

Abstract
Abstract
VM202 is a plasmid DNA encoding two isoforms of hepatocyte growth factor (HGF). A previous phase II study in subjects with painful diabetic peripheral neuropathy (DPN) showed significant reductions in pain. A phase III study was conducted to evaluate the safety and efficacy of VM202 in DPN. The trial was conducted in two parts, one for 9 months (DPN 3‐1) with 500 subjects (VM202: 336 subjects; and placebo: 164) and a preplanned subset of 101 subjects (VM202: 65 subjects; and placebo: 36) with a noninterventional extension to 12 months (DPN 3‐1b). VM202 or placebo was administered to calf muscles on days 0 and 14, and on days 90 and 104. The primary end point in DPN 3‐1 was change from baseline in the mean 24‐h Numerical Rating Scale (NRS) pain score. In DPN 3‐1b, the primary end point was safety, whereas the secondary efficacy end point was change in the mean pain score. VM202 was well‐tolerated in both studies without significant adverse events. VM202 failed to meet its efficacy end points in DPN 3‐1. In DPN 3‐1b, however, VM202 showed significant and clinically meaningful pain reduction versus placebo. Pain reduction in DPN 3‐1b was even greater in subjects not receiving gabapentin or pregabalin, confirming an observation noted in the phase II study. In DPN 3‐1b, symptomatic relief was maintained for 8 months after the last injection suggesting that VM202 treatment might change disease progression. Despite the perplexing discrepancy between the two studies, the safety and long‐lasting pain‐relieving effects of VM202 observed in DPN 3‐1b warrant another rigorous phase III study.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Current therapies for painful diabetic peripheral neuropathy (DPN) are palliative and do not target the underlying mechanisms. Moreover, symptomatic relief is often limited with existing neuropathic pain drugs. Thus, there is a great medical need for safer and effective treatments for DPN.
WHAT QUESTION DID THIS STUDY ADDRESS?
Can nonviral gene delivery of hepatocyte growth factor reduce pain in patients with DPN and potentially modify progression of the disorder?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Nonviral gene therapy can be used safely and practically to treat DPN.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
As the first gene medicine to enter advanced clinical trials for the treatment of DPN, this study provides the proof of concept of an entirely new potential approach to the disorder.
INTRODUCTION
Painful diabetic peripheral neuropathy (painful DPN) is a common and debilitating complication of diabetes mellitus that has a profound negative impact on quality of life, sleep, and mood. 1 , 2 , 3 Current therapies are palliative and do not target the mechanisms underlying painful DPN. Moreover, symptomatic relief is often limited with existing neuropathic pain drugs, and many patients still use opioids. 4 , 5 , 6 Thus, there is a great medical need for safer and effective treatments for painful DPN.
To overcome the limitations of currently used medicines, all based on small molecules, gene therapy technology has been explored with an aim to effectively manage pain. 7 , 8 Almost all studies used viral vectors, although a few nonviral approaches also have been tested. 9 , 10 , 11 Despite intense efforts, however, most investigations have been at the nonclinical level. VM202 (Engensis), a nonviral plasmid DNA product, is the first gene medicine to enter advanced clinical trials for treatment of painful DPN.
VM202 is designed to express two isoforms of HGF, HGF728 (cHGF) and HGF723 (dHGF), at biologically meaningful levels in vivo as well as in vitro. 12 HGF is a multifunctional protein with potent neurotrophic and angiogenic activities. HGF promotes neuronal survival and axonal outgrowth from sensory and motor neurons. 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 In addition, it induces formation of collateral vessels and increases blood flow in animal diabetic models. 24 , 25 , 26 , 27 However, HGF has a short serum half‐life of 5 min or less, 28 , 29 , 30 , 31 so the potential use of injectable recombinant HGF protein is unlikely to be a viable approach toward restoring sensory neurons damaged by progressive DPN. However, intramuscular injection of VM202 to hind limb muscles in rodents drives HGF expression for about 2 weeks with peak protein expression at day 7 and a gradual decrease over the next week. 12 In previous studies, VM202 produced therapeutic benefits in experimental models and clinical trials in a variety of diseases, including critical limb ischemia, myocardial infarction, and amyotrophic lateral sclerosis, as well as painful DPN. 10 , 40
Phase I and phase II clinical studies of VM202 in painful DPN demonstrated both safety and efficacy in reducing pain and improving quality of life. 10 , 32 In the phase II study, 10 administration of VM202 reduced pain for 3 to 9 months after treatment even though plasmid DNAs are rapidly degraded in plasma and intracellular protein expression lasts only for 2 weeks. The persistence of such long‐lasting therapeutic benefits suggested a possible disease‐modifying effect. Of particular interest was the finding in the phase II study that VM202 appeared to work more effectively in subjects not receiving concurrent pregabalin or gabapentin.
Encouraged by the efficacy and safety data from the phase I and II studies for painful DPN, a large scale double‐blind placebo‐controlled phase III study was conducted in two parts: (a) in the main study for 9 months (DPN 3‐1, N = 500 subjects), and (b) in a subset of later‐enrolling subjects from the main study who participated in a noninterventional 3‐month safety and efficacy extension (DPN 3‐1b, N = 101 subjects). The specific objective of the DPN 3‐1 study was to evaluate the safety and efficacy of intramuscular administration of VM202 in subjects with painful DPN in lower extremities. The primary statistical hypothesis was that VM202 administration would reduce average daily DPN scores more that placebo administration at the primary efficacy end point, the mean 24‐h Numerical Rating Scale (NRS) pain score on the daily pain and sleep diary at 3 months.
MATERIALS AND METHODS
DPN 3‐1 (http://www.clinicaltrials.gov registration number: NCT02427464; Phase 3 Gene Therapy for Painful Diabetic Neuropathy) was a phase III, double‐blind, randomized, parallel design, placebo‐controlled multicenter study with subjects recruited at 25 sites in the United States. Subjects were randomized by an unblinded study statistician via the EDC Master Randomization file in blocks of 6 subjects in a 2:1 ratio of VM202 to placebo, and further stratified by concomitant use of gabapentin and/or pregabalin. Two cycles of treatment were administered via intramuscular (i.m.) injections separated by 3 months (Figure 1). In each cycle, VM202 was delivered in equally divided doses administered 2 weeks apart. Subjects received VM202 or placebo by deep i.m. injections in the calf of both legs on days 0 and 14 (first treatment), and on days 90 and 104 (second treatment). All subjects received 16 injections of VM202 or placebo evenly distributed over each calf at each treatment visit. Subjects were followed from the first injection to day 270 to evaluate the efficacy and safety of treatment.
Figure 1.
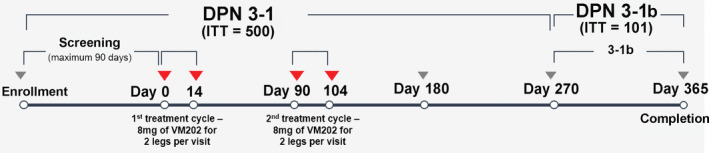
Study design. Consenting subjects who met eligibility criteria (ITT = 500) were treated with injections of VM202 or buffer into each leg on days 10 and 14 and then again on days 90 and 104. The DPN 3‐1 study ended on day 270. At that time, 101 subjects were consented for DPN 3‐1b for follow‐up, including continuation of the daily pain diary but no further treatments until day 365. DPN, diabetic peripheral neuropathy; ITT, intent‐to‐treat
DPN 3‐1b (registration number: NCT04055090; Extension of Phase 3 Gene Therapy for Painful Diabetic Neuropathy) was a noninterventional extension of DPN 3‐1 designed to explore the longer‐term safety profile of VM202 as well as the efficacy and durability of pain relief. One hundred one subjects among those who participated in DPN 3‐1 were followed for 3 additional months to 365 days after the first treatment. No further treatment was given during this period.
Supplementary Methods include details of the approvals, inclusion criteria, exclusion criteria, concurrent medications, description of the study participants, study material and administration, end points and assessments, safety evaluations, and statistical analyses.
RESULTS
Study design and subject disposition
This phase III study, composed of two parts, was conducted from June 2016 to June 2019. The full study participation lasted 9 months for all subjects (DPN 3‐1, N = 500 subjects). A subset of later‐enrolling subjects participated in a noninterventional 3‐month extension for a total duration of 12 months (DPN 3‐1b, N = 101 subjects; Figure 1). There were no selection criteria for this subset of patients, but rather all late enrolling patients were asked for permission to be followed for an additional 3 months.
One thousand one hundred ninety‐one subjects were consented and screened for possible enrollment. Six hundred eighty‐four subjects were screen failures and 507 eligible subjects were randomized into DPN 3‐1 (Figure 2). The intent‐to‐treat (ITT) population included all subjects that received any study drug (DPN 3‐1, N = 500), excluding seven subjects due to errors in randomizing subjects who were screen failures. Four hundred thirty‐three subjects completed the study, and 48 subjects in the VM202 group and 19 in the placebo group were lost to follow‐up or withdrew prior to the 9‐month study visit. DPN 3‐1b began in January 2019 with the first subjects entering an additional 3‐month study. One hundred one subjects consented to a total follow‐up of 12 months (ITT population). All 36 subjects in the placebo group and 63 subjects in the VM202 group completed the study. Two subjects in the VM202 group withdrew prior to study completion.
Figure 2.
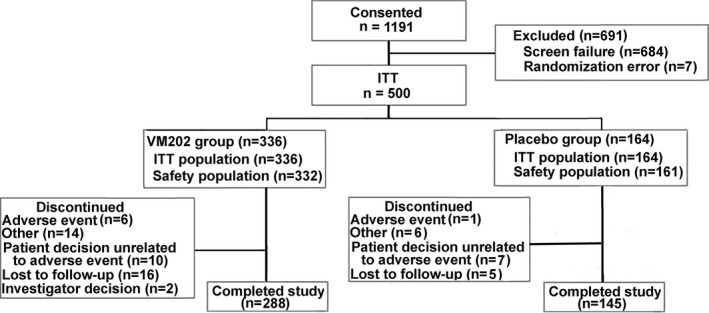
Analysis populations in the study
Baseline demographics and medical history
Baseline demographics and characteristics of subjects in DPN 3‐1 and DPN 3‐1b are summarized in the Table S1. The majority of the subjects in both parts of the study were White (74.4% and 80.2%, respectively) and the overall mean age was 61.5 years. There was little difference in demographics or baseline characteristics between the DPN 3‐1 and DPN 3‐1b studies or between active and placebo groups within each study. Further, there were no discernible differences between the 101 subjects in DPN 3‐1b and the 399 subjects who were not part of DPN 3‐1b other than a 0.5‐point difference in the baseline average daily pain scores between the 2 groups (NRS = 7.1 ± 1.4 for the 399 subjects who did not participate in DPN 3‐1b vs. NRS = 6.6 ± 1.3 for the 101 subjects in DPN 3‐1b; Table S2). For both treatment groups, the mean body mass index (BMI) was greater than 30 and most subjects (≥92%) had type 2 diabetes.
At the time of randomization, about half of the subjects were not receiving pregabalin or gabapentin (VM202: 50.6% and placebo: 49.4%) to manage the pain associated with DPN. A large percentage of the subjects had other baseline medical conditions, including hypertension, dyslipidemia, and obesity.
Safety and tolerability
Adverse events
Safety was assessed based on the incidence of treatment‐emergent adverse events (TEAEs) and serious adverse events (SAEs), and their relationship to the study drug. Because of the theoretical angiogenic potential of VM202 and the macro‐/microvascular complications of diabetes, adverse events of special interest (AESI) were also assessed. AESIs included five categories of TEAEs; injection site reactions, ophthalmologic events, acute cardiac events, foot ulcers, and symptoms of CNS depression commonly observed in gabapentinoid users.
Over the course of DPN 3‐1 (with 493 subjects in the total safety population), 241 of 332 (72.6%) VM202‐treated and 111 of 161 (68.9%) placebo‐treated subjects reported at least one TEAE (Table S3). The most common TEAEs categorized by system organ class (SOC) were infection and infestations, with similar incidences for VM202 (99/332, 29.8%) and placebo (43/161, 26.7%). A total of 57 of 332 (17.2%) VM202‐treated and 27 of 161 (16.8%) placebo‐treated subjects experienced AESI. The most frequent AESIs were diabetic retinopathy, peripheral edema, and skin ulcer (Table S4). Diabetic retinopathy was more common in the placebo group (10/161, 6.2%) than the VM202 group (7/332, 2.1%). The incidence of TEAEs and AESIs deemed potentially related to study drug was low, with no difference between the placebo and VM202 groups; the TEAEs for 13.3% and 12.4% of subjects, respectively, were considered possibly drug‐related and the AESIs for 2.4% and 3.1% of subjects were assessed as possibly drug‐related.
Among the AESIs, injection site reactions (ISRs) were infrequent at all four study visits when treatments were administered, similar between placebo and VM202‐treated groups, and mild in all but three cases (one VM202 treated subject had a moderate ISR at day 0; another VM202‐treated subject had a moderate ISR at day 14; one placebo‐treated subject had a moderate ISR at day 104; all 3 subjects recovered in <24 h).
SAEs were reported for 32 of 332 (9.6%) subjects in the VM202 group, and 16 of 161 (9.9%) subjects in the placebo group. The SAEs for two subjects in the VM202 group, adenocarcinoma and vitreous hemorrhage, were deemed possibly related to the study drug, although the vitreous hemorrhage subject had a history of similar events. Myocardial infarctions for two subjects in the VM202 group and one subject in the placebo group were considered not related to the study drug; all three subjects had histories of coronary or cerebrovascular disease.
In DPN 3‐1b (101 subjects in the safety population), 10 of 65 (15.4%) VM202‐treated and 8 of 36 (22.2%) placebo‐treated subjects experienced TEAEs, and AESI were experienced by 2 of 65 (3.1%) VM202‐treated subjects (peripheral edema and chest pain) and 1 of 36 (2.8%) placebo‐treated subjects (angina pectoris; Table S4). SAEs were reported for 1 of 65 (1.5%) subjects in VM202 group and 2 of 36 (5.6%) subjects in placebo group. All three subjects with SAEs had medical histories related to their respective adverse events (AEs), and none was considered drug related.
Laboratory testing
Retinal fundoscopic examinations of all subjects throughout the study showed no significant changes in either the VM202 or placebo groups. There were no significant changes in blood chemistries, hematologic examinations, or levels of HbA1c.
Nerve conduction studies
Nerve conduction studies were performed as a safety measure in a small subset of subjects at selected sites. Fifty‐eight subjects had nerve conduction studies with both baseline (prior to treatment) and follow‐up values at day 180 and/or day 270 post‐treatment. There were no significant differences from baseline among treatment groups on days 180 and 270 in nerve conduction velocity or the response amplitudes for the sural and peroneal nerves, and there were no differences in changes in patients receiving placebo versus VM202 (Table S5).
Serum protein levels of HGF
The normal level of serum HGF protein in humans is not fully established but appears to range between 300 pg and 2 ng/ml. 41 , 42 HGF levels were measured in DPN 3‐1 at 10 time points per subject between days 0 and 270. No subject showed a transient rise in HGF protein levels after i.m. injection of VM202. At each time point, ~ 90% of subjects showed less than 2 ng/ml of HGF. There were 5 placebo and 7 VM202 subjects with serum HGF levels greater than 2 ng/ml at 8 of 10 time points. There was no significant difference between treatment groups at any time point in the number of subjects with levels greater than or equal to 2 ng/ml of HGF.
Efficacy in DPN 3‐1
The main DPN 3‐1 study failed to meet its primary end points (Figure S1); differences between the VM202 and placebo groups in DPN 3‐1 were not statistically significant for any efficacy end points (daily pain and sleep interference diary for pain and sleep interference, Visual Analog Scale for pain, Brief Pain Inventory [BPI] for DPN for pain severity and pain interference, and Michigan Neuropathy Screening Instrument [MNSI]; Table S6). When the 251 subjects who were not receiving concurrent gabapentinoids were analyzed separately, again, no end points met statistical significance relative to placebo.
The ITT population (N = 500) included all randomized subjects with the exception of seven who were prospectively withdrawn before receiving study medication, without knowledge of treatment assignment, when it was discovered that they did not meet inclusion / exclusion criteria or who were randomized without PI oversight. The ITT population was used for all efficacy analyses. A modified ITT population (N = 440) also was analyzed that included all subjects who satisfied the inclusion / exclusion criteria, received at least one dose of study medication, and completed their pain and sleep diaries at baseline and at the 90‐day follow‐up visit. In addition, a per protocol (PP) population (N = 287) was analyzed that included all subjects who received all 4 protocol‐specified doses of study medication, did not use protocol‐specified prohibited concomitant medications for more than 14 consecutive days after the first dose of study drug, and who completed the study without major protocol deviations. The protocol specified that missing values would be imputed via multiple imputation methods. The results for the change in the average daily pain scores for all 3 study populations at the 90 day, 180 day, and 270 day visits using Mixed Model Repeated Measures (MMRMs) imputation are shown in Table S7. There was little difference among the ITT, mITT, and PP populations in calculations of the percent changes from baseline or in the p values.
Efficacy in DPN 3‐1b
Despite similar demographics and baseline characteristics, efficacy data differed strikingly between the two parts of the study, DPN 3‐1 and DPN 3‐1b. Of 101 later‐enrolling subjects belonging to the full ITT population who enrolled in DPN 3‐1b, complete pain diary data were available for all but 2 subjects. At screening, there was no significant difference in pain severity between the VM202 and placebo groups. However, at 12 months, there were significant reductions in the primary efficacy measure, mean pain score changes from baseline, for VM202 compared with placebo (Figure 3, Table S8).
Figure 3.
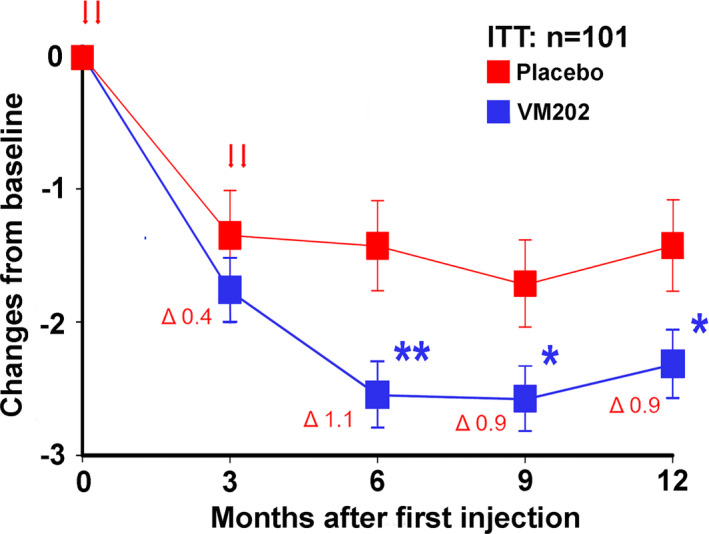
(a) Mean pain scores on the daily pain diary in DPN 3‐1b expressed as change from baseline ± SE for VM202 and placebo groups. The values for the means, standard errors and differences between groups are given in Table S8. Red arrows indicate times of treatment. The Δ values are differences at each timepoint between the groups. *p < 0.05 **p < 0.01. DPN, diabetic peripheral neuropathy; ITT, intent‐to‐treat
Analysis of earlier timepoints revealed no difference between the 2 groups at 3 months, but there were significant reductions in the VM202 group in the 24‐h average pain scores at 6 months and 9 months. There were no significant differences for VM202 compared with placebo for sleep interference.
Importantly, greater reductions in pain were found in subjects who were not on gabapentin or pregabalin during the 12‐month study, consistent with the phase II study results. Of 101 subjects, 53 subjects (52%) did not take gabapentinoids during the study. The subjects not on gabapentinoids experienced −1.34, −1.24, and −1.48 point reductions in the means of average 24‐h pain scores following VM202 compared with placebo at 6, 9, and 12 months (p = 0.0308, p = 0.0504, and p = 0.0155, respectively; Figure 4, Table S9). Subjects who were taking gabapentinoids also showed a trend toward pain reduction, although not statistically significant, and the difference in pain score between placebo and VM202 groups was much smaller than for nonusers.
Figure 4.
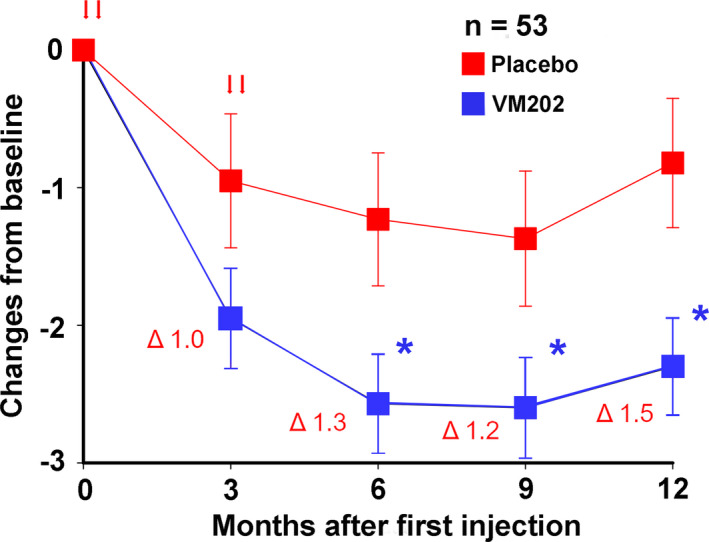
Mean pain scores on the daily pain diary for subjects not on gabapentin or pregabalin in DPN 3‐1b expressed as change from baseline ± SE for VM202 and placebo groups. The values for the means, standard errors, and differences between groups are given in Table S9. Red arrows indicate times of treatment. The Δ values are differences at each timepoint between the groups. *p < 0.05. DPN, diabetic peripheral neuropathy
Post hoc subgroup analysis of subjects enrolled by the second contract research organization
After nearly two‐thirds of the subjects were enrolled, the original contract research organization (CRO) resigned their responsibilities and discontinued operations. A second CRO was engaged to complete the study and subsequently enrolled the final 142 subjects in study DPN 3‐1 and most subjects who were continued into DPN 3‐1b. To evaluate the potential influence that this had on efficacy parameters in DPN 3‐1, and to understand whether this could have impacted the differences in efficacy outcomes in comparing the DPN 3‐1 to the original 9‐month phase II trial, the 142 subjects enrolled by the second CRO (n = 46 placebo and n = 96 VM202) were analyzed separately (Figure 5, Table S10). There were significant reductions in pain at 6 months measured by both the 24‐h NRS pain score (p = 0.047) as well as the BPI (p = 0.042). Further, reductions in pain were greater in subjects not on gabapentin or pregabalin, similar to the findings in both the phase II and the DPN 3‐1b studies. The significant reduction in pain in this group persisted until the end of the DPN 3‐1 study at 9 months (Table S11; NRS p = 0.036; BPI p = 0.044).
Figure 5.
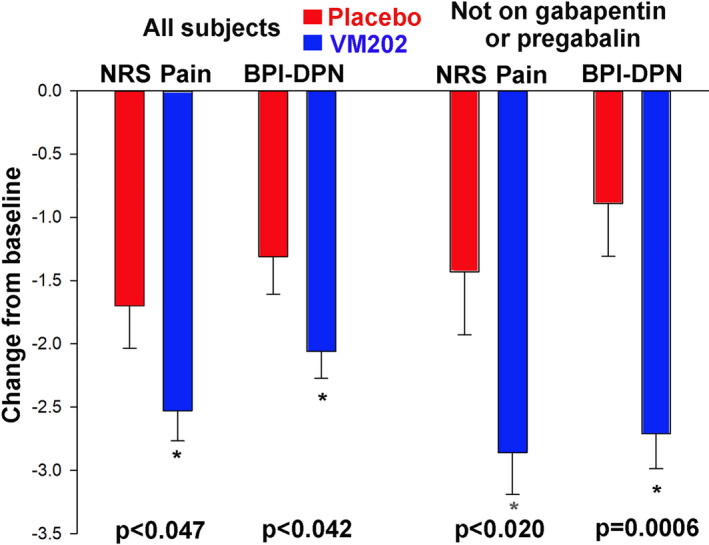
Mean pain scores at 6 months on both the daily pain diary (NRS‐Pain) and the Brief Pain Inventory for diabetic neuropathy (BPI‐DPN) expressed as change from baseline ± SE for VM202 and placebo groups for the 142 subjects enrolled by the second CRO. The first two sets of scores are for all subjects, and the second two sets of scores are for subjects not on gabapentin or pregabalin. CRO, contract research organization
DISCUSSION
The safety and efficacy of VM202 in reducing pain attributed to diabetic peripheral neuropathy were assessed in a large‐scale double‐blind, placebo‐controlled, randomized multicenter study. Intramuscular injections of VM202 to the calf muscles showed a good safety profile but produced puzzling and inconclusive efficacy results.
Safety
VM202 showed an excellent safety profile in both parts of this study as well as in the phase I and phase II studies. In the 9‐month DPN 3‐1 study, the incidences of TEAE, AESI, and SAE were similar for the VM202 and placebo groups. In the 12‐month DPN 3‐1b study, the incidences were even lower, and no drug‐related AEs were observed. In addition, there was a very low incidence of side effects, such as dizziness, somnolence, peripheral edema, or blurred vision, that are frequently observed in patients taking medicines commonly used for peripheral neuropathies.
There was a theoretical concern that the angiogenic activity of HGF protein expressed from VM202 DNA might result in vascular complications or cancers. However, there have been no such cases in phase I, II, and III trials involving VM202 for DPN or other indications, such as coronary artery disease, critical limb ischemia, and amyotrophic lateral sclerosis. The excellent safety profile for VM202 is not surprising, in that HGF expression from VM202 is short‐lived and likely restricted to the injection area. The N‐terminal of the HGF protein contains a domain that binds to heparin or heparan sulfate. Because heparan sulfate is abundant in extracellular matrix, diffusion of secreted HGF is limited. Further, the half‐lives of VM202 DNA and HGF protein in the circulation are both only a few minutes. 28 , 29 , 30 , 31 Thus, even if plasmid or HGF produced by the plasmid did reach the circulation, the possible biological effects would be minimal. More importantly, no noticeable changes in serum levels of HGF were observed after VM202 treatment in either the phase II or III study. Such transient, local expression of HGF protein from VM202 plasmid DNA provides an additional safety benefit compared with viral vectors.
Efficacy
In the full DPN 3‐1 population, VM202 did not meet the efficacy end points, in sharp contrast with results from the phase II study. This was surprising because subjects in the DPN 3‐1 study received 2 courses of treatment at days 0 and 14, followed by days 90 and 104, in contrast to subjects in the phase II study who only received one course of treatment (days 0 and 14). Both the phase II study and the full DPN 3‐1 study ended with 9‐month evaluations.
However, the subset of 101 subjects in DPN 3‐1b who continued to 12 months’ study participation demonstrated significant efficacy in pain reduction at 6, 9, and 12 months as determined by pain diaries. In contrast to the phase II study, subjects in DPN 3‐1b did not show significant analgesia at 3 months. This may represent either a too small sample size or a slower onset of efficacy in DPN 3‐1b compared with the phase II study because the peak magnitude of pain reduction at 6 and 9 months was similar to that observed in the phase II study.
Subjects in both DPN 3‐1 and DPN 3‐1b experienced a moderate‐to‐severe baseline level of pain (mean numerical pain scores = 7.03 ± 1.36 and 6.63 ± 1.31, respectively). Pain scores in VM202‐treated subjects were reduced to a mean range of 4.48 to 4.33 at 6 and 9 months in the full DPN 3‐1 study, and to a range of 3.99 to 4.21 at 6–12 months in the DPN 3‐1b study. These are clinically significant reductions of pain (−36% to −40% from baseline in both studies), but the difference was that the placebo effect was larger in DPN 3‐1 compared with DPN 3‐1b, so only the latter was statistically significant.
As part of the prespecified statistical analysis, VM202 effects were even more pronounced in subjects not receiving pregabalin or gabapentin in the DPN 3‐1b study, similar to the findings in the phase II study. The magnitude of the placebo effect was smaller in the DPN 3‐1b study than in the DPN 3‐1 study, and even smaller in the DPN 3‐1b subjects not taking gabapentinoids, suggesting that either differences in placebo‐response training and/or concomitant medications may, in part, have influenced the final results in DPN 3‐1 and DPN 3‐1b.
Thus, there was a serious discrepancy in results between DPN 3‐1 and DPN 3‐1b; only the latter study replicated results from the phase II study. Although it is unclear why efficacy was observed only in the latter group, there are several possibilities.
There was a change in the CRO during the study, and an exploratory post hoc subgroup analysis indicated that there were noticeable differences in efficacy data between the subjects enrolled by the first CRO compared with the second CRO. In the latter group, for example, VM202 showed significant pain‐reducing effects at 6 months, these effects were more prominent in subjects not receiving pregabalin or gabapentin, and significant effects persisted through the entire 9 months of the DPN 3‐1 study. The subjects in the DPN 3‐1b were largely recruited from the latter group, possibly explaining why significant efficacy was observed in that study. However, there is no conclusive evidence of substantial study conduct differences between those two CROs.
There was a large number (25) of centers in the study. The different results coming from the initial cohort of subjects versus the latter one could simply reflect the learning curve of the investigators.
Potential effects of variations in drug product quality by batch also were investigated, but subsequent analyses revealed that this was unlikely to be a factor in differences between the DPN 3‐1 and DPN 3‐1b studies. More than a dozen factors were evaluated for possible differences in study conduct, randomization, and data management; none revealed systemic errors. It is unlikely that the reasons for this discrepancy will be fully resolved. Despite the perplexing inconsistency in efficacy data from this study, the phase I multidose open‐label study showed dose‐related efficacy, and two double‐blind placebo‐controlled studies (phase II and DPN 3‐1b) have similarly showed efficacy, particularly in subjects not receiving concomitant gabapentinoids. This suggests that another study with rigorous pretrial training will be necessary to resolve this discrepancy.
One of the most clinically important findings in both the DPN 3‐1b and the phase II study is that VM202 was more effective in subjects not taking pregabalin or gabapentin, two of the most frequently prescribed medicines in the United States. In both the phase II and III studies, about 50% of study subjects did not take these medicines during the study period. Medical histories in this phase III study documented that 72% of subjects in both the placebo and VM202‐treated groups had previously tried gabapentinoids, but 22% had discontinued their use prior to enrollment in this study either because of lack of efficacy or side effects. The remaining 50% constituted the +gabapentinoid cohort in both treatment arms that was required per the stratification strategy. Thus, efficacy of VM202 for patients who discontinued use of gabapentinoids would provide a therapeutic option for a large group of patients with painful DPN who have limited treatment options.
Another interesting finding from DPN 3‐1b and the phase II study is the long‐term analgesic effect of VM202. In DPN 3‐1b, VM202 provided pain reduction for more than 8 months after the last cycle of treatment. The plasmid is rapidly degraded after injection, and local HGF production lasts only about 2 weeks. 31 Further, serum levels of HGF did not change after treatment with VM202, indicating that the effects of treatment reflected local effects of HGF. The prolonged effects long after disappearance of the plasmid suggests that VM202 treatment may change the course of disease progression. For example, in animal models, HGF produced from intramuscularly injected VM202 interacts with the c‐Met receptor present on Schwann cells or sensory neurons via ERK or AP‐1 signaling pathways, respectively, to promote axon outgrowth. 19 , 43 Current therapies are palliative and do not target the mechanisms underlying painful DPN. Thus, the availability of a potential therapy that changes pain‐generating circuits and/or regenerates damaged nerves would fundamentally change approaches to painful DPN.
To our knowledge, this is the first phase III gene therapy study for pain that has ever been done. VM202 treatment did not meet efficacy end points in the full DPN 3‐1 population, but VM202 demonstrated long‐term, clinically significant reductions in pain in the subset of subjects, particularly in subjects not on gabapentinoids, prospectively continued with no further treatment intervention into the 12‐month extension study, DPN 3‐1b. Similar findings were observed in the phase II study. Given the excellent safety profile of VM202, the potential for disease modifying effects, and the high unmet medical needs of the DPN patient population not on gabapentinoids, further study is warranted especially in patients not on gabapentinoids.
CONFLICT OF INTEREST
John A. Kessler declares no conflict of interest. Aziz Shaibani, Christine N. Sang, Mark Christiansen, David Kudrow, Aaron Vinik and the other members of the VM202 study group received funding from the sponsor, Helixmith, for performing the studies at their sites. Nari Shin, the statistician for the study, was paid by Helixmith.
AUTHOR CONTRIBUTIONS
J.A.K., A.S., C.N.S., M.C., D.K., and A.V. wrote the manuscript. N.S. and J.A.K. designed the research. J.A.K., A.S., C.N.S., D.K., A.V., and the VM202 Study Group performed the research. N.S. and J.A.K. analyzed the data.
Supporting information
Supplementary Methods S1
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Table S8
Table S9
Table S10
Table S11
Figure S1
Funding information
Helixmith Inc. provided funding for Aziz Shaibani, Christine Sang, Mark Christiansen, David Kudrow, Aaron Vinik, and Nari Shin. Helixmith paid the costs of performing the trial and paid the salary of the statistician, Nari Shin.
REFERENCES
- 1. Davies M, Brophy S, Williams R, Taylor A. The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes Care. 2006;29:1518‐1522. [DOI] [PubMed] [Google Scholar]
- 2. Jensen MP, Chodroff MJ, Dworkin RH. The impact of neuropathic pain on health‐related quality of life: review and implications. Neurology. 2007;68:1178‐1182. [DOI] [PubMed] [Google Scholar]
- 3. Vinik AI, Nevoret ML, Casellini C, Parson H. Diabetic neuropathy. Endocrinol Metab Clin North Am. 2013;42:747‐787. [DOI] [PubMed] [Google Scholar]
- 4. Hartsfield CL, Korner EJ, Ellis JL, et al. Painful diabetic peripheral neuropathy in a managed care setting: patient identification, prevalence estimates, and pharmacy utilization patterns. Population Health Manage. 2008;11:317‐328. [DOI] [PubMed] [Google Scholar]
- 5. Hoffman EM, Watson JC, St. Sauver J, Staff NP, Klein CJ. Association of long‐term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA Neurol. 2017;74(7):773‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Patil PR, Wolfe J, Said Q, Thomas J, Martin BC. Opioid use in the management of diabetic peripheral neuropathy (DPN) in a large commercially insured population. Clin J Pain. 2015;31(5):414‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guedon JM, Wu S, Zheng X, et al. Current gene therapy using viral vectors for chronic pain. Mol Pain. 2015;13(11):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumar S, Ruchi R, James SR, Chidiac EJ. Gene therapy for chronic neuropathic pain: how does it work and where do we stand today? Pain Med. 2011;12(5):808‐822. [DOI] [PubMed] [Google Scholar]
- 9. Dengler EC, Alberti LA, Bowman BN, et al. Improvement of spinal non‐viral IL‐10 gene delivery by D‐mannose as a transgene adjuvant to control chronic neuropathic pain. J Neuroinflammation. 2014;21(11):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kessler JA, Smith AG, Cha BS, et al. Double‐blind, placebo‐controlled study of HGF gene therapy in diabetic neuropathy. Ann Clin Transl Neurol. 2015;2(5):465‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsuchihara T, Ogata S, Nemoto K, et al. Nonviral retrograde gene transfer of human hepatocyte growth factor improves neuropathic pain‐related phenomena in rats. Mol Ther. 2009;17(1):42‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pyun W‐B, Hahn W, Kim D‐S, et al. Naked DNA expressing two isoforms of hepatocyte growth factor induces collateral artery augmentation in a rabbit model of limb ischemia. Gene Ther. 2010;17:1442‐1452. [DOI] [PubMed] [Google Scholar]
- 13. Cheng C, Guo GF, Martinez JA, Singh V, Zochodne DW. Dynamic plasticity of axons within a cutaneous milieu. J Neurosci. 2010;30:14735‐14744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ebens A, Brose K, Leonardo ED, et al. Hepatocyte growth factor scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron. 1996;17:1157‐1172. [DOI] [PubMed] [Google Scholar]
- 15. Funakoshi H, Nakamura T. Identification of HGF‐like protein as a novel neurotrophic factor for avian dorsal root ganglion sensory neurons. Biochem Biophys Res Commun. 2001;283:606‐612. [DOI] [PubMed] [Google Scholar]
- 16. Gascon E, Gaillard S, Malapert P, et al. Hepatocyte growth factor‐Met signaling is required for Runx1 extinction and peptidergic differentiation in primary nociceptive neurons. J Neurosci. 2010;30:12414‐12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hashimoto N, Yamanaka H, Fukuoka T, et al. Expression of HGF and cMet in the peripheral nervous system of adult rats following sciatic nerve injury. NeuroReport. 2001;12:1403‐1407. [DOI] [PubMed] [Google Scholar]
- 18. Kato N, Nemoto K, Nakanishi K, et al. Nonviral HVJ (hemagglutinating virus of Japan) liposome‐mediated retrograde gene transfer of human hepatocyte growth factor into rat nervous system promotes functional and histological recovery of the crushed nerve. Neurosci Res. 2005;52:299‐310. [DOI] [PubMed] [Google Scholar]
- 19. Ko KR, Lee J, Lee D, Nho B, Kim S. Hepatocyte growth factor (HGF) promotes peripheral nerve regeneration by activating repair schwann cells. Sci Rep. 2018;8(1):8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maina F, Hilton MC, Ponzetto C, Davies AM, Klein R. Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes Dev. 1997;11:3341‐3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson J, Dolcet X, Hilton M, Tolcos M, Davies AM. HGF promotes survival and growth of maturing sympathetic neurons by PI‐3 kinase‐ and MAP kinase‐dependent mechanisms. Mol Cell Neurosci. 2004;27(4):441‐452. [DOI] [PubMed] [Google Scholar]
- 22. Wong V, Glass DJ, Arriaga R, Yancopoulos GD, Lindsay RM, Conn G. Hepatocyte growth factor promotes motor neuron survival and synergizes with ciliary neurotrophic factor. J Biol Chem. 1997;272(8):5187‐5191. [DOI] [PubMed] [Google Scholar]
- 23. Yang XM, Toma JG, Bamji SX, et al. Autocrine hepatocyte growth factor provides a local mechanism for promoting axonal growth. J Neurosci. 1998;18(20):8369‐8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morishita R, Nakamura S, Hayashi S, et al. Therapeutic angiogenesis induced by human recombinant hepatocyte growth factor in rabbit hind limb ischemia model as cytokine supplement therapy. Hypertension. 1999;33:1379‐1384. [DOI] [PubMed] [Google Scholar]
- 25. Morishita R, Sakaki M, Yamamoto K, et al. Impairment of collateral formation in lipoprotein(a) transgenic mice: therapeutic angiogenesis induced by human hepatocyte growth factor gene. Circulation. 2002;105:1491‐1496. [DOI] [PubMed] [Google Scholar]
- 26. Taniyama Y, Morishita R, Aoki M, et al. Therapeutic angiogenesis induced by human hepatocyte growth factor gene in rat and rabbit hindlimb ischemia models: preclinical study for treatment of peripheral arterial disease. Gene Ther. 2001;8:181‐189. [DOI] [PubMed] [Google Scholar]
- 27. Taniyama Y, Morishita R, Hiraoka K, et al. Therapeutic angiogenesis induced by human hepatocyte growth factor gene in rat diabetic hind limb ischemia model: molecular mechanisms of delayed angiogenesis in diabetes. Circulation. 2011;104(19):2344‐2350. [DOI] [PubMed] [Google Scholar]
- 28. Appasamy R, Tanabe M, Murase N, et al. Hepatocyte growth factor, blood clearance, organ uptake, and biliary excretion in normal and partially hepatectomized rats. Lab Invest. 1993;68(3):270‐276. [PubMed] [Google Scholar]
- 29. Ido A, Moriuchi A, Kim I, et al. Pharmacokinetic study of recombinant human hepatocyte growth factor administered in a bolus intravenously or via portal vein. Hepatol Res. 2004;30(3):175‐181. [DOI] [PubMed] [Google Scholar]
- 30. Kawaida K, Matsumoto K, Shimazu H, Nakamura T. Hepatocyte growth factor prevents acute renal failure and accelerates renal regeneration in mice. Proc Natl Acad Sci USA. 1994;91(10):4357‐4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu KX, Kato Y, Narukawa M, et al. Importance of the liver in plasma clearance of hepatocyte growth factors in rats. Am J Physiol. 1992;263(5 Pt 1):G642‐G649. [DOI] [PubMed] [Google Scholar]
- 32. Ajroud‐Driss S, Christiansen M, Allen JA, Kessler JA. Phase 1/2 open‐label dose‐escalation study of plasmid DNA expressing two isoforms of hepatocyte growth factor in patients with painful diabetic peripheral neuropathy. Mol Ther. 2013;21(6):1279‐1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carlsson M, Osman NF, Ursell PC, Martin AJ, Saeed M. Quantitative MR measurements of regional and global left ventricular function and strain after intramyocardial transfer of VM202 into infarcted swine myocardium. Am J Physiol Heart Circ Physiol. 2008;295(2):H522‐H532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Henry TD, Hirsch AT, Goldman J, et al. Safety of a non‐viral plasmid‐encoding dual isoforms of hepatocyte growth factor in critical limb ischemia patients: a phase I study. Gene Ther. 2011;18(8):788‐794. [DOI] [PubMed] [Google Scholar]
- 35. Kibbe MR, Hirsch AT, Mendelsohn FO, et al. Perin EC Safety and efficacy of plasmid DNA expressing two isoforms of hepatocyte growth factor in patients with critical limb ischemia. Gene Ther. 2016;23(3):306‐312. [DOI] [PubMed] [Google Scholar]
- 36. Kim JS, Hwang HY, Cho KR, et al. Kim K‐B Intramyocardial transfer of hepatocyte growth factor as an adjunct to CABG: phase I clinical study. Gene Ther. 2013;20(7):717‐722. [DOI] [PubMed] [Google Scholar]
- 37. Nho B, Lee J, Lee J, Ko KR, Lee SJ, Kim S. Effective control of neuropathic pain by transient expression of hepatocyte growth factor in a mouse chronic constriction injury model. FASEB J. 2018;32(9):5119‐5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perin EC, Silva GV, Vela DC, et al. Human hepatocyte growth factor (VM202) gene therapy via transendocardial injection in a pig model of chronic myocardial ischemia. J Card Fail. 2011;17(7):601‐611. [DOI] [PubMed] [Google Scholar]
- 39. Saeed M, Martin A, Ursell P, et al. MR assessment of myocardial perfusion, viability, and function after intramyocardial transfer of VM202, a new plasmid human hepatocyte growth factor in ischemic swine myocardium. Radiology. 2008;249(1):107‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sufit RL, Ajroud‐Driss S, Casey P, Kessler JA. Open label study to assess the safety of VM202 in subjects with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3–4):269‐278. [DOI] [PubMed] [Google Scholar]
- 41. Cantón A, Burgos R, Hernández C, et al. Hepatocyte growth factor in vitreous and serum from patients with proliferative diabetic retinopathy. Br J Ophthalmol. 2000;84(7):732‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Malatino LS, Cataliotti A, Benedetto FA, et al. Hepatocyte growth factor and left ventricular geometry in end‐stage renal disease. Hypertension. 2003;41(1):88‐92. [DOI] [PubMed] [Google Scholar]
- 43. Ko KR, Lee J, Nho B, Kim S. c‐Fos is necessary for HGF‐mediated gene regulation and cell migration in Schwann cells. BBRC. 2018;503:2855‐2860. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods S1
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Table S8
Table S9
Table S10
Table S11
Figure S1