (D) or FLAG- (E) tagged MCL-1KR-overexpressing Eμ-Myc lymphoma cells determined by CHX chase after treatment with or without 5 nM ActD for 4 hours. Line graphs display FLAG-tagged MCL1 expression (arrowhead) over time determined from MCL-1 immunoblots (supplemental Figure 4F-G) as in Figure 3E (n = 2). (F) Representative western blot analysis of Trp53+/+;Eμ-Myc lymphoma cells treated for 30 minutes with 1.25 µM MG132 before treatment with ActD for 4 hours. Expression of antiapoptotic MCL-1 (arrowhead), calculated as in Figure 3B, is shown as relative to MCL-1 levels in NT cells in the absence of MG132 (n = 4). Data are presented as mean ± SEM. *P < .05; **P < .01 (2-way ANOVA test).
MCL-1 degradation is proteasome dependent
To ascertain the role of ubiquitination of MCL-1 in its decreased half-life, we stably introduced into Trp53+/+;Eμ-Myc cells a FLAG-tagged wt (MCL-1wt) or mutant (MCL-1KR) MCL-1 in which all lysines have been converted to arginines44 and then measured the half-life of both MCL-1 proteins in the presence of CHX, which were similar in untreated conditions (compare Figure 4D-E and supplemental Figure 4F-G). As observed for endogenous MCL-1 (Figure 3G-H), treatment with ActD led to a decrease in the half-life of MCL-1wt protein (Figure 4D; supplemental Figure 4F). Unexpectedly, MCL-1KR protein was not protected against degradation (Figure 4E; supplemental Figure 4G), suggesting that ActD-enhanced MCL-1 degradation is independent of direct ubiquitination. To test whether MCL-1 degradation was proteasome dependent, we treated Trp53+/+;Eμ-Myc cells with the proteasome inhibitor MG132. Such treatment protected against ActD-induced MCL-1 degradation and strongly suppressed PARP1 and caspase-3 cleavage (Figure 4F), consistent with the antiapoptotic role of MCL-1. Collectively, these results reveal that ActD treatment leads to the ubiquitin-independent but proteasome-dependent degradation of MCL-1, suggesting that MCL-1 may be indirectly targeted to the proteasome, potentially by NOXA binding.
ActD kills human BL and DLBCL cells
Our results clearly demonstrate that activation of the IRBC by ActD is a potent activator of apoptosis in Trp53+/+;Eμ-Myc cells. To test whether this is a mechanism common to human MYC-driven hematologic neoplasms, we first queried the curated Genomics of Drug Sensitivity in Cancer (GDSC) database.51 The 50% inhibitory concentration (IC50) values of ActD in 740 cancer cell lines revealed that hematologic (blood-borne) tumor cell lines are more sensitive to ActD than cell lines from other tumor types, with mean IC50 values of ∼5 nM or ∼75 nM, respectively (Figure 5A). Strikingly, an in silico analysis revealed that p53 status is the best predictor of response to ActD in hematologic malignancies (supplemental Figure 5A), that TP53mut tumors are less sensitive to the drug compared with TP53wt tumors overall (Figure 5B), and that mutations in TP53 are a common signature of the most resistant hematologic tumor cell lines (supplemental Figure 5B). We confirmed in a panel of BL and DLBCL cell lines in which MYC is overexpressed14,52 that ActD treatment killed TP53wt cells more effectively than TP53mut cell lines (Figure 5C-D), closely paralleled by p53-dependent PARP1 cleavage and the degradation of MCL-1, as suggested by the loss of the single MCL-1 band detected under these conditions (Figure 5E; see “Discussion”). These studies strongly support the potential for using ActD to treat TP53wt hematologic malignancies.
Figure 5.
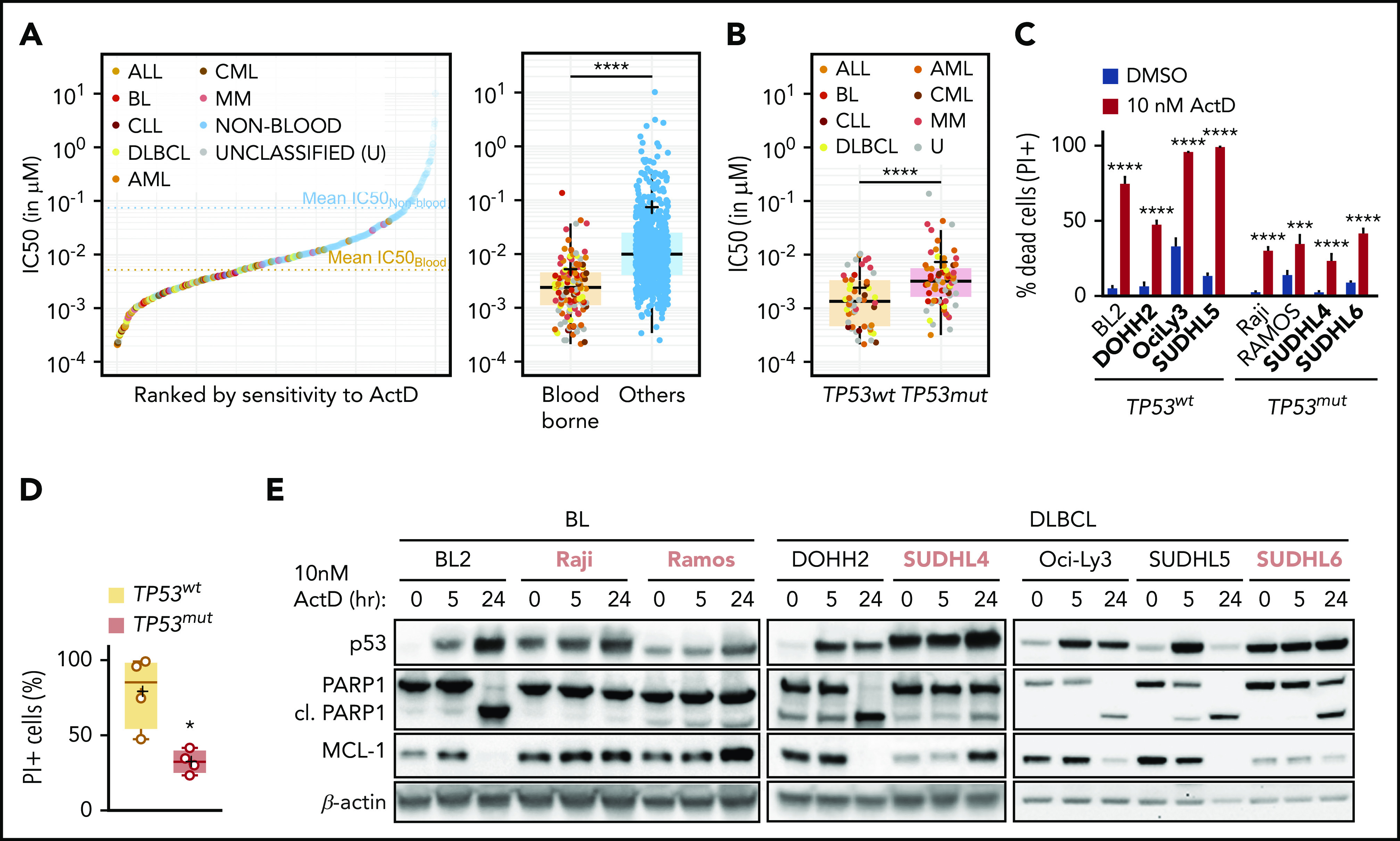
ActD treatment induces cell death in human TP53wtB-cell lymphomas. (A) ActD IC50 values of a data set with 740 tumor cell lines from the Genomics of Drug Sensitivity in Cancer (GDSC) database ranked by sensitivity to ActD (left) and box plot showing IC50 values of hematologic (blood-borne) and nonhematologic (others) tumors (right). Box plot represents median and first and third quartiles per group, with individual values for each cell line, and the mean IC50 (+) is indicated. The mean IC50 of ActD is 5.26 (95% confidence interval [CI], 2.96-7.56) in blood-borne tumor cell lines vs 74.12 (95% CI, 34.87-111.36) in other tumor type cell lines. ****P < .0001 (Mann-Whitney-Wilcoxon test). (B) Box plot showing IC50 values of blood-borne tumors as a function of their TP53 status. Box plot represents median and first and third quartiles per group with individual values for each cell line, and the mean IC50 (+) is indicated. ****P < .0001 (Mann-Whitney-Wilcoxon test). (C) Proportion of dead cells from a panel of wt TP53 (TP53wt) and mutant TP53 (TP53mut) BL- and DLBCL-derived cell lines (shown in boldface) determined by propidium iodide (PI) staining and flow cytometry analysis after treatment with 10 nM ActD for 48 hours (n = 3-4). Data are presented as mean ± SEM. ***P < .001; ****P < .0001 (2-way ANOVA test). (D) Scatter plot showing proportion of dead cells from panel C as a function of their TP53 status. Box plot represents median and first and third quartiles per group with individual values for each cell line, and the mean IC50 value (+) is indicated. *P < .05 (Mann-Whitney-Wilcoxon test). (E) Representative western blot analysis from lysates of the panel analyzed in panel C treated with 10 nM ActD for the indicated times (n = 3-4). TP53mut cell lines are shown in pink boldface. ALL, acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; AML, acute myeloid leukemia; CML, chronic myeloid leukemia; MM, multiple myeloma; U, unclassified.
ActD suppresses lymphoma growth in a p53-dependent manner
ActD is clinically used in multimodal treatment of rare pediatric tumors, such as Wilms tumor,53 rhabdomyosarcoma,54 and Ewing sarcoma,55 and for gestational trophoblastic neoplasia in women56 but at relatively high doses that presumably inhibit both Pol I- and Pol II-mediated transcription. To test the potential efficacy of low-dose ActD treatment against lymphoma progression, we transplanted Trp53+/+;Eμ-Myc and Trp53–/–;Eμ-Myc lymphoma cells into congenic C57BL/6J and C57BL/6J-Ly5.1 mice, respectively. After the disease was established, the mice were treated with either vehicle or with a single dose of 0.1 mg/kg ActD (Figure 6A), a dose fivefold lower than the equivalent standard dose given to human adults,56 with no effect on global transcription.57 After 6 hours, a single dose of ActD inhibited Pol I-dependent transcription (Figure 6B) and induced expression of p53 and its target genes Puma, Noxa, and p21 in lymph nodes collected from mice harboring Trp53+/+;Eμ-Myc lymphomas (Figure 6C; supplemental Figure 6A). In parallel, ActD rapidly reduced the number of Trp53+/+;Eμ-Myc cells in the spleen causing a modest reduction in spleen weight (Figure 6D; supplemental Figure 6B) but had no effect on Trp53–/–;Eμ-Myc cells (Figure 6E; supplemental Figure 6C). Consistent with these findings, ActD treatment induced p53 expression, led to PARP1 and caspase-3 cleavage, and led to a slight reduction in MCL-1 levels in the spleen of mice harboring Trp53+/+;Eμ-Myc (Figure 6F; supplemental Figure 6D) but not Trp53–/–;Eμ-Myc lymphoma cells (supplemental Figure 6E). Thus, ActD had an immediate on-target impact in Trp53+/+;Eμ-Myc lymphoma cells.
Figure 6.
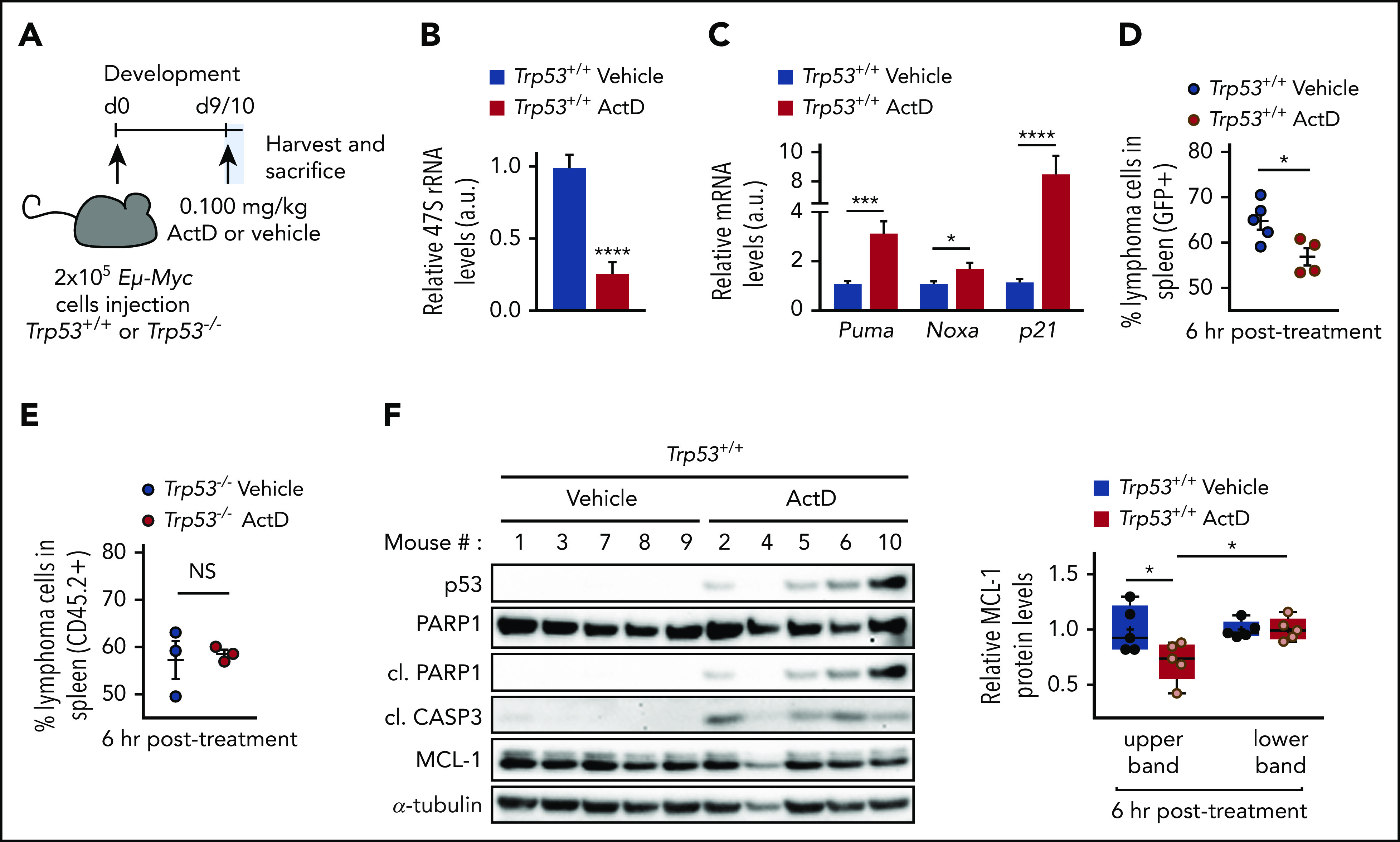
ActD treatment induces p53-dependent cell death in Eμ-Myc lymphomas in vivo. (A) C57BL/6J and C57BL/6J-Ly5.1 mice were intravenously injected with 200 000 Trp53+/+;Eμ-Myc or Trp53–/–;Eμ-Myc lymphoma cells, respectively, and were treated with a single dose of 0.1 mg/kg ActD or vehicle. Spleen, inguinal, and axillary lymph nodes were collected 6 hours after drug administration from 3-5 mice per group as described in supplemental Methods. (B-C) qRT-PCR analysis of Its1-containing 47S pre-rRNA (B) and of Puma, Noxa, and p21 (C) in axillary nodes from ActD-treated mice, normalized to β2m mRNA levels and compared with vehicle-treated axillary nodes. *P < .05; ***P < .001; ****P < .0001 (unpaired Student t test). (D-E) Percentage of viable lymphoma cells in the spleen of Trp53+/+;Eμ-Myc (D) or Trp53–/–;Eμ-Myc (E) lymphoma-bearing mice determined by flow cytometry as the percentage of PI-, B220+ (pan B-cell marker), and either green fluorescent protein (GFP+) for Trp53+/+;Eμ-Myc lymphoma cells or CD45.2+ cells for Trp53–/–;Eμ-Myc lymphoma cells, as detailed in supplemental Methods. Data are presented as individual values with error bars displaying mean ± SEM. *P < .05 (unpaired Student t test). (F) Western blot analysis of the spleen homogenates of each Trp53+/+;Eμ-Myc lymphoma-bearing mouse analyzed in panel D. Expression of the 2 MCL-1 forms (upper and lower bands) as determined in Figure 2A is shown as relative to MCL-1 expression in the vehicle-treated group. Data are presented as individual values with error bars displaying mean ± SEM. *P < .05 (2-way ANOVA test). a.u., arbitrary units; d0, day 0.
To test the therapeutic value of ActD on lymphoma regression, mice were administrated either vehicle or 0.1 mg/kg ActD once per day in 3 cycles (Figure 7A; supplemental Figure 7A). After 3 days of treatment, ActD reduced the number of circulating Trp53+/+;Eμ-Myc cells below detectable levels, but it had no effect on Trp53–/–;Eμ-Myc cells (Figure 7B-C). Consequently, ActD treatment restored the white blood cell and lymphocyte numbers to within the normal range in mice transplanted with Trp53+/+;Eμ-Myc lymphoma cells, whereas aberrant cell numbers persisted in those harboring Trp53–/–;Eμ-Myc lymphoma cells (supplemental Figure 7B-C). ActD treatment seems to selectively kill Trp53+/+;Eμ-Myc lymphoma cells, because no deleterious effects were detected on non–B-cell or wt–B-cell populations in the recipients bearing either Trp53+/+;Eμ-Myc or Trp53–/–;Eμ-Myc lymphomas (Figure 7D). Further analyses of spleen, bone marrow, and thymus showed that ActD significantly decreased the number of Trp53+/+;Eμ-Myc lymphoma cells within these tissues after 3 days of treatment (Figure 7E), effectively reversing the splenomegaly and thymus hyperplasia characteristics of the lymphoma-bearing mice (supplemental Figure 7D-E). Tissue weight reduction and Eμ-Myc lymphoma cell depletion correlated with a reduction in cell numbers per tissue and the restoration of white blood cell numbers to normal values (Figure 7F; supplemental Figure 7F).
Figure 7.
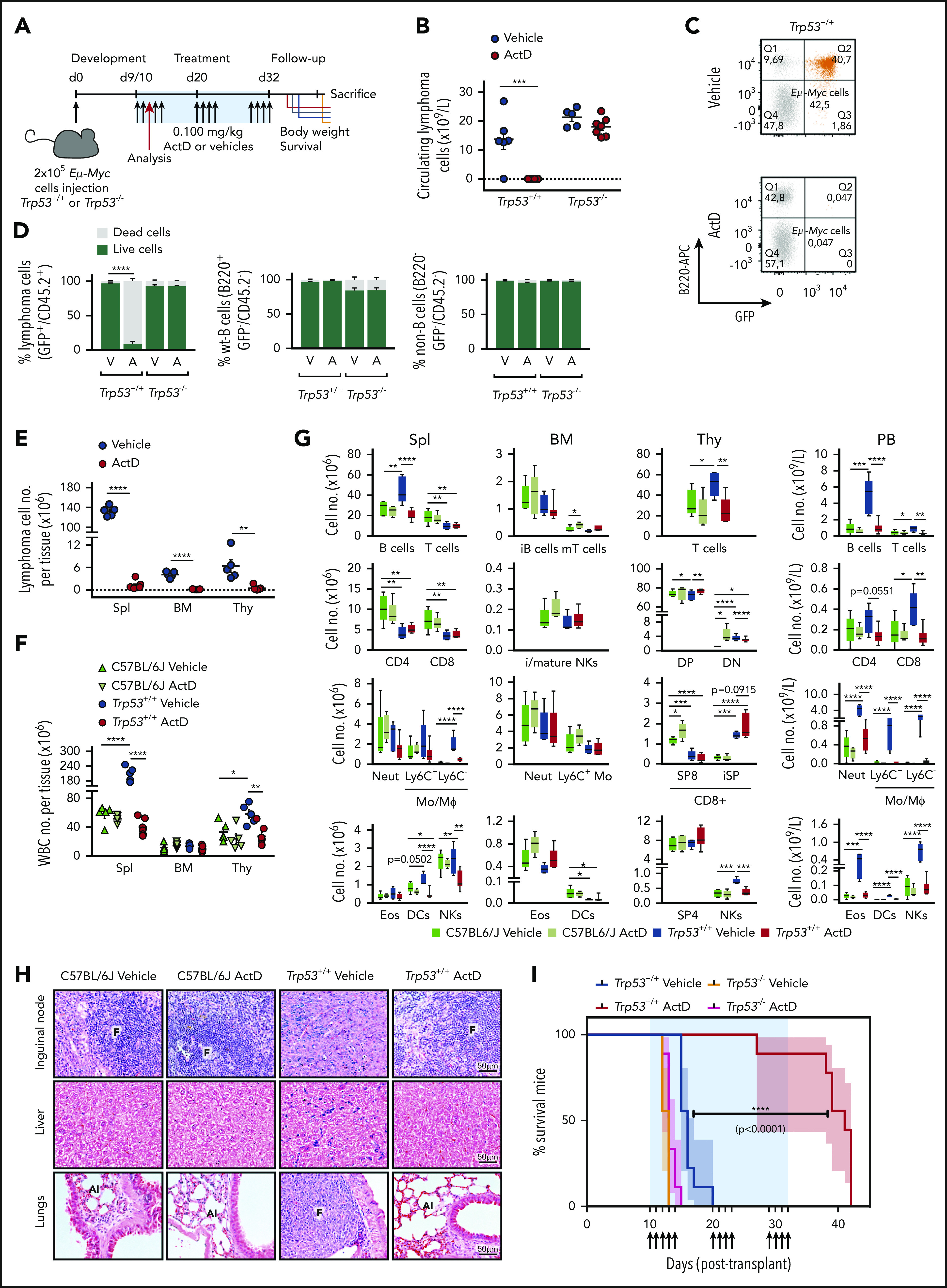
ActD treatment promotes Trp53+/+;Eμ-Myc lymphomas regression in vivo. (A) C57BL/6J and C57BL/6J-Ly5.1 mice intravenously injected with 200 000 Trp53+/+;Eμ-Myc or Trp53–/–;Eμ-Myc lymphoma cells, respectively, were treated with either ActD (0.1 mg/kg) or vehicle, starting 9 to 10 days after injection. Drug was delivered in 3 discontinuous cycles, with the dosing days indicated by the arrows, resuming at day 32 posttransplantation. Tumor burden was assessed by immunostaining, and flow cytometric analyses from peripheral blood and hematopoietic tissues were collected from 5 to 7 mice per group after the third administration (red arrow) as described in “Methods.” (B) Number of lymphoma cells in circulating blood from mice after 3 days of ActD or vehicle administration determined by flow cytometry as described in the legend for Figure 6D and calculated as detailed in “Methods.” ***P < .000 (2-way ANOVA test). Dotted line is set at y = 0. (C) Representative dot plots from peripheral blood of a vehicle-treated (top) and an ActD-treated (bottom) Trp53+/+;Eμ-Myc-bearing mouse from panel B showing the Trp53+/+;Eμ-Myc lymphoma cell population (GFP+B220+B220low) in orange. (D) Percentage of live and dead Trp53+/+;Eμ-Myc or Trp53–/–;Eμ-Myc lymphoma cells (left) wt-B cells (center), and non-B cells (right panel), from the peripheral blood of mice analyzed in panel B. The proportion of dead cells from each population was determined as described in the legend for Figure 6D, with the markers identifying each population indicated on the y-axis of the corresponding graph. A, ActD; V, vehicle. ****P < .0001 (2-way ANOVA test). (E-F) Number of Trp53+/+;Eμ-Myc lymphoma cells (E) and white blood cells (WBCs) (F) contained in the spleen (Spl), bone marrow (BM), and thymus (Thy) of Trp53+/+;Eμ-Myc lymphoma-bearing mice after 3 days of ActD or vehicle administration compared with ActD- or vehicle-treated C57BL/6J control mice (n = 5 mice per group), determined by multicolor flow cytometry panel evaluation as detailed in supplemental Figure 7G. Data are presented as individual values with error bars displaying mean ± SEM. *P < .05; **P < .01; ****P < .0001. Statistical analyses for unpaired Student t test (E) and 1-way ANOVA test (F) were performed separately in each tissue. Dotted line (E) is set at y = 0. (G) Number of major immune cell populations contained in the spleen, bone marrow, thymus, and peripheral blood (PB) of Trp53+/+;Eμ-Myc lymphoma-bearing mice (n = 5-7 mice per treatment group) and of C57BL/6J control mice (n = 5 mice per treatment group) treated as in panel E. Box plots represent the median and the 95% CI for each cell population, as indicated on the x-axis. DC, dendritic cell; CD4, CD4+ T cells; CD8, CD8+ T cells; DN, double-negative; DP, double-positive; Eos, eosinophils; i, immature; iSP, immature single-positive thymocytes; m, mature; Mo/MФ, monocytes/macrophages; Neut, neutrophils; NK, natural killer; SP4, single-positive CD4 thymocytes; SP8, single-positive CD8 thymocytes. *P < .05; **P < .01; ***P < .001; ****P < .0001 (1-way ANOVA test performed separately for each population). (H) Photomicrographs (original magnification ×40) of hematoxylin- and eosin-stained paraffin-embedded sections of inguinal lymph nodes, liver, and lungs from representative C57BL/6J control and ActD-treated mice and from lymphoma-burdened sick control and ActD-treated Eμ-Myc mice (n = 3 mice per group) treated as in panel E. Al, alveoli; F, follicle; T, infiltrating tumor mass. (I) Kaplan-Meier survival curves of C57BL/6J and C57BL/6J-Ly5.1 mice harboring Trp53+/+;Eμ-Myc and Trp53–/–;Eμ-Myc lymphomas, respectively, and treated with ActD or vehicle (n = 9 mice per group) as indicated in panel A. Data are presented as mean ± SEM. ****P < .0001 (log-rank Mantel-Cox and Gehan-Breslow-Wilcoxon text).
Flow cytometric analyses (supplemental Figure 7G) revealed that the myeloid and lymphoid populations present in the spleen, bone marrow, thymus, and blood were unaffected by 3 days of ActD administration, suggesting that acute Pol I inhibition does not alter the maturation of B cells, T cells, or myeloid cells (Figure 7G). Importantly, ActD treatment restored the number of most immune cell populations to the normal range in lymphoma-bearing mice (Figure 7G; supplemental Table 2). Moreover, elimination of lymphoma cells by ActD was not restricted to primary lymphoid tissues, spleen, or blood because its administration for 3 days restored the architecture of the inguinal nodes (Figure 7H, top panel) and promoted the clearance of infiltrated malignant cells from the liver and lungs (Figure 7H, middle and bottom panels). Of note, erythropoiesis was not affected by ActD treatment (supplemental Figure 7H).
Critically, although there was no benefit from ActD treatment in mice with Trp53–/–;Eμ-Myc lymphoma cells, 3 cycles of ActD treatment dramatically extended median survival of mice harboring Trp53+/+;Eμ-Myc lymphoma from 14 to 42 days (Figure 7I). When treatment was terminated, only 1 animal had succumbed to lymphoma, suggesting that prolonged ActD treatment might further increase survival. Therefore low-dose ActD caused major lymphoma regression and extended the survival of mice bearing Trp53+/+;Eμ-Myc but not Trp53–/–;Eμ-Myc lymphomas, revealing a potential therapeutic option for the treatment of MYC-driven B-cell lymphomas.
Discussion
Targeting RiBi in MYC-driven tumors has emerged as a potential avenue for cancer therapy,20,22 whereas new insights into the mechanisms involved in this response have indicated a potential role for the IRBC and p53.23,26,58,59 Here we show in Eμ-Myc lymphoma cells that depletion of RPL7a led to the activation of p53 through IRBC induction, similar to our findings in other cells types in which RP mRNAs were depleted.28,58,59 However, in contrast to the p53-dependent cell cycle arrest observed in previous studies, Eμ-Myc lymphoma cells partially depleted of RPL7a rapidly underwent apoptosis, a response not observed in Trp53–/–;Eμ-Myc lymphoma cells, suggesting that in Eμ-Myc lymphoma cells, the p53 response, rather than inducing cell cycle arrest, triggers apoptosis.60 Importantly, p53-mediated apoptosis was not observed in Eμ-Myc lymphoma cells partially depleted of RPL11, which fails to induce the IRBC. Although decreased rates of protein synthesis inhibit proliferation, the apoptotic response seems to play a major role in limiting Eμ-Myc lymphoma expansion. Indeed, ActD led to lymphoma regression and cleared secondary sites infiltrated with Trp53+/+;Eμ-Myc lymphoma cells but had no effect on the survival of mice transplanted with Trp53–/–;Eμ-Myc lymphoma cells. This is consistent with earlier findings showing that Rpl24 deletion has no additional effect on lymphoma regression in a TP53–/– background.22
Induction of apoptosis in RPL7a-depleted Eμ-Myc cells correlated with a selective loss of the slower migrating form of MCL-1. Neither form arises from alternative splicing,61,62 as confirmed in a recent study showing that ablation of the putative splice donor and acceptor sites does not alter the expression of the MCL-1 protein doublet.36 Instead, they demonstrated that the different isoforms were generated by differential proteolysis.36 The Mr 38K form, like the full-length MCL-1, has been reported to be located on the outer mitochondrial membrane where it co-immunoprecipitates with proapoptotic BIM and protects against apoptosis. In contrast, the Mr 36K form seems to be in the inner mitochondrial matrix where it is required for mitochondrial structure and function with no apparent role in protection from apoptosis.36 Our observation that cell death induced by the IRBC is associated with the selective loss of the upper band of MCL-1 implies that this most likely represents an antiapoptotic form, either full-length MCL-1 or the Mr 38K isoform. In contrast, the faster migrating form is most likely the Mr 36K isoform localized to the mitochondrial matrix. Interestingly, although our experimental conditions did not allow us to resolve several MCL1 isoforms in human BL or DLBCL cell lines, others have reported their existence, as well as an N-terminal truncated antiapoptotic form of MCL-1, which is more stable than full-length MCL-1 and is overexpressed in hematopoietic malignancies including BL.63 That the loss of the antiapoptotic MCL-1 protein is responsible for p53-induced cell death in RPL7a-depleted Eμ-Myc lymphoma cells is best supported by recent studies demonstrating that loss of a single allele of Mcl1 was sufficient to cause substantial regression of Eμ-Myc lymphomas in contrast to the deletion of BCL-XL, which had no impact on their growth.5
Unlike its antiapoptotic relatives BCL-XL and BCL-2, MCL-1 is an inherently short-lived protein,61,62 regulated at both the translational39 and posttranslational level.40,64-66 We show that the reduction of MCL-1 in RPL7a-depleted Eμ-Myc lymphoma cells depends on p53 activation and enhanced ubiquitin-independent proteasome degradation. Such a mode of degradation of antiapoptotic MCL-1 has been previously reported in unstressed or stressed conditions.44 A likely hypothesis is that the ActD-induced interaction of MCL-1 with NOXA targets MCL-1 to the proteasome in Trp53+/+;Eμ-Myc lymphoma cells. Indeed, NOXA was shown to bind more selectively to MCL-1 than other BH3-only proteins,67,68 and its overexpression leads to MCL-1 degradation.45 NOXA was also proposed to target MCL-1 to the proteasome.47,50 Interestingly, although NOXA can be ubiquitinated, this does not seem to affect its stability; instead, both its degradation and that of MCL-1 are mediated by a degron at the carboxy tail of NOXA.50 It cannot be excluded that another MCL-1–binding partner induced by p53 may be involved in MCL-1 degradation, or that MCL-1 could be ubiquitinated on non-lysine residues as shown for other proteins.69 Thus, it is important to better understand the relationship between the induction of BH3-only proteins and the stability of MCL-1 during early lymphomagenesis and whether the same mechanisms take place in human B-cell lymphomas.
The expression of MYC is dysregulated in ∼70% of human cancers,5 and in many cases, their survival depends on the upregulation of antiapoptotic BCL-2 family members, including MCL-1.5,70 Four MCL-1–specific BH3 mimetic drugs have entered clinical trials, including AZD5991,71 AMG-176,72 AMG-397, and S64315.19 However, deletion of MCL-1 in the adult heart leads to mitochondrial dysfunction and rapid heart failure,73,74 which may be associated with the loss of the Mr 36K isoform, which is required for normal mitochondrial activity.36 Recently the phase 1 clinical trials with AMG-397 and AMG-176 have been placed on hold by the FDA because of cardiotoxicity (ClinicalTrials.gov NCT03797261 and NCT03465540). Here we demonstrate an indirect approach for targeting MCL-1 in lymphoma cells, which does not affect the levels of the Mr 36K isoform or compromise normal immune cells, thus offering a therapeutic window to treat MYC-driven tumors different from that for MCL-1–specific BH3 mimetics.
Inhibition of RiBi has become an attractive target in cancer therapy, particularly with the recent development of Pol I inhibitors.20,75 However, their mechanism of action remains partially unclear, possibly involving the DNA damage response.76-78 Here, we show that the efficacy of ActD against B-cell lymphoma relies on a functional p53 protein, which argues in favor of stratifying patients receiving ActD on the basis of the genetic status of MDM2 and p53. Interestingly, compared with other tumor types, hematologic tumors present a lower frequency of TP53 mutations, corresponding to <20% vs the estimated 30% to 35% overall prevalence.79 In a chemical screen of 3000 small-molecule compounds, including 530 FDA-approved drugs, ActD ranked among the top 7 compounds reducing MCL-1 levels across a number of cancer cell lines.80 However, the effects of ActD, as well as the other 6 compounds were attributed to global transcriptional inhibition and the short half-life of MCL-1.80 In contrast, we show that a dose of ActD, ∼3 orders of magnitude lower than that used by Wei et al,80 selectively decreases the antiapoptotic MCL-1 form and that this seems to be mediated by the activation of the IRBC rather than by inhibition of transcription. Given the importance of targeting MCL-1 in cancer and the issues arising from drugs still in clinical trials, ActD may be an attractive therapeutic agent alone, in combination with lower doses of BH3 mimetics, or with inhibitors targeting other antiapoptotic BCL-2 members.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Joseph T. Opferman (St Jude Children’s Research Hospital, Memphis, TN) for kindly providing the murine stem cell virus-puromycin-resistant (pMSCV-puro)-3xFLAG-derived retroviral plasmids containing MCL-1wt or MCL-1KR, and the wt Mcl1 and Mcl1 knockout MEFs, and David Plas (University of Cincinnati, Cincinnati, OH) for providing the Platinum-E retroviral packaging cell lines. They also thank Scott W. Lowe, Michael D. Cole, Ross Hannan, Teng Teng, Megan Bywater, Jennifer Devlin, Lukas E. Dow, Siniša Volarevic, and Cristina Muñoz-Pinedo for providing materials and for their helpful discussions; Andreas Strasser for his advice and critical comments on earlier drafts of this manuscript; and the Elias Campos laboratory for providing SUDHL5, SUDHL6, and Oci-LY3 DLBCL-derived cell lines. The authors thank the Centres de Recerca de Catalunya (CERCA) Program/Generalitat de Catalunya for institutional support. Cell viability analysis by flow cytometry was performed at Shriners Hospitals for Children (Cincinnati, OH), and the in vivo experiments were performed at Peter MacCallum Cancer Center, (Melbourne, Australia) and at Centro de Medicina Regenerativa de Barcelona (Barcelona, Spain).
This work was supported by a grant from the National Institutes of Health, National Cancer Institute (R01-CA158768) and grants from the Spanish Ministry of Science and Innovation (SAF2011-24967, SAF2014-52162-P, and SAF2017-84301-P), the Marie Curie Action Career Integration Grant from European Commission (PCIG10-GA-2011-304160), the Asociación Española Contra el Cáncer (GCB14-2035), the Instituto de Salud Carlos III (ISCIII)–Red Temática de Investigación Cooperativa en Cáncer (RTICC) (RD12/0036/0049), the Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) (SGR 870 and SGR 01743), and the Spanish Ministry of Science and Innovation ISCIII (PIE13/00022) (all to G.T.); by fellowships from the Association pour la Recherche sur le Cancer (SAE20140601346) and Juan de la Cierva (FJCI-2014-20422) (J.P.); by project grants from the National Health and Medical Research Council of Australia (1053792 and 1102609) and from the Cancer Council of Victoria (1184873) (R.B.P.); by grants from the Spanish Ministry of Science and Innovation (SAF2017-84301-P) and Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) (SGR 01743) (A.G.); by funding from the Victorian Cancer Agency (MCRF 17028) and a Dyson Bequest Fellowship (G.L.K.); and by a Formación de Personal Investigador (FPI) grant from the Spanish Ministry of Science and Innovation (BES-2015-075840) (A.D.). The Spanish Ministry grants are cofunded by the European Regional Development Fund (FEDER).
Footnotes
Further information and requests for resources and reagents should be directed to and will be fulfilled by sending an e-mail to Joffrey Pelletier at joffrey.pelletier@irbbarcelona.org or George Thomas at gthomas@idibell.cat.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.D., S.P., G.T., and J.P. conceived and designed the study; A.D. and S.P. performed most of the experiments; F.I., M.L.R., M.G.-C., S.T.D., E.P.K., J.K., and A.G. contributed to individual experiments; A.D., S.P., C.A.M., S.C.K., R.B.P., G.T., and J.P. analyzed the data; A.D., S.P., C.A.M., G.T., and J.P. wrote the manuscript; and A.D., S.P., C.A.M., V.A., G.L.K., R.S., S.C.K., E.P.K., J.K., A.G., R.B.P., G.T., and J.P. provided intellectual support in the discussion of the results.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for J.P. is Colorectal Cancer Laboratory, Institute for Research in Biomedicine (IRB) Barcelona, Barcelona Institute of Science and Technology, Barcelona, Spain.
Correspondence: George Thomas, Laboratory of Cancer Metabolism, Molecular Mechanisms and Experimental Therapy in Oncology Program, Bellvitge Biomedical Research Institute, Gran Via de L’Hospitalet 199, 08908 Barcelona, Spain; e-mail: gthomas@idibell.cat; and Joffrey Pelletier, Colorectal Cancer Laboratory, Institute for Research in Biomedicine, Baldiri Reixac 10, 08028 Barcelona, Spain; e-mail: joffrey.pelletier@irbbarcelona.org.
REFERENCES
- 1.Kress TR, Sabò A, Amati B. MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer. 2015;15(10):593-607. [DOI] [PubMed] [Google Scholar]
- 2.Pelletier J, Thomas G, Volarević S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer. 2018;18(1):51-63. [DOI] [PubMed] [Google Scholar]
- 3.van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10(4):301-309. [DOI] [PubMed] [Google Scholar]
- 4.Brown SJ, Cole MD, Erives AJ. Evolution of the holozoan ribosome biogenesis regulon. BMC Genomics. 2008;9(1):442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly GL, Grabow S, Glaser SP, et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. 2014;28(1):58-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432(7015):307-315. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. [DOI] [PubMed] [Google Scholar]
- 8.Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets. 2005;5(1):27-41. [DOI] [PubMed] [Google Scholar]
- 9.Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Hematology Am Soc Hematol Educ Program. 2013;2013:575-583. [DOI] [PubMed] [Google Scholar]
- 10.Michalak EM, Jansen ES, Happo L, et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009;16(5):684-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004;101(16):6164-6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348(6299):331-333. [DOI] [PubMed] [Google Scholar]
- 13.Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delgado MD, León J. Myc roles in hematopoiesis and leukemia. Genes Cancer. 2010;1(6):605-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molyneux EM, Rochford R, Griffin B, et al. Burkitt’s lymphoma. Lancet. 2012;379(9822):1234-1244. [DOI] [PubMed] [Google Scholar]
- 16.Adams JM, Harris AW, Pinkert CA, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318(6046):533-538. [DOI] [PubMed] [Google Scholar]
- 17.Grabow S, Delbridge AR, Aubrey BJ, Vandenberg CJ, Strasser A. Loss of a single Mcl-1 allele inhibits MYC-driven lymphomagenesis by sensitizing pro-B cells to apoptosis. Cell Rep. 2016;14(10):2337-2347. [DOI] [PubMed] [Google Scholar]
- 18.Merino D, Whittle JR, Vaillant F, et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci Transl Med. 2017;9(401):eaam7049. [DOI] [PubMed] [Google Scholar]
- 19.Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477-482. [DOI] [PubMed] [Google Scholar]
- 20.Bywater MJ, Poortinga G, Sanij E, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22(1):51-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang F, Ni M, Chalishazar MD, et al. Inosine monophosphate dehydrogenase dependence in a subset of small cell lung cancers. Cell Metab. 2018;28(3):369-382.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barna M, Pusic A, Zollo O, et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008;456(7224):971-975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barkić M, Crnomarković S, Grabusić K, et al. The p53 tumor suppressor causes congenital malformations in Rpl24-deficient mice and promotes their survival. Mol Cell Biol. 2009;29(10):2489-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donati G, Peddigari S, Mercer CA, Thomas G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013;4(1):87-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gentilella A, Morón-Duran FD, Fuentes P, et al. Autogenous control of 5′TOP mRNA stability by 40S ribosomes. Mol Cell. 2017;67(1):55-70.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macias E, Jin A, Deisenroth C, et al. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 interaction. Cancer Cell. 2010;18(3):231-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morcelle C, Menoyo S, Morón-Duran FD, et al. Oncogenic MYC induces the impaired ribosome biogenesis checkpoint and stabilizes p53 independent of increased ribosome content. Cancer Res. 2019;79(17):4348-4359. [DOI] [PubMed] [Google Scholar]
- 28.Teng T, Mercer CA, Hexley P, Thomas G, Fumagalli S. Loss of tumor suppressor RPL5/RPL11 does not induce cell cycle arrest but impedes proliferation due to reduced ribosome content and translation capacity. Mol Cell Biol. 2013;33(23):4660-4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sloan KE, Bohnsack MT, Watkins NJ. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013;5(1):237-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perry RP, Kelley DE. Inhibition of RNA synthesis by actinomycin D: characteristic dose-response of different RNA species. J Cell Physiol. 1970;76(2):127-139. [DOI] [PubMed] [Google Scholar]
- 31.Valente LJ, Aubrey BJ, Herold MJ, et al. Therapeutic response to non-genotoxic activation of p53 by Nutlin3a is driven by PUMA-mediated apoptosis in lymphoma cells. Cell Rep. 2016;14(8):1858-1866. [DOI] [PubMed] [Google Scholar]
- 32.Diepstraten ST, Chang C, Tai L, et al. BCL-W is dispensable for the sustained survival of select Burkitt lymphoma and diffuse large B-cell lymphoma cell lines. Blood Adv. 2020;4(2):356-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zuber J, McJunkin K, Fellmann C, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol. 2011;29(1):79-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25(1):104-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Llambi F, Moldoveanu T, Tait SW, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44(4):517-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perciavalle RM, Stewart DP, Koss B, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14(6):575-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drygin D, Lin A, Bliesath J, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71(4):1418-1430. [DOI] [PubMed] [Google Scholar]
- 38.Peltonen K, Colis L, Liu H, et al. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell. 2014;25(1):77-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mills JR, Hippo Y, Robert F, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci U S A. 2008;105(31):10853-10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085-1095. [DOI] [PubMed] [Google Scholar]
- 41.Mojsa B, Lassot I, Desagher S. Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells. 2014;3(2):418-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weng C, Li Y, Xu D, Shi Y, Tang H. Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J Biol Chem. 2005;280(11):10491-10500. [DOI] [PubMed] [Google Scholar]
- 43.Herrant M, Jacquel A, Marchetti S, et al. Cleavage of Mcl-1 by caspases impaired its ability to counteract Bim-induced apoptosis. Oncogene. 2004;23(47):7863-7873. [DOI] [PubMed] [Google Scholar]
- 44.Stewart DP, Koss B, Bathina M, Perciavalle RM, Bisanz K, Opferman JT. Ubiquitin-independent degradation of antiapoptotic MCL-1. Mol Cell Biol. 2010;30(12):3099-3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willis SN, Chen L, Dewson G, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19(11):1294-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Day CL, Smits C, Fan FC, Lee EF, Fairlie WD, Hinds MG. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J Mol Biol. 2008;380(5):958-971. [DOI] [PubMed] [Google Scholar]
- 47.Czabotar PE, Lee EF, van Delft MF, et al. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci U S A. 2007;104(15):6217-6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song T, Wang Z, Ji F, et al. Deactivation of Mcl-1 by dual-function small-molecule inhibitors targeting the Bcl-2 homology 3 domain and facilitating Mcl-1 ubiquitination. Angew Chem Int Ed Engl. 2016;55(46):14250-14256. [DOI] [PubMed] [Google Scholar]
- 49.Gomez-Bougie P, Ménoret E, Juin P, Dousset C, Pellat-Deceunynck C, Amiot M. Noxa controls Mule-dependent Mcl-1 ubiquitination through the regulation of the Mcl-1/USP9X interaction. Biochem Biophys Res Commun. 2011;413(3):460-464. [DOI] [PubMed] [Google Scholar]
- 50.Pang X, Zhang J, Lopez H, et al. The carboxyl-terminal tail of Noxa protein regulates the stability of Noxa and Mcl-1. J Biol Chem. 2014;289(25):17802-17811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang W, Soares J, Greninger P, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41(D1):D955-D961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Blood. 2013;122(24):3884-3891. [DOI] [PubMed] [Google Scholar]
- 53.Malogolowkin M, Cotton CA, Green DM, et al. ; National Wilms Tumor Study Group . Treatment of Wilms tumor relapsing after initial treatment with vincristine, actinomycin D, and doxorubicin. A report from the National Wilms Tumor Study Group. Pediatr Blood Cancer. 2008;50(2):236-241. [DOI] [PubMed] [Google Scholar]
- 54.Skapek SX, Ferrari A, Gupta AA, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. 2019;5(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jaffe N, Paed D, Traggis D, Salian S, Cassady JR. Improved outlook for Ewing’s sarcoma with combination chemotherapy (vincristine, actinomycin D and cyclophosphamide) and radiation therapy. Cancer. 1976;38(5):1925-1930. [DOI] [PubMed] [Google Scholar]
- 56.Goldstein DP, Berkowitz RS. Current management of gestational trophoblastic neoplasia. Hematol Oncol Clin North Am. 2012;26(1):111-131. [DOI] [PubMed] [Google Scholar]
- 57.Siboni RB, Nakamori M, Wagner SD, et al. Actinomycin D specifically reduces expanded CUG repeat RNA in myotonic dystrophy models. Cell Rep. 2015;13(11):2386-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fumagalli S, Ivanenkov VV, Teng T, Thomas G. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 2012;26(10):1028-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fumagalli S, Di Cara A, Neb-Gulati A, et al. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol. 2009;11(4):501-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a license to kill. Nat Rev Mol Cell Biol. 2015;16(7):393-405. [DOI] [PubMed] [Google Scholar]
- 61.Shmueli A, Oren M. Life, death, and ubiquitin: taming the mule. Cell. 2005;121(7):963-965. [DOI] [PubMed] [Google Scholar]
- 62.Michels J, Johnson PW, Packham G. Mcl-1. Int J Biochem Cell Biol. 2005;37(2):267-271. [DOI] [PubMed] [Google Scholar]
- 63.De Biasio A, Vrana JA, Zhou P, et al. N-terminal truncation of antiapoptotic MCL1, but not G2/M-induced phosphorylation, is associated with stabilization and abundant expression in tumor cells. J Biol Chem. 2007;282(33):23919-23936. [DOI] [PubMed] [Google Scholar]
- 64.Schwickart M, Huang X, Lill JR, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463(7277):103-107. [DOI] [PubMed] [Google Scholar]
- 65.Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121(7):1071-1083. [DOI] [PubMed] [Google Scholar]
- 66.Inuzuka H, Shaik S, Onoyama I, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471(7336):104-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17(3):393-403. [DOI] [PubMed] [Google Scholar]
- 68.Kuwana T, Bouchier-Hayes L, Chipuk JE, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17(4):525-535. [DOI] [PubMed] [Google Scholar]
- 69.McClellan AJ, Laugesen SH, Ellgaard L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019;9(9):190147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Opferman JT. Attacking cancer’s Achilles heel: antagonism of anti-apoptotic BCL-2 family members. FEBS J. 2016;283(14):2661-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tron AE, Belmonte MA, Adam A, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. 2018;9(1):5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Caenepeel S, Brown SP, Belmontes B, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018;8(12):1582-1597. [DOI] [PubMed] [Google Scholar]
- 73.Thomas RL, Roberts DJ, Kubli DA, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013;27(12):1365-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo L, Eldridge S, Furniss M, Mussio J, Davis M. Role of Mcl-1 in regulation of cell death in human induced pluripotent stem cell-derived cardiomyocytes in vitro. Toxicol Appl Pharmacol. 2018;360:88-98. [DOI] [PubMed] [Google Scholar]
- 75.Khot A, Brajanovski N, Cameron DP, et al. First-in-human RNA polymerase I transcription inhibitor CX-5461 in patients with advanced hematologic cancers: results of a phase I dose-escalation study. Cancer Discov. 2019;9(8):1036-1049. [DOI] [PubMed] [Google Scholar]
- 76.Xu H, Di Antonio M, McKinney S, et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat Commun. 2017;8(1):14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hald OH, Olsen L, Gallo-Oller G, et al. Inhibitors of ribosome biogenesis repress the growth of MYCN-amplified neuroblastoma. Oncogene. 2019;38(15):2800-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quin J, Chan KT, Devlin JR, et al. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget. 2016;7(31):49800-49818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bouaoun L, Sonkin D, Ardin M, et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37(9):865-876. [DOI] [PubMed] [Google Scholar]
- 80.Wei G, Margolin AA, Haery L, et al. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell. 2012;21(4):547-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.