

Abstract
Positron emission tomography (PET) using 2‐deoxy‐2‐[18F]fluoro‐d‐glucose ([18F]FDG), a marker of energy metabolism and cell proliferation, is routinely used in the clinic to assess patient response to chemotherapy and to monitor tumor growth. Treatment with some tyrosine kinase inhibitors (TKIs) causes changes in blood glucose levels in both nondiabetic and diabetic patients. We evaluated the interaction of several classes of TKIs with human glucose transporter‐1 (hGLUT‐1) in FaDu and GIST‐1 cells by measuring [3H]2‐deoxy‐d‐glucose ([3H]2‐DG) and [3H]FDG uptake. Uptake of both was inhibited to varying extents by the TKIs, and representative TKIs from each class showed competitive inhibition of [3H]2‐DG uptake. In GIST‐1 cells, [3H]FDG uptake inhibition by temsirolimus and nilotinib was irreversible, whereas inhibition by imatinib, gefitinib, and pazopanib was reversible. Molecular modeling studies showed that TKIs form multiple hydrogen bonds with polar residues of the sugar binding site (i.e., Q161, Q282, Q283, N288, N317, and W388), and van der Waals interactions with the H‐pocket site. Our results showed interaction of TKIs with amino acid residues at the glucose binding site to inhibit glucose uptake by hGLUT‐1. We hypothesize that inhibition of hGLUT‐1 by TKIs could alter glucose levels in patients treated with TKIs, leading to hypoglycemia and fatigue, although further studies are required to evaluate roles of other SLC2 and SLC5 members. In addition, TKIs could affect tumor [18F]FDG uptake, increasingly used as a marker of tumor response. The hGLUT‐1 inhibition by TKIs may have implications for routine [18F]FDG‐PET monitoring of tumor response in patients.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Human glucose transporter‐1 transports glucose and 18‐flurodexoyglucose (FDG) into cells where it gets phosphorylated and trapped. A retrospective study of blood glucose concentrations in diabetic and nondiabetic patients with cancer treated with dasatinib, imatinib, sorafenib, or sunitinib showed a statistically significant decrease in blood glucose levels.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Inhibition of glucose uptake was examined and tyrosine kinase inhibitors (TKIs) inhibited [3H]2‐DG and [3H]FDG uptake to varying extents and competitively. Molecular modeling studies confirmed TKI interaction with amino acid residues at glucose binding sites.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ We hypothesized that TKI inhibition of glucose uptake might lead to changes in tissue and blood glucose levels and might explain the hypo/hyperglycemia and fatigue observed in TKI‐treated patients.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Because FDG uptake is increasingly used as a marker of tumor response, these results indicate careful evaluation of [18F]FDG‐positron emission tomography tumor response monitoring during TKI treatment, and in clinical trials validating new TKI effectiveness.
Clinical decision making involves cancer treatment response monitoring to prevent ineffective therapy and resulting side effects. Positron emission tomography (PET) is used for monitoring treatment effectiveness. PET tracers 2‐deoxy‐2‐[18F]fluoro‐d‐glucose ([18F]FDG) and 3′‐deoxy‐3′‐[18F]fluorothymidine have been used for noninvasive detection of tumor energy metabolism and cell proliferation, respectively. The glucose analog [18F]‐FDG is the most widely used PET tracer for tumor staging and diagnosis, treatment response, and routine treatment monitoring. 1 Human glucose transporter‐1 (hGLUT‐1) transports FDG, like glucose, into cells where it subsequently gets phosphorylated and trapped. 2
Many small molecule tyrosine kinase inhibitors (TKIs), such as erlotinib, gefitinib and vandetanib, and lapatinib, targeted against epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR) and EGFR, and EGFR and Her‐2, respectively, have been developed. Multitargeted TKIs include sorafenib, axitinib, pazopanib, and sunitinib. TKIs targeting intracellular BCR‐ABL kinase of chronic myeloid leukemia include bosutinib, dasatinib, imatinib, nilotinib, and ponatinib. Multiple TKIs are used successfully for treatment of renal cell carcinoma, leukemia, gastrointestinal tumors, lung cancer, and several other malignancies. A retrospective study of blood glucose concentrations in diabetic and nondiabetic patients with cancer treated with dasatinib, imatinib, sorafenib, or sunitinib showed statistically significant decrease in blood glucose levels. 3 Many preclinical studies have investigated use of [18F]FDG PET imaging for TKI treatment monitoring, and some studies showed early changes in [18F]FDG levels while others did not. 4 Depending on the TKI used, accumulating evidence show either increased or decreased glucose levels in patients during TKI therapy, although dasatinib, erlotinib, sunitinib, and sorafenib were shown to lower glucose levels and improve glycemic control. 5 PET tracer uptake is complex and depends on tumor type, targeted kinase, and tracer uptake by tumors. In general, higher standardized uptake values have been correlated with poorer outcomes. 6
Solute carrier (SLC) transporters for sugars include SLC2 and SLC5 families, which regulate cellular concentrations of sugars. 7 , 8 hGLUT‐1, a widely distributed member of the facilitative SLC2 family, transports glucose down its concentration gradient. 9 XylE, a structurally characterized xylose transporter of E. coli, shows 65% sequence identity and 88% sequence similarity with hGLUT‐1. 10 Residues important for binding of d‐glucose (DG) in the crystal structure of XylE (although XylE does not transport dg) are conserved in hGLUT‐1 with the exception of Gln415, which is Asn411 in hGLUT‐1. Gln‐282 contributes to sugar binding in all conformations of hGLUT‐1 via hydrogen bonding. 11 The sugar‐binding site is located at the interface of N‐terminal and C‐terminal domains. An earlier study showed competitive inhibition of glucose transport activity of hGLUT‐1 by flavones and isoflavones. 12 WZB117, a polyphenol compound, inhibits erythrocyte 3‐O‐methylglucose uptake competitively by binding at the exofacial binding site. 13
We examined whether hGLUT‐1 activity is inhibited by TKIs targeted against EGFR, VEGFR, Her‐2, BCR‐ABL, MTK, MEK, and mTOR using [3H]2‐deoxy‐d‐glucose ([3H]2‐DG) and [3H]FDG uptake inhibition experiments in cultured FaDu and GIST‐1 cells to understand underlying mechanisms of TKI‐induced changes in glucose levels of TKI‐treated patients. TKI inhibition of hGLUT‐1 was tested further in competition experiments using one representative TKI from BCR‐ABL, EGFR, MTK, and mTOR TKIs. Reversibility of TKI inhibition of [3H]FDG uptake after removing TKIs was examined in GIST‐1 cells. Further evidence for TKI interactions with hGLUT‐1 glucose binding site was obtained by molecular modeling using three models of outward‐facing partly occluded conformation (models 1–3), one model of inward‐open conformation (model 4), and one model of fully open outward‐facing conformation (model 5).
METHODS
Chemicals and reagents
Nitrobenzylmercaptopurine riboside, 2‐DG and other chemicals were from Sigma Chemical Company (Mississauga, ON, Canada). Tissue culture (12‐well) plates and flasks were from VWR International (Mississauga, ON, Canada). Cell culture media and fetal bovine serum (FBS) were from Gibco BRL (Burlington, ON, Canada). Ecolite was from ICN Pharmaceuticals (Montreal, PQ, Canada). The [3H]2‐DG was from Moravek Biochemicals (Brea, CA). The [3H]FDG was from American Radiolabeled Chemicals (St. Louis, MO). TKIs were from LC Laboratories (Woburn, MA). Mouse anti‐GLUT‐1 antibody was from Abcam and goat anti‐mouse Alexa 488 antibody was from Molecular Probes (Eugene, OR).
Cell culture
The human nasopharyngeal carcinoma cell line (FaDu) obtained from the American Type Culture Collection, was routinely passaged in growth media containing Dulbecco’s Modified Eagle Medium with high glucose, 25 mM HEPES, 1 mM sodium pyruvate, 2 mM l‐glutamine, 10% FBS, and penicillin‐streptomycin. Cultures were maintained at 37°C in 95% air and 5% CO2 humidified atmosphere. Plating densities were such that cells typically reached 80–90% confluence within 48 hours after plating.
The human GIST‐1 cell line from Cosmo Bio (Cedarlane, Burlington, ON, Canada) was routinely passaged in growth media containing Dulbecco’s Modified Eagle Medium with high glucose with 10 mM HEPES, 10% FBS, and penicillin‐streptomycin.
Analysis of hGLUT‐1 expression in FaDu cells
FaDu and GIST‐1 cells were seeded in T25 flasks at a density of 1 million cells/flask and 24 hours later were removed with 0.05% trypsin. Approximately 300,000 cells/tube were fixed in 4% paraformaldehyde for 30 minutes at room temperature (RT). Cells were washed with phosphate buffered saline (PBS) and centrifuged. Cells were permeabilized with PBS containing 0.5% saponin and 5% FBS for 10 minutes. hGLUT‐1 expression by FaDu and GIST‐1 cells was evaluated using anti‐hGLUT‐1 antibody (0.3 µg/tube) with 60‐minute incubations at RT. Cells were washed twice with PBS containing 0.1% saponin and 2% FBS. Secondary antibody (goat anti‐mouse Alexa 488) at a dilution of 1/300 was incubated with cells for 30 minutes at RT in the dark. After washing, 300 µL of PBS/tube was added, and fluorescence was measured by analytical flow cytometry.
[3H]2‐DG and [3H]FDG uptake assays in FaDu and GIST‐1 cells
FaDu or GIST‐1 cells were seeded in 24‐well plates, and grown for 48 or 72 hours. To screen for the type of glucose transporter/s present, the time course of uptake of 20 µM [3H]2‐DG for 20 minutes was measured in FaDu or GIST‐1 cells, in the absence or presence of sodium or phloretin (200 µM), an inhibitor of glucose uptake, which was used to completely inhibit mediated uptake. Uptake activity was measured with 20 µM [3H]2‐DG or [3H]FDG at RT in transport buffer (pH 7.4) containing 20 mM HEPES, 150 mM NaCl, 5 mM KCl, 1.2 mM K2HPO4, 5 mM MgSO4, and 2.5 mM CaCl2. At the end of uptake intervals, permeant‐containing solutions were aspirated, and plates were washed twice with ice‐cold buffer after which cells were solubilized with 5% Triton X‐100. Radioactivity in solubilized extracts was measured by liquid scintillation counting. Uptake values were expressed as pmol/106 cells, converted to percent (%) control activity (in the absence of inhibitor) and graphs were generated using GraphPad Prism (GraphPad Software, San Diego, CA).
Both FaDu and GIST‐1 cells were exposed to equal amount of DMSO in controls or 25 µM individual TKIs in DMSO and uptake of 20 µM The [3H]2‐DG or [3H]FDG was measured for 15 minutes at RT. Phloretin was used as the hGLUT‐1 inhibitor control. To determine half‐maximal inhibitory concentration (IC50) values (concentration of test compound that inhibited uptake by 50% relative to that of untreated cells) for TKI inhibition of [3H]2‐DG uptake, FaDu cells were incubated with 20 µM [3H]2‐DG in buffer for 15 minutes in absence (uninhibited controls) or presence of graded concentrations (0–100 µM) of various classes of TKIs. Concentration‐effect curves were subjected to nonlinear regression analysis using GraphPad Prism to obtain IC50 values. Each IC50 value determination was carried out with seven concentrations and three replicates per concentration and experiments were repeated three times.
We examined reversibility of TKI inhibition of [3H]FDG uptake in GIST‐1 cells. Cells were seeded in 24‐well plates, grown for 48 hours, and then exposed to DMSO or 25 µM individual TKIs under the following conditions: cells + DMSO (control (C)), cells + TKI (C + TKI) for 15 minutes, cells + TKI for 15 minutes and TKI removed (C + TKI‐), cells + TKI for 15 minutes and TKI removed and cells washed once (C + TKI + wash) and uptake of 20 µM [3H]FDG was measured for 15 minutes at RT. Results are presented as percent control.
For TKI effects on kinetics of 2‐DG uptake, uptake experiments (15 minutes) with graded concentrations of [3H]2‐DG (0–30 mM) in the absence or presence of fixed concentrations of 0, 25, 50, and 100 µM of bosutinib, gefitinib, pazopanib, or temsirolimus were conducted in FaDu cells. Each experiment was repeated three times with triplicates for each condition.
MOLECULAR MODELING METHODS
Homology models
We used five models for the outward‐facing open conformation (models 1–3), one for the inward‐open conformation (model 4), and one for the fully open outward‐facing conformation (model 5). The first three models were adopted from Ung et al., 14 in which homology models of hGLUT‐1 are constructed in a partially occluded outward open conformation (partly occluded) based on the X‐ray structure of XylE as the template (PDB: 4GBZ). 10 , 15 The inward‐open conformation (model 4) is adopted from the available crystal structure (PDB: 4PYP) 16 for hGLUT‐1. The fully open outward facing structure (model 5) is from Park et al. 17 and was prepared with homology modeling using XylE as the template (PDB: 4GBZ). Details on molecular modeling simulations and MM/PBSA and MM/GBSA methods is presented in the Supplementary Methods section.
RESULTS
Flow cytometry analysis of hGLUT‐1 expression in FaDu and GIST‐1 cells
We determined hGLUT‐1 protein expression on FaDu and GIST‐1 cells by staining with anti‐hGLUT‐1 antibody, as described in the Materials and Methods section. Fluorescence intensity data from flow cytometry analysis of unstained, secondary antibody‐stained, and anti‐GLUT‐1‐stained FaDu and GIST‐1 cells is presented in Figure 1 a. Results showed that both cell types possessed hGLUT‐1 with a higher abundance on FaDu than on GIST‐1 cells.
Figure 1.
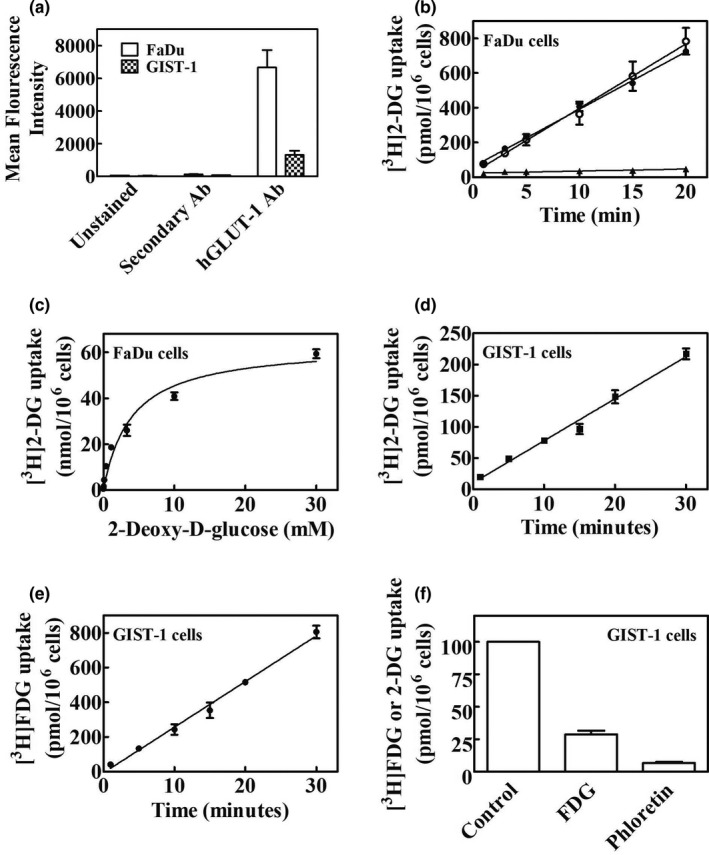
Analysis of hGLUT‐1 protein abundance and uptake of [3H]2‐DG and [3H]FDG in FaDU and GIST‐1 cells. (a) Flow cytometry analysis of hGLUT‐1 protein. FaDu and GIST‐1 cells were stained with hGLUT‐1 antibody for 60 minutes at RT followed by staining with secondary antibody at a dilution of 1/300. Cells were incubated for 30 minutes at RT in the dark. After washing, cells were analyzed for fluorescence by analytical flow cytometry. Mean results from three independent experiments with untreated cells and cells stained with secondary antibody only or hGLUT‐1 plus secondary antibody are shown. (b) [3H]2‐DG uptake in FaDu cells. Time courses of uptake of [3H]2‐DG in FaDu cells in presence of potassium or sodium with or without phloretin are shown (○, NaCl; ●, KCl; ▲, 200 µM Phloretin). (c) The concentration dependence (0–30 mM) of [3H]2‐DG uptake rates in FaDu cells. (d, e) Time courses of [3H]2‐DG and [3H]FDG uptake, respectively, in GIST‐1 cells. (f) Inhibition of [3H]2‐DG uptake by phloretin or excess FDG in GIST‐1 cells. Values with mean ± SE. are shown in each panel. All experiments were repeated three times with three to four replicates per condition. 2‐DG, 2‐deoxy‐d‐glucose; hGLUT, human glucose transporter; RT, room temperature.
[3H]2‐Deoxyglucose uptake characteristics
We examined glucose uptake activity in both FaDu and GIST‐1 cells in time course experiments (0–20 minutes) with 20 µM [3H]2‐DG in the absence or presence of sodium ions or in the presence of sodium and phloretin. Figure 1 b shows time courses of uptake of 20 µM [3H]2‐DG in FaDu cells, which were linear up to 20 minutes and independent of sodium, thus indicating the absence of any sodium‐dependent glucose uptake activity. Similar results were seen in GIST‐1 cells (data not shown). In experiments illustrated in Figure 1 c, the concentration dependence of 2‐DG uptake rates was determined in FaDu cells. Uptake rates exhibited saturation as concentrations of 2‐DG increased from 0 to 30 mM (Figure 1 c). Kinetic parameters kinetic metabolite (Km) and maximal rate of metabolism (Vmax) obtained from Michaelis–Menten analysis (mean ± SE) were 3.8 ± 0.2 mM and 62.7 ± 0.7 nmol/106 cells/15 minutes, respectively. In GIST‐1 cells uptake of both 2‐DG (Figure 1 d) and FDG (Figure 1 e) was linear and uptake of [3H]2‐DG was inhibited by excess FDG or phloretin (Figure 1 f), indicating hGLUT‐1 mediated FDG uptake activity.
Inhibition of hGLUT‐1 activity in FaDu and GIST‐1 cells by TKIs
The ability of several classes of TKIs to inhibit uptake of 20 µM [3H]2‐DG or [3H]FDG in FaDu and GIST‐1 cells, respectively, was tested in the presence and absence of 25 µM of each representative TKI. Uptake of [3H]2‐DG in FaDu (Figure 2a ) and [3H]FDG in GIST‐1 cells (Figure 2b ) was inhibited to varying extents by different TKIs, although the inhibitory profile of TKIs was similar between the two cell lines. We studied reversibility of inhibition of [3H]FDG uptake in GIST‐1 cells after treatment with bosutinib, gefitinib, pazopanib, temsirolimus, or nilotinib (Figure 2c ) and removal of TKIs followed by subsequent washing as described in the Methods section. Although imatinib, gefitinib, and pazopanib showed reversibility of [3H]FDG uptake inhibition, temsirolimus and nilotinib did not.
Figure 2.
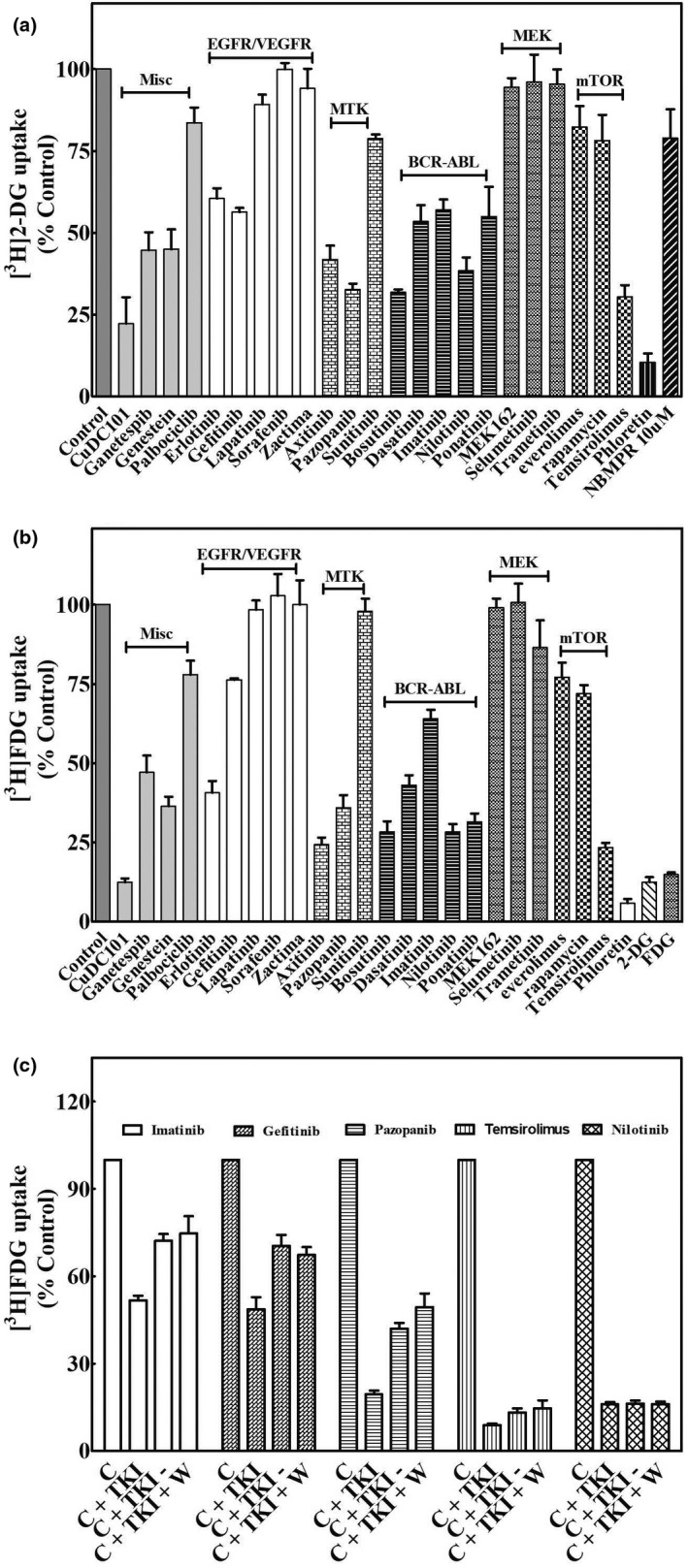
Inhibition of [3H]2‐DG and [3H]FDG uptake by TKIs in FaDU and GIST‐1 cells: reversibility of TKI inhibition. (a) The effect of TKIs on [3H]2‐DG uptake in FaDU cells. FaDu cells were incubated with 20 µM [3H]2‐DG for 15 minutes in the absence (control) or presence of 25 µM of each of the indicated TKIs or 200 µM phloretin and uptake was measured as described in the Methods section. (b) Effects of TKIs on [3H]FDG uptake in GIST‐1 cells. GIST‐1 cells were incubated with 20 µM [3H]FDG for 15 minutes in the absence (control) or presence of 25 µM of each of the indicated TKIs or 200 µM phloretin and uptake was measured as described in the Methods section. Data presented are means (± SE values) of three or more experiments each conducted with three replicates. Uptake values represent percentage of 2‐DG or FDG uptake in the presence of TKIs relative to that in its absence (control). Error bars are not shown where SE values are smaller than the size of the symbol. (c) Reversibility of TKI inhibition in GIST‐1 cells. GIST‐1 cells were incubated first for 15 minutes in the absence (control) or presence of 25 µM concentration of various TKIs as follows: control cells (c) in DMSO containing buffer for 15 minutes; cells + TKI (C + T) in buffer for 15 minutes; cells + TKI for 15 minutes, after which TKI containing buffer was removed (C + T−); and cells + TKI for 15 minutes, the TKI containing buffer was removed and washed once with fresh drug free buffer (C + T + W). After these manipulations, GIST‐1 cells were incubated further with 20 µM [3H]FDG in fresh buffer for 15 minutes and processed as described in the Methods section. Data presented are averages of three or more experiments each conducted with three replicates and are expressed as mean ± SE values. Uptake values represent percentage of FDG uptake in the presence of TKIs relative to that in its absence (controls). Error bars are not shown where SE values are smaller than the size of the symbols. 2‐DG, 2‐deoxy‐d‐glucose; FDG, fluoro‐d‐glucose; EGFR, endothelial growth factor receptor; TKI, tyrosine kinase inhibitor; VEGFR, vascular endothelial growth factor receptor.
TKIs that had the highest inhibitory effects in the above experiments were tested for their relative abilities to inhibit [3H]2‐DG uptake in FaDu cells, in concentration‐dependent inhibition experiments. The IC50 values for TKIs, cytochalasin B (CB), and phloretin (obtained from literature) 18 are summarized in Table 1 . Representative concentration‐effect curves for inhibition of hGLUT‐1 mediated glucose uptake are presented in Figure 3 a–d.
Table 1.
Summary of IC50 values for inhibition of 2‐DG uptake in FaDu cells
| TKI | IC50, µM |
|---|---|
| 2‐DG | 840 ± 79 |
| Cytochalasin B a | 0.52 ± 0.09 |
| Phloretin a | 61 ± 3 |
| Axitinib | 29 ± 4.8 |
| Bosutinib | 13 ± 1.2 |
| Gefitinib | 40 ± 1.7 |
| Imatinib | 25 ± 1.6 |
| Nilotinib | 6 ± 0.7 |
| Pazopanib | 44 ± 6.0 |
| Temsirolimus | 17 ± 3.3 |
Inhibition of 20 µM [3H]2‐DG uptake by TKIs was assessed in FaDu cells in concentration‐effect experiments as described in Methods. IC50 values (mean ± SE) are listed below.
2‐DG, 2‐deoxy‐d‐glucose; IC50, half‐maximal inhibitory concentration; TKI, tyrosine kinase inhibitor.
Reference 24.
Figure 3.
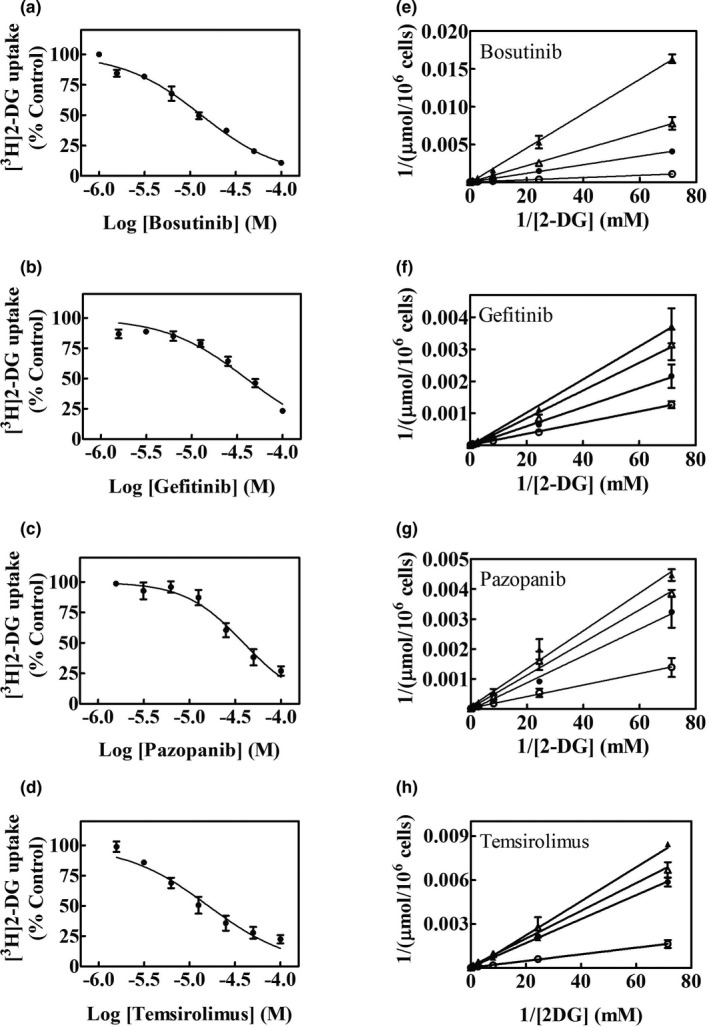
Concentration dependence of inhibition of hGLUT‐1 mediated [3H]2‐DG uptake by TKIs. Effects of increasing concentrations of bosutinib, gefitinib, pazopanib, and temsirolimus, respectively, on uptake of [3H]2‐DG in FaDu cells are shown in panels (a–d). Effects of 0 (○), 25 (●), 50 (∆), or 100 (▲) µM of bosutinib, gefitinib, pazopanib, or temsirolimus, respectively, on [3H]2‐DG uptake rates are shown in panels (e–h) as Lineweaver–Burk transformations of the data. Each experiment was repeated three times with three to four replicates for each experiment and values (means ± SE) are shown in each panel. 2‐DG, 2‐deoxy‐d‐glucose; hGLUT, human glucose transporter; TKI, tyrosine kinase inhibitor.
Effects of TKIs on kinetics of 2‐DG uptake in FaDu cells
Kinetic studies of hGLUT‐1 with one inhibitory TKI from each class were undertaken in FaDu cells by studying effects of fixed concentrations of bosutinib, gefitinib, pazopanib, or temsirolimus on varying concentrations of [3H]2‐DG (0–30 mM) uptake. Results analyzed by Lineweaver–Burk plots are shown in Figure 3 e–h, and all tested TKIs showed competitive inhibition of [3H]2‐DG uptake.
Molecular docking studies
As a first step, we docked CB to all conformations (models 1–5). The docking results reproduced the crystallographic binding to the inward‐open model, as published previously, 19 increasing the confidence in our model and approach. The confidence in the partly occluded models were evaluated before. 14 Figure 4 shows TKI structures, helical models used, and their binding affinities. We undertook molecular docking studies of bosutinib, gefitinib, pazopanib, temsirolimus, imatinib, nilotinib, axitinib, 2‐DG, and CB with different hGLUT‐1 conformations (models 1–5). For each TKI/protein‐model pair, we used the pose with the best score for molecular mechanics Gibbs–Boltzmann surface area MM/GBSA computations. Docking poses with the best binding affinity were chosen for final analysis. Predicted binding energies for bosutinib (model 3), gefitinib (model 3), pazopanib (model 1), temsirolimus, imatinib (model 2), nilotinib (model 2), axitinib (model 3), 2‐DG (model 1), and CB (model 4) are −78.74, −67.28, −63.99, −93.88, −90.45, −66.93, −54.34, −26.57, and −51.35 kcal/mol, respectively. Figure 4 m shows detailed binding energies for each TKI/protein‐model pair as well as 2‐DG and CB compounds.
Figure 4.
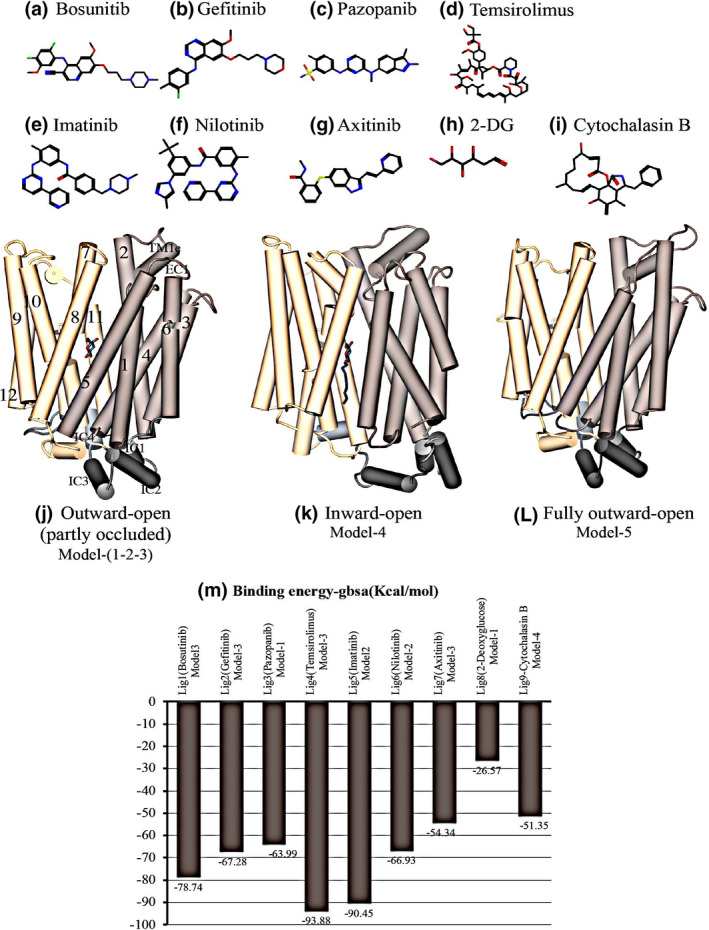
Chemical structures of inhibitors used in this study, the conformational models of hGLUT‐1 and the binding affinity for TKIs. The panels show structures of bosutinib (a), gefitinib (b), pazopanib (c), temsirolimus (d), imatinib (e), nilotinib (f), axitinib (g), 2‐DG (h), and CB (i). The panels e–g show outward‐open (partly occluded) (j), inward‐open (PDB: 4PYP) (k), and fully open outward facing (l) homology models. The structures are color coded as follows: N‐terminal domain, beige; C‐terminal domain, light brown; and the loop connecting the N and C domains, dark gray. Helices are shown as rods labeled using the conventional numbering scheme for transmembrane segments. IC and EC indicates intracellular and extracellular helix. TM1e is the extracellular part of the TM1 helix. Panel (m) shows binding affinities for the above‐mentioned compounds complexed with hGLUT‐1 protein models (model 1–5). Binding energies are predicted by the MM/GBSA method. The best ligand‐hGLUT‐1 pose is shown in dark brown colored bars. hGLUT, human glucose transporter; TKI, tyrosine kinase inhibitor.
Figure 5 shows that all TKI ligands extend from the sugar‐binding site to the nearby exofacial channel through the H‐pocket site, gated by the side chain of F291 (Figure 5 ). Bosutinib (ligand 1) forms a hydrogen bond with the polar residues of the sugar binding site (Q161 and N288), while also interacting with the H‐pocket site through F291 providing additional affinity and selectivity (Figure 5 Lig1). Similarly, gefitinib (ligand 2) interacts with polar residues of the sugar‐binding site through N317 and forms hydrogen bonds with the H‐pocket site of hGLUT‐1 (T30 and F291; Figure 5 Lig2). Pazopanib (ligand 3) forms a hydrogen bond with the indole group of W388 channel from sugar binding site at the opening of the cytosolic channel, where it completely blocks the channel entry. W388 is a key residue that coordinates sugar binding via a hydrogen bond taking part in the major conformational change from inward‐open to outward‐facing. This arrangement may provide a stronger binding affinity between the ligand and TM 10, potentially increasing the activation energy of a conformational transition, and blocking the bound transporter in an inactive, occluded state (Figure 5 Lig3). Temsirolimus (ligand 4) forms hydrogen bonds with (Q288) from sugar‐binding site and H‐pocket (T30, V165, and Y292). It also forms hydrogen bonds with S313 from TM 8 near the exofacial end of TM 8 (Figure 5 Lig4). Ligand 5, 6, and 7 show similar interactions as the other TKIs that were examined (ligand 1–4). They all interact with the sugar binding site and showed highest affinity to the outward‐facing partly occluded conformation. Active residues involved in binding are (F285 and S307) in ligand 5 (Figure 5 Lig5), F285 and W382 in ligand 6 (Figure 5 Lig6), and N282, F285, and N28 in ligand 7 (Figure 5 Lig7). In addition, 2‐DG interacts with residues N282, Q277, Q151, N409, and W382 at the sugar binding site (Figure 5 Lig8). The ligand interactions application of MOE 20 is used to visualize active sites of ligand‐protein complexes in 2D diagrammatic form (Figure 5 c). A selection of interacting entities, including hydrogen‐bonded residues and close but nonbonded residues, are drawn around the ligand. Additional properties, such as solvent accessible surface area and the ligand proximity outline, are shown. The hydrophobic region around the ligand is shown by the proximity of “greasy” residues, which are colored light green. Polar ends of ligands are closely affiliated with polar residues in pink, both H‐bonded and non‐H‐bonded (Figure 5 e).
Figure 5.
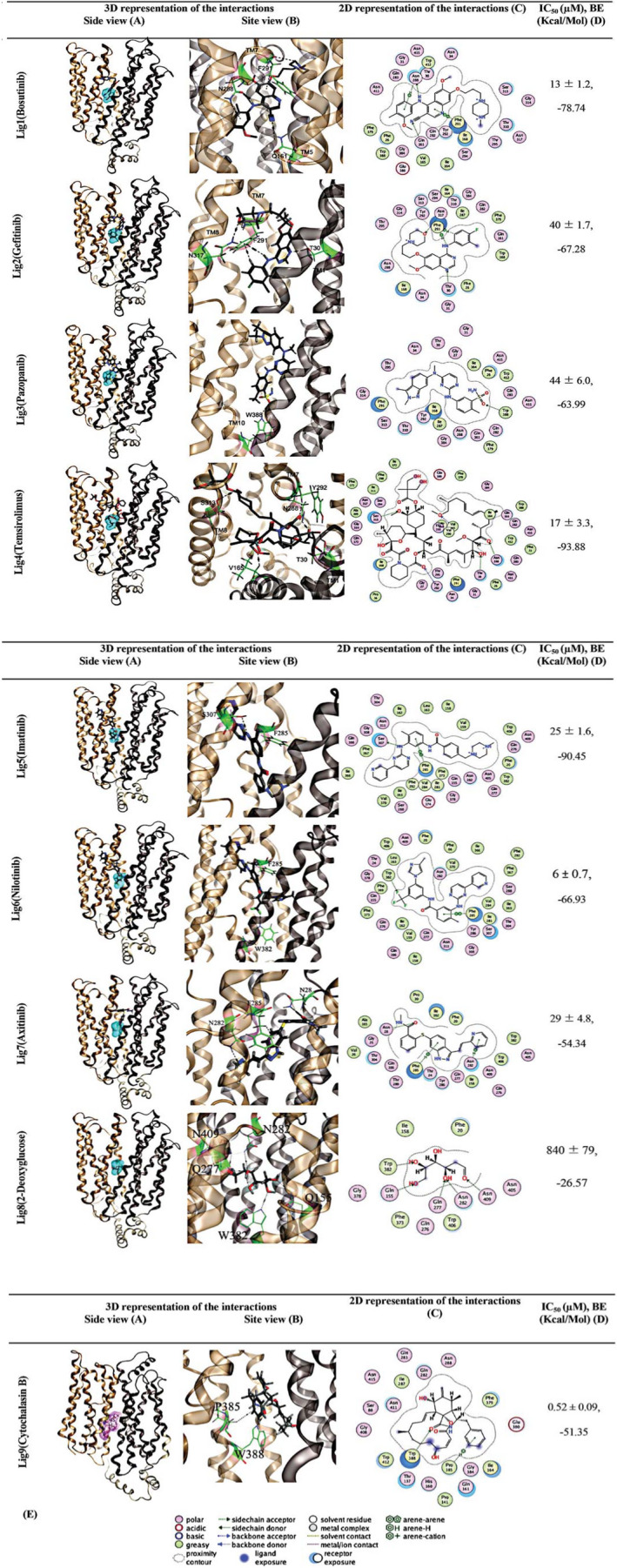
Molecular docking studies of ligand binding to hGLUT‐1. Homology modeled hGLUT‐1 outward‐open (partly occluded) complexed with bosutinib, gefitinib, pazopanib, temsirolimus, imatinib, nilotinib, axitinib, 2‐DG, and CB compounds. [3H]2‐DG binding site is represented with cyan binding pocket. CB is complexed with the inward open conformation. CB site is represented with the pink binding pocket. H‐bonds and their directionality are represented as black dashed arrows. The hGLUT‐1 is shown in cartoon representation, membrane spanning α‐helices (TM 1, 5, 7, 8, and 10) are indicated. Structures are color coded as follows: N‐terminal domain, gold; C‐terminal domain, brown; the loop connecting the N and C domains, gray. Ligands are displayed with stick and the atoms are colored as O (red), C (gray), N (blue), S (yellow), and Cl (green). Interacting residues in hGLUT‐1 are shown in stick presentation and their carbons are colored as green. A graphical key (e) is included to help interpret the 2D part of the ligand interactions panel (c). 2‐DG, 2‐deoxy‐d‐glucose; CB, cytochalasin B; hGLUT, human glucose transporter; IC50, half‐maximal inhibitory concentration.
DISCUSSION
A recent retrospective analysis of patients treated with TKIs 3 showed that TKI treatment results in hypoglycemia in both diabetic and nondiabetic patients. Because [18]FDG PET uptake is used as a marker of response, and has been correlated with outcomes, it is important to understand TKI effects on glucose uptake by transporters and those effects on [18]FDG tracer uptake during PET monitoring in patients receiving TKI‐based anticancer therapies. For example, changes in uptake and standardized uptake values have been incorporated into the Lugano criteria in patients with lymphoma. 21 In addition, PET response criteria in solid tumors (PERCIST study) have been proposed as functional markers of response comparable to response evaluation criteria in solid tumors (RECIST study). 6 , 22 A recent editorial in the Journal of Thoracic Disease 23 highlighted the technical rigor required to conduct comparative [18]FDG examinations in the setting of assessing response and commented on four different tools for looking at changes in [18]FDG in response to a therapeutic intervention. However, those investigations have assumed that there is a uniform molecular basis for in vivo changes in [18]FDG uptake in response to treatment.
Uptake of [18F]FDG was analyzed in several preclinical studies after treatment with TKIs. 4 In one study, [18F]FDG uptake was unchanged after erlotinib treatment, 24 whereas another study showed decreased uptake 1–2 days after erlotinib or gefitinib treatment. 25 , 26 Treatment with canertinib, a pan‐HER inhibitor, showed decreased [18F]FDG in 3–6 days after treatment initiation. 27 Imatinib treatment resulted in a dramatic decrease in [18F]FDG uptake in tumor models with c‐Kit mutations. 28 , 29 The mTOR inhibitors decreased [18F]FDG uptake from 1–2 days after treatment started. 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 Sunitinib, sorafenib, and axitinib reduced [18F]FDG uptake in PET. 38 Treatment with HDAC inhibitors decreased [18F]FDG uptake. 39 The above results suggest that individual TKIs may impact post treatment changes in [18]FDG uptake. In the current work, we examined effects of several classes of TKIs on hGLUT‐1 mediated uptake of [3H]2‐DG or [3H]FDG in FaDu and GIST‐1 cells. We examined the nature of inhibition by these TKIs and confirmed those results using molecular modeling studies.
Studies with human anti‐GLUT‐1 antibody in FaDu and GIST‐1 cells showed positive membrane staining for hGLUT‐1, consistent with the presence of hGLUT‐1 protein. We showed hGLUT‐1 functional activity using [3H]2‐DG or [3H]FDG uptake in both FaDu and GIST‐1 cells. The [3H]2‐DG uptake in both cell lines was inhibited by excess 2‐DG or FDG or phloretin, an inhibitor of hGLUT‐1. We demonstrated sodium‐independent hGLUT‐1 activity in FaDu cells showing that 2‐DG uptake was saturable with Km and Vmax values of 3.8 ± 0.2 mM and 62.7 ± 0.7 nmol/106 cells/15 minutes, respectively, and confirmed hGLUT‐1 activity in GIST‐1 cells.
Several classes of TKIs inhibited uptake of [3H]2‐DG or [3H]FDG in FaDu and GIST‐1 cells, although their effects differed substantially. CUDC‐101, an inhibitor of histone deacetylase, EGFR1, and EGFR2, inhibited uptake in FaDu and GIST‐1 cells by 80–90%. Ganetespib, a small‐molecule inhibitor of heat shock protein 90, and genistein, a soy isoflavone, also inhibited > 50% uptake in both FaDu and GIST‐1 cells. Among EGFR/VEGFR inhibitors, erlotinib and gefitinib were most inhibitory in both cell lines. MTK inhibitors pazopanib and axitinib inhibited uptake of [3H]2‐DG or [3H]FDG, whereas sunitinib inhibited to a smaller extent and sorafenib did not inhibit. All BCR‐ABL TKIs inhibited [3H]2‐DG or [3H]FDG uptake to varying extents in both cell lines and MEK inhibitors did not. Among mTOR inhibitors, temsirolimus inhibited [3H]2‐DG or [3H]FDG uptake the most. These results show that although many classes of TKIs inhibit [3H]2‐DG or [3H]FDG uptake in cells, not all TKIs behave the same, indicating that TKI structures, as well as target kinases, may have a role in inhibiting uptake by hGLUT‐1.
In concentration‐dependent inhibition experiments, BCR‐ABL TKIs showed the lowest IC50 values among all TKI classes tested. Bosutinib, gefitinib, pazopanib, and temsirolimus showed competitive inhibition of [3H]2‐DG uptake, suggesting TKIs and 2‐DG bind to the same or overlapping sites on hGLUT‐1. In addition, [3H]FDG uptake experiments in GIST‐1 cells showed that TKI inhibition was reversible with some TKIs but not others, an observation supported by their binding energies. It is likely that dissociation constants of respective TKIs could contribute to the observed reversible or irreversible inhibition of glucose uptake. In addition to the direct inhibition of glucose uptake, other regulatory effects on downstream kinase pathways might influence changes in glucose transporter levels or activities. Our results suggest that to avoid interference in the [18]FDG PET screening, patients should stop taking TKIs for at least a few days for complete removal of the inhibitory TKI from tumors.
Interaction of TKIs with hGLUT‐1 was confirmed with molecular docking studies of seven TKIs binding to hGLUT‐1 structures with different conformations (models 1–5), as described in the Methods section. The predicted binding energies for bosutinib (model 3), gefitinib (model 3), pazopanib (model 1), temsirolimus (model 3), imatinib (model 2), nilotinib (model 3), and axitinib (model 3) are −78.74, −67.28, −63.99, −93.88, −90.45, −66.93, −54.34, −26.57, and −51.35 kcal/mol, respectively.
Docking results showed highest affinity to the outward‐facing partly occluded conformation with TKIs forming multiple hydrogen bonds with the polar residues of the sugar binding sites as well as engaging in the van der Waals interaction with the H‐pocket site. All docked ligands extend from the sugar‐binding site to the nearby exofacial channel through the H‐pocket site to block free exchange of glucose between the cytoplasm and external environments of cells.
These results are similar to earlier studies that showed competitive inhibition of hGLUT‐1 activity by flavones and isoflavones 12 and, more recently, WZB117, a polyphenol compound competitively inhibited erythrocyte 3‐O‐methylglucose uptake by binding at the exofacial binding site. 13 Other studies with hGLUT‐1 showed interactions of a putative nucleotide‐binding site on the cytosolic face of hGLUT‐1 with purine nucleoside analogs, such as ATP, AMP, and caffeine. 40 Recently, a potent GLUT inhibitor, named rapaglutin A (RgA), was shown to inhibit GLUT1, GLUT3, and GLUT4 with low IC50 values in nanomolar range, 41 and showed inhibition of glycolysis and ATP biogenesis by RgA leading to AMPK activation, mTOR inhibition, cell‐cycle arrest, and apoptosis induction. We noticed structural similarities between temsirolimus and RgA, thus indicating that temsirolimus might inhibit GLUTs.
Although, our results from this study identified interaction of TKIs with hGLUT‐1 in modeling studies, TKI inhibition of hGLUT‐1 in addition to inhibition of other GLUTs, as well as SGLTs should be explored further in model systems expressing each individual transporter in isolation. Although it is difficult to extrapolate in vitro studies to the clinic, TKIs show extensive protein binding and accumulation in tumors, thus reaching levels in tissues that could inhibit GLUTs. In humans, steady‐state peak plasma concentration (Cmax) values of imatinib on day 8 were 3.13 µg/mL (i.e., 6.0 µM) , and after multiple doses imatinib levels in mice were 6‐fold to 8‐fold higher in tissues than in plasma. 42 Average peak plasma nilotinib concentrations are 3.6 µM, with steady‐state levels of 1.7 µM. 43 In mice, sunitinib tissue concentrations had high tissue distributions with high tissue‐to‐plasma ratios in the liver (17.8 ± 1.2), kidneys (14.6 ± 1.52), and brain (2.25 ± 0.18), 44 indicating that if similar distribution occurs in humans, sunitinib tissue concentrations vary from 1 to 5 µM. From four multiple dosing studies with sorafenib, plasma steady‐state levels (Cmax) in humans were 12–22 µM on the last day of dosing in each cycle, 45 suggesting that sorafenib concentrations in plasma are in the range of 10–30 µM. Pazopanib plasma concentration was 103 µmol/L, 46 and gefitinib tumor levels were 17–22.7 µmol/L. 47
The hGlut‐1 inhibition may help explain other TKI toxicities. Mutations of hGlut‐1 can occur and in their most severe form manifest as physical and mental growth delays, movement disorders, and intractable seizures. There is substantial phenotypic variation based on the nature and effects of mutations of hGlut‐1. Milder symptoms include confusion, lethargy, somnolence, and migraine headaches. Nilotinib’s side effects include headache (19%) and fatigue (19%), it is not impossible to imagine that hGlut‐1 inhibition at the blood brain barrier by TKIs could contribute to these side effects. Similarly, bosutinib, which is a potent inhibitor of hGlut‐1, causes headache (13%) and fatigue (13%).
In summary, our results support inhibition of hGLUT‐1 by TKIs, a transporter present in many cells and tissues. Inhibitory TKIs reduce 2‐DG uptake by acting as competitive inhibitors. Such interactions suggest that TKIs and glucose may compete for the same site on hGLUT‐1 and docking results supported interaction of TKIs with the glucose binding site of hGLUT‐1. These results offer insights to better understand mechanisms underlying hypoglycemic effects and fatigue observed in patients with cancer treated with certain TKIs. In addition, our study raises doubts about [18]FDG’s utility as a noninvasive measure of tumor response, because [18]FDG uptake inhibition by certain TKI classes might limit PET monitoring, thus resulting in false‐positive tumor response.
Based on our findings, PET screening should be done after patients stop taking TKIs for a few days, to yield reliable data and avoid false‐positive results. To do so requires a greater understanding of temporal relationships between TKI administration and changes in glucose levels. Therefore, caution is advised in the use of [18]FDG for monitoring PET tumor response during TKI treatment especially if [18]FDG is to be used in a setting of clinical trials to validate new drug effectiveness. Although some TKIs inhibited glucose uptake mediated by GLUT‐1, roles for other SLC2 and SLC5 members cannot be ruled out in modulating glucose levels and thus warrants further research. Future efforts directed toward examining inhibition of other GLUT‐isoforms by TKIs may shed further light on mechanisms of changes in glucose levels in patients.
Funding
The Alberta Cancer Foundation funded this research (M.B.S). J.T. acknowledges funding for his research from NSERC (Canada).
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
V.L.D., M.A., P.W., J.P., J.T., A.M., and M.S. wrote the manuscript. V.L.D., M.A., and M.S. designed the research. M.K. and M.A. performed the research. V.L.D., M.A., M.K., P.W., J.P., and J.T. analyzed the data. J.P. contributed new reagents/analytical tools.
Supporting information
Supplementary Material
Supplementary Material
Acknowledgment
The authors thank Dr. Min‐Sun Park for providing the pdb file of the fully outward open structure.
References
- 1. Kelloff, G.J. et al. Progress and promise of FDG‐PET imaging for cancer patient management and oncologic drug development. Clin. Cancer Res. 11, 2785–2808 (2005). [DOI] [PubMed] [Google Scholar]
- 2. Bos, R. et al. Biologic correlates of (18)fluorodeoxyglucose uptake in human breast cancer measured by positron emission tomography. J. Clin. Oncol. 20, 379–387 (2002). [DOI] [PubMed] [Google Scholar]
- 3. Agostino, N.M. et al. Effect of the tyrosine kinase inhibitors (sunitinib, sorafenib, dasatinib, and imatinib) on blood glucose levels in diabetic and nondiabetic patients in general clinical practice. J. Oncol. Pharm. Pract. 17, 197–202 (2011). [DOI] [PubMed] [Google Scholar]
- 4. Jensen, M.M. & Kjaer, A. Monitoring of anti‐cancer treatment with (18)F‐FDG and (18)F‐FLT PET: a comprehensive review of pre‐clinical studies. Am. J. Nucl. Med. Mol. Imaging 5, 431–456 (2015). [PMC free article] [PubMed] [Google Scholar]
- 5. Lin, M. & Jin, J. Cancer, obesity, and diabetes: TKIs exert multiple effects on glucose homeostasis. Nat. Rev. Clin. Oncol. 14, 268 (2017). [DOI] [PubMed] [Google Scholar]
- 6. Wahl, R.L. , Jacene, H. , Kasamon, Y. & Lodge, M.A. From RECIST to PERCIST: evolving considerations for PET response criteria in solid tumors. J. Nucl. Med. 50 (suppl. 1), 122S–150S (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kasahara, M. & Hinkle, P.C. Reconstitution and purification of the D‐glucose transporter from human erythrocytes. J. Biol. Chem. 252, 7384–7390 (1977). [PubMed] [Google Scholar]
- 8. Thorens, B. & Mueckler, M. Glucose transporters in the 21st century. Am. J Physiol. Endocrinol. Metab. 298, E141–E145 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carruthers, A. Facilitated diffusion of glucose. Physiol. Rev. 70, 1135–1176 (1990). [DOI] [PubMed] [Google Scholar]
- 10. Sun, L. et al. Crystal structure of a bacterial homologue of glucose transporters GLUT1‐4. Nature 490, 361–366 (2012). [DOI] [PubMed] [Google Scholar]
- 11. Henderson, P.J.F. , Maiden, M.C.J. , Kornberg Hans, L. & Henderson, P.J.F. Homologous sugar transport proteins in Escherichia coli and their relatives in both prokaryotes and eukaryotes. Phil. Trans. Royal Soc. London B Biol. Sci. 326, 391–410 (1990). [DOI] [PubMed] [Google Scholar]
- 12. Vera, J.C. et al. Direct inhibition of the hexose transporter GLUT1 by tyrosine kinase inhibitors. Biochemistry 40, 777–790 (2001). [DOI] [PubMed] [Google Scholar]
- 13. Ojelabi, O.A. , Lloyd, K.P. , Simon, A.H. , De Zutter, J.K. & Carruthers, A. WZB117 (2‐Fluoro‐6‐(m‐hydroxybenzoyloxy) Phenyl m‐Hydroxybenzoate) inhibits GLUT1‐mediated sugar transport by binding reversibly at the Exofacial sugar binding site. J. Biol. Chem. 291, 26762–26772 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ung, P.M. et al. Inhibitor discovery for the human GLUT1 from homology modeling and virtual screening. ACS Chem. Biol. 11, 1908–1916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sen, R. et al. The novel BCR‐ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR‐associated ATP‐binding cassette transporter ABCG2. Mol. Cancer Ther. 11, 2033–2044 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deng, D. et al. Crystal structure of the human glucose transporter GLUT1. Nature 510, 121–125 (2014). [DOI] [PubMed] [Google Scholar]
- 17. Park, M.S. Molecular dynamics simulations of the human glucose transporter GLUT1. PLoS One 10, e0125361 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kasahara, T. & Kasahara, M. Expression of the rat GLUT1 glucose transporter in the yeast Saccharomyces cerevisiae. Biochem. J. 315(Pt 1), 177–182 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kapoor, K. et al. Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. U S A 113, 4711–4716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chemical Computing Group . (2012). Molecular Operating Environment (MOE), S.S.W., Suite #910, Montreal, QC, Canada, H3A 2R7.
- 21. Cheson, B.D. et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non‐Hodgkin lymphoma: the Lugano classification. J. Clin. Oncol. 32, 3059–3068 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pinker, K. , Riedl, C. & Weber, W.A. Evaluating tumor response with FDG PET: updates on PERCIST, comparison with EORTC criteria and clues to future developments. Eur. J. Nucl. Med. Mol. Imaging 44, 55–66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ordu, C. Are the metabolic evaluation criteria sufficient for FDG PET/CT after chemo‐radiotherapy in non‐small cell lung cancer? J. Thorac. Dis. 11, S1263–S1266 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ullrich, R.T. et al. Early detection of erlotinib treatment response in NSCLC by 3'‐deoxy‐3'‐[F]‐fluoro‐L‐thymidine ([F]FLT) positron emission tomography (PET). PLoS One 3, e3908 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Su, H. et al. Monitoring tumor glucose utilization by positron emission tomography for the prediction of treatment response to epidermal growth factor receptor kinase inhibitors. Clin. Cancer Res. 12, 5659–5667 (2006). [DOI] [PubMed] [Google Scholar]
- 26. Vergez, S. et al. Preclinical and clinical evidence that Deoxy‐2‐[18F]fluoro‐D‐glucose positron emission tomography with computed tomography is a reliable tool for the detection of early molecular responses to erlotinib in head and neck cancer. Clin. Cancer Res. 16, 4434–4445 (2010). [DOI] [PubMed] [Google Scholar]
- 27. Dorow, D.S. et al. Multi‐tracer small animal PET imaging of the tumour response to the novel pan‐Erb‐B inhibitor CI‐1033. Eur. J. Nucl. Med. Mol. Imaging 33, 441–452 (2006). [DOI] [PubMed] [Google Scholar]
- 28. Cullinane, C. et al. An in vivo tumor model exploiting metabolic response as a biomarker for targeted drug development. Cancer Res. 65, 9633–9636 (2005). [DOI] [PubMed] [Google Scholar]
- 29. Prenen, H. et al. Establishment of a mouse gastrointestinal stromal tumour model and evaluation of response to imatinib by small animal positron emission tomography. Anticancer Res. 26, 1247–1252 (2006). [PubMed] [Google Scholar]
- 30. Brepoels, L. et al. (18)F‐FDG and (18)F‐FLT uptake early after cyclophosphamide and mTOR inhibition in an experimental lymphoma model. J. Nucl. Med. 50, 1102–1109 (2009). [DOI] [PubMed] [Google Scholar]
- 31. Cejka, D. et al. FDG uptake is a surrogate marker for defining the optimal biological dose of the mTOR inhibitor everolimus in vivo. Br. J. Cancer 100, 1739–1745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Honer, M. et al. Anti‐angiogenic/vascular effects of the mTOR inhibitor everolimus are not detectable by FDG/FLT‐PET. Transl. Oncol. 3, 264–275 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li, Z. et al. FLT‐PET is superior to FDG‐PET for very early response prediction in NPM‐ALK‐positive lymphoma treated with targeted therapy. Cancer Res. 72, 5014–5024 (2012). [DOI] [PubMed] [Google Scholar]
- 34. Saint‐Hubert, M.D. et al. Molecular imaging of therapy response with (18)F‐FLT and (18)F‐FDG following cyclophosphamide and mTOR inhibition. Am. J. Nucl. Med. Mol. Imaging 2, 110–121 (2012). [PMC free article] [PubMed] [Google Scholar]
- 35. Saint‐Hubert, M.D. et al. Bioluminescence imaging of therapy response does not correlate with FDG‐PET response in a mouse model of Burkitt lymphoma. Am. J. Nucl. Med. Mol. Imaging 2, 353–361 (2012). [PMC free article] [PubMed] [Google Scholar]
- 36. Thomas, G.V. et al. Hypoxia‐inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 12, 122–127 (2006). [DOI] [PubMed] [Google Scholar]
- 37. Wei, L.H. et al. Changes in tumor metabolism as readout for Mammalian target of rapamycin kinase inhibition by rapamycin in glioblastoma. Clin. Cancer Res. 14, 3416–3426 (2008). [DOI] [PubMed] [Google Scholar]
- 38. Yang, M. et al. PET imaging of early response to the tyrosine kinase inhibitor ZD4190. Eur. J. Nucl. Med. Mol. Imaging 38, 1237–1247 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jensen, M.M. et al. [18F]FDG and [18F]FLT positron emission tomography imaging following treatment with belinostat in human ovary cancer xenografts in mice. BMC Cancer 13, 168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sage, J.M. , Cura, A.J. , Lloyd, K.P. & Carruthers, A. Caffeine inhibits glucose transport by binding at the GLUT1 nucleotide‐binding site. Am. J. Physiol. Cell Physiol. 308, C827–C834 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guo, Z. et al. Discovery of a potent GLUT inhibitor from a library of rapafucins by using 3D microarrays. Angew. Chem. 131, 17318–17322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Teoh, M. et al. HPLC determination of imatinib in plasma and tissues after multiple oral dose administration to mice. Pak. J. Pharm. Sci. 23, 35–41 (2010). [PubMed] [Google Scholar]
- 43. Weisberg, E. , Manley, P.W. , Cowan‐Jacob, S.W. , Hochhaus, A. & Griffin, J.D. Second generation inhibitors of BCR‐ABL for the treatment of imatinib‐resistant chronic myeloid leukaemia. Nat. Rev. Cancer 7, 345–356 (2007). [DOI] [PubMed] [Google Scholar]
- 44. Chee, E.L. , Lim, A.Y. , Modamio, P. , Fernandez‐Lastra, C. & Segarra, I. Sunitinib tissue distribution changes after coadministration with ketoconazole in mice. Eur. J. Drug Metab. Pharmacokinet. 41, 309–319 (2016). [DOI] [PubMed] [Google Scholar]
- 45. Strumberg, D. et al. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 12, 426–437 (2007). [DOI] [PubMed] [Google Scholar]
- 46. Hurwitz, H.I. et al. Phase I trial of pazopanib in patients with advanced cancer. Clin. Cancer Res. 15, 4220–4227 (2009). [DOI] [PubMed] [Google Scholar]
- 47. Haura, E.B. , Sommers, E. , Song, L. , Chiappori, A. & Becker, A. A pilot study of preoperative gefitinib for early‐stage lung cancer to assess intratumor drug concentration and pathways mediating primary resistance. J. Thorac. Oncol. 5, 1806–1814 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material