

Abstract
Cone dystrophies are a rare subgroup of inherited retinal dystrophies and hallmarked by color vision defects, low or decreasing visual acuity and central vision loss, nystagmus and photophobia. Applying genome-wide linkage analysis and array comparative genome hybridization, we identified a locus for autosomal dominant cone dystrophy on chromosome 16q12 in four independent multigeneration families. The locus is defined by duplications of variable size with a smallest region of overlap of 608 kb affecting the IRXB gene cluster and encompasses the genes IRX5 and IRX6. IRX5 and IRX6 belong to the Iroquois (Iro) protein family of homeodomain-containing transcription factors involved in patterning and regionalization of embryonic tissue in vertebrates, including the eye and the retina. All patients presented with a unique progressive cone dystrophy phenotype hallmarked by early tritanopic color vision defects. We propose that the disease underlies a misregulation of the IRXB gene cluster on chromosome 16q12 and demonstrate that overexpression of Irx5a and Irx6a, the two orthologous genes in zebrafish, results in visual impairment in 5-day-old zebrafish larvae.
Introduction
Cone dystrophies (CD) represent a rare subgroup of inherited retinal dystrophies characterized by color vision defects, low or decreasing visual acuity and central visual field loss and can be accompanied by nystagmus and photophobia (1). Visual impairment progresses with age and often into cone-rod dystrophy (CRD) with additional symptoms including night blindness and affection of peripheral vision. All modes of Mendelian inheritance are observed, and more than 20 genes have been reported to be associated with CRD but far less with isolated CD (RetNet database (https://sph.uth.tmc.edu/Retnet/) (1,2).
IRX5 and IRX6 belong to the Iroquois (Iro) protein family of homeodomain-containing transcription factors involved in patterning and regionalization of embryonic tissue in vertebrates and invertebrates. The six IRX genes are organized in two evolutionary highly conserved clusters: the IRXA and the IRXB gene clusters, each consisting of three genes. The IRXB gene cluster is located on human chromosome 16 and contains IRX3, IRX5 and IRX6 (3–5).
All IRX genes are expressed in the human developing eye (eyeIntegration (https://eyeIntegration.nei.nih.gov) (6,7). In the mouse developing eye, Irx6 expression is confined to the marginal zone of the neural layer of the retina (8), specifically in type 2 and 3a OFF bipolar interneurons (9). In comparison, Irx5 is expressed during neurulation in the neuroepithelium in the mouse and expression is first detected at E12.5 at the onset of retinal neuron differentiation (10,11). In the adult mouse retina, Irx5 is expressed in at least three cell types in the inner nuclear layer of the mature mouse retina: Müller glia, a subset of type 3 and/or type 5 CaB5-positive cone bipolar cells, and Vsx1-positive/Rcv1-positive type 2 OFF cone bipolar cells (12). In Irx5-deficient mice, defects were observed in the expression of some, but not all immunohistological markers that define mature type 2 and type 3 OFF cone bipolar cells, indicating a role for Irx5 in bipolar cell differentiation (12). Irx knockout mice are smaller in size and have smaller eyes, but no electroretinographic defects were observed (12).
Here we describe a large German multigeneration family in which CD segregates as an autosomal dominant trait. By linkage analysis and copy number variation (CNV) analysis, we identified a large 608 kb duplication affecting the IRXB cluster in this family and subsequently overlapping 733 and 848 kb duplications in a Chinese and a Dutch family with autosomal dominant CD (adCD), respectively. The smallest region of overlap (SRO) is described by the initially identified 608 kb duplication and covers the genes IRX5 and IRX6 completely, and the proximal exons of MMP2. We hypothesized that overexpression of IRX5 and IRX6 may be the cause of the disease, which is corroborated by expression analysis in patient-derived fibroblasts and zebrafish experiments.
Results
Genetic analysis
Using genotypes obtained with SNP arrays, we performed genome-wide parametric linkage analysis (easyLINKAGE package) (13) in 14 subjects of a large five-generation German family with a clinical diagnosis of adCD (Family ZD3; Fig. 1B), and obtained increased LOD scores for a series of SNPs on chromosomes 8, 10 and 16. Only the LOD score for chromosome 16 was >3, and a maximum LOD score of 3.0103 for a series of SNPs on chromosome 16q12.2-q21 was considered significant (Supplementary Material, Fig. S1). Haplotype reconstruction defined a non-recombinant interval of 9.77 cM/8.2 Mb flanked by SNPs rs12929895 and rs8049061. Sanger sequencing of the coding exons of several retinal expressed candidate genes (i.e. IRX5, IRX6, MMP2, NUP93, AMFR, POLR2C, KIFC3) did not result in the identification of putative disease-causing variants.
Figure 1 .
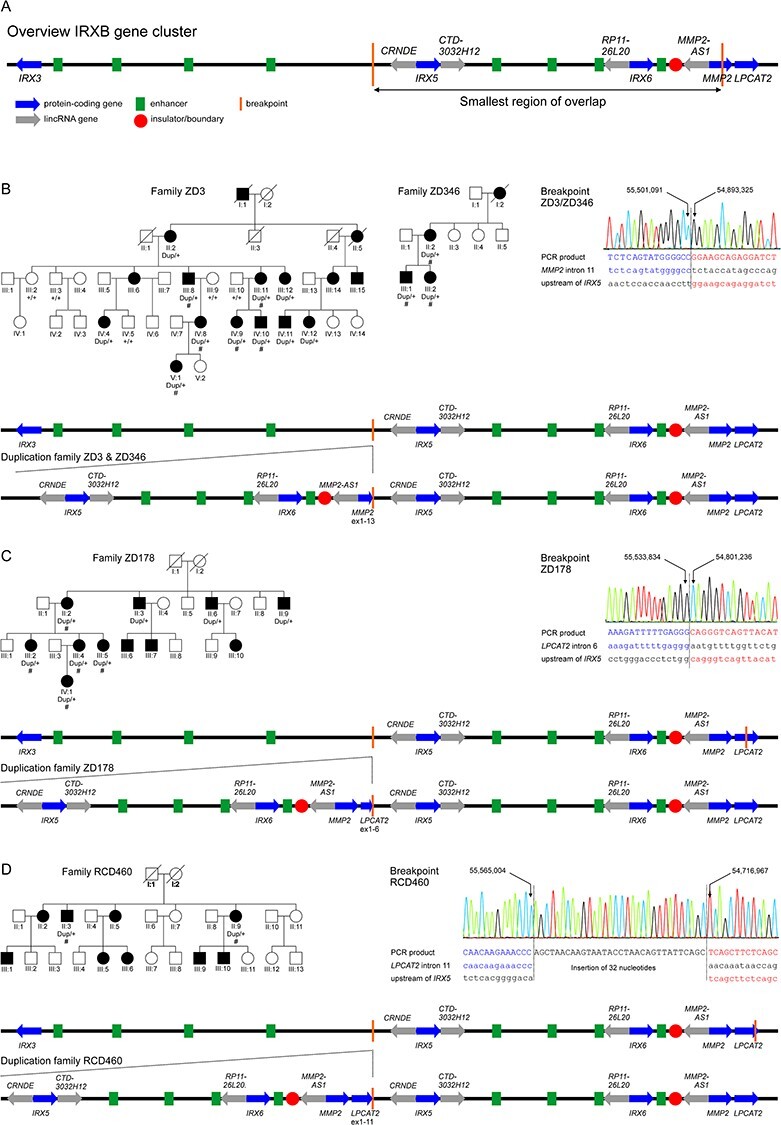
Tandem duplications at the IRXB gene cluster on chromosome 16q12 segregate with autosomal dominant cone dystrophy in four families. (A) Genomic organization of the IRXB gene cluster and the other two genes involved in the duplications, MMP2 and LPCAT2. Blue arrows indicate the protein-coding genes IRX3, IRX5, IRX6, MMP2 and LPCAT2, and gray arrows the non-coding RNAs CRNDE, CTD-3032H12, RP11-26 L20 and MMP2-AS1. The IRXB locus is highly conserved throughout evolution, not only in its genes but also its regulatory elements. Conserved enhancer elements are depicted in green and insulators/boundaries are represented with a red circle (4). The smallest region of overlap of the duplications found in this study is depicted as by vertical orange lines. (B–D) Pedigrees and segregating genotypes for the analyzed subjects and families in this study, breakpoint mapping and genomic organization of the duplication. Pedigrees: Patients for which DNA was available for CNV analysis are indicated by the identified genotype beneath the individual identifier. The presence of the duplication is indicated by ``dup'', while ``+'' indicates normal wild-type alleles. Patients for which clinical data was available are presented in Figure 3, Table 2 and Supplementary Material, Table S1 and are indicated by ``#''. Circles depict females and squares males, affected individuals by filled and unaffected individuals by open symbols. Already deceased individuals are marked by a diagonal slash. Breakpoint mapping in the investigated families: Electropherograms of the breakpoint sequences as obtained from Sanger sequencing of PCR products covering the breakpoints. Left and right junction sequences are given in blue and red, respectively. The reference sequences are given in lowercase letters below the sequence for the PCR product. Genomic positions refer to GRCh38/hg38. Schematic representation of the tandem repeated duplication as observed in the families in this study in comparison to the genomic organization of the wild-type IRXB gene cluster in families ZD3 and ZD346 (B), family ZD178 (C) and family RCD460 (D). The tandem repeated sequence is expected to create a new topological associated domain (TAD) flanked by the insulators as boundaries.
ArrayCGH (comparative genome hybridization) analysis in two family members (ZD3-III:8 and ZD3-III:11) identified an approximately 600 kb copy number gain within the linkage interval (Supplementary Material, Fig. S2). This duplication covered IRX5 (MIM 606195) and IRX6 (MIM 606196) in their entirety, as well as the proximal exons of MMP2 (MIM 120360) (Fig. 1A).
Applying a customized qPCR assay for IRX6, we screened for CNVs at this locus in a cohort of 25 genetically undefined autosomal dominant cone and cone-rod dystrophy families. Hereby, we identified two additional families (Family ZD178—Chinese descent, Family RCD460—Dutch descent, Fig. 1C and D) with copy number gain for this target sequence (Supplementary Material, Fig. S3). ArrayCGH for individuals ZD178-III:2 and RCD460-II:3 confirmed distinct copy number gains of ~700 and ~850 kb, respectively, indicating duplications larger than the one identified in family ZD3, and extending proximally further into the upstream regulatory sequences of IRX5 and distally to the LPCAT2 gene (MIM 612040), respectively (Supplementary Material, Fig. S2).
The exact extent and orientation of the duplication in each family was established by breakpoint PCR and sequencing of subcloned PCR products by primer walking. The duplication in family ZD3 is 607 766 bp in size and covers the protein-coding genes IRX5 and IRX6 completely, as well as exons 1–11 of the MMP2 gene (Fig. 1B, Table 1). In addition, the duplicated sequence contains the lincRNAs CRNDE, CTD-3032H12.2, RP11-226 L20 and MMP2-AS1. The duplications in families ZD178 (732 598 bp) and RCD460 (848 069 bp) are even larger, encompassing the entire MMP2 and extending toward the q-arm telomer into intron 6 and intron 11 of LPCAT2, respectively (Fig. 1C and D, Table 1). An additional 32 bp sequence is inserted into the breakpoint of the duplicated sequence in family RCD460 (Fig. 1D, Table 1). All duplications are oriented head-to-tail in tandem.
Table 1.
Breakpoint mapping results and gene content of the duplications
| Family | Structural variant and breakpoint (GRCh38/hg38) |
Size (bp) |
Genes included in or affected by the duplication |
|---|---|---|---|
| ZD3/ZD346 | NC_000016.10:g.54893325_55501091dup | 607 766 | protein-coding genes: IRX5, IRX6, MMP2 (exons 1–11/13) lincRNAs: CRNDE, CTD-3032H12, RP11-26 L20, MMP2-AS1 |
| ZD178 | NC_000016.10:g.54801236_55533834dup | 732 598 | protein-coding genes: IRX5, IRX6, MMP2, LPCAT2 (exons 1–6/14) lincRNAs: CRNDE, CTD-3032H12, RP11-26 L20, MMP2-AS1 |
| RCD460 | NC_000016.10:g.54716967_54716968ins [54716967_55565004; AGCTAACAAGTAATACCTAACAGTTATTCAGC] | 848 069 | protein-coding genes: IRX5, IRX6, MMP2, LPCAT2 (exons 1–11/14) lincRNAs: CRNDE, CTD-3032H12, RP11-26 L20, MMP2-AS1 |
While the duplications in families ZD178 and RCD460 were unique, we identified another adCD family of German descent harboring the same duplication as observed in family ZD3 by CNV analysis upon exome sequencing in both siblings ZD346-III:1 and ZD346-III:2, which was confirmed by subsequent breakpoint PCR and sequencing (Fig. 1B). Of note, exome sequencing did not reveal any other likely pathogenic and causative variants sufficient to explain the retinal phenotype in these patients. The duplication in families ZD3 and ZD346 represents the SRO that is encompassed by all duplications. Whether these families are distantly related was not established.
Segregation analysis was performed for all available individuals of these families either using qPCR-based CNV analysis targeting IRX6 (Fig. 1B–D, Supplementary Material, Fig. S3) or duplication-specific breakpoint PCR (data not shown), and confirmed that all affected family members harbored the heterozygous duplication segregating in their families, while none of the unaffected was a carrier of the duplication (Fig. 1B–D).
Expression of IRX5 and IRX6 in retinal tissue
The SRO comprises the genes IRX5 and IRX6 completely, as well as exons 1–11 of the MMP2 gene and the lincRNAs CRNDE, CTD-3032H12.2, RP11-226L20 and MMP2-AS1. Evaluation of RNA-seq data out of human retinal RNA established that all three protein coding genes in the SRO, IRX5, IRX6 and MMP2 are expressed in human adult retina (14) (data not shown). By order, MMP2 shows the highest expression levels followed by IRX5 and IRX6, these latter showing similar expression levels. Of all IRX genes, IRX6 is expressed weakest in the adult human retina as evaluated by analysis of our RNA-seq data (data not shown). Furthermore, several lincRNA within the locus are more abundantly expressed than their protein-coding counterparts, with CRNDE displaying higher expression levels than CTD-3032H12.2 and RP11–26220 (data not shown).
Increased expression of IRX5 and IRX6 in patient-derived fibroblast
As the phenotype observed in our patients is rather homogeneous, we hypothesized that the adCD phenotype is caused by a misregulation of the genes located in the SRO, most likely IRX5 and IRX6. The effect of the chromosome 16q12 duplication on the relative expression level of IRX5 and IRX6 was assessed by pyrosequencing on cDNA synthesized from total RNA extracted out of patient-derived fibroblast cell lines (ZD3-IV:8 and ZD178-III:2) as well as of a control subject. Both genes, IRX5 and IRX6 showed increased expression levels of the duplicated allele compared to the normal (Supplementary Material, Fig. S4).
Overexpression of IRX5 and IRX6 results in impaired visual performance in zebrafish larvae
These results prompted us to assess the effect of overexpression of irx5a and irx6a onto the functionality of the developing zebrafish retina. The zebrafish genome presents a single orthologous conserved gene cluster including irx5a, irx6a, mmp2 and lpcat2 on zebrafish chromosome 7 (Fig. 2A). To mimic overexpression of irx5a and/or irx6a, we injected one-cell zebrafish oocytes with 50 pg of cRNA of the irx5a and/or irx6a orthologs. We introduced a nucleotide substitution that enabled relative quantification of cRNA to endogenous transcripts at 1 and 5 dpf (days postfertilization) by means of pyrosequencing. The ratio of persisting cRNA to endogenous transcripts at 1 dpf corresponded to a gain of two extra gene copies for irx5a and one extra gene copy for irx6a (Fig. 2B). At 5 dpf, levels of injected cRNA were hardly detectable, in line with degradation and a dilution effect due to cell division in the developing and growing larvae.
Figure 2 .
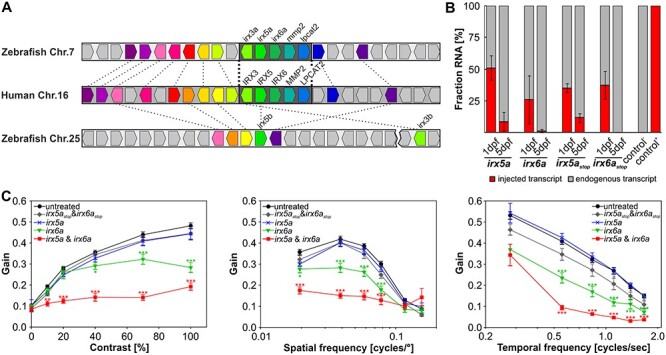
Overexpression of Irx6a and Irx6a & Irx5a cause reduced optokinetic reflex response in zebrafish larvae. (A) Gene synteny between the human chromosome 16 locus containing the IRXB cluster and the zebrafish chromosomes 7 and 25. Using the Genomicus database (http://www.genomicus.biologie.ens.fr/genomicus-97.01/cgi-bin/search.pl) and the UCSC genome browser (http://genome-euro.ucsc.edu/index.html), the conservation of the IRXB cluster between human and zebrafish was analyzed. The schematic comparative gene organization is shown and homologous genes are coded by the same color and are connected by dashed lines between the two analyzed species, whereas nonhomologous genes are depicted in gray. There is a high degree of synteny between human chromosome 16 and zebrafish chromosome 7, whereas the overall synteny between human chromosome 16 and zebrafish chromosome 25 is low for the analyzed 25 human chromosome 16 genes within and flanking the IRXB cluster. Strikingly, the gene cluster spanning IRX3, IRX5, IRX6, MMP2 and LPCAT2 is completely conserved between human and zebrafish. The genes irx5 and irx3 are duplicated in the zebrafish genome with the ‘a’-paralogs being located on chromosome 7 and the ‘b’-paralogs on chromosome 25—the distance between irx5b and irx3b is around 340 kb (GRCz11/danRer11) as depicted by the break on chromosome 25. irx6a, mmp2 or lpcat2 are not duplicated in the zebrafish genome. Thus, the zebrafish genome possesses one highly conserved IRXB cluster on chromosome 7. (B) Relative comparison between endogenously expressed irx5a and ixr6a versus injected transcripts (50 pg cRNA per oocyte) in 1 and 5 dpf old zebrafish embryos/larvae using pyrosequencing. In 1 dpf embryos, we detected an allelic ratio compatible with four copies of irx5a or three copies of irx6a. In 5 dpf larvae, the injected irx5a and irx6a transcripts were barely detectable. A second set of transcripts carrying a premature stop codon (irx5astop and irx6astop) was injected (50 pg cRNA per oocyte) and was detected in similar amounts compared to the injected irx5a and ixr6a transcripts at both time points analyzed. The irx5astop and irx6astop served later on as controls for the functional assay (OKR). RNA of untreated larvae (control −) or plasmid DNA containing a variant for pyrosequencing detection (control +) was used as controls for pyrosequencing. Data are presented as mean ± SEM. (C) Optokinetic response (OKR) analysis with respect to contrast sensitivity and spatial and temporal frequency measured in 5 dpf old treated larvae in comparison to untreated controls. Overexpression of Irx5a did not affect OKR for any of the analyzed stimulus conditions in 5 dpf larvae. Overexpression of Irx6a (equivalent to approximately three gene copies at 1 dpf) induced significant OKR alteration in comparison to untreated larvae at high contrasts and at a variety of spatial and temporal frequencies. Simultaneous overexpression of both Irx6a and Irx5a resulted in an even more reduced OKR when comparing to overexpression of Irx6a alone. Additionally, irx5a and ixr6a transcripts harboring a premature stop codon (irx5astop and irx6astop) were injected to verify that the reduced OKR in irx6a and irx6a & irx5a larvae is indeed due to Irx overexpression. OKR with respect to contrast sensitivity and spatial frequency measured in 5 dpf old larvae treated with 50 pg irx5astop or irx6astop did not show any effect for different contrast or spatial stimuli in comparison to untreated controls. For the tested temporal frequencies, reduced OKR responses were detected at high frequencies. For clarity, statistical significance of only treated versus untreated samples are depicted. Two-way ANOVA with Bonferroni multiple comparison was performed and the results of the statistical test within all groups are summarized in Supplementary Material, Tables S4–S7. Data are presented as mean ± SEM. Nuntreated = 60 larvae. Ntreated = 24 larvae per group. P ≤ 0.001 is indicated with ***, P ≤ 0.01 with ** and P ≤ 0.05 is displayed with *.
Visual function testing for contrast sensitivity and spatial and temporal frequency was carried out to detect whether augmented dosages of these two genes result in visual impairment manifesting by alterations of the optokinetic response (OKR) in zebrafish at 5 dpf (Fig. 2C). At this developmental stage, the OKR is well established and mainly relies on cone function (15,16). We observed that larvae mimicking overexpression of irx6a displayed impaired visual performance for a wide range of stimuli of spatial and temporal frequencies and varying contrasts compared to untreated larvae (Fig. 2C). In contrast, larvae mimicking overexpression of irx5a did not show major modifications in the OKR at none of three analyzed parameters (Fig. 2C). Finally, co-injections of irx5a and irx6a severely reduced contrast, spatial and temporal sensitivity in the treated larvae. Since these effects were even more pronounced compared to injection of irx6a alone, a potential synergistic effect might be suggested (Fig. 2C). The specificity of this observation was confirmed by injection of transcripts of irx5a and irx6a harboring a premature termination codon variant. Larvae treated with those irx5astop and irx6astop transcripts showed similar OKR compared to untreated larvae at all tested stimuli—except for high temporal frequencies—indicating that the observed reduced OKR following injection of irx6a and irx5a&irx6a is indeed a result of Irx5 and Irx6 overexpression (Fig. 2C).
Ophthalmological examination and patient phenotype
Sixteen affected individuals (female n = 12, male n = 4) from four independent families carrying large duplications at the IRXB gene locus were examined by psychophysical and functional testing, and multimodal imaging (Table 2, Supplementary Material, Table S1, Figs 1 and 3). Mean age at the last ophthalmological examination was 39 years (range, 8–67). The first ophthalmological symptom reported in almost all of the patients was a decreased visual acuity in their first decade of life. Other frequently reported ophthalmological symptoms were difficulties in differentiating colors, glare sensitivity and difficulties seeing at night (4/16). In terms of refractive error, 5 of the 16 patients were highly myopic. Anterior segment findings as well as intraocular pressure were within normal limits in all examined eyes with a tendency to develop early cataract as previously reported in patients with inherited retinal diseases (17).
Table 2.
Visual acuity, optical coherence tomography and color vision in cone dystrophy patients displaying a duplication at the IRXB gene cluster
| Patient Sex/Agea | Visual acuity | Optical coherence tomography | Color vision |
|---|---|---|---|
| ZD3-V:1 F/8 |
OD = 20/32 OS = 20/40 |
OU central thinning of the ONL, paracentral hyporeflectivity of the RPE | Panel D15 desaturated: OU tritan defect |
| ZD178-IV:1 F/8 |
OU = 20/20 | OU central thinning of the ONL, OS central RPE irregularities | ChromaTest: protan OD 2%, OS 4%, tritan OU 21% |
| ZD3-IV:10 M/19 |
OU = 20/63 | n.a. | Panel D15 desaturated: OU tritan/scotopic defect |
| ZD178-III:5 F/24 |
OU = 20/25 | OU central thinning of the ONL, OD paracentral hyporeflectivity of the RPE, OS central hyporeflectivity of the RPE, paracentral subretinal cleft | ChromaTest: protan OD 4%, OS 10%, tritan OD 32%, OS 50% |
| ZD346-III:1 M/26 |
OU = 20/100 | n.a. | n.a. |
| ZD346-III:2 F/31 |
OU = 20/100 | OU central thinning of the ONL, OD central hyporeflectivity of the RPE, OS central subretinal cleft | Panel D15 saturated: OU tritan defect |
| ZD178-III:4 F/38 |
OU = 20/80 | OU central atrophy | ChromaTest: protan OD 5%, OS 6%, tritan OD 34%, OS 51% |
| ZD178-III:2 F/39 |
OU = 20/100 | OU central atrophy | ChromaTest: protan OD 4%, OS 5%, tritan OD 56%, OS 64% |
| ZD3-IV:9 F/40 |
OU = 20/100 | OU central subretinal cleft | Panel D15 saturated: OU tritan defect |
| RCD460-II:9 F/41 |
OU = 20/25 | n.a. | Panel D15 saturated and desaturated: OU tritan defect; Lanthony tritan plates: only test plate is seen |
| ZD3-IV:8 F/43 |
OU = 20/100 | OD central subretinal cleft | Panel D15 desaturated: OU tritan defect |
| RCD460-II:3 M/46 |
OD = 20/200 OS = 20/32 |
n.a. | 28 Hue: OU outspoken tritan defect |
| ZD3-III:8 M/53 |
OU = 20/200 | n.a. | Panel D15 desaturated: OU tritan defect |
| ZD346-II:2 F/62 |
OU = 20/200 | OU central thinning of the ONL, central subretinal cleft, hyporeflectivity of the RPE elsewhere | Panel D15 saturated: OU tritan defect |
| ZD178-II:2 F/65 |
OU = 20/400 | OU central atrophy, OS central epiretinal membrane | ChromaTest: protan OD 12%, OS 20%, tritan OU >100% |
| ZD3-III:11 F/67 |
OD = 20/200 OS = 20/630 |
OU central atrophy | Panel D15 saturated: OD tritan defect, OS tritan/scotopic defect |
F: female, M: male, n.a.: not available, OD: right eye, ONL: outer nuclear layer, OS: left eye, OU: both eyes, RPE: retinal pigment epithelium, ChromaTest: protan <6% = norm, tritan <8% = norm.
aAge referring to age at examination.
Figure 3 .
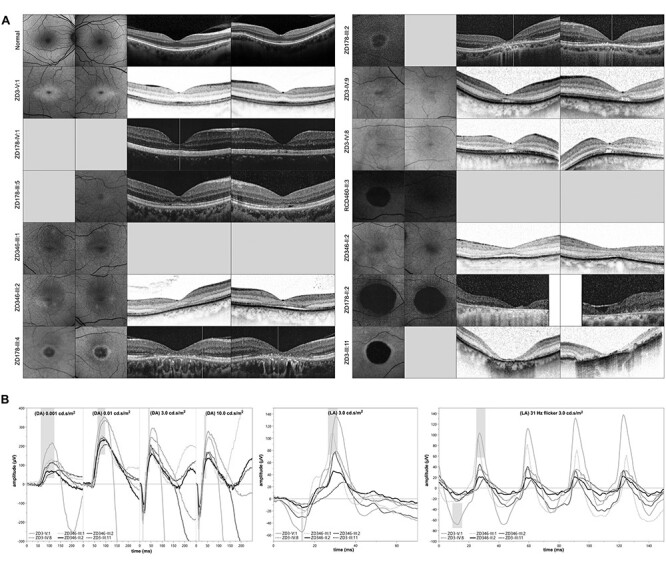
Clinical presentation of patients affected by autosomal dominant cone dystrophy caused by duplications at the IRXB gene cluster: (A) Fundus autofluorescence (FAF) and optical coherence tomography (OCT) images of 13 patients with a duplication at the IRXB gene cluster, sorted by the age at examination (compare Fig. 1, Table 2 and Supplementary Material, Table S1). Note the central hypoautofluorescence and paracentral hyperautofluorescence on FAF imaging in the younger patients and the well-demarcated central hypoautofluorescence in the older patients, as well as the central thinning of the outer nuclear layer, the paracentral hyporeflectivity of the retinal pigment epithelium, and/or a subretinal cleft on OCT imaging in the younger patients and the central atrophy in the older patients. (B) Electroretinography (ERG) of six patients sorted by age, commencing with the youngest in light gray and ending with the oldest in black. The normative data are highlighted in gray bars. Under scotopic light conditions, the ERG (indicated by ‘DA’, left) shows some decline, which may be explained by an ageing effect. The photopic ERG (indicated by ‘LA’, middle: single flash; right: 30 Hz flicker) shows a decline in the amplitude of the a- and b-wave.
Past medical history was remarkable for hearing difficulties/otosclerosis in four patients from family ZD3 (Patients ZD3-V:1, ZD3-IV:8, ZD3-III:8, ZD3-III:11), presenting early in childhood or later. Hearing impairment and otosclerosis was not reported in the other families. Yet, it was not specifically asked for. The presence of hearing impairment/otosclerosis in members from family ZD3 suggests a genetic background. However, no data are available on analysis of variants in genes associated with hearing impairment in this family (Supplementary Material, Table S1).
Visual acuity was decreased in almost all eyes and fellow eyes symmetrically, ranging from 20/20 to 20/630, and declining with age. Kinetic visual field testing was performed in 12 of the 16 patients. External borders of the visual field were unremarkable in all eyes except for the eye that had undergone scleral buckling for retinal detachment (Patient ZD3-III:11). Static visual field testing was performed in 7 of the 16 patients and revealed symmetrical defects centrally in all tested eyes and fellow eyes.
Panel D-15 color vision testing was performed in nine patients, if possible using desaturated (five patients), if not using saturated color cups (four patients) (18). A ChromaTest (19) was performed in four patients. 28 Hue color vision testing (20) was performed in one patient. All tested eyes showed tritan defects.
Imaging studies showed alterations in color fundus photography, FAF and OCT, namely central hypoautofluorescence and paracentral hyperautofluorescence on FAF imaging in younger patients and well demarcated central hypoautofluorescence as in areolar atrophy in older patients (Fig. 3A). OCT imaging showed central thinning of the outer nuclear layer, paracentral hyporeflectivity of the retinal pigment epithelium and/or a subretinal cleft in the younger patients and central atrophy in older patients (Fig. 3A). Findings were bilaterally symmetrically, except for in two patients (RCD460-II:3 and ZD3-III:11), which was also reflected by their visual acuity (Table 2, Supplementary Material, Table S1).
Full field ERG testing was performed in 14 of the 16 patients, showing a range in responses from within normal limits to markedly reduced, both under scotopic and photopic lighting conditions but intra-individually more so under photopic than under scotopic lighting conditions, and declining with age (Fig. 3B). Findings were bilaterally symmetrical, except for the patient that had undergone surgery for retinal detachment in one eye (Patient ZD3-III:11).
Discussion
Interestingly, family ZD3 was first described by Hammerstein in 1991 as a family with adCD and circular defects of the retinal pigment epithelium at the macula (21). Our molecular and genetic data provide strong evidence that duplications at the IRXB gene cluster—covering IRX5 and IRX6—cause an early onset autosomal dominant inherited progressive CD with early blue cone involvement. The latter may be the most characteristic feature of the condition, and we could show that in our cohort, tritan defects were present in all patients that underwent color vision testing (14 patients), and already at a very young age. Generally, color vision defects in CD affect all three color axes due to parallel degeneration of the three cone photoreceptor subtypes (22). Other causes of tritan defects may well be excluded to be causative for the tritan color vision defects in our cohort, which has been thoroughly examined (18).
Our classification of the inherited retinal dystrophy described herein being a CD is derived from the fact that in our cohort, we could show a decline in ERG responses to light stimuli under photopic lighting conditions that exceeds what one would expect to be an ageing effect (Fig. 3B). Additionally, a progressive decrease in visual acuity starting early in life as well as the frequently reported glare sensitivity are also in line with the diagnosis of a CD (22). Despite the fact that some patients also reported difficulties seeing at night, the outer borders of the kinetic visual field exams were within normal limits, and the ERG responses to light stimuli under scotopic lighting conditions showed some decline, however in a range that could be an ageing effect (Fig. 3B). Nonetheless, the rod ERG did show some features suggestive for rod involvement, i.e. the a-wave amplitude was more reduced when compared to the b-wave amplitude, and the oscillatory potentials had a somewhat lumpy aspect. The imaging studies in our cohort are also in line with what has been described in patients with CD: In early stages, one may find a reduction in the thickness of the parafoveal retinal tissue, as well as irregularities with regards to the central photoreceptors, whereas in advanced disease, one would expect outer retinal atrophy including the retinal pigment epithelium (Fig. 2A) (22,23).
While no microhomologies were evident at the breakpoints, chromosome 16 and also the SRO and flanking sequences are rich in repetitive elements as retrieved by RepeatMasker analysis (https://www.repeatmasker.org) that may have contributed to the duplication events observed in our families. A review of CNV databases [i.e. DECIPHER (https://decipher.sanger.ac.uk/) (24) and Database of Genomic Variants (DGV) (http://dgv.tcag.ca/dgv/app/home)] (25) for the SRO revealed no comparable duplications as observed in our families with adCD. In these databases, the region presents with several small duplications located between the genes of the duplicated region [the International Genome Sample Resource (IGSR) (http://www.1000genomes.org/phase-3-structural-variant-dataset)] (26), and four very large duplications covering 8–39 Mb of genomic sequence on chromosome 16. Inquiries sent to the DECIPHER online tool indicated that none of the responding cases had symptoms comparable to the severe CD phenotype observed in our families. These patients ranged from 5 to 17 years of age. Two cases were reported to be hyperopic but ophthalmologic examination was normal. One case is already published and presents with visual acuity of 0.8, normal color vision, normal perimetry but abnormal macular pigmentation (27). A single 9.5 kp duplication covering IRX6 alone was predicted from short read whole genome sequencing in an anamnestically healthy but anonymous Turkish male subject (28); if this patient truly does not have a retinal phenotype, this would rule out that the duplication of IRX6 alone is the cause of the disease in our patients but would favor the hypothesis that the duplications observed in our families result in a misregulation of part or the whole the IRXB cluster.
To the best of our knowledge no human disease has been associated with variants or aberrations of IRX6 or CNVs covering the SRO, whereas loss-of-function missense and small indel variant in IRX5 have been associated with autosomal recessive Hamamy syndrome (MIM 611174) characterized by craniofacial dysmorphology, osteopenia, severe myopia, hearing loss and mild intellectual disability (29–31). No such disease features were observed in our adCD families with duplications at the IRXB gene cluster, except for the high myopia in 5/16 subjects and the hearing difficulties in four subjects of family ZD3. Notably, we did not find any putative pathogenic point mutation in IRX5 and IRX6 in our cohort of unsolved adCD/adCRD patients, but in total four families with large overlapping duplications, suggesting that the disease mechanism underlying this form of adCD is linked to these duplications, and possibly an increase in gene dosage of genes located at this locus.
Interpreting the genomic and phenotypic consequences of CNVs can be challenging. Whereas deletion CNVs often lead to haploinsufficiency, duplications may cause disease through triplosensitivity, gene disruption or gene fusion at breakpoints (32). Actually, Newman and coworkers showed that most duplications are in tandem in direct orientation adjacent to the original locus (32), as it is also the case in all three independent duplications identified in this study. Although our approach of breakpoint PCR, subcloning and sequencing by primer walking cannot be considered an unbiased approach, we cannot completely exclude a more complex embedding of the duplication in the genome. This could only be elucidated by whole genome long-read sequencing.
The SRO encompasses three protein coding genes, IRX5, IRX6 and MMP2, the first two completely, the last partially, but also some lincRNAs as well as regulatory elements [i.e. enhancers, topologically associating domain (TAD) boundaries and promoters] and is interesting from the structural point of view (Fig. 1) (4,5). The IRXB gene cluster of Iroquois transcription factors comprises IRX3, IRX5 and IRX6. The composition and order of genes is conserved in vertebrates, and evolutionary convergent clustering of IRX genes has been observed in lower metazoans including Drosophila (33,34). A hallmark of the IRX gene clusters is the complexity in genomic architecture with a large number of ultraconserved non-coding sequence elements (UCNEs) interspersed between the genes (UCNEbase; http://genome-euro.ucsc.edu/index.html) (35) (Supplementary Material, Table S2). Most of these UCNEs are conserved throughout evolution. Some of the UCNEs have validated enhancer activity and eventually interact to govern expression of the IRX genes within functional genomic territories. A strong enhancer element located between the head-to-head oriented IRX3 and IRX5 genes dominates expression of these two genes (Fig. 1A) (5). This likely explains the similar expression profiles for IRX3 and IRX5, while the expression of IRX6 is more distinct (8,11). As presented in Figure 1A, apart from these protein coding genes, there is a complex interdependent array of regulatory elements, i.e. enhancers, lincRNAs and one ultraconserved domain that at least to some extent is conserved throughout evolution (4,5).
The SRO of the duplications described herein encompasses IRX5, IRX6, several lincRNAs and parts of the MMP2 gene. We consider the latter unlikely causative for the disease because while MMP2 is duplicated in its entirety in families ZD178 and RCD460, it is only partially duplicated in families ZD3 and ZD346. We rather suggest that increased gene dosage or the disruption of regulatory domains and elements governing spatiotemporal expression of IRX6, or both IRX5 and IRX6 (and the flanking lincRNA genes) underlie the phenotype of adCD with early blue cone involvement in our families, possibly due to the creation of a new TAD flanked by the insulators as boundaries (Fig. 1A).
We demonstrated that the IRX5 and IRX6 genes are expressed in the retina and that the allelic transcript proportions reflect the increased gene dosage due to the duplication (Supplementary Material, Fig. S4). Moreover, our zebrafish experiments support that increased expression of Irx genes can result in impaired visual function in 5 dpf larvae (Fig. 2C). Dose-adjusted overexpression of irx6a demonstrated significantly reduced OKR for stimuli of different contrasts and temporal and spatial frequencies, while overexpression of irx5a alone did not have a significant effect on the OKR. Yet there may be a synergistic effect when both irx6a and irx5a are overexpressed as suggested by the severely impaired OKR in this situation.
It is plausible that an extra copy of the SRO-encompassed genes leads to a pathologic increase in expression. Such gene dosage mechanisms of disease are well described, and nowadays, reports on the identification of CNVs and in particular on duplications are increasingly being published and also associated with inherited retinal disease: Only recently, the genetic cause of autosomal dominant North Carolina macular dystrophy (MCDR) was finally identified (36–38). In most MCDR1 families, the disease is caused by single nucleotide variants within a DNase 1 hypersensitivity site upstream of both the PRDM13 and CCNCC genes on chromosome 6q16.2 (MCDR1 locus, MIM 136550) (38–40), but also tandem duplications have been described (38–41). Manes and coworkers showed that overexpression of CG13296 (PRDM13) leads to severe loss of imaginal eye-antennal disc of Drosophila (41). In addition, duplications at the MCDR3 locus (MIM 608850) on chromosome 5p15.33-p13.1 affect the IRX1 gene (38,42), IRX1 belonging to the IRXA cluster.
Recently complex structural variants including duplications, triplications and inversions at the RP17 locus on chromosome 17q22 have been shown to cause autosomal dominant retinitis pigmentosa with the SRO affecting the protein coding genes YPEL2 and GDPD1 and the lincRNA LINC0147 (43). Within this comprehensive study, impairment of the TAD has been demonstrated in patient’s material (i.e. photoreceptor precursor cells or retinal organoids). The structural variants lead to the creation of new TADs with ectopic contacts between GDPD1 and retinal enhancers, which in turn lead to increased expression of GDPD1 and increased expression of the retinal enhancer, which is the likely mechanism of disease, consistent with a dominant gain of function (43).
All these findings may be considered an additional important link to our findings, implying that the IRX genes are not only important for eye and retinal development but also specifically play a role in cone photoreceptor and macula development, maintenance and survival.
Yet, it also cannot be excluded that the chromosomal rearrangement in our probands not only results in a duplication of several genes, but also in repositioning of enhancer elements that alter expression of other downstream target genes (5), or alteration of the expression of genes neighboring the breakpoints. We have here mimicked the effect of an increased expression by injecting Irx5a and Irx6a cRNA in zebrafish oocytes—the two IRX genes duplicated in our patients—and assessing the effect of these increased amounts on development of the zebrafish retina and visual function. But it has to be noted that the true effect of this disease-associated duplication might also involve or disrupt correct expression of all three genes in the IRXB cluster.
In conclusion, we provide compelling clinical, genetic, molecular and functional data supporting that tandem duplications affecting the IRXB gene locus on chromosome 16q12 are associated with an autosomal dominantly inherited specific form of CD with early tritanopic color vision defects.
Material and Methods
Patients and families
The study was conducted pro- and retrospectively (1989–2020) in accordance with the tenets of the World Medical Association Declaration of Helsinki and approval was obtained from the respective local research and ethical boards or dependent on the local regulatory bodies at the time the patients were seen. Specifically, the study was approved by the Ethics Board of the Medical Faculty, Eberhard Karls University, Tübingen, under the study no. 349/2003V and 116/2015BO2. Venous blood was taken from patients and family members after informed consent. Total genomic DNA was extracted according to standard procedures.
Ophthalmological examination
Patients were examined at the Ophthalmological Centers in Tübingen and Munich, Germany, and Utrecht, the Netherlands. Comprehensive ophthalmological examinations were performed, including psychophysical testing (best-corrected visual acuity, visual field and color vision tests), full field electroretinography (ERG) and multimodal imaging [color fundus photography, fundus autofluorescence (FAF) and spectral domain optical coherence tomography (OCT)] depending on the local setup and availability.
Linkage analysis and arrayCGH
Genotyping was performed using SNP arrays (250k Nsp Array, Affymetrix, Santa Clara, CA, USA). Genome-wide parametric linkage analysis was calculated with the easyLINKAGE package (13). ArrayCGH (comparative genome hybridization) analysis was performed using human CGH 385 K chromosome 16 tiling arrays (Roche NimbleGen Inc., Madison, WI, USA).
Breakpoint mapping
Breakpoint mapping was performed by long-range PCR using TaKaRa LA Taq Kit (Thermo Fisher Scientific, Waltham, MA USA) or Long Amp Taq DNA polymerase (NEB, New England Biolabs GmbH, Ipswich, MA, USA) and primers were selected dependent on the most likely physical breakpoint position deduced from the arrayCGH breakpoints according to the rise in the log2 ratios. Amplified PCR products were cloned in pCR2.1 using the TA Cloning Kit (Thermo Fisher Scientific) and the breakpoint identified by primer walking. Primer pairs closely flanking the breakpoints were designed and used for breakpoint-specific segregation analysis (for primers see Supplementary Material, Table S3).
Quantitative PCR assay to search for copy-number variants
Allele quantification for IRX6 by qPCR was performed applying the QuantiTec SYBR Green PCR Kit (QIAGEN, Hilden, Germany) on an Applied Biosystems 7500 Real-Time PCR system using autosettings (cycle threshold (Ct) = 0.2) and primers IRX6-Ex4_SYBR 5′-AAACCCCTACCCCACTAAGG-3′ and 5′-GTCCTCCTCTCCTCCCTC-3′ and SDC4 5′-CAGGGTCTGGGAGCCAAGT-3′ and 5′-GCACAGTGCTGGACATTGACA-3′. The obtained data were normalized against a reference gene (SDC4) (44). To each run, a control sample was added. All samples were measured as triplicates, and for data analysis, the ΔΔCt value and the standard deviation were calculated.
Segregation analysis
Segregation analysis was performed either by breakpoint PCR or allele quantification applying quantitative real-time PCR (qPCR) as described above.
Exome sequencing
We performed exome sequencing on the siblings III:1 and III:2 of family ZD346 as described previously (45).
Database submission
Novel variants have been submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) (46) with accession numbers SCV001479359, SCV001479360 and SCV001479361.
RNA-seq
We used RNA-seq data from retinal RNA of two independent eye donors as described previously (14).
Pyrosequencing for allele-specific quantification of IRX transcripts in patient-derived fibroblasts
Fibroblast cell cultures of patients ZD3-IV:8 and ZD178-III:2 were used to perform allele-specific expression quantification of IRX5 and IRX6, respectively, in comparison to a healthy control cell line by pyrosequencing. Genomic DNA and RNA from the different fibroblast cell lines were isolated at a passage of p7–9, and RNA reverse transcribed to cDNA. For statistical data evaluation, technical replicates were conducted. Out of each fibroblast sample, one RNA sample was isolated and two independent cDNA samples were synthesized. From each cDNA sample, two independent PCRs were conducted and four independent pyrosequencing experiments. The following primers were used to quantify the expression levels of IRX5 and IRX6 transcripts by pyrosequencing: IRX6_EX2_Hs_rs74412242 primer pair 5′-GTAGGGTGCGCAGCAGAGAG-3′ and 5′-[Biotin]-TTCTGGCGTCGGCAAGTT-3′; IRX5_EX3_Hs_rs13336114 primer pair 5′-[Biotin]-CTCCGGCGGGGTAGTGAG-3′ and 5′-AGCAGAAGGCGGCTTCGG-3′. Pyrosequencing was carried out using a PyroMark Q96 Workstation (QIAGEN) and a PyroMark Q96 ID (QIAGEN) analogous to previous description (14).
Zebrafish strain and husbandry
Zebrafish were bred and maintained at the aquarium of the Institute for Ophthalmic Research, Centre for Ophthalmology, University Clinics, Tübingen. All procedures were performed with permission of local authorities (Regierungspräsidium Tübingen, Tübingen, Germany) and conducted in accordance with German legislation on the protection of animal welfare.
Specifically, zebrafish of the Tuebingen strain were used and kept under 14 h light/10 h dark cycle at 27.5°C under standard conditions (47). Zebrafish larvae were kept in E3 medium (5 mm NaCl, 0.17 mm KCl, 0.33 mm CaCl2, 0.33 mm MgSO4, 0.00001% (w/v) methylene blue) at 28°C and staged according to Kimmel and coworkers (48) up to 5 dpf (days postfertilization).
Genetic engineering in zebrafish experiments
Total RNA was extracted from 5 dpf zebrafish with RNeasy Mini Kit (Qiagen, Hilden, Germany). Single strand cDNA synthesis was performed with random hexamers using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche, Mannheim, Germany). Primers containing overhangs for PmeI restriction site were designed for irx5a and irx6a, spanning from most of the 5′ to the 3′ untranslated regions (irx5a: NM_001045227.1; irx6a: NM_001023575.1). PCR amplifications were carried out with PfuUltra II Fusion HS DNA Polymerase (Agilent Technologies, Santa Clara, California, USA). Prior to cloning into pcDNA3.1/Zeo(+) vector (Invitrogen; Thermo Fisher Scientific, Inc.), the plasmid underwent site-directed mutagenesis to create an EcoRI restriction site at the end of the multiple cloning site for later linearization. DpnI (NEB) digestion was used to eliminate original vectors. Plasmid was subjected to blunt-end enzymatic digestion with PmeI (NEB), and CIP (NEB) was used to dephosphorylate the 5′ ends of DNA. MinElute Gel Extraction Kit (Qiagen, Hilden, Germany) was used to purify digested fragments. Ligation of inserts and plasmid was carried on with T4 DNA Ligase (NEB). Escherichia coli 10β competent cells (NEB) were transformed with the ligation products and then plated onto petri dishes (Agar-LB-Amp). Isolated colonies were picked and vectors amplified by colony PCR to detect correctly inserted constructs. Same isolated colonies were grown in 15 ml tubes with 5 ml of LB and 1.5 μl/ml of ampicillin (Carl Roth GmbH & Co. KG, Karlsruhe, Germany). Finally, minipreps (Peqlab, Erlangen, Germany) were carried out to obtain purified constructs.
irx5a and irx6a constructs were subjected to site-directed mutagenesis with primers designed with the online tool PrimerX (http://www.bioinformatics.org/primerx/cgi-bin/DNA_1.cgi) to generate silent/missense variants (for irx5a: c.288G>A and irx6a: c.345T>A) to be able to distinguish between endogenously expressed and injected mRNA in the pyrosequencing assays. A second transcript set (referred to as irx5astop und irx6astop) was generated that represents mutated inactive forms of these transcripts due to introduction of premature termination codons and stop mutations (irx5a: c.255T>A and irx6a: c.78C>A). PfuUltra II Fusion HS DNA Polymerase (Agilent Technologies, Santa Clara, California, USA) was used for PCR amplification. Original construct template was digested with DpnI (NEB). Confirmation of correctly performed insertions and site-directed mutagenesis was carried out with Sanger sequencing as follows: dye-terminator chemistry (Big Dye Termination chemistry; Applied Biosystems [ABI], Weiterstadt, Germany) was used for sequencing reaction. Products were separated on a DNA capillary sequencer (3100 Genetic Analyzer; ABI, Weiterstadt, Germany) and analyzed with the Sequence Analysis software (Vers. 5.1; ABI) and sequence trace alignment software (SeqMan; DNASTAR, Madison, WI, USA).
Limitation: The variant c.345T>A introduced a missense change (p.S115R) into Irx6a. Similar and comparable missense variants are observed in the general healthy human population (gnomAD). The amino acid residue is not conserved and located downstream of the homeobox domain. To the best of our knowledge, we believe that this missense variant does not have an impact onto Irx6a function, stability or our zebrafish experiments and results, as missense mutations in IRX6 have not been linked to any human (ocular) disease till this day.
Constructs were linearized with EcoRI (NEB) for the in vitro transcription of irx5a and irx6a (silent and stop modifications) with MessageMAX™ T7 ARCA-Capped Message and A-Plus™ Poly(A) Polymerase Tailing Kit (CELLSCRIPT, Wisconsin, USA). Copy RNA (cRNA) was purified using the RNeasy Mini Kit (Qiagen, Hilden, Germany).
Microinjection into zebrafish oocytes
The cRNA microinjections into zebrafish oocytes were performed using a stereo microscope Olympus SZX7 (Olympus, Valley Center, PA, USA) and a Microinjector FemtoJet 5247 (Eppendorf, Hamburg, Germany). Injection needles were generated from borosilicate glass capillaries (1B100F-4; World Precision Instruments, Sarasota, FL, USA) with a Narishige PC-10 Micropipette/Microelectrode Puller (NARISHIGE, Tokyo, Japan). Zebrafish oocytes at the one-cell stage were injected with 50 pg of in vitro transcribed cRNA in a total volume of 3 nl per oocyte. The cRNA solution was supplemented with 0.1% (v/v) Phenol Red (Sigma-Aldrich, MO, USA) for better visualization during microinjection. From each clutch, a portion of the eggs was kept as untreated control. To assess toxic effects due to cRNA injection, the fraction of dead, deformed and morphological normal larvae was determined for treated and untreated larvae up to 5 dpf. We found that after injection of 50 pg of irx5a, irx6a or irx6astop cRNA per oocyte >47% of the resulting larvae developed normal (Supplementary Material, Fig. S5). The irx5astop transcript did not show any toxic effect compared to untreated larvae. We did not observe additive effects of combined injection of 50 pg irx5a and 50 pg irx6a transcripts. Only morphologically normal larvae were subjected to further experiments (i.e. pyrosequencing and functional evaluation using the OKR).
Pyrosequencing for relative comparison between endogenously expressed irx5a and irx6a versus injected transcripts
irx5a and irx6a sequence modifications were used to distinguish between the endogenously expressed and injected transcripts—at 1 and 5 dpf—by means of pyrosequencing. RNA was extracted from pools of 1 dpf embryos or 5 dpf larvae using Lysing matrix D (FastPrep®, MP Biomedicals, LLC, Solon, Ohio, USA) and homogenization with Precellys 24 (Bertin Technologies, France) and purifying the RNA with RNeasy Mini Kit (Qiagen; Hilden, Germany). For injected transcript quantification, the pyrosequencing allele quantification method was selected. All primers and assays were designed with PyroMark Assay Design Software 2.0 (QIAGEN, Hilden, Germany) using allele quantification (AQ) settings. Primers were checked for off-target pairing with the BLAST-like alignment tool (BLAT, http://genome.ucsc.edu/). PCRs from single-strand cDNA syntheses (Transcriptor High Fidelity cDNA Synthesis Kit; Roche, Mannheim, Germany) obtained from three independent injected clutches per condition were performed. Streptavidin Sepharose™ High Performance beads were used for biotinylated strand separation. Pyrosequencing reaction was carried out with PyroMark® Gold Q96 Reagents (QIAGEN, Hilden, Germany) using a PyroMark Q96 Workstation (QIAGEN, Hilden, Germany) and a PyroMark Q96 ID (QIAGEN, Hilden, Germany).
Optokinetic response analysis on zebrafish larvae overexpressing irx5a or/and irx6a
To check visual impairment due to increased dosages of IRX5 and IRX6, we injected their respective zebrafish irx5a and irx6a orthologous cRNAs into one-cell stage zebrafish oocyte. Biological triplicates of single cRNA injections of irx5a and irx6a and combined injections of modified mRNAs of Irx5a and Irx6a were performed (with 50 pg of mRNA for each gene). Each triplicate consisted in series of eight treated larvae and five untreated controls from the same clutch. Likewise, controls consisting in combined injections of inactive 50 pg of irx5a and irx6a each were analyzed to rule out that cRNA injections by themselves might damage the OKR.
Zebrafish eye detection and tracking as well as visual stimulation with moving bars were carried out with ZebEyeTrack, a LabVIEW-based software for real-time measurement of angular eye position based on a precursory software (49). Its source code is available for free, and hands-on protocols have been published (50). The stimulus protocol for the moving bars was adapted from previous publications (51,52) and generated in ZebEyeTrack. Stimuli with varying contrast (from 0 to 1, 0–89.2% of Michelson contrast) as well as temporal and spatial frequencies were displayed on four 3.5″ computer screens arranged in a square around the animal. The stimulus protocol consisted of several motion phases, which differed in contrast, temporal frequency, spatial frequency and direction (Supplementary Material, Table S4). Stimuli were shown in the order and timing given in Supplementary Material, Table S4. The contrast, temporal and spatial stimulus protocol parts were separated by a control condition phase in which a strong OKR stimulus was presented, serving as a positive control in-between different experimental stimulus phases of reduced strength. To prevent body movement, except from eye rotation, the analyzed 5 dpf larvae were embedded into 3% methyl-cellulose and recorded with a CCD camera using infrared (850 nm) illumination. The software allowed us to semiautomatically analyze optokinetic response (OKR) performance, including the gain of the OKR by first detecting saccade time points and then fitting a line to the postsaccadic slow phase.
Supplementary Material
Contributor Information
Susanne Kohl, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany.
Pablo Llavona, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany.
Alexandra Sauer, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany.
Peggy Reuter, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany.
Nicole Weisschuh, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany.
Melanie Kempf, University Eye Hospital, Centre for Ophthalmology, University of Tübingen, Universitätsklinikum Tübingen, Tübingen 72076, Germany; Center for Rare Eye Diseases, University of Tübingen, Tübingen 72076, Germany.
Florian Alexander Dehmelt, Werner Reichardt Centre for Integrative Neuroscience and Institute of Neurobiology, University of Tübingen, Tübingen 72076, Germany.
Aristides B Arrenberg, Werner Reichardt Centre for Integrative Neuroscience and Institute of Neurobiology, University of Tübingen, Tübingen 72076, Germany.
Ieva Sliesoraityte, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany.
Eberhart Zrenner, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany; Werner Reichardt Centre for Integrative Neuroscience and Institute of Neurobiology, University of Tübingen, Tübingen 72076, Germany.
Mary J van Schooneveld, Department of Ophthalmology, Amsterdam University Medical Centre, Amsterdam 1100 DD, The Netherlands; Bartiméus Diagnostic Department, Zeist, The Netherlands.
Günther Rudolph, Department of Ophthalmology, University Hospital, LMU Munich, München 80336, Germany.
Laura Kühlewein, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany; University Eye Hospital, Centre for Ophthalmology, University of Tübingen, Universitätsklinikum Tübingen, Tübingen 72076, Germany.
Bernd Wissinger, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany.
Funding
The Deutsche Forschungsgemeinschaft (grant no. KFO134-Ko2176/1); Werner Reichardt Centre for Integrative Neuroscience (EXC307); the European Union’s Seventh Framework Programme for research, technological development and demonstration (317472).
Conflict of Interest statement. The authors declare no conflict of interest. S.K. has been a consultant to Novartis with respect to another project.
References
- 1. Michaelides, M., Hardcastle, A.J., Hunt, D.M. and Moore, A.T. (2006) Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv. Ophthalmol., 51, 232–258. [DOI] [PubMed] [Google Scholar]
- 2. Roosing, S., Thiadens, A.A., Hoyng, C.B., Klaver, C.C., den Hollander, A.I. and Cremers, F.P. (2014) Causes and consequences of inherited cone disorders. Prog. Retin. Eye Res., 42, 1–26. [DOI] [PubMed] [Google Scholar]
- 3. Peters, T., Dildrop, R., Ausmeier, K. and Rüther, U. (2000) Organization of mouse Iroquois homeobox genes in two clusters suggests a conserved regulation and function in vertebrate development. Genome Res., 10, 1453–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pennacchio, L.A., Ahituv, N., Moses, A.M., Prabhakar, S., Nobrega, M.A., Shoukry, M., Minovitsky, S., Dubchak, I., Holt, A., Lewis, K.D. et al. (2006) In vivo enhancer analysis of human conserved non-coding sequences. Nature, 444, 499–502. [DOI] [PubMed] [Google Scholar]
- 5. Tena, J.J., Alonso, M.E., de la Calle-Mustienes, E., Splinter, E., de Laat, W., Manzanares, M. and Gómez-Skarmeta, J.L. (2011) An evolutionarily conserved three-dimensional structure in the vertebrate Irx clusters facilitates enhancer sharing and coregulation. Nat. Commun., 2, 310. [DOI] [PubMed] [Google Scholar]
- 6. Bryan, J.M., Fufa, T.D., Bharti, K., Brooks, B.P., Hufnagel, R.B. and McGaughey, D.M. (2018) Identifying core biological processes distinguishing human eye tissues with precise systems-level gene expression analyses and weighted correlation networks. Hum. Mol. Genet., 27, 3325–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Swamy, V. and McGaughey, D. (2019) Eye in a disk: eyeIntegration human pan-eye and body transcriptome database version 1.0. Invest. Ophthalmol. Vis. Sci., 60, 3236–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mummenhoff, J., Houweling, A.C., Peters, T., Christoffels, V.M. and Rüther, U. (2001) Expression of Irx6 during mouse morphogenesis. Mech. Dev., 103, 193–195. [DOI] [PubMed] [Google Scholar]
- 9. Star, E.N., Zhu, M., Shi, Z., Liu, H., Pashmforoush, M., Sauve, Y., Bruneau, B.G. and Chow, R.L. (2012) Regulation of retinal interneuron subtype identity by the Iroquois homeobox gene Irx6. Development, 139, 4644–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bosse, A., Stoykova, A., Nieselt-Struwe, K., Chowdhury, K., Copeland, N.G., Jenkins, N.A. and Gruss, P. (2000) Identification of a novel mouse Iroquois homeobox gene, Irx5, and chromosomal localisation of all members of the mouse Iroquois gene family. Dev. Dyn., 218, 160–174. [DOI] [PubMed] [Google Scholar]
- 11. Cohen, D.R., Cheng, C.W., Cheng, S.H. and Hui, C.C. (2000) Expression of two novel mouse Iroquois homeobox genes during neurogenesis. Mech. Dev., 91, 317–321. [DOI] [PubMed] [Google Scholar]
- 12. Cheng, C.W., Chow, R.L., Lebel, M., Sakuma, R., Cheung, H.O., Thanabalasingham, V., Zhang, X., Bruneau, B.G., Birch, D.G., Hui, C.C. et al. (2005) The Iroquois homeobox gene, Irx5, is required for retinal cone bipolar cell development. Dev. Biol., 287, 48–60. [DOI] [PubMed] [Google Scholar]
- 13. Lindner, T.H. and Hoffmann, K. (2005) easyLINKAGE: a PERL script for easy and automated two−/multi-point linkage analyses. Bioinformatics, 21, 405–407. [DOI] [PubMed] [Google Scholar]
- 14. Llavona, P., Pinelli, M., Mutarelli, M., Marwah, V.S., Schimpf-Linzenbold, S., Thaler, S., Yoeruek, E., Vetter, J., Kohl, S. and Wissinger, B. (2017) Allelic expression imbalance in the human retinal transcriptome and potential impact on inherited retinal diseases. Genes (Basel), 8, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brockerhoff, S.E., Hurley, J.B., Janssen-Bienhold, U., Neuhauss, S.C., Driever, W. and Dowling, J.E. (1995) A behavioral screen for isolating zebrafish mutants with visual system defects. Proc. Natl. Acad. Sci. USA., 92, 10545–10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bilotta, J., Saszik, S. and Sutherland, S.E. (2001) Rod contributions to the electroretinogram of the dark-adapted developing zebrafish. Dev. Dyn., 222, 564–570. [DOI] [PubMed] [Google Scholar]
- 17. Pruett, R.C. (1983) Retinitis pigmentosa: clinical observations and correlations. Trans. Am. Ophthalmol. Soc., 81, 693–735. [PMC free article] [PubMed] [Google Scholar]
- 18. Melamud, A., Hagstrom, S. and Traboulsi, E. (2004) Color vision testing. Ophthalmic Genet., 25, 159–187. [DOI] [PubMed] [Google Scholar]
- 19. Wong, R., Khan, J., Adewoyin, T., Sivaprasad, S., Arden, G.B. and Chong, V. (2008) The Chroma test, a digital color contrast sensitivity analyzer, for diabetic maculopathy: a pilot study. BMC Ophthalmol., 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amos, J.F. and Piantanida, T.P. (1977) The Roth 28-hue test. Am. J. Optom. Physiol. Optic, 54, 171–177. [DOI] [PubMed] [Google Scholar]
- 21. Hammerstein, W. (1991) Dystrophien der Retina. Thieme, Stuttgart, ISBN-10: 3432992610/ISBN-13: 978-3432992617, pp. 51–53 Figures 112, 113, 114. [Google Scholar]
- 22. Gill, J.S., Georgiou, M., Kalitzeos, A., Moore, A.T. and Michaelides, M. (2019) Progressive cone and cone-rod dystrophies: clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol., 103, 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kellner, U. and Kellner, S. (2009) Clinical findings and diagnostics of cone dystrophy. Ophthalmologe, 106, 99–108. [DOI] [PubMed] [Google Scholar]
- 24. Firth, H.V., Richards, S.M., Bevan, A.P., Clayton, S., Corpas, M., Rajan, D., Van Vooren, S., Moreau, Y., Pettett, R.M. and Carter, N.P. (2009) DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet., 84, 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mac Donald, J.R., Ziman, R., Yuen, R.K., Feuk, L. and Scherer, S.W. (2014) The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res., 42, D986–D992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park, H., Kim, J.I., Ju, Y.S., Gokcumen, O., Mills, R.E., Kim, S., Lee, S., Suh, D., Hong, D., Kang, H.P. et al. (2010) Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat. Genet., 42, 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hansson, K., Dauwerse, H., Gijsbers, A., van Diepen, M., Ruivenkamp, C. and Kant, S. (2010) Interstitial duplication in the proximal long arm of chromosome 16. Am. J. Med. Genet. Part A, 152A, 1858–1861. [DOI] [PubMed] [Google Scholar]
- 28. Dogan, H., Can, H. and Out, H.H. (2014) Whole genome sequence of a Turkish individual. PLoS One, 9, e85233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamamy, H.A., Teebi, A.S., Oudjhane, K., Shegem, N.N. and Ajlouni, K.M. (2007) Severe hypertelorism, midface prominence, prominent/simple ears, severe myopia, borderline intelligence, and bone fragility in two brothers: new syndrome? Am. J. Med. Genet. Part A, 143A, 229–234. [DOI] [PubMed] [Google Scholar]
- 30. Bonnard, C., Strobl, A.C., Shboul, M., Lee, H., Merriman, B., Nelson, S.F., Ababneh, O.H., Uz, E., Güran, T., Kayserili, H. et al. (2012) Mutations in IRX5 impair craniofacial development and germ cell migration via SDF1. Nat. Genet., 44, 709–713. [DOI] [PubMed] [Google Scholar]
- 31. Posey, J.E., Harel, T., Liu, P., Rosenfeld, J.A., James, R.A., Coban Akdemir, Z.H., Walkiewicz, M., Bi, W., Xiao, R., Ding, Y. et al. (2017) Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med., 376, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Newman, S., Hermetz, K.E., Weckselblatt, B. and Rudd, M.K. (2015) Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am. J. Hum. Genet., 96, 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Irimia, M., Maeso, I. and Garcia-Fernàndez, J. (2008) Convergent evolution of clustering of Iroquois homeobox genes across metazoans. Mol. Biol. Evol., 25, 1521–1525. [DOI] [PubMed] [Google Scholar]
- 34. Kerner, P., Ikmi, A., Coen, D. and Vervoort, M. (2009) Evolutionary history of the Iroquois/Irx genes in metazoans. BMC Evol. Biol., 9, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dimitrieva, S. and Bucher, P. (2013) UCNEbase—a database of ultraconserved non-coding elements and genomic regulatory blocks. Nucleic Acids Res., 41, D101–D109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Small, K.W., Weber, J.L., Roses, A., Lennon, F., Vance, J.M. and Pericak-Vance, M.A. (1992) North Carolina macular dystrophy is assigned to chromosome 6. Genomics, 13, 681–685. [DOI] [PubMed] [Google Scholar]
- 37. Small, K.W., Puech, B., Mullen, L. and Yelchits, S. (1997) North Carolina macular dystrophy phenotype in France maps to the MCDR1 locus. Mol. Vis., 3, 1. [PubMed] [Google Scholar]
- 38. Small, K.W., DeLuca, A.P., Whitmore, S.S., Rosenberg, T., Silva-Garcia, R., Udar, N., Puech, B., Garcia, C.A., Rice, T.A., Fishman, G.A. et al. (2016) North Carolina macular dystrophy is caused by dysregulation of the retinal transcription factor PRDM13. Ophthalmology, 123, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bowne, S.J., Sullivan, L.S., Wheaton, D.K., Locke, K.G., Jones, K.D., Koboldt, D.C., Fulton, R.S., Wilson, R.K., Blanton, S.H., Birch, D.G. and Daiger, S.P. (2016) North Carolina macular dystrophy (MCDR1) caused by a novel tandem duplication of the PRDM13 gene. Mol. Vis., 22, 1239–1247. [PMC free article] [PubMed] [Google Scholar]
- 40. Silva, R.S., Arno, G., Cipriani, V., Pontikos, N., Defoort-Dhellemmes, S., Kalhoro, A., Carss, K.J., Raymond, F.L., Dhaenens, C.M., Jensen, H. et al. (2019) Unique noncoding variants upstream of PRDM13 are associated with a spectrum of developmental retinal dystrophies including progressive bifocal chorioretinal atrophy. Hum. Mutat., 40, 578–587. [DOI] [PubMed] [Google Scholar]
- 41. Manes, G., Joly, W., Guignard, T., Smirnov, V., Berthemy, S., Bocquet, B., Audo, I., Zeitz, C., Sahel, J., Cazevieille, C. et al. (2017) A novel duplication of PRMD13 causes North Carolina macular dystrophy: overexpression of PRDM13 orthologue in drosophila eye reproduces the human phenotype. Hum. Mol. Genet., 26, 4367–4374. [DOI] [PubMed] [Google Scholar]
- 42. Cipriani, V., Silva, R.S., Arno, G., Pontikos, N., Kalhoro, A., Valeina, S., Inashkina, I., Audere, M., Rutka, K., Puech, B. et al. (2017) Duplication events downstream of IRX1 cause North Carolina macular dystrophy at the MCDR3 locus. Sci. Rep., 7, 7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Bruijn, S.E., Fiorentino, A., Ottaviani, D., Fanucchi, S., Melo, U.S., Corral-Serrano, J.C., Mulders, T., Georgiou, M., Rivolta, C., Pontikos, N. et al. (2020) Structural variants create new topological-associated domains and ectopic retinal enhancer-gene contact in dominant retinitis Pigmentosa. Am. J. Hum. Genet., 107, 802–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Preter, K., Speleman, F., Combaret, V., Lunec, J., Laureys, G., Eussen, B.H., Francotte, N., Board, J., Pearson, A.D., De Paepe, A. et al. (2002) Quantification of MYCN, DDX1, and NAG gene copy number in neuroblastoma using a real-time quantitative PCR assay. Mod. Pathol., 15, 159–166. [DOI] [PubMed] [Google Scholar]
- 45. Weisschuh, N., Mayer, A.K., Strom, T.M., Kohl, S., Glöckle, N., Schubach, M., Andreasson, S., Bernd, A., Birch, D.G., Hamel, C.P. et al. (2016) Mutation detection in patients with retinal dystrophies using targeted next generation sequencing. PLoS One, 11, e0145951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Landrum, M.J., Lee, J.M., Riley, G.R., Jang, W., Rubinstein, W.S., Church, D.M. and Maglott, D.R. (2014) ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res., 42, D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nüsslein-Volhard, C. and Dahm, R. (2002) Zebrafish. In A practical approach. UK. Oxford University Press, Oxford. [Google Scholar]
- 48. Kimmel, C.B., Ballard, W.W., Kimmel, S.R., Ullmann, B. and Schilling, T.F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn., 203, 253–310. [DOI] [PubMed] [Google Scholar]
- 49. Schoonheim, P.J., Arrenberg, A.B., Del Bene, F. and Baier, H. (2010) Optogenetic localization and genetic perturbation of saccade-generating neurons in zebrafish. J. Neurosci., 30, 7111–7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dehmelt, F.A., von Daranyi, A., Leyden, C. and Arrenberg, A.B. (2018) Evoking and tracking zebrafish eye movement in multiple larvae with ZebEyeTrack. Nat. Protoc., 13, 1539–1568. [DOI] [PubMed] [Google Scholar]
- 51. Rinner, O., Rick, J.M. and Neuhauss, S.C. (2005) Contrast sensitivity, spatial and temporal tuning of the larval zebrafish optokinetic response. Invest. Ophthalmol. Vis. Sci., 46, 137–142. [DOI] [PubMed] [Google Scholar]
- 52. Huber-Reggi, S.P., Mueller, K.P. and Neuhauss, S.C. (2013) Analysis of optokinetic response in zebrafish by computer-based eye tracking. Methods Mol. Biol., 935, 139–160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.