

Abstract
Bruton’s tyrosine kinase (BTK) is a target for treatment of hematologic malignancies and autoimmune diseases. TAK‐020 is a highly selective covalent BTK inhibitor that inhibits both B cell receptor and fragment crystallizable receptor signaling. We assessed the safety/tolerability and pharmacokinetics/pharmacodynamics (PDs) of TAK‐020 in healthy subjects. Each cohort of the single‐rising dose (n = 72; 9 cohorts) and the multiple‐rising dose (n = 48; 6 cohorts) portions of the study comprised six TAK‐020‐treated and two placebo‐treated, subjects aged 18–55 years (inclusive). The PD effects were assessed by measuring BTK occupancy and the inhibition of fragment crystallizable epsilon receptor 1 (FcεRI)‐mediated activation of basophils. Overall, treatment‐emergent adverse events (TEAEs) were similar to placebo; there were no serious TEAEs or no TEAEs leading to discontinuation. TAK‐020 was rapidly absorbed (median time to maximum plasma concentration (Tmax) 45–60 minutes) with a half‐life of ~ 3–9 hours at doses ≥ 2.5 mg. TAK‐020 exposure was generally dose proportional for single doses ≤ 70 mg and after multiple doses of ≤ 60 mg once daily. Target occupancy was dose dependent, with doses ≥ 2.5 mg yielding maximum and sustained occupancy > 70% for > 96 hours. Single doses ≥ 4.4 mg reduced FcεRI‐mediated activation of basophils by > 80% and comparable inhibition was observed with daily dosing ≥3.75 mg for 9 days. Inhibition persisted for 24–72 hours postdose and the duration generally increased with dose. TAK‐020 was generally well‐tolerated in healthy subjects after single and multiple doses and demonstrated target engagement and pathway modulation. The PD effects outlasted drug exposures, as expected for covalent inhibition of BTK.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ First‐generation Bruton’s tyrosine kinase inhibitors (BTKis; e.g., ibrutinib and acalabrutinib) are effective therapeutics for hematologic malignancies (e.g., chronic lymphocytic leukemia and mantle cell lymphoma); however, treatment‐emergent adverse events that resulted from off‐target effects have restricted the application of these drugs to nonmalignant indications.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ A new generation of BTKis (TAK‐020, evobrutinib, etc.) with greater selectivity was developed with the hope that their safety and tolerability profiles would enable treatment of nonmalignant conditions.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ BTK saturation and inhibition by TAK‐020 was safe and well‐tolerated by healthy subjects, supporting additional investigation in nonmalignant conditions.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ These results indicate that the selective inhibition of BTK could be utilized to safely treat nonmalignant pathologies mediated by hematopoietic cells of the B and myeloid lineages.
TAK‐020 is an orally available, small‐molecule, irreversible inhibitor of Bruton’s tyrosine kinase (BTK), the expression of which is restricted to B cells, neutrophils, basophils, monocytes, mast cells, and osteoclasts. 1 , 2 , 3 , 4 BTK plays a critical role in B cell receptor activation, which results in proliferation and phenotypic differentiation of immature B cells into antibody‐secreting cells and in Fc gamma and fragment crystallizable epsilon (Fcε) receptor signaling by mediating activation of immunoreceptor tyrosine‐based activation motifs in multiple cell types. 1 , 2 , 5 The complete absence of BTK activity is associated with primary immunodeficiency, X‐linked agammaglobulinemia, and no other dominant phenotype, 6 indicating that the effect of BTK inhibition could be well‐tolerated by patients.
First‐generation BTK inhibitors (BTKis; e.g., ibrutinib and acalabrutinib) are effective therapeutics for hematologic malignancies, such as chronic lymphocytic leukemia and mantle cell lymphoma. Because of adverse reactions (e.g., atrial fibrillation and cytopenia), which are uncharacteristic of patients with X‐linked agammaglobulinemia and perceived to result from off‐target effects, BTKis have limited application to nonmalignant indications. Therefore, a new generation of BTKi (e.g., TAK‐020 and evobrutinib) is being developed on the therapeutic premise that greater selectivity for BTK would yield tolerability profiles that permitted the exploration of their use in nonmalignant indications. 7 , 8 A first‐in‐human, phase I, double‐blind, placebo‐controlled study demonstrated that evobrutinib was well‐tolerated, showed linear and time‐independent pharmacokinetics (PKs), and induced long‐lasting BTK inhibition in healthy subjects; studies were initiated for the treatment of rheumatoid arthritis, systemic lupus erythematosus, and relapsed‐remitting multiple sclerosis.
TAK‐020 engages its target rapidly and remains covalently bound, even after plasma concentrations are undetectable, and is efficacious in a rat collagen‐induced arthritis model of autoimmune disease with a median effective dose of 0.31 mg/kg per day. At the human equivalent of the median effective dose (3 mg/day), it was predicted that therapeutic levels of > 90% BTK occupancy could be achieved in the peripheral blood for the majority of the dosing interval.
In nonclinical studies, TAK‐020 (as an oral solution) was absorbed rapidly and extensively after oral administration, with peak plasma concentrations generally occurring within 2 hours postdose. The oral bioavailability of TAK‐020 in animals was low (13.2% in rats and 8.17% in dogs). Absorption could be modulated by P‐glycoprotein. Terminal disposition phase elimination half‐life (t1/2z) of TAK‐020 after oral administration was 4.4 to 7.3 hours in rats and 3.8 to 6.7 hours in dogs.
The nonclinical safety, tolerability, and pharmacodynamic (PD) profiles of TAK‐020 suggest that it may have the potential to address the unmet needs of patients suffering from autoantibody‐mediated diseases, such as myasthenia gravis and immune thrombocytopenia.
Objectives
The objectives of this study were to characterize the safety and tolerability of TAK‐020 after single‐rising and multiple‐rising oral doses (SRD and MRD, respectively) in healthy subjects. The secondary objectives were to characterize the PKs and PDs of TAK‐020.
METHODS
Study design
This was a phase I, randomized, double‐blind, placebo‐controlled, SRD (part 1) and MRD (part 2) study in healthy subjects aged 18–55 years (inclusive), who weighed ≥ 45 kg, with a body mass index between 18 and 32 kg/m2 (inclusive).
Part 1/SRD comprised nine cohorts (Figure S1 ). Each cohort comprised eight randomized subjects (6 subjects received TAK‐020 and 2 subjects received placebo in the fasted state). Sentinel dosing was used for cohort 1 with two subjects dosed on the morning of day 1 (1 subject received TAK‐020 and 1 subject received placebo). The starting dose of TAK‐020 for cohort 1 was 0.1 mg, orally administered as a solution. Dose selection was based on both the US Food and Drug Administration guidance on estimating the maximum safe starting dose and estimated pharmacologically active dose/exposure in humans. PK/PD modeling predicted 20% BTK occupancy at this starting dose, which was considered to represent a minimum anticipated biological effect level. Furthermore, the rat was identified as the most sensitive toxicological species. In order to adequately assess the potential benefit to risk profile of BTK inhibition in patients (e.g., protection against bone erosion and cartilage damage), the rat no observed adverse effect level exposure limit (total area under the plasma concentration‐time curve (AUC): 63 ng·hour/mL) was increased to approximately twice the rat low‐observed‐adverse‐effect‐level based on unbound exposure (unbound AUC: 8.62 ng·hour/mL; total AUC: 431 ng·hour/mL). Consequently, for each dose escalation, exposures could be increased by a maximum of fivefold for cohorts 2–4 and a maximum of 2‐fold for cohorts 5–9. Dosing for the subsequent cohorts occurred after review of the safety, tolerability, and PK data (minimum of 24 hours) from the previous cohort. Overall, a total of 72 subjects were randomized into 9 SRD cohorts.
Part 2/MRD comprised six cohorts (Figure S1 ). Each cohort comprised less than or equal to eight randomized subjects (6 subjects received TAK‐020 and 2 subjects received placebo). The first MRD cohort received TAK‐020 3.75 mg, with a predicted exposure at steady‐state estimated to be below the rat no observed adverse effect level. In each successive cohort, the dose increments increased the predicted exposures by no more than twofold. The predicted steady‐state exposure in MRD did not exceed the maximum exposure observed in SRD. Overall, a total of 48 subjects were randomized into 6 MRD cohorts.
Potential cytopenia and atrial fibrillation were specifically examined because these events are associated with exposure to the BTK inhibitor ibrutinib. Hematograms were analyzed at screening, day −1, and once daily prior to check‐out (e.g., day 5 for subjects receiving a single dose and day 10 for multiple doses) or early termination. In addition, triplicate 12‐lead echocardiograms (ECGs) printed in standard format were collected at screening, check‐in (day −1), and day 1 predose (0 hours) and at 1, 4, 12, 24, 48, 72, and 96 hours postdose or early termination after a single dose of TAK‐020. Similarly, ECGs were collected at screening and check‐in (day −1), predose on day 1 (0 hours), and at 1, 4, 12, and 24 hours postdose; predose on days 3, 5, 9, and 10 (0 hours) or early termination with multiple dosing.
Bioanalytical methods
Plasma concentrations of TAK‐020 were measured by a validated high‐performance liquid chromatography with tandem mass spectrometry assay with a lower limit of quantification of 0.10 ng/mL.
The BTK occupancy assay directly measured unoccupied BTK receptor (i.e., not bound to TAK‐020 in whole blood cell lysates). It used a biotin‐modified version of TAK‐020 as a covalent occupancy probe to capture the proportion of BTK receptor that was unoccupied; therefore, it could be used to quantify BTK occupancy by TAK‐020. Human B cells were incubated with increasing concentrations of BTKi for 60 minutes. Cells were washed and lysed, and free BTK was captured using the covalent probe. Free BTK was quantified by enzyme‐linked immunosorbent assay.
The basophil activation test (BAT) is a flow cytometry‐based assay that directly measured CCR3‐positive basophil activation and degranulation based on CD63 surface expression. Blood cells were collected and evaluated by flow cytometry per instructions (BÜHLMANN Laboratories AG, Switzerland). 9 , 10 , 11 Those cells were treated ex vivo with an antibody that cross‐links the Fcε receptor 1 (FcεRI) and the high‐affinity immunoglobulin E receptor on basophils. Anti‐FcεRI induced signaling through a pathway requiring BTK activity, resulting in the upregulation of CD63 expression at the cell surface.
Statistical analysis plan
Data for the SRD and MRD parts were summarized separately for all randomized subjects in each study part. Continuous data were summarized using descriptive statistics, including the number of subjects, mean, SD, median, minimum, and maximum. The percent coefficient of variation (%CV) and geometric mean were included in the summary of continuous data where indicated. Categorical data were summarized using the number and percentage of subjects for each category. Missing values were categorized separately where deemed appropriate and necessary. The baseline value for a variable was defined as the last observation collected before the first dose of study drug unless stated otherwise.
Plasma concentrations of TAK‐020 for the SRD and MRD parts were summarized by dose for each scheduled sampling time using descriptive statistics. The plasma PK parameters of TAK‐020 were estimated from the individual concentration‐time data for all evaluable subjects using standard noncompartmental methods (Phoenix WinNonlin, version 6.4) and summarized descriptively by treatment (SRD, MRD, and dose). Actual sampling times were used in all computations. Geometric mean and %CV were computed for peak plasma concentration (Cmax) and AUCs.
BTK’s occupancy was derived for each postdose collection time using the following formula: 100 − [100 × (BTK concentration ÷ predose BTK concentration)]. Baseline normalized values from the BAT (%CD63‐positive basophils) were also derived for each postdose collection time using the following formula: 100 × [(BAT concentration − predose concentration) ÷ predose BAT concentration]. BTK occupancy was summarized for SRD cohorts 1–4 for each postdose sample collection time using descriptive statistics. Baseline normalized BAT was summarized for SRD and MRD cohorts for each postdose sample collection time using descriptive statistics.
This study (NCT02413255) was conducted at Parexel in Glendale, CA, received institutional review board approval, and adhered to the ethical principles of the Declaration of Helsinki, the International Council for Harmonisation, and Good Clinical Practice. All subjects provided written informed consent.
RESULTS
Subject disposition
Of the 497 subjects screened, 120 were eligible for randomization. In the SRD part, a total of 72 subjects were randomized, and all subjects completed the study. In the MRD part, a total of 48 subjects were randomized; all placebo‐treated subjects and 35 of 36 TAK‐020–treated subjects (97.2%) completed the study and all planned study visits.
Overall (SRD part and MRD part combined), subjects were 55.8% white or 31.7% black and 66.7 men between 18 and 55 years of age who had never smoked (88.3%; Table S1 ). The subjects in the overall pooled TAK‐020 group were 73.3% men and 60% white, and the overall pooled placebo group were 46.7% men and 43.3% white.
In the SRD part, 54 subjects (6 subjects per cohort) received a single TAK‐020 dose of 0.1, 0.5, 2.5, 4.4, 8.8, 17.5, 35, 70, or 105 mg, and 18 subjects (2 subjects per cohort) received placebo.
In the MRD part, 36 subjects (6 subjects per cohort) received multiple once‐daily (q.d.) TAK‐020 doses of 3.75, 5.75, 13, 25, 45, or 60 mg, and 12 subjects (2 subjects per cohort) received placebo. One subject in the TAK‐020 25‐mg dose group voluntarily withdrew from the study and was prematurely discontinued.
Safety/tolerability
In the SRD part, the same percentage of subjects (11.1% each) in the pooled placebo group and the overall TAK‐020 group experienced treatment‐emergent adverse events (TEAEs; Table 1 ), all of which were considered nonserious and mild in intensity. In the pooled placebo group, subjects experienced headache, dizziness, ecchymosis, and oropharyngeal pain. The TEAEs that occurred in the overall TAK‐020 group were abdominal distension, upper abdominal pain, decreased appetite, headache, nausea, phlebitis, and superficial phlebitis ( Table S2 ). One subject each in the TAK‐020 2.5‐mg, 8.8‐mg, and 105‐mg dose groups had one or more TEAEs. Study treatment‐related TEAEs were reported in 3 subjects given TAK‐020 2.5, 8.8, and 70 mg (one in each group). Abdominal distension, upper abdominal pain, nausea, and headache were reported in one subject each, all of which were mild in intensity.
Table 1.
Overview of TAK‐020 TEAEs for SRD
| TEAE category, n (%) | Pooled placebo (n = 18) | 0.1 mg (n = 6) | 0.5 mg (n = 6) | 2.5 mg (n = 6) | 4.4 mg (n = 6) | 8.8 mg (n = 6) | 17.5 mg (n = 6) | 35 mg (n = 6) | 70 mg (n = 6) | 105 mg (n = 6) | Overall (N = 54) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TEAEs | 2 (11.1) | 0 | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 3 (50.0) | 1 (16.7) | 6 (11.1) |
| Related | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 1 (16.7) | 0 | 3 (5.6) |
| Not related | 2 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (33.3) | 1 (16.7) | 3 (5.6) |
| Mild | 2 (11.1) | 0 | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 3 (50.0) | 1 (16.7) | 6 (11.1) |
| Moderate | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Serious TEAEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Related | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Not related | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Leading to DC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Deaths | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
A TEAE was defined as an adverse event (AE) or serious AE that started or worsened after first study drug administration and within 30 days of the last dose of study drug. Subjects with more than one AE were counted only once using the most related or severe event. Percentages are based on the total number of subjects in the safety set for each treatment group.
DC, discontinuation; SRD, single‐rising dose; TEAEs, treatment‐emergent adverse events.
In the MRD part, a higher percentage of subjects in the pooled placebo group (41.7%) had TEAEs compared with the overall TAK‐020 group (30.6%; Table 2 ). All TEAEs were considered mild, and most TEAEs occurred in only one subject. In the pooled placebo group, the TEAEs that occurred in two subjects each were upper respiratory tract infection, dizziness, and cough; three subjects reported headache. In the overall TAK‐020 group, the TEAEs that occurred in two subjects each were diarrhea (1 subject each in the 3.75‐mg and 5.75‐mg dose groups), upper respiratory tract infection (1 subject each in the 13‐mg and 60‐mg dose groups), dizziness (1 subject each in the 13‐mg and 45‐mg dose groups), and headache (both subjects in the 5.75‐mg dose group); three subjects reported oropharyngeal pain (2 subjects in the 5.75‐mg dose group and 1 subject in the 13‐mg dose group). All three TEAEs of oropharyngeal pain were described as a sore throat and were of mild intensity. In the overall TAK‐020 group, four subjects (11.1%) had study treatment‐related TEAEs, which included nausea, diarrhea, dizziness, and headache; one subject (8.3%) in the pooled placebo group had study treatment‐related TEAEs of nausea and headache ( Table S3 ). System organ class and preferred term listings for TEAEs in the SRD and MRD parts are included in Tables [Link] , [Link]
Table 2.
Overview of TAK‐020 TEAEs for MRD
| TEAE category, n (%) | Pooled placebo (n = 12) | 3.75 mg (n = 6) | 5.75 mg (n = 6) | 13 mg (n = 6) | 25 mg (n = 6) | 45 mg (n = 6) | 60 mg (n = 6) | Overall (N = 36) |
|---|---|---|---|---|---|---|---|---|
| TEAEs | 5 (41.7) | 1 (16.7) | 3 (50.0) | 3 (50.00) | 2 (33.3) | 1 (16.7) | 1 (16.7) | 11 (30.6) |
| Related | 1 (8.3) | 1 (16.7) | 2 (33.3) | 1 (16.7) | 0 | 0 | 0 | 4 (11.1) |
| Not related | 4 (33.3) | 0 | 1 (16.7) | 2 (33.3) | 2 (33.3) | 1 (16.7) | 1 (16.7) | 7 (19.4) |
| Mild | 5 (41.7) | 1 (16.7) | 3 (50.0) | 3 (50.0) | 2 (33.3) | 1 (16.7) | 1 (16.7) | 11 (30.6) |
| Moderate | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Serious TEAEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Related | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Not related | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Leading to DC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Deaths | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
A TEAE was defined as an adverse event (AE) or serious AE that started or worsened after first study drug administration and within 30 days of the last dose of study drug. Subjects with more than one AE were counted only once using the most related or severe event. Percentages are based on the total number of subjects in the safety set for each treatment group.
DC, discontinuation; MRD, multiple‐rising dose; TEAEs, treatment‐emergent adverse events.
In both SRD and MRD parts, no subject had a serious TEAE or a TEAE leading to study drug discontinuation. There were no dose‐related increases in TEAEs, and no apparent trends were observed. There were no deaths during the study. No subject had clinical laboratory test results (including liver function tests), vital sign findings, cytopenias, or ECG results reported as a TEAE. Isolated markedly abnormal values were reported for clinical laboratory tests, vital signs, and ECG results; however, no apparent temporal or dose‐related trends were observed. No subject had a clinically significant physical examination finding.
Pharmacokinetics
SRD part
After single doses of TAK‐020 ranging from 0.1 to 105 mg, TAK‐020 was rapidly absorbed, with measurable concentrations detected at the first postdose sampling time point (0.25 hours) in most subjects (Figure 1a,b ). The median time to peak plasma concentration (Tmax) ranged from 0.63 to 1.01 hours postdose. After achieving a maximum, plasma concentrations declined with a mean t1/2z that ranged ~ 1–3 hours at doses ≤ 4.4 mg and 4–9 hours at higher doses up to 105 mg. The shorter t1/2z estimates at lower doses may be related to the observed plasma concentrations falling below the lower limit of quantification of the assay early in the time course, thus preventing accurate characterization of the terminal elimination phase. Comparison of the mean dose‐normalized Cmax and AUC over 24 hours (AUC24) values showed that TAK‐020 systemic exposure generally increased in a dose‐proportional manner up to 70 mg, whereas it plateaued between 70 and 105 mg. The %CV for most plasma PK parameters generally ranged from a low of ~ 15% to a high of ~ 53%, which was considered low‐to‐moderate variability.
Figure 1.
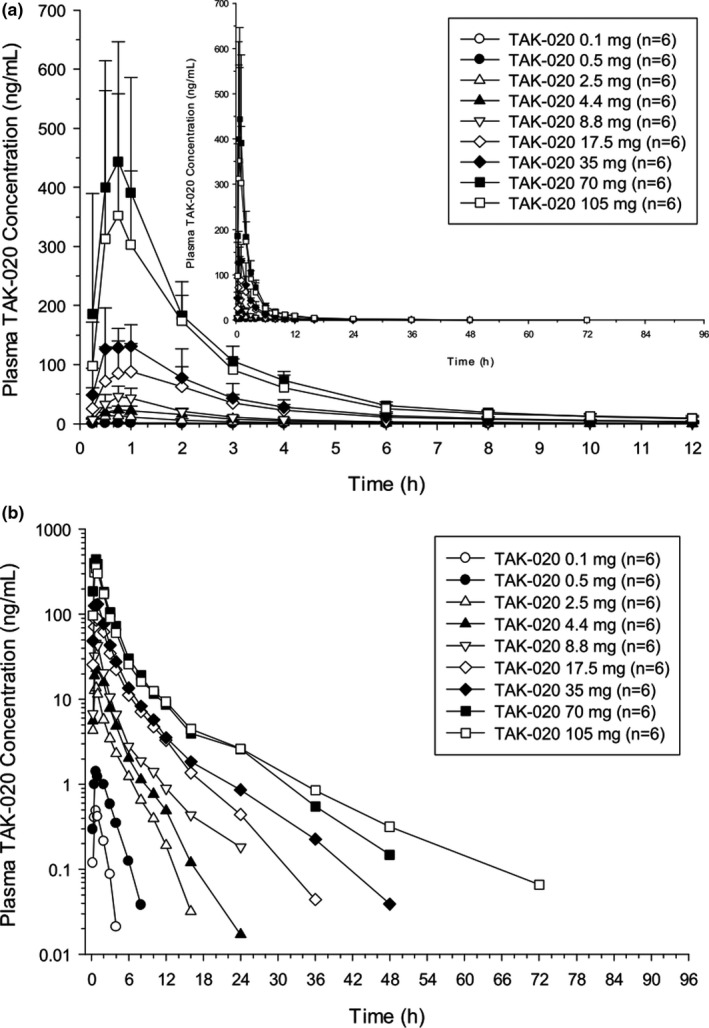
Single‐rising dose. Mean (SD) plasma concentration (ng/mL) of TAK‐020 vs. time (12 and 96 hours (inset)) by dose on a linear scale (a) and on a log‐linear scale (b).
MRD part
After multiple q.d. doses of TAK‐020 ranging from 3.75 to 60 mg, the median Tmax ranged from 0.50 to 0.75 hours after a single dose on day 1, and the range was 0.75 to 1.00 hours on day 9 after q.d. dosing (Figure 2a,b ). The mean t1/2z ranged from 2.59 hours for the 3.75‐mg dose to 6.30 hours for the 60‐mg dose after a single dose on day 1. On day 9 after q.d. dosing, the mean t1/2z ranged from 2.15 hours for the 3.75‐mg dose to 5.46 hours for the 60‐mg dose. There was little to no drug accumulation after multiple dosing, as evidenced by geometric mean values for the accumulation ratio based on Cmax (Rac(Cmax)) and AUC24 (Rac(AUC24)) near unity. This lack of drug accumulation was consistent with the estimated short t1/2z of TAK‐020. Comparison of the mean dose‐normalized Cmax and AUC24 values on days 1 and 9 showed that steady‐state TAK‐020 systemic exposure generally increased in a dose‐proportional manner up to 60 mg. Low‐to‐moderate PK variability was observed with %CV for most of the plasma PK parameters generally ranging from ~ 10% to ~ 50%. In addition, TAK‐020 PK appeared to be time‐independent over the dose range studied, as shown by the least‐squares means ratio of AUC24 (day 9):AUC∞ (day 1) near unity.
Figure 2.
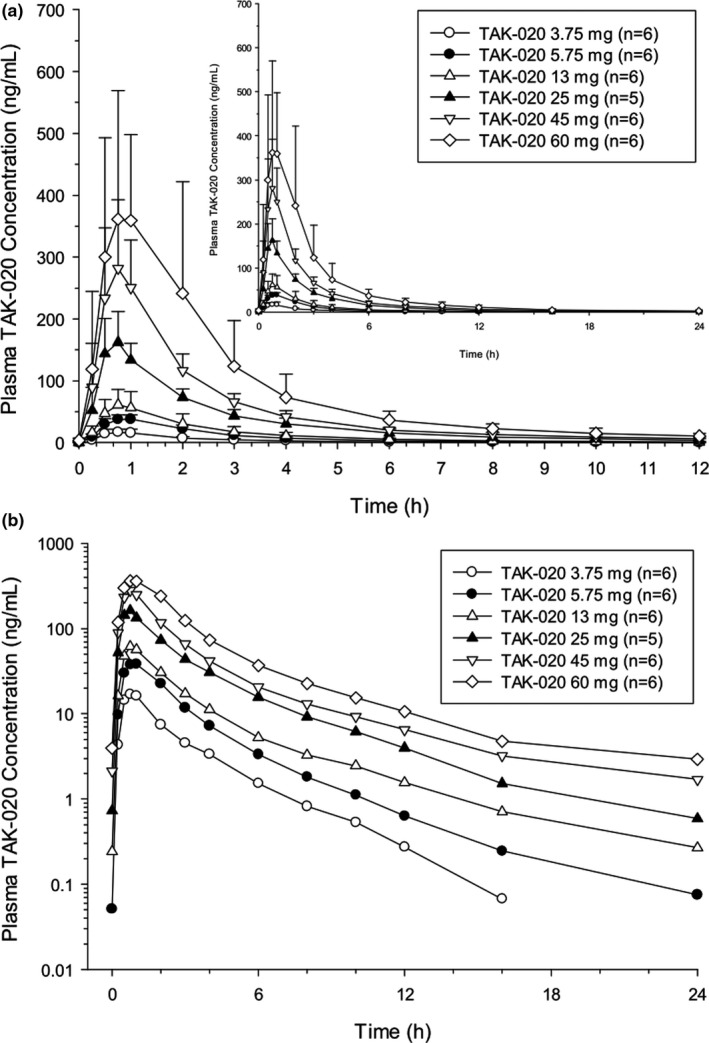
Multiple‐rising dose on day 9. Mean (SD) plasma concentration (ng/mL) of TAK‐020 vs. time (12 and 24 hours (inset)) by dose on a linear scale (a) and on a log‐linear scale (b).
Pharmacodynamics
Target occupancy in the blood was dose‐dependent, with the 2.5‐mg and 4.4‐mg doses yielding saturation and maintaining occupancy > 70% for > 96 hours postdose, which was the longest duration investigated (Figure 3 ). There was essentially no change in the mean BTK occupancy (%) observed in the placebo group. BTK occupancy (%) was not calculated for TAK‐020 doses above 4.4 mg in the SRD part and for any subjects in the MRD part because saturation was maintained > 24 hours at single doses ≥2.5 mg (Figure 3 ). Because of the lack of BTK data for the majority of the dose groups in both the SRD and MRD parts, the noncompartmental BTK analysis was not conducted.
Figure 3.
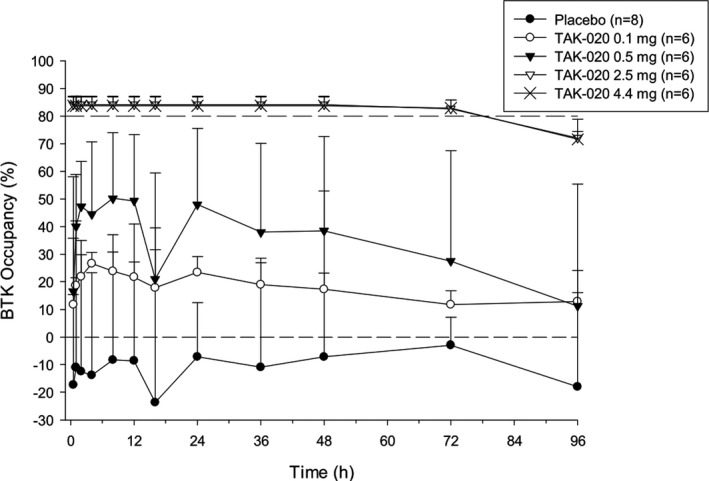
Mean (SD) Bruton’s tyrosine kinase (BTK) occupancy (%) vs. time by treatment: single‐rising dose.
Pathway inhibition was dose‐dependent, with single doses of TAK‐020 ≥ 4.4 mg, which inhibited the activation of blood basophils > 80% and returned to baseline activity within 36–72 hours postdose (Figure 4a ). The duration of inhibition generally increased with dose (Figure 4a ). There was essentially no reduction in the %CD63‐positive basophils observed in the placebo group and in the 0.1‐mg and 0.5‐mg TAK‐020 dose groups. By day 9 after q.d. administration, TAK‐020 doses from ≥3.75 mg produced a > 80% reduction from baseline in the %CD63‐positive basophils (Figure 4b ). There was essentially no reduction in the %CD63‐positive basophils observed in the placebo group. The ≥80% reduction in the %CD63‐positive basophils was sustained throughout the entire dosing interval (predose to 24 hours postdose) for ≥ 45‐mg doses of TAK‐020 after multiple dosing for 9 days (Figure 4b ) and was not observed 1 week after the last dose (data not shown).
Figure 4.
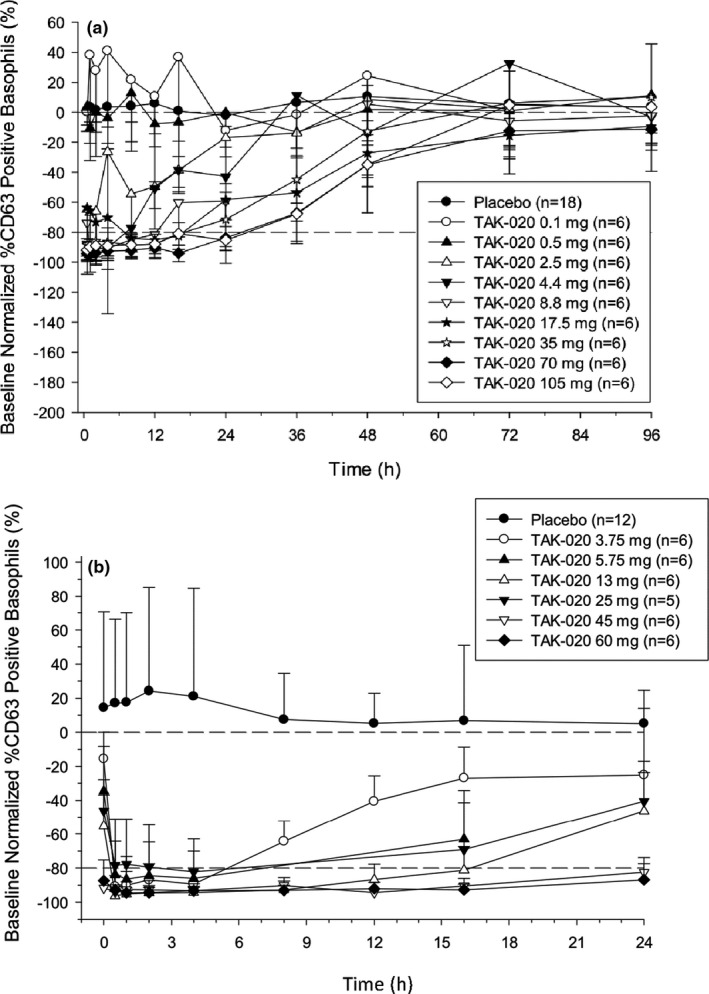
Basophil activation test baseline normalized %CD63‐positive basophils (%+SD). After fragment crystallizable epsilon receptor 1 stimulation in the single‐rising dose (a) and the multiple‐rising dose on day 9 (b).
DISCUSSION
Following oral administration of single (0.1–105 mg) and multiple (3.75–60 mg) doses, TAK‐020 was rapidly absorbed (median Tmax was ~ 45 minutes to 1 hour) and mean systemic exposure was, in general, proportional up to 70 mg single dosing and up to 60 mg after q.d. dosing. There was little to no drug accumulation following repeat dosing, which is consistent with the mean half‐life estimates of TAK‐020 ranging from ~ 2–6 hours in plasma with q.d. dosing. In addition, TAK‐020 PK appears to be concentration and time independent.
The percent occupancy of TAK‐020 binding sites covalently bound to BTK was calculated at each timepoint after the first four TAK‐020 dose levels and placebo in the SRD part. BTK occupancy (%) was not calculated for TAK‐020 doses above 4.4 mg in the SRD part, and it was not calculated for any doses in the MRD part because target saturation had been demonstrated with a single dose ≥ 4.4 mg. Single doses of 2.5 and 4.4 mg of TAK‐020 produced levels of BTK occupancy (%) of TAK‐020 binding sites of > 80%. This level of binding was sustained for 72 hours postdose and outlasted exposure to free TAK‐020 as assessed by PK. There was essentially no change in mean BTK occupancy (%) observed in the placebo group. FcεR1‐activated CCR3‐positive basophil degranulation was measured by the upregulation of surface CD63 expression and used to measure pathway inhibition by TAK‐020.
Notably, it has been discovered that basophils contribute to the development of certain allergic (e.g., atopic dermatitis) and autoimmune pathologies (e.g., RA). In addition, depletion of basophils has been associated with clinical benefits in these disorders. 12 The sustained binding of BTK demonstrated with single‐dose administration of TAK‐020 resulted in a reduction from baseline of > 80% in the %CD63‐positive FcER1‐activated basophils after single doses of 4.4–105 mg, and is consistent with the covalent ligand‐receptor interaction predicted from the structure of TAK‐020 and the relatively slow turnover of BTK. It is noteworthy that comparing target occupancy within the 2.5 or 4.4 mg cohorts (e.g., > 80% at 72 hours postdose; Figure 3 ), to corresponding pathway activity within these cohorts (e.g., baseline levels of CD63‐expressing basophils; Figure 4a ) is confounded by differences in the constituents of these populations; the occupancy assay assesses basophils present during dosing exclusively, whereas the pathway assay assesses these and basophils, which were generated since dosing and consequently were not exposed directly to TAK‐020. After multiple q.d. administrations, TAK‐020 doses from 3.75 to 60 mg produced a > 80% reduction from baseline in the %CD63‐positive basophils, which was sustained throughout the entire dosing interval (predose to 24 hours postdose) after the two highest doses of TAK‐020 (45 and 60 mg). As with the occupancy of TAK‐020 binding sites, this effect outlasted the plasma PK exposure to the free drug, emphasizing the sustained duration of action despite a relatively short plasma elimination half‐life. There was essentially no reduction from baseline in the %CD63‐positive basophils observed in the placebo group for the SRD and MRD parts.
In the SRD part, the same percentage of subjects (11.1% each) in the pooled placebo group and the overall TAK‐020 group experienced TEAEs. In the MRD part, a higher percentage of subjects in the pooled placebo group (41.7%) had TEAEs compared with the overall TAK‐020 group (30.6%). All TEAEs were considered mild, and no significant changes in neutrophil, platelets, or red blood cells counts were observed. No subject had a serious TEAE or TEAE leading to study drug discontinuation, and no subject died during the study.
In conclusion, first‐in‐human studies of TAK‐020 yielded safety and tolerability profiles that are adequate for supporting the investigation of their activity in nonmalignant pathologies while saturating BTK and completely inhibiting its activity. The prolonged receptor occupancy and extended PD effects suggest the potential for q.d. dosing despite the relative short plasma t1/2 of TAK‐020. Furthermore, evobrutinib has exhibited promising initial activity in patients with relapsed‐remitting multiple sclerosis. These collective data indicate that this generation of highly selective BTKi, including TAK‐020, could emerge as safe and effective therapies for novel indications and support further investigations in phase II and III studies.
Funding
Funding for this manuscript was provided by Takeda Pharmaceuticals Inc.
Conflicts of Interest
E.E., M.C., G.S., D.B., H.F., J.W., L.M., and E.R.F. are/were employees of Takeda Pharmaceuticals Inc. during the conduct of this study. As Editor‐in‐Chief of Clinical and Translational Science, John Wagner was not involved in the review or decision process for this paper. [Correction added on 18th June 2021, after first online publication: COI section has been updated in this version.]
Author Contributions
E.E., M.C., G.S., H.F., J.W., L.M., and E.R.F. wrote the manuscript. M.C., G.S., D.B., J.W., L.M., and E.R.F. designed the research. E.E., M.C., G.S., D.B., and L.M. performed the research. E.E., M.C., G.S., D.B., H.F., J.W., L.M., and E.R.F. analyzed the data. G.S. and D.B. contributed new reagents/analytical tools.
Supporting information
Figure S1
Table S1
Table S2
Table S3
Acknowledgment
Medical writing assistance was provided by Leonard Lionnet, PhD, CMPP of Synchrogenix, a Certara company.
References
- 1. Kawakami, Y. , Kitaura, J. , Hata, D. , Yao, L. & Kawakami, T. Functions of Bruton's tyrosine kinase in mast and B cells. J. Leukoc. Biol. 65, 286–290 (1999). [DOI] [PubMed] [Google Scholar]
- 2. Krupa, A. et al. Bruton's tyrosine kinase mediates FcgammaRIIa/Toll‐like receptor‐4 receptor crosstalk in human neutrophils. Am. J. Resp. Cell Mol. Biol. 48, 240–249 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonami, R.H. et al. Bruton's tyrosine kinase promotes persistence of mature anti‐insulin B cells. J. Immunol. 192, 1459–1470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luo, W. et al. A balance between B cell receptor and inhibitory receptor signaling controls plasma cell differentiation by maintaining optimal Ets1 levels. J. Immunol. 193, 909–920 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chang, B.Y. et al. The Bruton tyrosine kinase inhibitor PCI‐32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res. Ther. 13, R115 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Picard, C. et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency 2015. J. Clin. Immunol. 35, 696–726 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Montalban, X. et al. Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N. Engl. J. Med. 380, 2406–2417 (2019). [DOI] [PubMed] [Google Scholar]
- 8. Becker, A. et al. Safety, tolerability, pharmacokinetics, target occupancy, and concentration‐QT analysis of the novel BTK inhibitor evobrutinib in healthy volunteers. Clin. Transl. Sci. 13, 325–336 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McGowan, E.C. & Saini, S. Update on the performance and application of basophil activation tests. Curr. Allergy Asthma Rep. 13, 101–109 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Advani, R.H. et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J. Clin. Oncol. 31, 88–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coffey, G. et al. PRT062607 achieves complete inhibition of the spleen tyrosine kinase at tolerated exposures following oral dosing in healthy volunteers. J. Clin. Pharmacol. 57, 194–210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karasuyama, H. , Miyake, K. , Yoshikawa, S. & Yamanishi, Y. Multifaceted roles of basophils in health and disease. J. Allergy Clin. Immunol. 142, 370–380 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Table S2
Table S3