

Abstract
Abstract
Liver microphysiological systems (MPSs) are promising models for predicting hepatic drug effects. Yet, after a decade since their introduction, MPSs are not routinely used in drug development due to lack of criteria for ensuring reproducibility of results. We characterized the feasibility of a liver MPS to yield reproducible outcomes of experiments assaying drug toxicity, metabolism, and intracellular accumulation. The ability of the liver MPS to reproduce hepatotoxic effects was assessed using trovafloxacin, which increased lactate dehydrogenase (LDH) release and reduced cytochrome P450 3A4 (CYP3A4) activity. These observations were made in two test sites and with different batches of Kupffer cells. Upon culturing equivalent hepatocytes in the MPS, spheroids, and sandwich cultures, differences between culture formats were detected in CYP3A4 activity and albumin production. Cells in all culture formats exhibited different sensitivities to hepatotoxicant exposure. Hepatocytes in the MPS were more functionally stable than those of other culture platforms, as CYP3A4 activity and albumin secretion remained prominent for greater than 18 days in culture, whereas functional decline occurred earlier in spheroids (12 days) and sandwich cultures (7 days). The MPS was also demonstrated to be suitable for metabolism studies, where CYP3A4 activity, troglitazone metabolites, diclofenac clearance, and intracellular accumulation of chloroquine were quantified. To ensure reproducibility between studies with the MPS, the combined use of LDH and CYP3A4 assays were implemented as quality control metrics. Overall results indicated that the liver MPS can be used reproducibly in general drug evaluation applications. Study outcomes led to general considerations and recommendations for using liver MPSs.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Microphysiological systems (MPSs) have been designed to recreate organ‐ or tissue‐specific characteristics of extracellular microenvironments that enhance the physiological relevance of cells in culture. Liver MPSs enable long‐lasting and stable culture of hepatic cells by culturing them in three‐dimensions and exposing them to fluid flow.
WHAT QUESTION DID THIS STUDY ADDRESS?
What is the functional performance relative to other cell culture platforms and the reproducibility of a liver MPS for assessing drug development and evaluation questions, such as toxicity, metabolism, and pharmacokinetics?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The liver MPS systematically detected the toxicity of trovafloxacin. When compared with spheroids and sandwich cultures, this system had a more stable function and different sensitivity to troglitazone, tamoxifen, and digoxin. Quantifying phase II metabolism of troglitazone and intracellular accumulation of chloroquine demonstrated the potential use of the liver MPS for studying drug metabolism and pharmacokinetics. Quality control criteria for assessing chip function were key for reliably using the liver MPS.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Due to its functional robustness and physiological relevance (3D culture, cells expose to fluid flow and co‐culture of different cell types), the liver MPS can, in a reproducible manner: (i) detect inflammatory‐induced drug toxicity, as demonstrated with trovafloxacin, (ii) detect the toxicity of other drugs, such as troglitazone, tamoxifen, and digoxin, with different effects than those detected in spheroids and sandwich cultures, (iii) enable studies of hepatic function that rely on prolonged cellular activity, and (iv) detect phase II metabolites and drug accumulation to potentially support the interpretation of clinical data. The integration of MPSs in drug development will be facilitated by careful evaluation of performance and reproducibility as performed in this study.
INTRODUCTION
Microphysiological systems (MPSs) are micro‐engineered cell culture platforms used to study physiologically relevant cell function by simulating key properties of the extracellular microenvironment within a specific tissue type. When assessing drug toxicity, metabolism, and transport, the liver is the central organ of study, and among the most common MPS modalities for drug development applications. 1 , 2 , 3 Liver MPSs are designed to maintain hepatocyte‐like cells or primary human hepatocytes (PHHs) in three‐dimensional (3D) culture, under continuous fluid exchange, and in co‐cultures with other cell types, such as primary human Kupffer cells (PHKCs); conditions known to collectively improve function and physiological relevance of these cells. 1 As reviewed elsewhere regarding contexts of drug evaluation, 1 , 2 , 3 , 4 , 5 liver MPSs have been demonstrated to potentially improve the estimation of drug pharmacokinetics, 6 , 7 , 8 model drug‐drug interactions, 9 predict human‐specific toxicity, 10 and detect toxicity that depends on drug metabolism. 11 Despite recent advancements in the field, data from MPSs are not routinely used by the pharmaceutical industry in the regulatory stages or drug evaluation. 12 This is primarily due to a lack of available quality control and performance criteria for consistent use of the devices and reproducibility of results between experimental sites, which are major limitations for translating experiments with MPSs into contexts of use in drug development. 13 , 14 Proof of concept results from pilot studies performed upon the prototyping of MPSs upheld their potential utility in drug discovery and motivated drug development stakeholders to start investigating performance standards for risk assessment uses or for predicting drug absorption, distribution, metabolism, and excretion (ADME). 2 , 3 This study addresses the need for performance standards of liver MPSs for general uses in drug pharmacology and toxicology, as applications in these fields rely on the metabolic function of cells, the stability and robustness of their function, and on their response to hepatotoxic compounds. 1
Prior to establishing specific contexts of use for MPSs, where the technology may demonstrate clear advantages over current approaches and tools, the reproducibility of a systems’ performance should be tested to ensure adequate reliability for drug evaluation, especially when considering applications in the later stages of drug development, where MPSs are acknowledged to hold great potential in replacing, reducing, or refining animal tests or clinical trials. 1 , 15 Here, a liver MPS was evaluated for its ability to yield reproducible results, following quality control criteria that enforced consistency across multiple test sites and MPS batches. Previous investigations using the liver MPS have involved a variety of end point assays of cell death and hepatic function, 7 , 8 , 9 , 11 , 16 , 17 , 18 , 19 of which some are also used here. In addition to lack of criteria to reduce inter‐batch or inter‐site variability in MPS results, the complexity in operating MPSs can also introduce challenges in generating reproducible results. 1 , 14 , 15 When using MPSs, ensuring cellular stability dependent on system assembly, priming, and more complex cell seeding methods can require levels of experimental planning and expertise far beyond what is required for maintaining two‐dimensional (2D) cultures. Based on the reported results and discussion, general considerations and recommendations are provided in Supplementary Materialfor liver MPSs to be used in a reproducible manner. For specific contexts of use, Fowler et al. 3 comprehensively described unmet needs in ADME studies that could be addressed with MPSs, whereas Baudy et al. 2 established detailed criteria for developing systems for predicting drug risk with liver MPSs. Along with these recent contributions, additional expert perspectives further described opportunities in the drug development field, where liver MPSs may eventually play paradigm shifting roles, 5 , 12 , 20 , 21 as tools that yield reproducible results and have cellular functional stability.
The liver MPS used in the current study was developed at the Massachusetts Institute of Technology, 22 leveraging microfluidic techniques, 23 and has been used in multiple laboratory sites and in different pilot studies for varied potential applications in drug testing, 7 , 8 , 9 , 11 , 19 suggesting its operational robustness and replicability. In addition to the characterization done so far with this system, 8 , 9 , 11 , 16 , 17 , 18 , 19 the liver MPS may also be used to expand on the capabilities of current cell culture approaches generally used in pharmacology 5 and toxicology/safety applications. 12 To explore this possibility, we tested the utility of the liver MPS for general drug development applications related to detecting drug toxicity, metabolism, and intracellular accumulation, which are key pharmacological determinants for drug development studies. 20 Reproducibility of MPS results was tested considering previously published work and the experimental outcomes from two test sites, from using different cell batches and from other cell culture platforms (spheroids and sandwich cultures).
METHODS
Detailed methods are provided as Supplementary Material, where a dedicated section details the execution of each experiment and Figures S1–S4 graphically represent the sequence of steps for each described experimental execution. The liver MPS was assembled (Figures S5, S6) according to manufacturer recommendations (CN Bio Innovations Ltd.). Viability of PHHs was ensured to be above 85% (Figure S7). Reproducibility of results was verified by detecting trovafloxacin toxicity (Figure S1) between different MPS batches. PHKCs were activated with lipopolysaccharide (LPS; Figures S8, S9). Toxicity of troglitazone, tamoxifen, and digoxin on spheroids and sandwich cultures were compared with their effects on liver MPS cultures (Figures S2, S10). Troglitazone phase II metabolites and chloroquine intracellular accumulation in the MPS were also quantified after days of incubation (Figure S4) to further test its use in applications that rely on characterizing phase II metabolism or drugs with slow clearance. Execution of experimental goals is also described in detail in Supplementary Materials.
RESULTS
The overall goal of this study was to test the performance of the liver MPS in reproducibly predicting drug hepatotoxicity, analyzing drug metabolism and intracellular drug accumulation. We first demonstrated the reproducibility of the liver MPS in detecting hepatotoxicants using trovafloxacin, before comparing it with other hepatic culture formats regarding their response to other toxicants and their functionality. The liver MPS was subsequently assessed for its ability to be used for drug metabolism, clearance, and distribution studies using troglitazone, diclofenac, and chloroquine. Finally, the appropriate use of quality control criteria was assessed to ensure that reproducible data was generated from MPS studies.
Consistency in MPS results across sites and with different batches of Kupffer cells
We sought to test if using the liver MPS in two sites or with two different batches of PHKCs (lot number of each batch in Supplementary Materials) could lead to differences in results between identical experiments. The experimental plan (Figure S1) consisted of exposing PHHs co‐cultured with PHKCs in the liver MPS 9 to trovafloxacin, which induces idiosyncratic hepatotoxicity in humans, 24 or to levofloxacin, 25 which does not cause hepatotoxicity. 26
Lactate dehydrogenase (LDH) production (Figure 1a–c) and cytochrome P450 3A4 (CYP3A4) activity (Figure 1d–f) were quantified following exposure of the MPSs to compounds at concentrations of 25 and 100 µM, and in the presence or absence of LPS. Increased levels of LDH to above 4‐fold of baseline were observed for all treatments with 100 µM trovafloxacin with LPS, and no significant differences relative to control were detected in LDH production when LPS was not co‐dosed with trovafloxacin. With or without levofloxacin, LPS reduced CYP3A4 activity to levels below 50% of baseline. However, 100 µM trovafloxacin, independently of LPS, further reduced CYP3A4 activity to levels below 10% of baseline. These results were observed in both experimental sites and independently of the batch of PHKCs.
Figure 1.
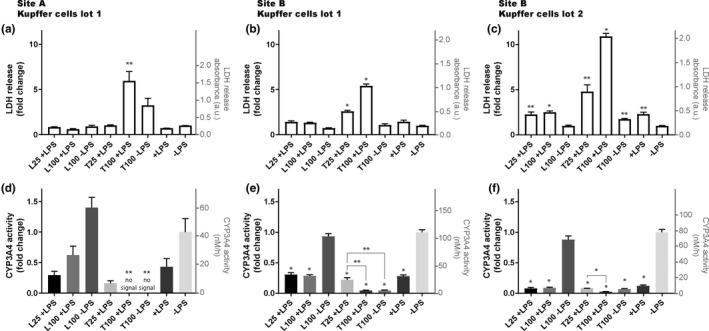
Toxic effects of trovafloxacin detected with PHHs co‐cultured with PHKCs in the liver MPS in two different experimental sites and using two validated lots of PHKCs. Site A was CN Bio Innovations in Cambridge, UK, and site B was FDA laboratories in Silver Spring, MD, USA. Trovafloxacin (T) and levofloxacin (L) were added to the cell culture medium of the liver MPS at a concentration of 25 or 100 µM in the presence (+) or absence (−) of lipopolysaccharide (LPS) at a concentration of 1 µg/ml to activate PHKCs. After 2 days of exposure, cell death was estimated by measuring LDH (a–c) and CYP3A4 activity (d–f) in the cell culture medium. Boxes represent average values and error bars represent the SEM. Identical lots of PHHs and PHKCs were used between site A (a, d) and site B (b, e). A separate lot of PHKCs was also used in site B (c, f). For each panel, *p < 0.005 and **p < 0.05 by unpaired t‐test with Welch’s correction. * and ** above column indicate statistical significance of difference relative to the control ‐LPS. Unless indicated otherwise, differences between values for presented conditions are not statistically different from the control ‐LPS. For (e) and (f), the statistical significance of differences between values of T25 +LPS condition and other conditions are also presented, such as T100 +LPS (e, f) and T100 ‐LPS (e). Three different wells were used per condition, except for experiments performed in site A, where 4 wells were used for condition L100 +LPS and T100 +LPS. LDH, lactate dehydrogenase; MPS, microphysiological system; PHHs, primary human hepatocytes; PHKCs, primary human Kupffer cells
Prior to use, PHKCs were characterized based on their response to LPS when co‐cultured with PHHs in the liver MPS (Figures S8, S9). Following LPS exposure for 24 h, PHHs in co‐cultures with PHKCs in the MPS exhibited reduced albumin production but without increasing LDH release (Figure S8a,b). Activation of PHKCs by LPS, which lead to impaired hepatic function without cell death, was confirmed by measuring the production of interleukin 6, which increased above 8‐fold of baseline (Figure S8c). The batch of PHKCs used in site B also showed impaired hepatic function once activated by exhibiting reduced CYP3A4 activity (Figure S9) without a major change in LDH production (Figure 1c). These results qualified the batches of PHKCs to be quiescent in the MPS at baseline, unless activated with LPS.
Levofloxacin had no effect on LDH production or CYP3A4 activity (Figure 1). A twofold increase in LDH induced by 25 µM trovafloxacin co‐dosed with LPS was detected in site B (Figure 1b), whereas such an effect was not detected in the same PHKCs in site A. However, similar effects of 25 µM trovafloxacin with LPS on CYP3A4 activity were observed in both sites (Figure 1d,e). In summary, experiments performed in both sites and with different batches of PHKCs yielded similar results.
PHHs cultured in the liver MPS exhibit different sensitivity to hepatotoxicants
To determine whether PHHs maintained in a liver MPS exhibited altered sensitivity to hepatotoxicants relative to cells in spheroids or sandwich cultures, all three platforms were exposed to varying concentrations of troglitazone, tamoxifen, and digoxin (Figure S2). Concentration‐dependent effects of these hepatotoxicants on LDH production, CYP3A4 activity, and albumin production are shown for each platform in Figure 2 (half‐maximal effective concentration [EC50] values shown in Table 1). Overall, results showed that the sensitivity of PHHs to these hepatotoxicants differed depending on the cell culture platform used. Cells cultured using liver MPS exhibited higher resistance to troglitazone (MPS, EC50 = 210.9 ± 107.8 μM; spheroid, EC50 = 67.1 ± 13.6 μM; sandwich culture, EC50 = 35.1 ± 22.2 μM) and tamoxifen (MPS, EC50 = 36.7 ± 3.0 μM; spheroid EC50 = 13.7 ± 2.4 μM; and sandwich culture, EC50 = 23.8 ± 14.8 μM) relative to other platforms tested. Divergently, cells maintained in liver MPS exhibited a level of resistance to digoxin that was similar to those in sandwich culture (MPS, EC50 = 84.4 ± 30.8 μM; sandwich culture, EC50 = 122.3 ± 17.3 μM), whereas cells cultured in spheroids were more sensitive to digoxin (spheroid, EC50 = 6.4 ± 0.9 μM).
Figure 2.
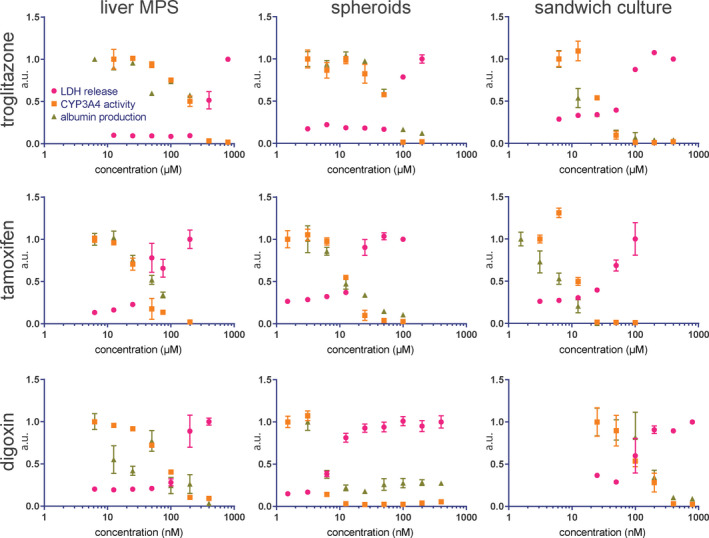
LDH release (pink circles), CYP34A activity (orange squares), and albumin production (green triangles) responses to 48 h of exposure to a range of concentrations of troglitazone, tamoxifen, and digoxin, in all 3 platforms: liver MPS, spheroid, and sandwich culture. All concentration ranges cover approximately three orders of magnitude, and upper and lower concentration limits were selected to encompass the half‐maximal inhibitory concentration for all three responses in each platform‐drug condition. Responses were normalized to the recorded value for the lowest drug concentration (highest, for LDH) and presented as a decimal value in arbitrary units. Each data point represents the average of three biological replicates, and error bars indicate the SEM, not presented if shorter than the size of the symbol. Results of three technical replicates from one MPS well are presented for 800 µM troglitazone. Results of three technical replicates of albumin measurements from one MPS well are presented for 6.25 nM digoxin. LDH, lactate dehydrogenase; MPS, microphysiological system
Table 1.
Estimated concentration values that yield EC50 for variations in LDH production, CYP3A4 activity, and albumin secretion upon exposure of hepatocytes cultured in different platforms (liver MPS, spheroids, and sandwich cultures) to varied concentrations of troglitazone, tamoxifen, and digoxin
| μM | Liver MPS | Spheroids | Sandwich cultures | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Method | LDH | CYP3A4 | Albumin | LDH | CYP3A4 | Albumin | LDH | CYP3A4 | Albumin |
| Troglitazone | |||||||||
| EC50 | 406.6 | 191.6 | 34.6 | 94.2 | 55.1 | 51.9 | 77.7 | 25.0 | 2.6 |
| Mean | 210.9 | 67.1 | 35.1 | ||||||
| SEM | 107.8 | 13.6 | 22.2 | ||||||
| R | – | – | – | 0.20 | 0.80 | 0.71 | 0.09 | 0.77 | 0.83 |
| Tamoxifen | |||||||||
| EC50 | 41.3 | 31.1 | 37.7 | 18.3 | 12.7 | 10.1 | 53.2 | 12.3 | 5.9 |
| Mean | 36.7 | 13.7 | 23.8 | ||||||
| SEM | 3.0 | 2.4 | 14.8 | ||||||
| R | – | – | – | 1.0 | 1.0 | 0.80 | 1.0 | 1.0 | 0.5 |
| Digoxin | |||||||||
| EC50 | 146.5 | 77.1 | 41.6* | 8.1 | 5.6 | 5.4 | 102.3 | 108.0 | 156.7 |
| Mean | 88.4 | 6.4 | 122.3 | ||||||
| SEM | 30.8 | 0.9 | 17.3 | ||||||
| R | – | – | – | 0.79 | −0.04 | −0.04 | 0.80 | 1.0 | 0.80 |
SEM of EC50 values were calculated from the variations of each functional endpoint to varying concentrations of hepatotoxicants. *‐ maximum set to 1 to calculate this value. The presented correlation parameter r was calculated for each type of response (LDH, CYP3A4, and albumin) from nonparametric Spearman correlation test relative to the respective type of response in the liver MPS.
Abbreviations: EC50, half‐maximal effective concentration; LDH, lactate dehydrogenase; MPS, microphysiological system.
In liver MPS and spheroid cultures, reductions in CYP3A4 activity and albumin production occurred at lower hepatotoxicant concentrations than those at which LDH production was increased (Table 1). These results suggest that loss of hepatic function can precede cell death. The dose‐toxicity curves obtained for LDH, CYP3A4, and albumin end points generally correlated well between liver MPS and the other two platforms. Of all toxicants tested, the effects of troglitazone varied most widely between platforms, as well as between end points within the same platform. These results suggested that microenvironments unique to each platform may differentially affect hepatic function and toxicity.
PHH cell function is more stable in liver MPS cultures
To determine whether altered sensitivity of liver MPS PHHs to hepatotoxicants could be due to a change in their functional stability over time, CYP3A4 activity and albumin production were continuously monitored in the absence of hepatotoxicants for each culture platform (Figure S3). As expected, CYP3A4 activity (Figure 3a,b) decreased over time following initial measurements for all platforms tested. However, the fold change of variation was different between platforms and CYP3A4 activity in spheroids diverged in relation to other platforms when normalized to protein content quantified from the cellular lysate (Figure 3a) or to the total number of seeded cells (Figure 3b). CYP3A4 activity in spheroids was higher in relation to sandwich cultures and within the range of MPS activity when normalized to number of seeded cells (Figure 3b), instead of amount of lysed protein (Figure 3a). Despite divergencies between normalization strategies in relative spheroid activity, CYP3A4 activity in the liver MPS decreased at a lower rate compared with other platforms. CYP3A4 activity decreased most rapidly in sandwich cultures. The activity observed in sandwich cultures on day 3 was followed by a drop to ~ 50% of that value on day 5, and eventual loss of detectable CYP3A4 activity by day 12. By contrast, sustained CYP3A4 activity was observed through day 29 in the liver MPS, where it remained above 20% of day 3 activity, in addition to being greater than that observed in sandwich cultures. In spheroids, CYP3A4 activity was sustained for longer than in sandwich cultures. On day 10, CYP3A4 activity in spheroids was ~ 50% of day 3 activity, followed by a decrease below 20% of day 3 activity after day 20. Overall and independently of normalization strategies, CYP3A4 activity in PHHs was more stable in liver MPS cultures when compared with that observed in spheroids or sandwich cultures.
Figure 3.
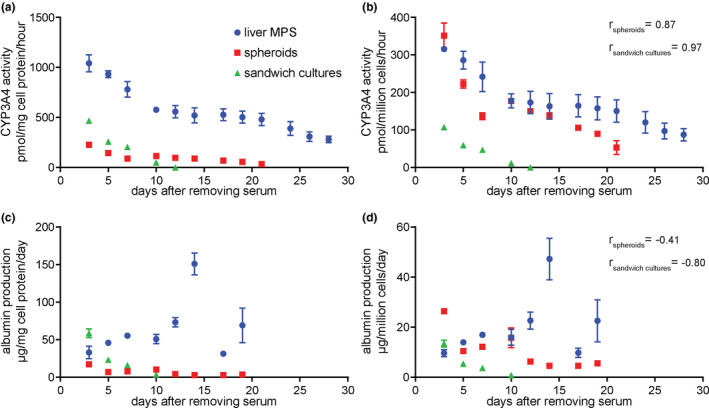
Hepatic function lasts longer and is more stable when cells are cultured in the liver MPS than when cultured as spheroids or sandwich cultures. PHHs were cultured in the liver MPS, within spheroids and as sandwich cultures and the function of these platforms was analyzed. (a, b) Average CYP3A4 activity was measured for each culture platform at discrete times and (a) normalized to the total protein of cellular material or (b) normalized to the number of cells used for each platform. (c, d) Albumin production at different times and (c) divided by the total cellular protein lysed from platforms and the number of days between media changes or (d) divided by the used number of cells and the number of days between media changes. Three wells of the liver MPS, three spheroids, and six wells with sandwich cultures were analyzed. Error bars correspond to SEM and are not presented if shorter than the size of the symbol. One‐way ANOVA was performed to evaluate the variance between means up to 10 days of culture. For CYP3A4 activity (b), ANOVA pvalue was 0.022 for liver MPS, < 0.0001 for spheroids and < 0.0001 for sandwich cultures. For albumin production (d), p value was 0.1 for the liver MPS, 0.002 for spheroids, and < 0.0001 for sandwich cultures. Correlation parameter r calculated from nonparametric Spearman correlation test relative to functional variations in the liver MPS is presented in (b) and (d) for functional variations of spheroids (r spheroids) and sandwich cultures (r sandwich cultures). ANOVA, analysis of variance; MPS, microphysiological system; PHHs, primary human hepatocytes
To further assess the functional stability of PHH cultures, albumin production was measured from each type of cell culture platform until day 19 of culture and normalized to the amount of lysed cellular protein after experiments (Figure 3c) or number of seeded cells (Figure 3d). In albumin production, spheroids and sandwich cultures presented higher values at day 3 relatively to the MPS when normalized to the number of seeded cells. Independently of normalization strategy, albumin values of spheroids and sandwich cultures decreased over time in culture, while increasing in the MPS. Albumin in sandwich cultures decreased to a minimum level within 10 days and to below 50% of day 3 level within 12 days in spheroids. In contrast, albumin production proceeded to increase above 100% over time in the MPS after day 3, reaching a peak value around day 15.
In summary, the stability of PHH function, defined by CYP3A4 activity and albumin production, was greater in the MPS, as these signals persisted for more than 2 weeks of maintenance. In comparison, PHHs in spheroid and sandwich cultures exhibited earlier deterioration of hepatic function over time.
Quantification of phase II drug metabolites and drug intracellular accumulation
Characterizing human‐specific drug metabolism and intracellular accumulation are among the most relevant applications of liver MPSs in pharmacology. 1 Given that troglitazone can generate phase II metabolites, we used this compound to explore the utility of the liver MPS in detecting and quantifying phase II metabolites, 27 in addition to phase I metabolism (Figure S4). Following application to the liver MPS, troglitazone was almost completely metabolized after 48 h of incubation for both concentrations tested (Figure 4a). As expected for modeling physiologically relevant metabolism, phase II metabolites of troglitazone (glucuronidation and sulfation) were quantified in the cell culture medium, with higher amounts detected in 50 µM than in 100 µM troglitazone, possibly due to toxic effects on metabolic pathways when dosed at the highest concentration. Next, the metabolism of diclofenac was quantified by analyzing culture medium samples from the liver MPS, and its concentration was observed to decrease over time (Figure S11a), showing that it was metabolized. However, glucuronidated diclofenac was shown to become unstable in solution at 37°C within 2 h of incubation (Figure S11b) and therefore was not detected in the MPS medium after 48 h, as detected with troglitazone. Consequently, phase II metabolites produced by PHHs in the liver MPS could be detected if stable in cell culture medium.
Figure 4.
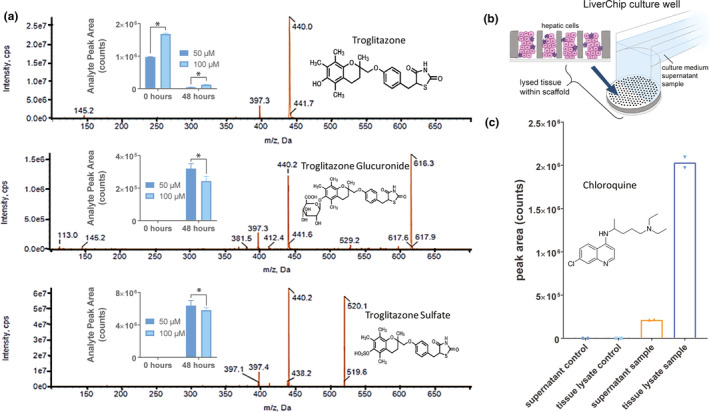
Application of liver MPS to drug metabolism and pharmacokinetics studies. (a) Detection of phase II metabolites of troglitazone: troglitazone glucuronide and troglitazone sulfate. Culture medium containing 50 or 100 µM of troglitazone was added to 3 wells per concentration of the liver MPS and maintained for 2 days to be metabolized. Troglitazone and metabolites in the medium were detected with liquid chromatography–mass spectrometry before and after being added to the liver MPS. *p < 0.03 by the unpaired Mann‐Whitney nonparametric test. Error bars correspond to SD. (b) In addition to analyzing the supernatant culture medium in the liver MPS, cells within scaffolds were lysed to evaluate intra‐tissue accumulation of drugs. (c) Culture medium containing 31.5 µM of chloroquine was added to 2 wells of a liver MPS and incubated for 2 days to measure its intra‐tissue accumulation with liquid chromatography–mass spectrometry after digesting the cell‐containing scaffolds. For both samples, a 10‐fold higher concentration of chloroquine was observed in the tissue lysate relative to the supernatant sample. MPS, microphysiological system
To assess the potential of the liver MPS in quantifying the intracellular accumulation of compounds relative to the extracellular milieu, we exposed PHHs cultured in the MPS to chloroquine (Figure 4b,c), as it is known to accumulate in the liver in vivo. 28 , 29 Following treatment, chloroquine was detected in cellular lysates in amounts fivefold higher than those of the extracellular medium. To ensure that these results were not impacted by compound adsorption to the MPS materials, concentrations for lidocaine, phenacetin, propranolol, prednisolone, diclofenac, and ibuprofen were measured in the culture medium before and after 48‐h circulation in the liver MPS. As shown in Figure S12, no significant change in concentration was observed for any of these compounds when measuring before and after circulation in the liver MPS. These data suggest minimal involvement of nonspecific interactions between compounds and microfluidic materials when modeling intracellular drug accumulation. However, other compounds with differing physical‐chemical properties may bind to the device and may therefore require a similar evaluation prior to testing.
Quality control for individual MPS wells by measuring CYP3A4 activity and LDH production
To validate the use of nondestructive and minimally invasive assays as reliable quality control metrics of MPS replicates, CYP3A4 activity and LDH production were measured from perfusate material (Figure 5a,b) in conjunction with scaffold imaging (after experiments) and gene expression analysis (after imaging: Figure 5c). These parameters were studied for individual wells within a single liver MPS plate (Figure 5), where imaging and gene expression analysis were used as confirmatory measurements for quality control parameters. Analysis of one liver MPS plate (12 wells) showed that LDH production in a particular well (B6) was statistically greater than the amount produced in other wells (Figure 5a). In accordance with this result, images of the scaffold in well B6 revealed a lower cell count compared with the other wells (Figure 5c), thus confirming its unsuitability for use based on this criterion. Although a higher degree of inter‐well variability was observed when assaying CYP3A4 activity (Figure 5b), this measurement appeared to coincide with abnormal cellular density, as demonstrated with scaffold A3, which exhibited low cell density as well as nonuniform cell distribution. However, a relationship between CYP3A4 activity and visual features of microtissues was not evident in some cases, as exemplified by scaffolds A6 and B5, which visually resembled scaffold B4 (Figure 5c). For each plate used in experiments, we disqualified data collected from wells associated with high LDH production values that were statistical outliers or CYP3A4 activity below 50% of the maximum measured value. Wells A3 and B6 would clearly match these exclusion criteria, and wells A6 and B5 would be flagged for later consideration if identified as statistical outliers.
Figure 5.
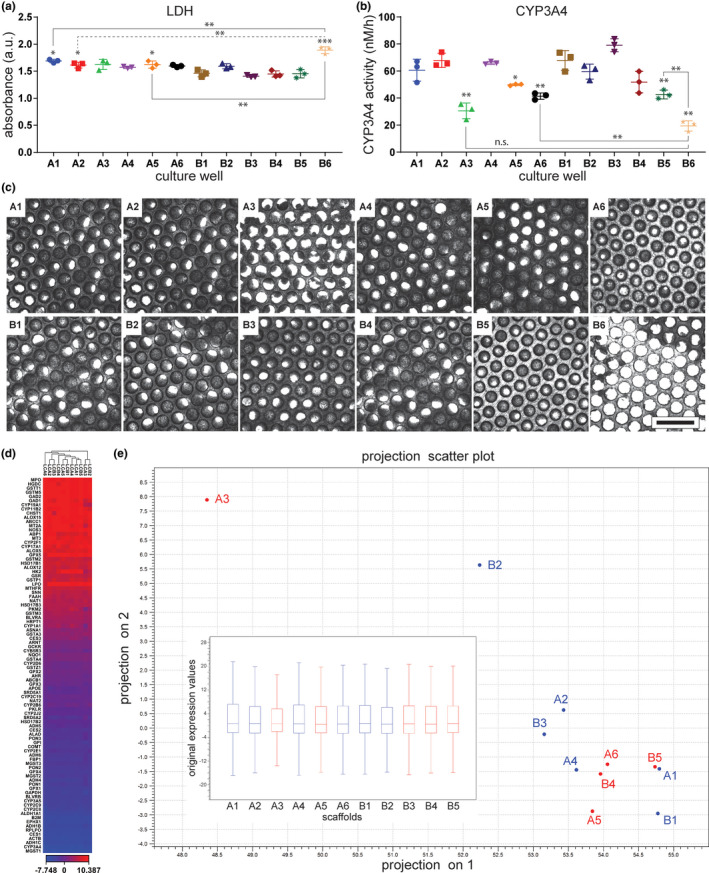
Assessing LDH production and CYP3A4 activity as quality control measures for individual wells. Before changing the cell culture medium on day 4 of operation after seeding cells, LDH was assayed in medium collected from each well (a) and CYP3A4 activity (b) was measured to evaluate the quality of cellular function in the wells of a liver MPS plate. Error bars represent the SD of three measured technical replicates. *0.05 < p < 0.02 and **0.02 < p < 0.002 and ***p < 0.002 by unpaired t‐test with Welch’s correction. *, **, and *** above the presented data sets indicate statistical significance of difference in LDH or CYP3A4 production relative to the mean of values from well B1. Unless indicated otherwise, presented mean values were not statistically different from the mean values obtained from well B1. (c) Each scaffold corresponding to a liver MPS well was imaged with brightfield microscopy after being removed from the plate. Scale bar represents a length of 1 mm. For gene expression analysis (d, e), Qiagen’s RT2 Profiler PCR Array for Human Drug Metabolism was used to assess a panel of 84 genes involved in drug metabolism. Sample B6 was excluded from the gene expression analysis for having a lower yield than required by the assay. The mean of the five least variant targets on the panel array was used as the endogenous control value for the denominator of the normalized Ct values (ΔCt) calculation. (d) Hierarchical clustering plot of gene expression. Heat map represents ΔCt of expression of a panel of genes (rows) for each isolated scaffold (columns). Euclidean distance and average linkage were used as parameters for the clustering. (e) Principal component plot from software CLC Genomics Workbench (Qiagen) depicting the projection of the samples onto a two‐dimensional space spanned by the first and second principal component of the covariance matrix. Box plot inside of main plot presents the distribution of ΔCt values. LDH, lactate dehydrogenase; MPS, microphysiological system; n.s. indicates not significant
To further test whether these criteria are effective in distinguishing functional wells from defective wells, we analyzed inter‐well variability in the expression of 84 genes screened using a pathway‐specific polymerase chain reaction array used to detect human drug metabolism. The total RNA yield from well B6 was lower than the suggested 400 ng input level for the assay and was therefore excluded from analysis. The tested samples were hierarchically clustered based on the expression of the 84 genes in the drug metabolism panel (Figure 5d). A principal component analysis of gene expression profiling data was performed to better evaluate well clustering (Figure 5e). Overall, no major differences in gene expression were observed between wells, however, wells A3 and B2 clustered separately from the other wells. Well B2 clustered more closely with the other wells than A3. Based on unbiased cluster analysis of samples (Figure 5d, Figure S13), wells A3, A6, and B2 were classified as outliers within a control group consisting of wells A1, A2, A4, A5, B1, B3, B4, and B5. Fold change in gene expression of the outlier group relative to the control group are presented in Figure S14, and genes that varied significantly between groups are listed in Table S2. Of the 84 genes tested, 8 were upregulated and 11 were downregulated in the outlier group relative to control. Altogether, these results supported the use of LDH production and CYP3A4 activity as reliable, noninvasive, quality control indicators for experiments conducted with the liver MPS. As these, other hepatic biomarkers or metabolism assays quantified from the perfusate could also be equally useful quality control metrics, which may depend on context of use.
DISCUSSION
Reproducibility of the liver MPS was characterized regarding its potential to predict hepatotoxicity, estimate drug metabolism and accumulation, and study drug effects that require prolonged functionality of cells. Cell‐based methods for drug development must be robust, yield reproducible results, have well‐defined quality control criteria, and rely on materials that can be easily accessed or manufactured. 1 , 30 , 31 , 32 In the current study, we followed this guideline by using commercially available cells, devices, instrumentation, and supplies. One challenge facing widespread MPS implementation is the difficulty in transferring MPSs to drug development laboratories from academic groups, which often have the advantage of unique expertise in microfabrication and instrumentation methods, or semi‐exclusive access to cell types. In addition to efforts from industry, 2 , 3 testing centers have been created to address such translational hurdles, and initial results demonstrated the need of different drug development stakeholders for generating key characterization data on MPS use. 15 , 33 , 34
In the current study, experiments conducted in PHHs used the same batch of cells to ensure a primary focus on the reliability of MPS operation and to eliminate batch effects. 35 Previous studies using the liver MPS have demonstrated that batch effects can impact performance, and future evaluations should further investigate cell qualification criteria. 8 , 9 In our trovafloxacin experiments, drug responses were compared from two distinct batches of PHKCs, as well as from multiple test sites to confirm the reproducibility of data collected (Figure 1). Based on prior work published using the liver MPS, as well as past performance of other cell culture platforms used to study hepatic function (i.e., spheroids and sandwich cultures), the following MPS performance properties were addressed: (i) reproducibility of results, (ii) functional stability, and (iii) ability to develop future contexts of use in drug evaluation.
Given the complexity of MPS operation, inconsistency between experimental replicates can arise due to several factors, including cells, devices, and handling of instrumentation. Overall, our data allowed us to examine variability between MPS wells (Figure 5), MPS plates, distinct batches of qualified PHKCs, and between test sites (Figure 1). The standard error of the mean in LDH production or CYP3A4 activity obtained with experimental replicates was low enough to allow detection of differences between drug concentration groups, as well as between groups treated or untreated with LPS. Concentration‐dependent effects induced by tamoxifen, digoxin, and troglitazone on CYP3A4 activity, and LDH and albumin production were also clearly detected with qualified replicates from different MPS batches per condition (Figure 2). By performing quality control assays (LDH and CYP3A4) prior to testing, outlier samples were successfully excluded from experiments due to impaired cellular activity. In addition to this procedure, the quality of liver MPS assembly was also assessed. Replicating previously published results further confirmed the possibility to use the liver MPS with reproducibility. The quantified diclofenac clearance (Figure S11a), phase I and phase II metabolism of troglitazone (Figure 4a), the ability to maintain the liver MPS for weeks (Figure 3), and the observed microtissue morphology within scaffolds (Figure S15) were all observed in a manner consistent with that of previous reports. 9 , 17 , 18 , 19 Despite potential differences between sites or cell batches, 100 µM trovafloxacin with LPS induced a significant increase in LDH (Figure 1), and trovafloxacin and levofloxacin have previously been used to test liver systems. 36 However, despite the convergence in results between test conditions, the magnitude of drug effect was variable between experiments. Discrepancies in levels of LDH and CYP3A4 activity (Figure 1) between test sites illustrate the need to standardize operations and procedures concerning MPSs. 37 , 38 In summary, our results indicated that experimentation with the liver MPS is likely to generate reproducible results when supported by quality control metrics based on LDH and CYP3A4 activity.
The functional stability of PHHs was studied by comparing the longevity of signaling responses while maintained in MPSs, spheroids, and sandwich cultures (Figure 3). Overall, different toxicant responses were observed between culture platforms, while using the same batch of PHHs. At later time points after seeding PHHs, baseline CYP3A4 activity and albumin production were also observed to be greater and therefore more prolonged in liver MPS cells. Albumin production above 37 μg/day/million PHHs, as noted by Baudy and colleagues 2 to represent an acceptable liver activity, was observed in the MPS around day 15 after seeding cells (Figure 3d), suggesting this time point to be appropriate for liver safety applications. In addition, a more stable function in the tested MPS and the observed CYP3A4 activity and expression of ADME genes (Figure 5d,e, Figures S13, S14) met additional requirements for liver MPSs. 2 , 3 Use of a media volume of 1.6 ml (Table S3), low nonspecific binding of compounds to the MPS (Figure S12), recirculation of media, and use of sufficient cellular mass were other characteristics that met criteria for ADME applications. 3 The observed disparities in toxicant responses using the same batch of cells (Figure 2, Table 1) may indicate divergent functional activity that is determined by microenvironments particular to each platform (Tables S3, S4) or relate to their unique pharmacokinetics as reflected by differences in the stability of CYP3A4 activity (Figure 3). For developing predictive assays of drug hepatotoxicity, future work should further optimize physiologically relevant dosing, time of exposure, relevant functional end points, measurement schedules, and wider ranges of drugs that represent diverse mechanisms and severity levels of drug‐induced liver injury. 39 Since CYP3A4 activity is affected by a variety of cellular properties, it was difficult to identify differences between platforms that may relate to specific drug toxicity mechanisms. Future work should investigate direct targets of tested drugs to better understand differences between platforms. The function of PHHs in sandwich cultures is known to significantly decline within a few days after plating, 40 and their function is prolonged when cultured as spheroids. 41 Hepatic function is also partially determined by cell polarity in PHHs, 42 and future work should elucidate the MPS‐specific factors that might regulate polarity.
For each platform, the functional end points presented in Figure 3 were normalized to media volume and quantified cellular protein content (Figure 3a,c) or number of seeded cells (Figure 3b,d), and all experiments were conducted in the same medium. However, the protein quantification method (Supplementary Materials) did not seem to be sensitive enough to determine the total protein in spheroids (Figure 3). Future work should investigate how the protein quantification method can fail to estimate the amount of cellular material. Differences in cell to volume ratio between platforms (Table S3), and in cell density could have contributed to inter‐platform variation. Table S4 details differences in operational and practical features between culture platforms. The disparity in EC50 values between the liver MPS and other platforms was largest for troglitazone (Figure 2). The response of hepatocytes to this compound can depend on culture format (2D vs. 3D). 43 The mechanisms driving troglitazone toxicity remain unclear, but more stable metabolic activity and function of MPS cultures may increase resistance to toxicity. These results support the importance of understanding the relationships between drug exposure/pharmacokinetics and efficacy/toxicity when comparing between culture platforms or when trying to perform in vitro to in vivo translation. 12 , 39 With trovafloxacin, we showed how similar results could be obtained with the MPS in multiple sites, whereas the experiments with troglitazone, tamoxifen, or digoxin demonstrated how the same cells could yield divergent sensitivities to toxicants if cultured in the MPS. Future studies are also required to investigate the translation of MPS data to in vivo and demonstrate the benefits of using MPSs for specific contexts in relation to simpler alternatives. Our results suggest that the used MPS could benefit applications where co‐culturing of PHHs with PHKCs is relevant, or to evaluate chronic drug effects or multiple exposures due to improved functional stability.
The potential use of the liver MPS for evaluating drug metabolism and intracellular accumulation, which can support drug pharmacokinetics studies, 44 , 45 was also investigated. The formation of phase II metabolites can often differ between animals and humans. 46 Given that MPSs may confer stable cellular function and heightened resilience to toxicity, they could be used to refine predictions of drug ADME in humans. Troglitazone phase II metabolites were detected after 2 days of incubation in the liver MPS (Figure 4a). These metabolites were detected in higher amounts with exposure to 50 µM troglitazone when compared with 100 µM, which was closer to the range that induced toxicity (Figure 2) and may have impaired metabolism. The activity of enzymes involved in liver metabolism has been previously identified in the liver MPS, 7 , 8 , 17 , 19 , 47 and these studies extensively quantified the activity of CYP3A4 (Figures 1, 2, 3, 5), which is the CYP isoform most commonly expressed in human hepatocytes 48 and metabolizes up to 50% of drugs. 49 In the current study, the metabolism of diclofenac was quantified (Figure S11), also confirming previous results. 18 However, we found that diclofenac phase II metabolites were unstable in culture (Figure S11b), and accurate quantification may require collecting the perfusate within the first hours of incubation. Our data demonstrated that whereas the liver MPS enables analysis of drug metabolism, careful attention should be given to metabolite stability and timing for sample collection. Our detection of chloroquine within PHHs cultured in the MPS (Figure 4c) was consistent with in vivo results 28 , 29 and demonstrated the MPS potential for drug distribution studies. 44 Cellular transport is central in regulating accumulation, and MPS studies have reported robust transport activity through drug clearance, gene expression, and imaging assays. 7 , 8 , 17 , 18 In line with published results, our data confirmed the potential for MPS to predict drug metabolism and pharmacokinetics.
Additional applications for drug development should be explored with the liver MPS, 12 , 50 as multiple types of liver systems have been developed, and future work should compare them to assess strengths and weaknesses of each model. Collectively, the results from this study, in agreement with published data, show that the liver MPS can reproduce results. The use of quality control criteria to yield functional microtissues was key in enabling reproducibility. Moreover, the liver MPS stabilized hepatic function relative to other PHH culture platforms. In addition to this study, future work should evaluate liver MPSs as drug development tools for specific contexts of use.
DISCLAIMER
The opinions expressed in this manuscript are those of the authors and should not be interpreted as the position of the US Food and Drug Administration.
CONFLICT OF INTEREST
Dr. Hughes owns stock in and is employed by CN Bio Innovations. Dr. Kostrzewski is employed by CN Bio Innovations. Ms. Miedzik was employed by CN Bio Innovations. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
A.R., B.R., A.A., K.D., D.G.S., D.J.H., T.K., and A.J.S.R. wrote the manuscript. B.R., N.H., D.A.V., M.K.M., D.J.H., D.G.S., T.K., and A.J.S.R. designed the research. A.R., A.I., R.Y., A.M., B.R., A.A., C.M.M., K.D., T.K., and A.J.S.R. performed the research. A.R., A.I., R.Y., B.R., M.K.M., and A.J.S.R. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Jessica A. Bonzo from the US Food and Drug Administration (FDA) for helpful discussions on the quality of hepatocytes.
Funding information
The authors acknowledge support from the Defense Advanced Research Projects Agency via an Interagency Agreement with the FDA.
REFERENCES
- 1. Ribeiro AJS, Yang X, Patel V, Madabushi R, Strauss DG. Liver microphysiological systems for predicting and evaluating drug effects. Clin Pharmacol Ther. 2019;106:139‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baudy AR, Otieno MA, Hewitt P, et al. Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip. 2020;20:215‐225. [DOI] [PubMed] [Google Scholar]
- 3. Fowler S, Chen WLK, Duignan DB, et al. Microphysiological systems for ADME‐related applications: current status and recommendations for system development and characterization. Lab Chip. 2020;20:446‐467. [DOI] [PubMed] [Google Scholar]
- 4. Beckwitt CH, Clark AM, Wheeler S, et al. Liver ‘organ on a chip'. Exp Cell Res. 2018;363:15‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Isoherranen N, Madabushi R, Huang SM. Emerging role of organ‐on‐a‐chip technologies in quantitative clinical pharmacology evaluation. Clin Transl Sci. 2019;12:113‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maass C, Stokes CL, Griffith LG, Cirit M. Multi‐functional scaling methodology for translational pharmacokinetic and pharmacodynamic applications using integrated microphysiological systems (MPS). Integr Biol (Camb). 2017;9:290‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsamandouras N, Chen WLK, Edington CD, et al. Integrated gut and liver microphysiological systems for quantitative in vitro pharmacokinetic studies. AAPS J. 2017;19:1499‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsamandouras N, Kostrzewski T, Stokes CL, et al. Quantitative assessment of population variability in hepatic drug metabolism using a perfused three‐dimensional human liver microphysiological system. J Pharmacol Exp Ther. 2017;360:95‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Long TJ, Cosgrove PA, Dunn RT, et al. Modeling therapeutic antibody‐small molecule drug‐drug interactions using a three‐dimensional perfusable human liver coculture platform. Drug Metab Dispos. 2016;44:1940‐1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jang KJ, Otieno MA, Ronxhi J, et al. Reproducing human and cross‐species drug toxicities using a Liver‐Chip. Sci Transl Med. 2019;11(517):eax5516. [DOI] [PubMed] [Google Scholar]
- 11. Rowe C, Shaeri M, Large E, et al. Perfused human hepatocyte microtissues identify reactive metabolite‐forming and mitochondria‐perturbing hepatotoxins. Toxicol In Vitro. 2018;46:29‐38. [DOI] [PubMed] [Google Scholar]
- 12. Weaver RJ, Blomme EA, Chadwick AE, et al. Managing the challenge of drug‐induced liver injury: a roadmap for the development and deployment of preclinical predictive models. Nat Rev Drug Discov. 2020;19:131‐148. [DOI] [PubMed] [Google Scholar]
- 13. Cirit M, Stokes CL. Maximizing the impact of microphysiological systems with in vitro‐in vivo translation. Lab Chip. 2018;18:1831‐1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dehne EM, Hasenberg T, Marx U. The ascendance of microphysiological systems to solve the drug testing dilemma. Future Sci OA. 2017;3:Fso185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Low LA, Tagle DA. Microphysiological systems ("organs‐on‐chips") for drug efficacy and toxicity testing. Clin Transl Sci. 2017;10:237‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clark AM, Wheeler SE, Taylor DP, et al. A microphysiological system model of therapy for liver micrometastases. Exp Biol Med (Maywood). 2014;239:1170‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sarkar U, Rivera‐Burgos D, Large EM, et al. Metabolite profiling and pharmacokinetic evaluation of hydrocortisone in a perfused three‐dimensional human liver bioreactor. Drug Metab Dispos. 2015;43:1091‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sarkar U, Ravindra KC, Large E, et al. Integrated assessment of diclofenac biotransformation, pharmacokinetics, and omics‐based toxicity in a three‐dimensional human liver‐immunocompetent coculture system. Drug Metab Dispos. 2017;45:855‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ortega‐Prieto AM, Skelton JK, Wai SN, et al. 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat Commun. 2018;9:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Avila AM, Bebenek I, Bonzo JA, et al. An FDA/CDER perspective on nonclinical testing strategies: classical toxicology approaches and new approach methodologies (NAMs). Regul Toxicol Pharmacol. 2020;114:104662. [DOI] [PubMed] [Google Scholar]
- 21. Walker PA, Ryder S, Lavado A, Dilworth C, Riley RJ. The evolution of strategies to minimise the risk of human drug‐induced liver injury (DILI) in drug discovery and development. Arch Toxicol. 2020;94:2559‐2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Domansky K, Inman W, Serdy J, et al. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip. 2010;10:51‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inman W, Domansky K, Serdy J, et al. Design, modeling and fabrication of a constant flow pneumatic micropump. J Micromech Microeng. 2007;17:891‐899. [Google Scholar]
- 24. Roth RA, Ganey PE. Intrinsic versus idiosyncratic drug‐induced hepatotoxicity–two villains or one? J Pharmacol Exp Ther. 2010;332:692‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ernst ME, Ernst EJ, Klepser ME. Levofloxacin and trovafloxacin: the next generation of fluoroquinolones? Am J Health Syst Pharm. 1997;54:2569‐2584. [DOI] [PubMed] [Google Scholar]
- 26. De Sarro A, De Sarro G. Adverse reactions to fluoroquinolones. An overview on mechanistic aspects. Curr Med Chem. 2001;8:371‐384. [DOI] [PubMed] [Google Scholar]
- 27. Smith MT. Mechanisms of troglitazone hepatotoxicity. Chem Res Toxicol. 2003;16:679‐687. [DOI] [PubMed] [Google Scholar]
- 28. Adelusi SA, Salako LA. Tissue and blood concentrations of chloroquine following chronic administration in the rat. J Pharm Pharmacol. 1982;34:733‐735. [DOI] [PubMed] [Google Scholar]
- 29. MacIntyre AC, Cutler DJ. Role of lysosomes in hepatic accumulation of chloroquine. J Pharm Sci. 1988;77:196‐199. [DOI] [PubMed] [Google Scholar]
- 30. Hillier C, Bunton D. Functional human tissue assays. Drug Discov Today. 2007;12:382‐388. [DOI] [PubMed] [Google Scholar]
- 31. Perego P, Hempel G, Linder S, et al. Cellular pharmacology studies of anticancer agents: recommendations from the EORTC‐PAMM group. Cancer Chemother Pharmacol. 2018;81:427‐441. [DOI] [PubMed] [Google Scholar]
- 32. Capula M, Corno C, El Hassouni B, Li Petri G, Arandelovic S. A Brief guide to performing pharmacological studies in vitro: reflections from the EORTC‐PAMM course "preclinical and early‐phase clinical pharmacology". Anticancer Res. 2019;39:3413‐3418. [DOI] [PubMed] [Google Scholar]
- 33. Sakolish C, Weber EJ, Kelly EJ, et al. Technology transfer of the microphysiological systems: a case study of the human proximal tubule tissue chip. Sci Rep. 2018;8:14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maass C, Sorensen NB, Himmelfarb J, et al. Translational assessment of drug‐induced proximal tubule injury using a kidney microphysiological system. CPT Pharmacometrics Syst Pharmacol. 2019;8:316‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alexandre E, Baze A, Parmentier C, et al. Plateable cryopreserved human hepatocytes for the assessment of cytochrome P450 inducibility: experimental condition‐related variables affecting their response to inducers. Xenobiotica. 2012;42:968‐979. [DOI] [PubMed] [Google Scholar]
- 36. Nguyen DG, Funk J, Robbins JB, et al. Bioprinted 3D primary liver tissues allow assessment of organ‐level response to clinical drug induced toxicity in vitro. PLoS One. 2016;11:e0158674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Young AT, Rivera KR, Erb PD, Daniele MA. Monitoring of microphysiological systems: integrating sensors and real‐time data analysis toward autonomous decision‐making. ACS Sens. 2019;4:1454‐1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dehne E‐M, Erfurth H, Muhsmann A‐K, Marx U. Chapter 14 – Automation and opportunities for industry scale‐up of microphysiological systems. In Hoeng J, Bovard D, Peitsch MC, eds. Organ‐on‐a‐chip. Salt Lake City, UT: Academic Press; 2020:441‐462. [Google Scholar]
- 39. Schadt S, Simon S, Kustermann S, et al. Minimizing DILI risk in drug discovery – a screening tool for drug candidates. Toxicol In Vitro. 2015;30:429‐437. [DOI] [PubMed] [Google Scholar]
- 40. Swift B, Pfeifer ND, Brouwer KL. Sandwich‐cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter‐based drug interactions and hepatotoxicity. Drug Metab Rev. 2010;42:446‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bell CC, Hendriks DFG, Moro SML, et al. Characterization of primary human hepatocyte spheroids as a model system for drug‐induced liver injury, liver function and disease. Sci Rep. 2016;6:25187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Treyer A, Müsch A. Hepatocyte polarity. Compr Physiol. 2013;3:243‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kostadinova R, Boess F, Applegate D, et al. A long‐term three dimensional liver co‐culture system for improved prediction of clinically relevant drug‐induced hepatotoxicity. Toxicol Appl Pharmacol. 2013;268:1‐16. [DOI] [PubMed] [Google Scholar]
- 44. Li L, Li X, Xu L, et al. Systematic evaluation of dose accumulation studies in clinical pharmacokinetics. Curr Drug Metab. 2013;14:605‐615. [DOI] [PubMed] [Google Scholar]
- 45. Nishidate M, Hayashi M, Aikawa H, et al. Applications of MALDI mass spectrometry imaging for pharmacokinetic studies during drug development. Drug Metab Pharmacokinet. 2019;34:209‐216. [DOI] [PubMed] [Google Scholar]
- 46. Sharer JE, Shipley LA, Vandenbranden MR, Binkley SN, Wrighton SA. Comparisons of phase I and phase II in vitro hepatic enzyme activities of human, dog, rhesus monkey, and cynomolgus monkey. Drug Metab Dispos. 1995;23:1231‐1241. [PubMed] [Google Scholar]
- 47. Vivares A, Salle‐Lefort S, Arabeyre‐Fabre C, et al. Morphological behaviour and metabolic capacity of cryopreserved human primary hepatocytes cultivated in a perfused multiwell device. Xenobiotica. 2015;45:29‐44. [DOI] [PubMed] [Google Scholar]
- 48. Zhou Y, Ingelman‐Sundberg M, Lauschke VM. Worldwide distribution of cytochrome P450 alleles: a meta‐analysis of population‐scale sequencing projects. Clin Pharmacol Ther. 2017;102:688‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9:310‐322. [DOI] [PubMed] [Google Scholar]
- 50. Chen M, Suzuki A, Borlak J, Andrade RJ, Lucena MI. Drug‐induced liver injury: Interactions between drug properties and host factors. J Hepatol. 2015;63:503‐514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material