

Abstract
We evaluated safety, tolerability, pharmacokinetics (PKs), and pharmacodynamics of AZD4831, a novel oral myeloperoxidase (MPO) inhibitor, in a randomized, single‐blind, placebo‐controlled study, following once‐daily multiple ascending dosing to steady‐state in healthy subjects. Target engagement was measured as specific MPO activity in plasma following ex vivo zymosan stimulation of whole blood. Except for generalized maculopapular rash in 4 of 13 subjects receiving the 2 highest doses, 15 and 45 mg AZD4831, no clinically relevant safety and tolerability findings were observed. AZD4831 was rapidly absorbed and plasma concentrations declined slowly with an elimination half‐life of ~ 60 hours. A dose/concentration‐effect relationship between MPO inhibition vs. AZD4831 exposure was established with > 50% MPO inhibition in plasma at concentrations in the low nanomolar range. Steady‐state levels were achieved within 10 days. Taken together, the PK profile, the sustained dose/concentration‐dependent MPO inhibition, and available clinical data support further clinical development of AZD4831 in patients with heart failure with preserved ejection fraction.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Myeloperoxidase (MPO) is reported to play a role in atherogenesis in humans and MPO plasma levels predict outcome of cardiovascular disease. In patients with chronic heart failure (HF), elevated plasma MPO levels are associated with more advanced HF. Additionally, elevated plasma MPO levels within an HF subject seem to be predictive of increased adverse clinical outcomes.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This was the first once‐daily multiple ascending dosing study of the MPO inhibitor AZD4831 in healthy subjects to explore safety, tolerability, pharmacokinetics, and pharmacodynamics at steady‐state conditions.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ AZD4831 has a long half‐life consistent with once daily dosing. Except for incidence of generalized maculopapular rash at the highest doses, no clinically relevant safety and tolerability findings were observed
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Target engagement was established and a dose/concentration‐effect relationship between MPO inhibition vs. AZD4831 exposure was observed with half‐maximal inhibitory concentration values in the lower nanomolar range. The study showed that AZD4831 is a suitable drug candidate for further evaluation in patients with heart failure with preserved ejection fraction.
Cardiovascular disease (CVD) remains the major cause of death in the industrialized countries, despite significant advancement in clinical cardiology during the past 2 decades. Beyond optimal control of traditional risk factors, such as lipids, blood pressure, and glucose, targeting novel molecular pathways may further improve outcome in high‐risk patients with CVD by exerting direct beneficial effects on macrovascular and microvascular structure and function.
Recently, the PROMIS study in heart failure with preserved ejection fraction (HFpEF) was reported and provided evidence that coronary microvascular dysfunction is highly prevalent in patients with HFpEF and that it correlates with several key features in this patient population, including disease severity, systemic endothelial dysfunction, as well as diastolic dysfunction. 1 To date, no or limited pharmacological therapies are available for treatment of patients with HFpEF and a novel pharmacological intervention improving both macrovascular and microvascular status in these patients would be beneficial.
Myeloperoxidase (MPO) is mainly present in granules of neutrophils, constituting 5% of the dry weight of the cells. In addition to neutrophils, there are also data suggesting the presence of MPO in monocytes and macrophages. MPO generates reactive chlorinating species, such as hypochlorous acid, the active component of bleach, which possesses potent bactericidal and viricidal activities and reacts with electron‐rich moieties of a large range of biomolecules. 2 These actions mainly occur in the phagolysosome, but also in the extracellular compartment, as MPO can be released following neutrophil degranulation.
Multiple lines of evidence suggest that MPO may play a role in atherogenesis in humans 2 , 3 , 4 and MPO plasma levels predict outcome of CVD. 3 , 4 , 5 In chronic heart failure (HF), elevated plasma MPO levels are associated with more advanced disease. Additionally, elevated plasma MPO levels within an HF subject seem to be predictive of increased adverse clinical outcomes. 6
In addition, individuals with inherited low MPO activity are protected from leukocyte activation‐induced deterioration of vascular function. 7 Direct MPO administration in anaesthetized pigs increased the tone of conductance and resistance vessels and adversely affected myocardial blood flow, thereby strengthening the concept that MPO indeed acts as a modulator of vascular tone in vivo and identifying MPO as a systemic regulator of vasomotion in humans and thus a potential therapeutic target. Furthermore, the degree of MPO‐deficiency was correlated to improved vascular function, such that 50% difference in MPO activity was associated with a 5% absolute difference in flow mediated dilatation. 7 As published by Rudolph et al. in 2010, MPO is also involved in structural remodeling of the myocardium, leading to an increased vulnerability to atrial fibrillation. 8 Overall, recent evidence suggests that MPO may provide a mechanistic link among inflammation, oxidative stress, vascular dysfunction, and impaired cardiac remodeling. Thus, it is hypothesized that an MPO inhibitor (MPOi) will improve both macrovascular and microvascular status in patients with CVD.
There is an offset between the MPOi concentration required to inhibit the MPO in the phagolysosome vs. that required to inhibit extracellular MPO activity. 9 From a biological perspective, we hypothesize that it is the extracellular MPO activity that causes the pathological microvascular dysfunction observed in CVD, whereas the intragranular MPO plays a physiological role in host defense.
AZD4831 is a novel, potent, and selective MPOi with an in vitro half‐maximal inhibitory concentration of 0.7 nM for human MPO. The pharmacokinetics (PKs), safety, and tolerability, and the effect on uric acid, a down‐stream biomarker of MPO inhibition, following single ascending doses (SADs) in healthy volunteers has been reported. 10 The human plasma protein binding is 65%, and in vitro studies have indicated that CYP3A4/CYP3A5 is the predominant CYP isoform involved in the metabolism of AZD4831, although other elimination pathways are predicted to contribute to the overall elimination (i.e., non‐CYP450 mediated metabolism as well as renal clearance). 10
Here, we report the results from the first multiple ascending dose study with the objective to evaluate safety, tolerability, PKs, and pharmacodynamics of AZD4831 in healthy subjects. As an exploratory objective, plasma 4ß‐hydroxycholesterol levels were measured for assessment of potential CYP3A4/A5 induction.
METHODS
Study design
This was a randomized, single‐blind, placebo‐controlled, phase I, multiple ascending dose study to evaluate safety and tolerability of the MPO inhibitor AZD4831 in healthy male volunteers (NCT03136991). A single‐blind design was selected as personnel needed to be unblinded for the dose escalation decisions made by the Safety Review Committee (SRC). Study personnel and subjects were blinded to treatment allocation. In each cohort, the aim was to randomize two subjects to placebo and eight subjects to AZD4831.
Eligibility criteria for the study included healthy male subjects aged 18–50 years, a body mass index between 18 and 30 kg/m2 and a body weight between 50 and 100 kg. Subjects were excluded if they had any clinically significant illness or disease that would have influenced the results of the study, specifically infections (particularly fungal infection), skin disorders, allergy, or thyroid disease. The study was conducted in healthy subjects to avoid interference from disease processes or other drugs. The subjects stayed at the study center during the whole dosing period until 48 hours post‐final dose and underwent 3 follow‐up visits before the final follow up visit 7–10 days after the last dose.
The study was submitted to the institutional review board (Aspire) for ethical review and approval by the investigator, and all subjects gave written informed consent before participating in the study.
Clinical safety
The tolerability and safety assessments involved evaluation of vital signs, echocardiogram (ECG), telemetry, routine clinical laboratory examinations, and adverse events. These assessments were taken before and at scheduled times after dosing as well as at the final follow‐up visit.
Starting dose, escalation, and dosing
The starting dose of 5 mg in cohort 1 was the same starting dose as previously evaluated in a SAD study and, although some accumulation was predicted following once‐daily dosing to steady‐state, exposures were predicted to be well below the highest doses/exposures explored in that study. 10 A period of 10 days was deemed sufficient to reach steady‐state conditions given the half‐life of AZD4831 as determined in the SAD study.
The subsequent dose levels were 15 and 45 mg of AZD4831 in cohorts 2 and 3, respectively, the condition being that the maximum dose escalation was not allowed to be higher than 3 times the previous dose level. Based on the emerging data on 45 mg, with 2 cases of generalized maculopapular rash on 15 mg and an additional 3 cases on 45 mg (2 cases on active drug and 1 on placebo), it was decided to stop further dose escalation. Instead, an extra cohort was included and evaluated at 10 mg to obtain additional PK and pharmacodynamic measurements to further explore the dose/concentration‐MPO inhibition relationship. In addition, study duration was increased to 14 days for the 10 mg cohort to evaluate the safety and tolerability at steady‐state conditions for a longer period of time compared with the 10 days once‐daily dosing of the 5, 15, and 45 mg AZD4831 cohorts.
Before frequent PK sampling (after the first and the last day of dosing) subjects fasted for 10 hours overnight before the morning dose of AZD4831. A total volume of 240 mL was administered, which included the volume of the oral suspension and water. Water (< 150 mL) was allowed up to 1 hour prior to each morning dose and could be resumed 1 hour after dosing. A meal was served 4 hours after the morning dose. On other study days during the once‐daily repeated dosing, subjects fasted for 10 hours overnight prior to the morning dose and breakfast was delayed until 2 hours after dosing in order to reduce the overall fasting period for the subjects. Water was allowed up to 1 hour before and from 1 hour after the morning dose.
Pharmacokinetic and Pharmacodynamic Assessments
Plasma pharmacokinetics of AZD4831 and 4β‐hydroxycholesterol measurements
Blood samples were collected for plasma PK analyses predose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 hours postdose on the first and the last day of dosing. In addition, predose samples were collected before all doses and after the last dose samples were also collected at 36, 48, 96, 144, 240, and 336 hours post last dose. When dosed for 14 days, predose samples were only collected on study days 2, 3, 4, 6, 8, 10, 12, and 13. Blood samples for measurement of 4ß‐hydroxycholesterol levels was collected before dosing on day 1 and before dosing on the last day. All samples were analyzed by Covance Laboratories Ltd. (Harrogate, UK).
AZD4831 and the stable labeled internal standard [13C3 15N2]AZD4831 were extracted from plasma by liquid–liquid extraction and analyzed by liquid chromatography tandem mass spectrometry. This method was validated prior to sample analysis in the range 2–2,000 nM in plasma, using a 25 µL sample aliquot. 10
Study samples were received in good condition and stored in a freezer set to maintain nominal −80°C and all samples were analyzed within the validated stability period. At a minimum, each analytical run included a calibration curve, a matrix blank, a control zero sample (matrix blank containing internal standard), a reagent blank, and duplicate quality control (QC) samples at four concentrations within the calibration range (6, 70, 800, and 1,600 nM). The results from the QC samples and calibration standards were evaluated and it was concluded that the method performed acceptably for this study. The inter‐run accuracy for the study QC samples ranged from 96.8–100.7% and the inter‐run precision was < 6.1%. In addition, incurred sample reproducibility analyses were performed during the study. Of the 101 samples reanalyzed, 98 (97.0%) were within 20% of the mean of the 2 values. Plasma samples for analysis of 4β‐hydroxycholesterol levels were analyzed as previously reported by van de Merbel et al. 11
Urinary pharmacokinetics of AZD4831
Urine was collected for PK analyses as a spot predose sample before the first dose and as pooled urine 0–3, 3–6, 6–9, 9–12, and 12–24 hours after the first and last day of dosing. Samples for determination of AZD4831 in urine were analyzed by Covance Laboratories Ltd. AZD4831 and the stable labeled internal standard [13C3 15N2]AZD4831 were prepared from urine by sample dilution and analyzed by liquid chromatography tandem mass spectrometry. This method was validated prior to sample analysis in the range 20–20,000 nM in urine, using a 25 µL sample aliquot. 10
Study samples were received in good condition and stored in a freezer set to maintain nominal −80°C and all samples were analyzed within the validated stability period. At a minimum, each analytical run included a calibration curve, a matrix blank, a control zero sample (matrix blank containing internal standard), a reagent blank, and duplicate QC samples at three concentrations within the calibration range (60, 1,000, and 16,000 nM). The results from the QC samples and calibration standards were evaluated and it was concluded that the method performed acceptably for this study. The inter‐run accuracy for the study QC samples ranged from 98.7–104.4% and the inter‐run precision was < 10.1%. In addition, incurred sample reproducibility analyses were performed during the study. Of the 29 samples reanalyzed, 27 (93.1%) were within 20% of the mean of the 2 values.
Blood sampling for MPO activity assay and uric acid measurements
Blood samples anticoagulated with K2EDTA were collected at day –1, at baseline, and at designated timepoints after dosing AZD4831 up to the final follow‐up at 24 ± 2 days. To promote neutrophil degranulation and release of MPO, 4.5 mL blood was stimulated with 125 µL of a 20 mg/mL zymosan (Sigma; cat #Z‐4250) suspension prepared in 0.9% saline solution. After gentle mixing by inversion of tubes, blood samples were incubated for 30 ± 2 minutes in a shaking incubator set at 37°C with rotation between 80 and 120 rpm. Plasma was prepared by centrifugation at 1,500 g, 4°C for 10 minutes and carefully removed to a fresh collection tube without disturbing the cell layer. The 500 µL plasma was then transferred to separate collection tubes and stored at −80°C prior to measurement of MPO activity and concentration according to the method described by Russell et al. 12
Serum samples for measurement of uric acid were collected predose, 2, 4, 6, 8, and 12 hours post first and last dose. In addition, predose day 2 to day 9 (to day 13 at 10 mg) and 24, 36, 48, 96, 144, 240, and 336 hours post last dose.
Rationale for MPO assessment, and quantification of MPO activity and uric acid
MPO concentration and activity were analyzed using 100 µL undiluted plasma samples in duplicate in a single plate assay according to the method described by Russell et al. 12 MPO is a heme‐containing enzyme in which the iron is oxidized from the resting ferric state to an activated two‐electron oxidized form by reacting with H2O2. AZD4831 acts as a substrate for this activated form, creating a covalent bond to the heme moiety, 13 whereas it is not active on the resting, ferric state of the enzyme. A fraction of the released MPO, and the circulating MPO pool is not active and thus not targeted by AZD4831. When MPO activity is quantified ex vivo by activation with H2O2, this nonactive fraction will constitute a residual MPO activity. Target engagement quantification can be refined by limiting the fraction of nonactive MPO by activating whole blood with zymosan. The remaining fraction can be estimated by spiking the plasma samples with a supra optimal concentration of AZD4831.
To evaluate the effect of AZD4831 on level of MPO, specific activity, that is MPO activity divided by MPO concentration, was studied. Before comparisons were made, the mean of the specific activity for the spiked samples collected on day −1, and predose on day 1 was subtracted from all observations to enable observing full inhibition. This correction was done on an individual level. In the subsequent text, the term activity is used for specific activity.
All uric acid samples were analyzed with validated standard methods.
The assay corrected MPO and uric acid data relative to baseline were explored graphically and plotted vs. exposure (MPO) and time (MPO and uric acid).
Pharmacokinetic analysis
Actual blood sampling time were used in all analysis and plasma concentration vs. time data of AZD4831 were analyzed by noncompartmental analysis with Phoenix WinNonlin (version 6.2). The maximum plasma concentration (Cmax) and the time to reach this concentration (Tmax), elimination half‐life (t½), and the total area under the plasma concentration time‐curve (AUC) were estimated. The AUC was calculated by log‐linear trapezoidal rule from time zero to the time for the last measurable concentration (Tlast) plus the extrapolated residual area to infinity. The residual area after Tlast was calculated as Clast,pred/λZ, where Clast,pred was the predicted concentration at Tlast and λZ was the terminal rate constant determined by linear regression analysis of log transformed plasma concentration vs. time, using the last plasma concentrations from each subject. The t½ was calculated as ln2/λZ. The apparent oral plasma clearance was estimated as dose/AUC and renal clearance as amount excreted unchanged divided by the corresponding AUC. Dose linearity was explored by fitting a power model to AUC and Cmax after the last dose, and time to steady‐state was explored by visual inspection of the data.
Statistical analysis
The sample size was chosen to obtain reasonable evidence of safety and tolerability without exposing undue numbers of healthy subjects to the compound at this stage of clinical drug development. Previous experience from similar studies have shown that the sample size used was reasonable to accomplish the objectives of this study.
RESULTS
Subjects
A total of 37 healthy male subjects between the ages of 23 and 50 years were included in all cohorts. The body mass index ranged between 18.8 and 30.0, and a summary of demographic data is presented in Table 1 . All subjects were judged to be in good health based on the results of medical history, physical examination, clinical laboratory evaluations, and ECG obtained within 28 days of the initial study drug administration.
Table 1.
Participant demographics (all subjects)
| Variable/category | Placebo (N = 8) |
5 mg AZD4831 (N = 8) |
15 mg AZD4831 (N = 8) |
45 mg AZD4831 (N = 5) |
10 mg AZD4831 (N = 8) |
Total AZD4831 (N = 29) |
|---|---|---|---|---|---|---|
| Age, years | ||||||
| Median | 28.0 | 33.5 | 34.5 | 31.0 | 30.5 | 33.0 |
| Min–Max | 23–40 | 29–47 | 25–41 | 27–35 | 23–50 | 23–50 |
| Sex n (%) | ||||||
| Male | 8 (100) | 8 (100) | 8 (100) | 5 (100) | 8 (100) | 29 (100) |
| Race n (%) | ||||||
| White | 5 (62.5) | 3 (37.5) | 4 (50.0) | 4 (80.0) | 5 (62.5) | 16 (55.2) |
| Black or African American | 2 (25.0) | 4 (50.0) | 3 (37.5) | 0 | 2 (25.0) | 9 (31.0) |
| Asian | 1 (12.5) | 1 (12.5) | 1 (12.5) | 0 | 0 | 2 (6.9) |
| Other | 0 | 0 | 0 | 1 (20.0) | 1 (12.5) | 2 (6.9) |
| Ethnicity n (%) | ||||||
| Hispanic or Latino | 3 (37.5) | 3 (37.5) | 1 (12.5) | 1 (20.0) | 1 (12.5) | 6 (20.7) |
| Not Hispanic or Latino | 5 (62.5) | 5 (62.5) | 7 (87.5) | 4 (80.0) | 7 (87.5) | 23 (79.3) |
| Height, cm | ||||||
| Median | 171.0 | 174.0 | 174.0 | 174.0 | 179.5 | 175.0 |
| Min–Max | 164–185 | 168–187 | 165–186 | 168–179 | 165–187 | 165–187 |
| Weight, kg | ||||||
| Median | 72.05 | 79.25 | 78.40 | 73.00 | 82.15 | 77.10 |
| Min–Max | 63.0–82.9 | 61.5–90.5 | 71.6–87.8 | 67.6–82.5 | 60.8–84.9 | 60.8–90.5 |
| BMI, kg/m2 | ||||||
| Median | 24.10 | 26.20 | 25.20 | 24.00 | 25.25 | 25.30 |
| Min–Max | 18.8–29.3 | 21.5–28.0 | 23.0–30.0 | 22.0–28.5 | 18.8–28.4 | 18.8–30.0 |
BMI, body mass index.
Safety and tolerability
There were no serious adverse events and except for generalized maculopapular rash in 4 of 13 subjects receiving the 2 highest doses, no clinically relevant safety and tolerability findings were observed, as presented in Table 2 . Subjects with rash were taken off treatment and were monitored and the rash was reversible in all cases. Due to the incidence of generalized maculopapular rash, dosing at the highest dose, 45 mg, was prematurely stopped and no further dose escalation was done. Based on this, a new cohort was studied at 10 mg with a dosing period of 14 days in order to obtain additional safety and tolerability data.
Table 2.
AEs reported by two or more subjects
| AE category | Number (%) of subjects | |||||
|---|---|---|---|---|---|---|
| Placebo (N = 8) |
5 mg AZD4831 (N = 8) |
10 mg AZD4831 (N = 8) |
15 mg AZD4831 (N = 8) |
45 mg AZD4831 (N = 5) |
Total AZD4831 (N = 29) |
|
| Subjects with any AE | 4 (50.0) | 2 (25.0) | 3 (37.5) | 3 (37.5) | 2 (40.0) | 10 (34.5) |
| Contact dermatitis | 0 | 0 | 2 (25.0) | 1 (12.5) | 0 | 3 (10.3) |
| Generalized maculopapular rash | 1 (12.5) | 0 | 0 | 2 (25.0) | 2 (40.0) | 4 (13.79) |
| Headache | 2 (25.0) | 1 (12.5) | 1 (12.5) | 0 | 1 (20.0) | 3 (10.3) |
| Dry throat | 0 | 0 | 0 | 1 (12.5) | 1 (20.0) | 2 (6.9) |
AE, adverse event.
Generalized maculopapular rash started 8–10 days after first dose except for 1 subject that developed rash already after 2 days. This subject was on placebo in the 45 mg cohort. The general duration of rash was 6–8 days but in the subject that received placebo it was 2 days. For 1 subject in the 45 mg cohort, the rash was self‐reported to be resolved after 28 days.
There were no apparent clinically significant trends observed in vital signs assessments, ECG measurements, telemetry, or any laboratory results.
Pharmacokinetics
AZD4831 was rapidly absorbed after oral administration, with a median tmax of 1 h (range 0.2–4 hours) on day 1, and last day of dosing across all dose groups (Figure 1 and Table 3 ). The t1/2λz of AZD4831 was similar at all dose levels with a mean of ~ 60 hours (Table 3 ). The mean renal clearance of AZD4831 ranged from 10.5 L/hour to 13.6 L/hour (Table 3 ), and was similar after the first and last day of dosing. The AUCτ and Cmax were approximately proportional to dose on the last day of dosing, with modeled slope estimates of 1.11 (90% confidence interval, 0.91–1.31) and 1.25 (1.06–1.44), respectively. Following once‐daily dosing, steady‐state condition was reached within 10 days, with an approximate threefold accumulation. Visual inspection of Figure 1 indicates a slight increase of the Ctrough after the last dose in all cohorts. This is likely explained by the longer fasting period compared with the preceding dose. Due to the early discontinuation and subjects treated for different days on 45 mg, the PK parameters were only estimated after the first day of dosing (Table 3 , Figure 2 ).
Figure 1.
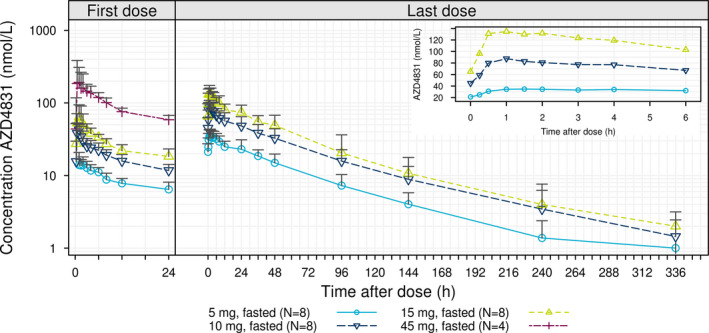
Geometric mean + SD* for plasma concentration‐time profiles by treatment following multiple ascending doses of AZD5718 (after first and last dose). Insert; first 6 hours after the last dose. *Geometric mean + SD is to be interpreted as exp(mean of log transformed values + SD of log transformed values).
Table 3.
Summary of the pharmacokinetic parameters (geometric mean and percentage of coefficient of variation, except for Tmax median (range)) following single and repeated once daily dosing of AZD4831 in healthy volunteers
| Parameter | First day | Last day | |||||
|---|---|---|---|---|---|---|---|
| 5 mg (n = 8) | 10 mg (n = 8) | 15 mg (n = 8) | 45 mg (n = 5) a | 5 mg (n = 8) | 10 mg (n = 8) | 15 mg (n = 8) | |
| AUCτ, hour·nM | 206.1 (17.0) | 438.1 (19.5) | 660.8 (11.1) | 2,381 a (9.7) | 655.5 (19.6) | 1,450 (24.7) | 2,208 (18.7) |
| Cmax, nM | 16.5 (17.7) | 48.9 (24.7) | 66.2 (37.1) | 236.2 (52.7) | 37.3 (19.1) | 93.0 (21.0) | 145.7 (19.7) |
| t½ λz, hour | NE | NE | NE | NE | 53.3 (16.4) | 72.6 (14.6) | 71.8 (10.1) |
| CL/F, L/hour | NE | NE | NE | NE | 22.8 (19.6) | 20.6 (24.7) | 20.3 (18.7) |
| Tmax, hour | 1.0 (0.2‐2.0) | 1.0 (0.4‐1.5) | 1.0 (0.2‐1.5) | 0.6 (0.5‐3.0) | 1.8 (0.5‐4.0) | 1.0 (0.5‐4.0) | 1.0 (0.5‐3.0) |
| CLR, L/hour | 11.3 (27.7) | 11.8 (45.6) | 13.6 (20.2) | 13.4 (23.4) | 11.1 (19.2) | 10.5 (33.3) | 10.7 (12.5) |
AUC, area under the concentration‐time curve; CL/F, oral plasma clearance; CLR, renal clearance; Cmax, peak plasma concentration; NE, not estimated; t½λz, terminal half‐life; Tmax, time of maximum plasma concentration.
n = 4. One subject withdrew informed consent prior to 24 hours measurement.
Figure 2.
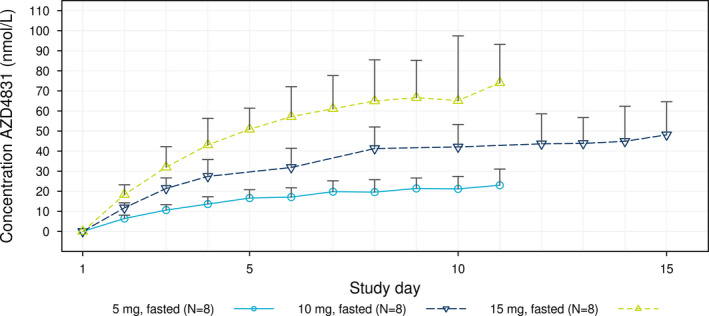
Geometric mean + SD* trough concentration‐time profiles by treatment following multiple ascending doses of AZD4831. *Geometric mean + SD is to be interpreted as exp(mean of log transformed values + SD of log transformed values).
No increases in 4β‐hydroxycholesterol levels were observed, with mean changes from before dosing on day 1 to last day of dosing of − 0.2 ng/mL (SD, 3.0), −1.0 ng/mL (SD, 4.2), and 6.2 ng/mL (SD, 4.7) for AZD4831 doses of 5, 10, and 15 mg, respectively.
MPO inhibition and effect on uric acid
A dose‐dependent target engagement, as assessed by measuring ex vivo zymosan stimulated inhibition of MPO activity, was observed (Figure 3 a; where activity relative to baseline is shown). In the highest dose, an inhibition of above 80% was reached by the end of treatment. An exposure response was also evident (Figure 4 ).
Figure 3.
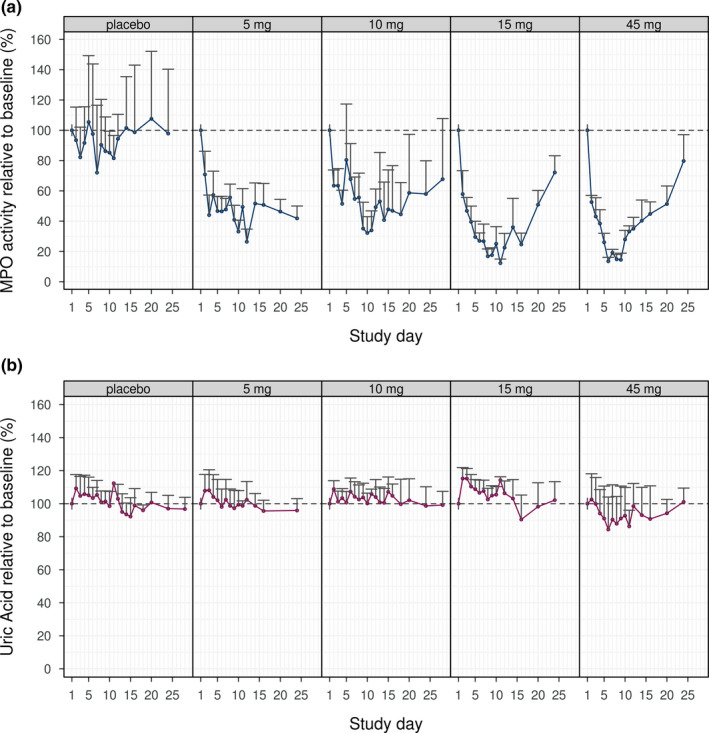
Geometric mean + SD* of spike corrected zymosan stimulated myeloperoxidase (MPO) activity relative to baseline (a) and of uric acid relative to baseline (b). *Geometric mean + SD is to be interpreted as exp(mean of log transformed values + SD of log transformed values).
Figure 4.
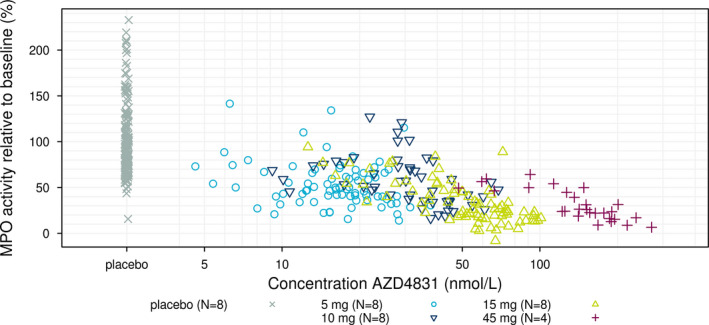
Spike corrected zymosan stimulated myeloperoxidase (MPO) activity relative to baseline vs. trough concentration. MPO values for placebo subjects included at an arbitrary position.
No statistically significant effect on uric acid was observed for any of the doses (Figure 3 b), although observed uric acid relative to baseline for the highest dose was below 100%.
DISCUSSION
In this phase I study, multiple oral doses of AZD4831 up to 45 mg dosed once daily for 10 or 14 days, was, except for reversible generalized maculopapular rash in 4 subjects, well‐tolerated by healthy male volunteers, with no other safety or tolerability concerns identified.
After the first and the last dose, AZD4831 was rapidly absorbed followed by an apparent biphasic decline in plasma concentrations with t½ estimates ranging between 53 and 73 hours. Consistent with this long half‐life steady‐state, conditions were achieved within 10 days and approximate dose‐proportional increases were observed in the dose range explored. Opposite to a report from another MPO inhibitor, 14 there was no indication of auto induction or CYP3A4/A5 induction because Ctrough levels remained constant following attainment of steady‐state conditions and no significant change in 4β‐hydroxycholesterol levels was detected. In addition, the higher potency of AZD4831 will reduce the risk for AZD4831 being a CYP3A4/A5‐inducer at clinically relevant doses/exposures.
A phase II metabolite was detected using an exploratory bioanalysis method. It may be speculated that this metabolite contributes to the longer than expected half‐life of AZD4831 based on scaling of in vitro data to man. Despite the above‐mentioned metabolism, renal clearance of AZD4831 was high (> 30% of total apparent oral plasma clearance), consistent across the dose range evaluated and contributed to the overall elimination of AZD4831.
We observed a clear dose‐response and exposure‐response relationship toward MPO activity, but not toward uric acid. This is in contrast to the SAD study of AZD4831 and also in other clinical studies with MPO inhibitors, where reduction of serum uric acid has been reported. 15 Uric acid is generated by oxidation of xanthine, a reaction that is enzymatically driven by xanthine oxidase, but can also be mediated by MPO (unpublished data). In the current study, no dose‐dependent or exposure‐dependent reduction of uric acid levels was observed. The reason for this discrepancy in relation to the SAD study of AZD4831 is not known but may relate to a difference of exposure levels explored, supported by the trend for a reduction of uric acid in the highest dose group in the current study. However, a 50% reduction in MPO activity, as shown in the present study, has been reported to result in clinically relevant improvement in flow mediated dilatation. 7 This level of inhibition was seen at steady‐state already at the lowest dose evaluated, 5 mg. In addition, the 15 mg dose yielded, on average, > 80% inhibition of MPO activity suggesting potential for therapeutic doses/exposures in the low mg/nM ranges.
Taken together, generated data including the PKs and the dose/concentration‐effect relationship between MPO inhibition vs. AZD4831 exposure support continued clinical development and the ongoing studies in patients with HFpEF. There are currently two ongoing patient studies with AZD4831 where one is aiming at assessing the acute effect on hemodynamics, exercise capacity, and endothelial function after a single dose of AZD4831 (NCT03611153) and where the other will determine the effect on coronary flow velocity reserve following 3‐month treatments with AZD4831 (NCT03756285).
Funding
This study was sponsored by AstraZeneca.
Conflicts of Interest
K.N., M.L.‐F, C.A, E.M, M.H, M.K., M.R., E.‐L.L., C.W., L.‐M.G., and H.E. are all employees of AstraZeneca. D.H. is employed at Parexel. AstraZeneca provided funding to Parexel for the conduct of this study.
Author Contributions
K.N., E.M., M.H., C.W., and H.E. wrote the manuscript. K.N., M.L.‐F., C.A., E.M., M.H., M.K., M.R., D.H., E.‐L.L., C.W., L.‐M.G., and H.E. designed and performed the research. K.N., M.L.‐F., C.A., E.M., M.H., M.K., M.R., D.H., E.‐L.L., C.W., L.‐M.G., and H.E. analyzed data. M.H., M.R., and C.W. contributed new reagents/analytical tools.
Acknowledgments
The authors thank the subjects participating and the personal involved in the conduct of the study.
References
- 1. Shah, S.J. et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS‐HFpEF. Eur. Heart J. 39, 3439–3450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nicholls Stephen, J. & Hazen Stanley, L. Myeloperoxidase and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 25, 1102–1111 (2005). [DOI] [PubMed] [Google Scholar]
- 3. Brennan, M.‐L. et al. Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 349, 1595–1604 (2003). [DOI] [PubMed] [Google Scholar]
- 4. Baldus, S. et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation 108, 1440–1445 (2003). [DOI] [PubMed] [Google Scholar]
- 5. Zhang, R. et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA 286, 2136–2142 (2001). [DOI] [PubMed] [Google Scholar]
- 6. Tang, W.H.W. et al. Prognostic value and echocardiographic determinants of plasma myeloperoxidase levels in chronic heart failure. J. Am. Coll. Cardiol. 49, 2364–2370 (2007). [DOI] [PubMed] [Google Scholar]
- 7. Rudolph, T.K. et al. Myeloperoxidase deficiency preserves vasomotor function in humans. Eur. Heart J. 33, 1625–1634 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rudolph, V. et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat. Med. 16, 470–474 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bjornsdottir, H. et al. Neutrophil NET formation is regulated from the inside by myeloperoxidase‐processed reactive oxygen species. Free Radic. Biol. Med. 89, 1024–1035 (2015). [DOI] [PubMed] [Google Scholar]
- 10. Gan, L.‐M. et al. Safety, tolerability, pharmacokinetics and effect on serum uric acid of the myeloperoxidase inhibitor AZD4831 in a randomized, placebo‐controlled, phase I study in healthy volunteers. Br. J. Clin. Pharmacol. 85, 762–770 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van de Merbel, N.C. et al. A validated liquid chromatography–tandem mass spectrometry method for the quantitative determination of 4β‐hydroxycholesterol in human plasma. J. Pharm. Biomed. Anal. 55, 1089–1095 (2011). [DOI] [PubMed] [Google Scholar]
- 12. Russell, M. et al. Determining myeloperoxidase activity and protein concentration in a single assay: utility in biomarker and therapeutic studies. J. Immunol. Methods 449, 76–79 (2017). [DOI] [PubMed] [Google Scholar]
- 13. Tidén, A.‐K. et al. 2‐thioxanthines are mechanism‐based inactivators of myeloperoxidase that block oxidative stress during inflammation. J. Biol. Chem. 286, 37578–37589 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dong, J.Q. et al. Examination of the human cytochrome P4503A4 induction potential of PF‐06282999, an irreversible myeloperoxidase inactivator: integration of preclinical, in silico, and biomarker methodologies in the prediction of the clinical outcome. Drug Metab. Dispos. 45, 501 (2017). [DOI] [PubMed] [Google Scholar]
- 15. Jucaite, A. et al. Effect of the myeloperoxidase inhibitor AZD3241 on microglia: a PET study in Parkinson’s disease. Brain 138, 2687–2700 (2015). [DOI] [PubMed] [Google Scholar]