

Abstract

Melamine oligomers composed of repeating triazine-piperidine units and equipped with phenol and phosphine oxide side-chains form H-bonded duplexes. The melamine backbone provides sufficient rigidity to prevent intramolecular folding of oligomers up to three recognition units in length, leading to reliable duplex formation between sequence complementary oligomers. NMR spectroscopy and isothermal titration calorimetry (ITC) were used to characterize the self-assembly properties of the oligomers. For length-complementary homo-oligomers, duplex formation in toluene is characterized by an increase in stability of an order of magnitude for every base-pair added to the chain. NMR spectra of dilute solutions of the AD 2-mer show that intramolecular H-bonding between neighboring recognition units on the chain (1,2-folding) does not occur. NMR spectra of dilute solutions of both the AAD and the ADD 3-mer show that 1,3-folding does not take place either. ITC was used to characterize interactions between all pairwise combinations of the six different 3-mer sequences, and the sequence complementary duplexes are approximately an order of magnitude more stable than duplexes with a single base mismatch. High-fidelity duplex formation combined with the synthetic accessibility of the monomer building blocks makes these systems attractive targets for further investigation.
Introduction
The molecular nanotechnology found in nature is based on linear oligomers, where function is encoded by the sequence of monomer building blocks. The solid-phase methods originally developed to make synthetic polypeptides and oligonucleotides have also been used to prepare nonbiological oligomers with a defined sequence of different monomer building blocks.1−9 However, these synthetic oligomers are largely devoid of the kind of functional properties found in biopolymers. The properties of polypeptides are determined by the folded three-dimensional structure, which is in turn encoded by sequence. Although progress has been made in the prediction of folding patterns based on sequence, prediction of the function associated with a particular folded structure remains a major challenge in protein chemistry.10,11 We have therefore focused our attention on motifs inspired by nucleic acids. The ladder structure found in the DNA double helix is a relatively straightforward supramolecular target. Moreover, this structure is intrinsically linked to function and forms the basis for the template-directed synthesis used in molecular replication, transcription, and translation in biological systems.12 The synthesis of oligomers that form duplexes has turned out to be a tractable problem,13−46 and we have found that some of these compounds also fold and catalyze reactions in a manner reminiscent of nascent RNA function.47,48
We have described a number of noncovalent base-pairing systems that have been used for duplex formation between sequence-complementary oligomers via H-bonding interactions.41−46 We have also used a covalent base-pairing system for replication of sequence information from a parent template oligomer to a daughter copy strand.49 The main factor which limits the sequence selectivity achieved in both these systems is related to the conformational flexibility of the backbone. Here we describe a new class of oligomers with a more rigid backbone that leads to high-fidelity sequence-selective duplex formation using a noncovalent base-pairing system.
Figure 1 illustrates our approach to duplex-forming oligomers.41 The base-pairing system uses formation of a single H-bond between complementary donor (blue) and acceptor (red) side-chains attached to a nonpolar backbone. In nonpolar solvents, H-bonding interactions between the recognition units shown in Figure 1 are strong enough to ensure that the stability of the duplex increases by about an order of magnitude for every base-pair formed. The use of a single H-bond as the base-pairing motif minimizes the chances of mispairing, and the lack of polar groups on the backbone avoids any competitive H-bond equilibria. Length-complementary homo-oligomers form stable duplexes in predicable manner for a wide variety of different backbones.41−46 In contrast, duplex formation between the hetero-oligomers can be compromised by the competing intramolecular folding equilibria illustrated in Figure 1.50 The flexibility of the polythioether backbone shown in Figure 1a leads to interactions between neighboring recognition units (1,2-folding). The polyaniline backbone in Figure 1b is rigid enough to prevent 1,2-folding, but 1,3-folding significantly reduces the fidelity of sequence-selective duplex formation in these systems. Here we describe a new backbone that is sufficiently rigid to prevent 1,3-folding, leading to high-fidelity duplex formation between mixed-sequence 3-mers.
Figure 1.
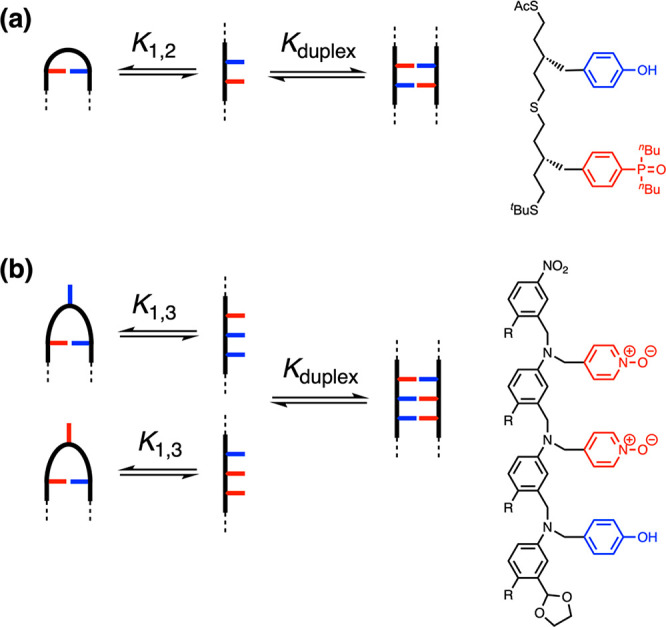
Duplex-forming oligomers. Intramolecular folding lowers duplex stability and reduces sequence-selectivity: (a) 1,2-folding on a polythioether backbone50 and (b) 1,3-folding on a polyaniline backbone.43
The approach to a rigid recognition-encoded melamine oligomer is illustrated in Figure 2. The monomer building blocks required for synthesis of oligomers must be equipped with a recognition unit and two reactive sites that allow polymerization. The oligomer in Figure 2 is an attractive target, because cyanuric chloride provides straightforward synthetic access to trifunctional monomers.51−54 Sequential temperature-controlled nucleophilic aromatic substitution reactions with secondary amines allow the stepwise functionalization of the triazine core in high yield (Figure 3a). Although the melamine backbone in Figure 2 appears to be relatively polar, the use of secondary amines ensures that the triazine nitrogen atoms are all sterically blocked from acting as H-bond acceptors (Figure 3b). The backbone is likely to adopt an extended conformation, because the piperazine linker has a strong preference for the chair conformation. A search of the Cambridge Structural Database found 79 crystal structures of nonmacrocyclic compounds which contain a piperazine linker where both nitrogen atoms are functionalized with sp2 carbons and where there is no disorder. The piperazine never adopts the boat conformation; the chair conformation is found in 77 of the 79 structures, and there are only two examples of a twist boat conformation.
Figure 2.
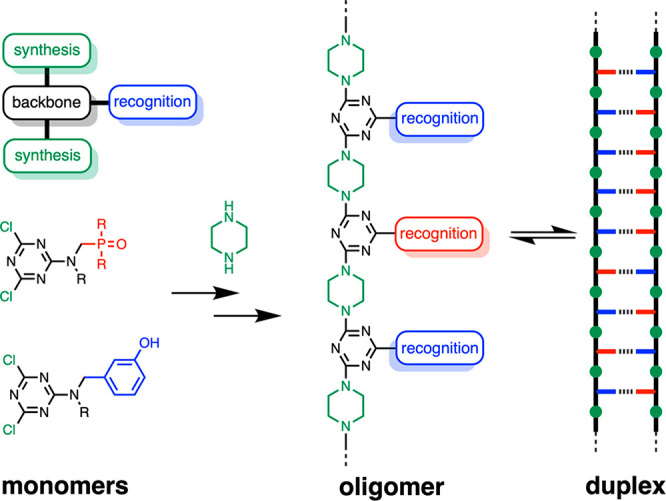
A blueprint for duplex-forming molecules. There are three key structural elements: recognition modules for base-pair formation (red and blue), functionality used for the synthesis of oligomers (green), and the backbone module which connects the three components of a monomer. Implementation in melamine oligomers equipped with a phenol·phosphine oxide base-pairing system is illustrated (R are solubilizing groups).
Figure 3.
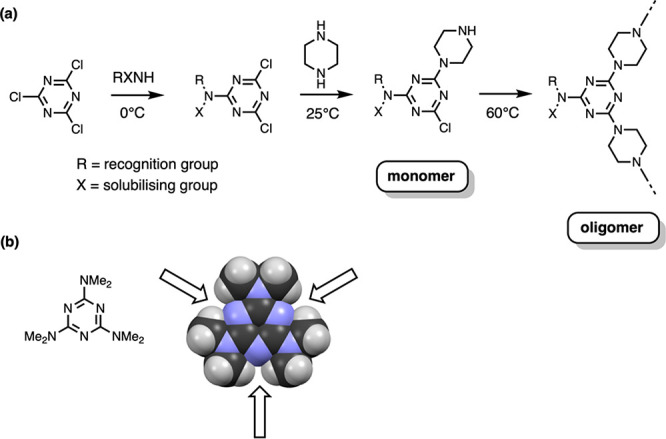
(a) Stepwise substitution of cyanuric chloride to give monomers required for the formation of melamine oligomers. (b) The X-ray crystal structure of 2,4,6-tris(dimethylamino)-1,3,5-triazine (CCDC ref code HMELAM) shows that access to the nitrogen lone pairs is sterically blocked in peralkylated melamines.
Results and Discussion
Synthesis
The H-bond donor and acceptor building blocks were synthesized from secondary amines 2 and 4, which have isobutyl solubilizing groups. Secondary amine 2 was obtained by protection of 3-hydroxybenzaldehyde using triisopropylsilyl chloride, followed by reductive amination with isobutylamine (Scheme 1). Secondary amine 4 was prepared from di-tert-butylchlorophosphine, which was treated with formaldehyde in strong aqueous acid to give phosphine oxide 3. Reaction of 3 with mesyl chloride, followed by microwave-assisted nucleophilic substitution with isobutylamine gave 4 (Scheme 1).
Scheme 1. Synthesis of 2 and 4: (a) TIPSCl, imidazole; (b) 1. i-butylamine, 2. NaBH4; (c) H2CO, HCl; (d) 1. MsCl, 2. i-butylamine.

Scheme 2 shows the synthesis of two different H-bond donor monomers. Monomer 5 has two reactive chlorotriazine sites for coupling with amine groups and was used to build up oligomer chains. Monomer 8 has a single reactive amine group and was used to terminate the chain end of an oligomer. Cyanuric chloride was reacted with secondary amine 2 in the presence of DIPEA at −10 °C to give 5. A second nucleophilic aromatic substitution with 1-Boc piperazine in the presence of DIPEA at 25 °C gave 6, which was then heated with excess piperidine in refluxing THF to afford 7. Deprotection with TFA gave 8.
Scheme 2. Synthesis of H-Bond Donor Monomers 5 and 8: (a) 2, DIPEA; (b) 1-Boc piperazine, DIPEA; (c) piperidine; (d) TFA.

A similar strategy was used to obtain the corresponding H-bond acceptor monomers 9 and 13 (Scheme 3). Reaction of cyanuric chloride with secondary amine 4 in the presence of DIPEA at −10 °C gave 9. Successive nucleophilic aromatic substitutions of cyanuric chloride with 1-Boc piperazine at −10 °C, secondary amine 4 at 25 °C, and piperidine at 66 °C gave 12. Deprotection with TFA gave 13.
Scheme 3. Synthesis of H-Bond Acceptor Monomers 9 and 13: (a) 4, DIPEA; (b) 1-Boc piperazine, DIPEA; (c) piperidine; (d) TFA.

Melamine derivative 15, which has a benzyl side-chain in place of the recognition units present in the other monomers, was synthesized as a reference compound to investigate the H-bonding properties of the backbone (Scheme 4). Secondary amine 14 was prepared from benzaldehyde and isobutylamine by reductive amination and reaction with cyanuric chloride at −10 °C, followed by refluxing with piperidine in THF gave 15.
Scheme 4. Synthesis of 15: (a) 1. Isobutylamine, 2. NaBH4; (b) 1. Cyanuric Chloride, DIPEA, 2. Piperidine.

A family of 13 recognition-encoded melamine oligomers of different length and sequence were synthesized as outlined in the Supporting Information. The 1-mers D and A were synthesized directly from 5 or 9 by reaction with piperidine in refluxing THF. Longer oligomers were synthesized using different combinations of the monomer building blocks 5, 8, 9, and 13, and Scheme 5 illustrates the general approach for the synthesis of mixed-sequence oligomers. Sequential piperazine-chlorotriazine coupling reactions were carried using DIPEA, and after assembly of the oligomer, any TIPS protecting groups present on phenol side chains were removed using TBAF.
Scheme 5. Synthetic Strategy for the Synthesis of a Mixed-Sequence 3-mer, Where R1, R2, and R3 Represent Different Combinations of the D and A Side Chains.

Duplex Formation by Homosequence Oligomers
NMR titration experiments were used to investigate the binding properties of the different components of the melamine oligomers in toluene. Addition of 1-mer D to 1-mer A led to large changes in the 31P NMR chemical shift of the signal due to the phosphine oxide group. The titration data fit well to a 1:1 binding isotherm with an association constant of 361 ± 9 M–1. The limiting complexation-induced change in 31P NMR chemical shift was 5.3 ppm, which is characteristic of formation of a phenol·phosphine oxide H-bond.41−46 In contrast, titration of melamine derivative 15 into m-cresol in toluene resulted in small changes in 1H NMR chemical shift, confirming that the alkyl substituents effectively shield the melamine nitrogen H-bond acceptor sites on the backbone, as suggested by the X-ray crystal structure shown in Figure 3b.
Interactions between length complementary homo-oligomers were investigated using isothermal titration calorimetry (ITC) in toluene. The data fit well to a 1:1 binding isotherm in all cases, and the results are summarized in Table 1. The complex formed by AA and DD is an order of magnitude more stable than the A·D complex, which indicates cooperative formation of two intermolecular H-bonds in the AA·DD complex. There is a significant increase in the stability of the complex for every recognition unit added to the oligomer, which shows that cooperative H-bonding interactions propagate with increasing chain length. In other words, the duplexes are fully assembled with H-bonds between each of the complementary recognition groups (Figure 4). Table 1 shows that there are compensating increases in the magnitudes of the enthalpy and entropy changes for duplex formation as the number of H-bonding interactions increases.
Table 1. Association Constants for Duplex Formation between Length-Complementary Homo-oligomers Measured by ITC Titration Experiments in Toluene at 298 Ka.
| complex | log K (M–1) | ΔG° (kJ mol–1) | ΔH° (kJ mol–1) | ΔS° (J K–1 mol–1) |
|---|---|---|---|---|
| A·Db | 2.6 | –14.6 | ||
| AA·DD | 3.7 | –20.9 | –29 | –27 |
| AAA·DDD | 5.2 | –29.5 | –57 | –92 |
| AAAA·DDDD | 5.9 | –33.8 | –92 | –195 |
Errors based on repeat experiments are 0.1 in log K, 0.5 kJ mol–1 in ΔG°, 5 kJ mol–1 in ΔH°, and 20 J K–1 mol–1 in ΔS°.
Measured by 31P NMR titration.
Figure 4.
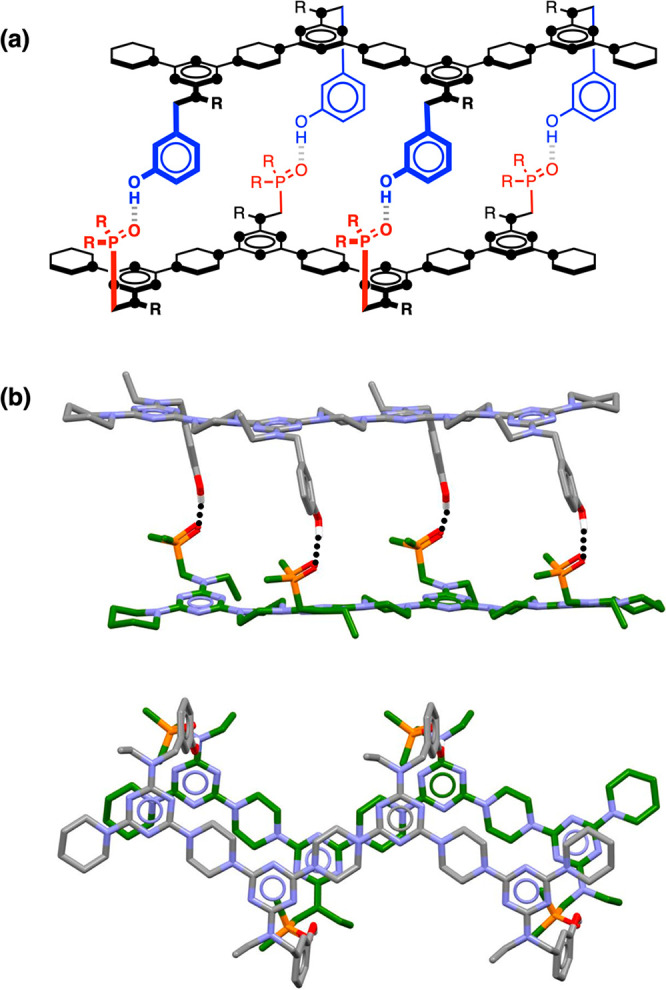
(a) Structure of the AAAA·DDDD duplex. The melamine nitrogen atoms are represented as black dots, and R’s are the solubilizing groups. (b) Side and top views of the energy minimum structure of the AAAA·DDDD duplex (MMFFs with chloroform solvation). H-bonds are indicated by black dotted lines. The solubilizing groups and hydrogen atoms are omitted for clarity.
Figure 5 shows the relationship between the association constant for duplex formation (log K) and the number of base-pairs (N). The linear relationship indicates that the stepwise effective molarities for sequential formation of each intramolecular H-bond in the duplex (EM) are approximately constant. The overall stability of the duplex is related to EM by eq 1, so the slope of the line of best fit in Figure 5 can be used to estimate the average effective molarity for intramolecular H-bonding in the duplex as 40 mM. This value is similar to the values of EM that we have reported previously for duplex formation by several different kinds of recognition-encoded oligomer.41−46
| 1 |
where Kref is the association constant for formation of a single intermolecular H-bond, which is measured using the A·D complex, and the statistical factor of 2 represents the degeneracy of the duplex.
Figure 5.

Relationship between the association constants for duplex formation between length-complementary homo-oligomers measured in toluene at 298 K (K) and the number of intermolecular H-bonds formed (N). The line of best fit shown is log K = 1.1N + 1.5.
Folding of Mixed-Sequence Oligomers
The self-assembly properties of mixed-sequence oligomers are generally more complicated, because there are multiple competing equilibria. In contrast to homo-oligomers, hetero-oligomers can fold because of intramolecular interactions between complementary recognition sites on the same molecule, and they can self-associate because of intermolecular interactions between complementary recognition sites on different molecules. The AD 2-mer was used to investigate the 1,2-folding process illustrated in Figure 1a. The 1H NMR spectra of the melamine oligomers are complicated because of the presence of multiple rotamers, which are in slow exchange on the NMR time scale. Slow exchange rotamers are also apparent in the 31P NMR spectrum of AD, but there is only one signal, and the weighted average value can be used to simplify the analysis. Figure 6 shows a 31P NMR dilution experiment for AD in chloroform. At low concentrations of AD, the chemical shift is similar to the values recorded for AA and for the TIPS-protected AD 2-mer, neither of which can form H-bonds. This observation indicates that there are no intramolecular H-bonds in the single stranded monomeric state of AD; that is, there is no 1,2-folding. At higher concentrations of AD, there is a significant increase in chemical shift, which is characteristic of formation of intermolecular H-bonds. The dilution data fit well to a dimerization isotherm, and Table 2 compares the results with the corresponding values for the AA·DD duplex measured in a titration experiment in chloroform. The limiting bound chemical shifts of the two complexes are very similar, which indicates that AD·AD also forms a duplex with both phosphine oxide groups involved in intermolecular H-bonds. The association constant for formation of the AD·AD duplex is approximately four times lower than the value for AA·DD, because there is a 4-fold difference in degeneracy between the two duplexes, which have different symmetry.50
Figure 6.

202 MHz 31P NMR spectra in CDCl3 at 298 K for dilution of the AD 2-mer (0.1–100 mM). The corresponding spectra of the TIPS-protected AD 2-mer and the AA 2-mer are shown for comparison.
Table 2. Association Constants and Limiting Chemical Shifts Measured by 31P NMR Dilution and Titration Experiments in CDCl3 at 298 Ka.
| complex | log K (M–1) | δfree(ppm) | δbound(ppm) |
|---|---|---|---|
| AA·DD | 2.6 | 58.6 | 61.6 |
| AD·AD | 2.0 | 58.6 | 61.8 |
| AAD·AAD | 2.0 | 58.5 | 61.1 |
| ADD·ADD | 2.5 | 58.5 | 62.2 |
Errors based on repeat experiments are 0.1 in log K.
The relative stabilities of the duplexes in toluene were investigated using 31P NMR melting experiments. The behavior of AD is compared with a 1:1 mixture of AA and DD and with a 1:1 mixture of the 1-mers A and D in Figure 7. At low temperatures, the 31P NMR chemical shifts are all around 61 ppm, which is characteristic of complexes where all of the phosphine oxide groups are fully H-bonded (the limiting bound chemical shift of the A·D complex measured at 298 K in titration experiments in toluene was also 61 ppm, see the Supporting Information). At higher temperatures, the chemical shifts decrease toward the value observed for the free phosphine oxide in toluene (56 ppm). Dissociation of the AD·AD and AA·DD duplexes occurs over the same temperature range, which indicates similar stability. In contrast, the 1:1 mixture of A and D dissociates at much lower temperatures. These results are consistent with the titration experiments, which indicate the absence of intramolecular H-bonding in the monomeric single-stranded form of AD and formation of two H-bonds in the AD·AD and AA·DD duplexes.
Figure 7.
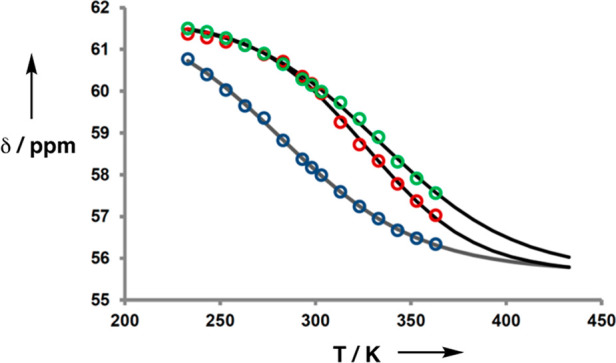
31P NMR chemical shifts in d8-toluene plotted as a function of temperature for the AD 2-mer (green), a 1:1 mixture of AA and DD (red), and a 1:1 mixture of A and D (blue). The concentration of AD was 6 mM, and the concentrations of AA, DD, A, and D were 3 mM, so that the concentration of fully bound duplex was the same in all experiments.
The AAD and ADD 3-mers were used to investigate the 1,3-folding process illustrated in Figure 1b. The 31P NMR spectra of these oligomers are very similar to the spectra recorded for the AD 2-mer (see the Supporting Information). At low concentrations, the chemical shifts are similar to the values recorded for the TIPS-protected analogues, which cannot form H-bonds. At higher concentrations, there is a significant increase in the chemical shift of the signal due to the phosphine oxides, which is characteristic of formation of intermolecular H-bonds. The dilution data fit well to a dimerization isotherm in both cases, and the results are reported in Table 2. For both AAD and ADD, the limiting chemical shift for the single-stranded monomeric state is very similar to the free chemical shifts of AA and AD. This result indicates that there is no 1,3-folding in these systems. The limiting chemical shifts for the dimeric states indicate the formation of intermolecular H-bonds with the phosphine oxide groups. The values of the association constants for all four complexes in Table 2 are similar, which suggests that they all form duplexes with two intermolecular H-bonds.
Self-Association of Mixed-Sequence Oligomers
The self-association observed for mixed-sequence oligomers complicates the analysis of duplex formation between sequence complementary oligomers, so the properties of the individual oligomers were characterized first. Association constants for self-association of all of the mixed-sequence oligomers were determined using ITC dilution experiments in toluene. In all cases, the data fit well to a dimerization isotherm, and the results are summarized in Table 3. The AD 2-mer and the four mixed-sequence 3-mers have similar self-association constants, which suggests that they all form duplexes with two phenol·phosphine oxide H-bonds. Each of the 3-mers contains the self-complementary AD motif, which leads to formation of a two base-pair duplex, with the third base on each strand left unpaired.
Table 3. Dimerization Constants Measured by ITC Dilution Experiments in d8-Toluene at 298 Ka.
| sequence | log K(M–1) | ΔG° (kJ mol–1) | ΔH° (kJ mol–1) | ΔS° (J K–1 mol–1) |
|---|---|---|---|---|
| AD | 3.2 | –18.3 | –40 | –77 |
| AAD | 3.1 | –17.8 | –39 | –70 |
| ADA | 3.5 | –20.2 | –36 | –54 |
| ADD | 3.3 | –19.0 | –36 | –57 |
| DAD | 3.4 | –19.2 | –39 | –67 |
Errors based on repeat experiments are 0.1 in log K, 0.5 kJ mol–1 in ΔG°, 5 kJ mol–1 in ΔH°, and 20 J K–1 mol–1 in ΔS°.
Sequence Selectivity of Duplex Formation
Interactions between all pairwise combinations of 3-mers were investigated using ITC in toluene. For duplex formation between two mixed-sequence oligomers, self-association of both compounds competes with formation of the 1:1 complex. This leads to four different types of behavior as illustrated in Figure 8.
Figure 8.
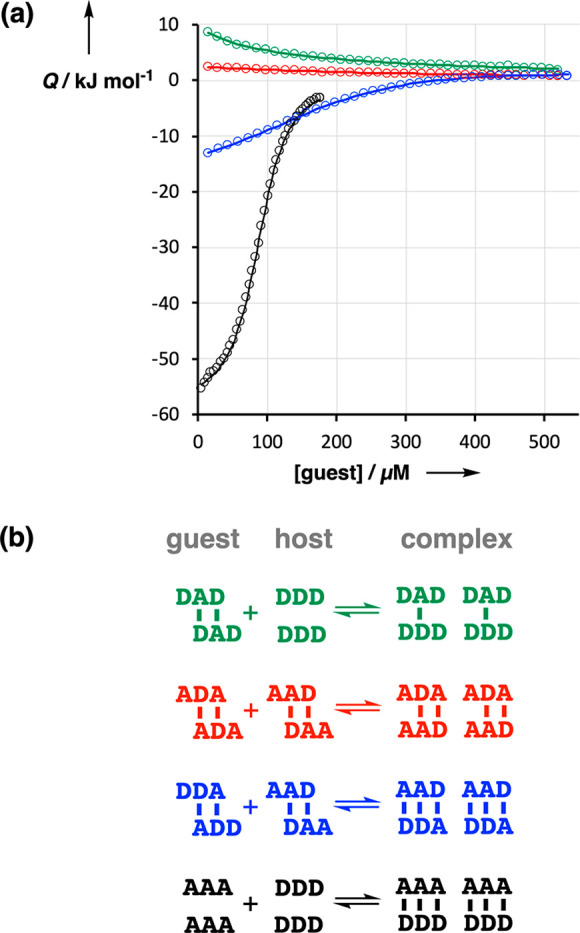
(a) Isothermal titration calorimetry data (circles) for titration of DAD into DDD (green), ADA into AAD (red), ADD into AAD (blue), and AAA into DDD (black). The lines represent the best fits to an isotherm that allows for formation of a 1:1 complex as well as dimerization of the two components. (b) Structures of the duplexes formed by self-association of the guest and the host used in the ITC experiments, and the structures of the duplexes formed in the mixture (color coding as in panel a).
When two complementary 3-mers that do not self-associate are mixed (AAA and DDD, black data in Figure 8), duplex assembly leads to formation of three new intermolecular H-bonds, which results in a large release of heat (Q).
When two complementary 3-mers that self-associate are mixed (AAD and ADD, blue data in Figure 8), two intermolecular H-bonds are broken in each of the components, and three intermolecular H-bonds are formed in the new duplex. Thus, there is a net gain of one H-bond per AAD·ADD duplex formed, and the amount of heat released is roughly one-third of that observed for AAA·DDD.
Even less heat is released when 3-mers that are not complementary are mixed, because the products form less than three H-bonds. For example, ADA and AAD are noncomplementary 3-mers that both contain the self-complementary AD motif. Although these two 3-mers can interact to form a duplex with two H-bonds, both also self-associate to form duplexes with two H-bonds. Thus, there is no net change in H-bonding when these 3-mers are mixed, and the heat change measured by ITC is close to zero (red data in Figure 8).
The other type of behavior observed in the ITC experiments is an endothermic response (green data in Figure 8). When a 3-mer that self-associates is mixed with a noncomplementary 3-mer that does not self-associate, there is a net decrease in the amount of H-bonding. The example shown in Figure 8 is addition of DAD to DDD. DAD self-associates as a duplex with two H-bonds but can form only a singly H-bonded complex with DDD, which is not sufficiently stable to be significantly populated at the concentrations used in these experiments.
The titration data were fit to an isotherm that accounts for formation of a 1:1 complex between the two different 3-mers, as well as self-association of the host and the guest (see the Supporting Information).55 The thermodynamic parameters for self-association of the individual oligomers were measured separately in the dilution experiments, as reported in Table 3, so there are only two variables that need to be optimized in the fitting procedure, the association constant and the enthalpy change for formation of the new complex. The agreement between the fitted lines and the experimental data points illustrated in Figure 8 is excellent, despite the complexity of these systems. Sequence complementary 3-mers form the most stable complexes with association constants of the order 104–105 M–1 in toluene (Table 4). These values are an order of magnitude greater than the corresponding values for formation of two base-pair duplexes reported in Table 3, providing good evidence for the formation of fully assembled duplexes with three phenol·phosphine oxide H-bonds in all three systems.
Table 4. Association Constants for Duplex Formation between Sequence-Complementary 3-mers Measured by ITC Titration Experiments in Toluene at 298 Ka.
| complex | log K (M–1) | ΔG° (kJ mol–1) | ΔH° (kJ mol–1) | ΔS° (J K–1 mol–1) |
|---|---|---|---|---|
| AAA·DDD | 5.2 | –29.5 | –57 | –92 |
| AAD·ADD | 4.2 | –23.9 | –45 | –71 |
| ADA·DAD | 4.3 | –24.6 | –50 | –85 |
Errors based on repeat experiments are 0.1 in log K, 0.5 kJ mol–1 in ΔG°, 5 kJ mol–1 in ΔH°, and 20 J K–1 mol–1 in ΔS°.
Duplex formation by sequence complementary 3-mers was confirmed using NMR melting experiments. Figure 9 shows the average 31P NMR chemical shift measured in d4-1,1,2,2-tetrachloroethane for a 1:1 mixture of AAA and DDD, a 1:1 mixture of AAD and ADD, and a 1:1 mixture of ADA and DAD. The behavior of all three duplexes is practically identical. The chemical shift decreases from a value of 64 ppm at low temperatures, which is characteristic of H-bonded phosphine oxide groups, to 60 ppm at high temperatures, which is characteristic of free phosphine oxide groups.
Figure 9.
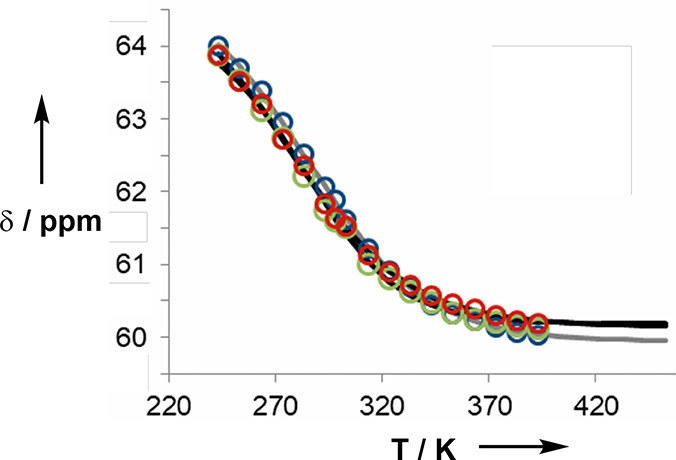
Average 31P NMR chemical shifts in d4-1,1,2,2-tetrachloroethane plotted as a function of temperature for 0.1 mM 1:1 mixtures of sequence-complementary 3-mers: AAA·DDD (blue), AAD·DDA (green), and ADA·DAD (red).
Figure 10 illustrates the results for the introduction of single base mismatches into the 3-mer duplexes in toluene. In every case, there is a significant drop in the stability of the duplex compared with the matched sequence. The melamine backbone therefore provides much higher-fidelity sequence-selective duplex formation than the polyaniline backbone that we reported previously. The reason is illustrated in Figure 1b. 1,3-Folding of the polyaniline backbone reduces the stability of the AAD·ADD duplex, which results in an increase in the relative stability of duplexes formed by these oligomers with mismatched sequences that do not fold. The melamine backbone abolishes 1,3-folding and leads to the excellent sequence-selectivity illustrated in Figure 10.
Figure 10.
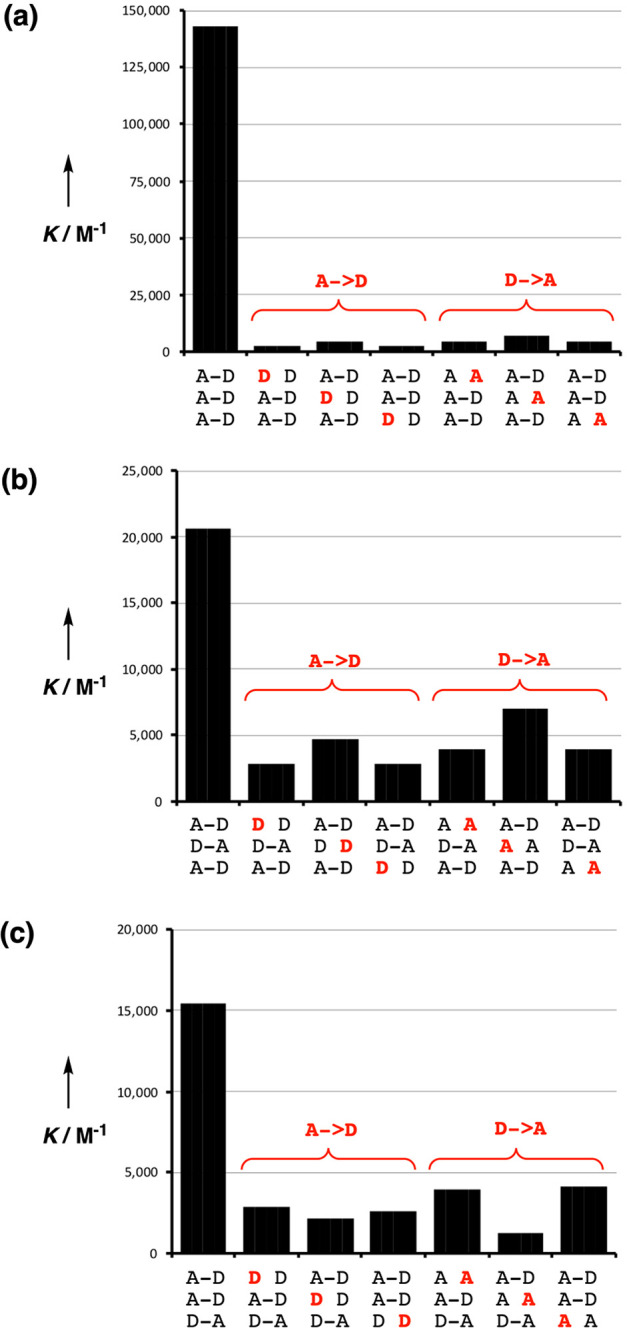
Effects of single A → D and D → A mutations (red) on the stabilities of sequence-complementary duplexes: (a) AAA·DDD, (b) ADA·DAD, and (c) DDA·AAD..
The duplexes that have a single base mismatch have association constants of the order 103 M–1 in toluene. For most combinations of 3-mer that feature two base mismatches, it was not possible to accurately determine association constants, because the 1:1 complexes are not sufficiently stable. The green data in Figure 8 shows one example: titration of DAD into DDD. This complex can form only one H-bond, but DAD dimerizes via formation of two H-bonds. Thus, the 1:1 complex formed by the two mismatched oligomers is never significantly populated in the titration. The ITC data for these systems fit well to an isotherm that considers only dimerization of the guest, albeit with a dimerization constant approximately 2-fold lower than the value obtained in dilution experiments in the absence of a competing host.
There are two cases where the stability of the complex formed by noncomplementary 3-mers with two base mismatches is anomalously high: DAD·AAD and ADA·DDA have values of log K of 4.0 and 3.9, respectively. This result implies that three H-bonds are formed in each of these complexes. All four of the 3-mers involved contain the self-complementary AD motif and so would be expected to form duplexes with two H-bonds, both with themselves and with one another. However, what the DAD·AAD and ADA·DDA duplexes have in common is that the unpaired bases on opposite ends of the duplex are complementary (Figure 11a). Figure 11b shows how the structure of the ADA·DDA duplex can be reorganized to allow the unpaired terminal bases to fold back to make a third intermolecular H-bond. This feature is likely to be unique to the 3-mer duplexes, because the arrangement of the two backbones is not compatible with propagation of the base-pairing interactions in a longer duplex. The only other combination of oligomers where this arrangement of base-pairing interactions is possible is homosequence duplex AAA·DDD. The population of multiple isomeric complexes would account for the unusually high stability observed for this system compared with the other sequence complementary duplexes.
Figure 11.

(a) Two mismatched duplexes that are expected to make two H-bonds when the sequences are arranged in a linear fashion but actually form three H-bonds based on ITC measurements. (b) Structure of the DAD·AAD duplex showing how the terminal dangling bases (left) can fold back to make a third H-bond (right). The melamine nitrogen atoms are represented as black dots, and the terminal piperidine and solubilizing groups are not shown. (c) Top and side views of the energy minimum structure of the DAD·AAD duplex (MMFFs with chloroform solvation). H-bonds are indicated by black dotted lines. The solubilizing groups and hydrogen atoms are omitted for clarity
Conclusion
A new family of recognition-encoded oligomers has been developed. Complementary phenol and phosphine oxide side-chains provide the recognition units, which lead to duplex formation through the formation of intermolecular H-bonds. The backbone is composed of repeating triazine-piperidine units, providing sufficient rigidity to prevent intramolecular folding of oligomers three recognition units in length. NMR spectroscopy and ITC were used to characterize the self-assembly properties of the oligomers. For length-complementary homo-oligomers, duplex formation in toluene is characterized by an increase in stability of an order of magnitude for every base-pair added to the chain. NMR spectra of dilute solutions of the AD 2-mer show intramolecular H-bonding between neighboring recognition units on the chain (1,2-folding) does not occur. Similarly, NMR spectra of dilute solutions of both the AAD and the ADD 3-mer show that 1,3-folding does not take place in this system either. ITC was used to characterize interactions between all pairwise combinations of the six different 3-mer sequences, and the sequence complementary duplexes are approximately an order of magnitude more stable than duplexes with a single base mismatch. High-fidelity duplex formation combined with the synthetic accessibility of the monomer building blocks makes these systems attractive targets for the investigation of the properties of longer oligomers.
Acknowledgments
We thank the European Research Council for Grant ERC-2012-AdG 320539-duplex, Marie Skłodowska-Curie Actions for Individual Fellowship H2020-MSCA-IF-2014 656902, and the Herchel Smith Fund for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c02275.
Materials and methods, detailed synthetic procedures, full characterization including 1H and 13C NMR spectra of all compounds, and the NMR and ITC titration data (PDF)
Author Contributions
‡ P. Troselj and P. Bolgar contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Merrifield R. B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85 (14), 2149. 10.1021/ja00897a025. [DOI] [Google Scholar]
- Köster H. Polymer support oligonucleotide synthesis VI: Use of inorganic carriers. Tetrahedron Lett. 1972, 13, 1527. 10.1016/S0040-4039(01)84677-3. [DOI] [Google Scholar]
- Gough G. R.; Brunden M. J.; Gilham P. T. Recovery and recycling of synthetic units in the construction of oligodeoxyribonucleotides on solid supports. Tetrahedron Lett. 1981, 22, 4177. 10.1016/S0040-4039(01)82097-9. [DOI] [Google Scholar]
- Köster H.; Stumpe A.; Wolter A. Polymer support oligonucleotide synthesis 13: Rapid and efficient synthesis of oligodeoxynucleotides on porous glass support using triester approach. Tetrahedron Lett. 1983, 24, 747. 10.1016/S0040-4039(00)81515-4. [DOI] [Google Scholar]
- Wurtz N. R.; Turner J. M.; Baird E. E.; Dervan P. B. Fmoc Solid Phase Synthesis of Polyamides Containing Pyrrole and Imidazole Amino Acids. Org. Lett. 2001, 3, 1201. 10.1021/ol0156796. [DOI] [PubMed] [Google Scholar]
- Lutz J.-F.; Ouchi M.; Liu D. R.; Sawamoto M. Sequence-Controlled Polymers. Science 2013, 341, 1238149. 10.1126/science.1238149. [DOI] [PubMed] [Google Scholar]
- Trinh T. T.; Oswald L.; Chan-Seng D.; Lutz J.-F. Synthesis of Molecularly Encoded Oligomers Using a Chemoselective “AB + CD” Iterative Approach. Macromol. Rapid Commun. 2014, 35, 141. 10.1002/marc.201300774. [DOI] [PubMed] [Google Scholar]
- Lutz J.-F.; Lehn J.-M.; Meijer E.; Matyjaszewski K. From precision polymers to complex materials and systems. Nat. Rev. Mater. 2016, 1, 16024. 10.1038/natrevmats.2016.24. [DOI] [Google Scholar]
- Grate J. W.; Mo K.-F; Daily M. D. Triazine-Based Sequence-Defined Polymers with Side-Chain Diversity and Backbone–Backbone Interaction Motifs. Angew. Chem., Int. Ed. 2016, 55, 3925. 10.1002/anie.201509864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D.; Sali A. Protein Structure Prediction and Structural Genomics. Science 2001, 294, 93. 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- Kuhlman B.; Dantas G.; Ireton G. C.; Varani G.; Stoddard B. L.; Baker D. Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Science 2003, 302, 1364. 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- Orgel L. E. Unnatural Selection in Chemical Systems. Acc. Chem. Res. 1995, 28, 109. 10.1021/ar00051a004. [DOI] [PubMed] [Google Scholar]
- Kramer R.; Lehn J.-M.; Marquis-Rigault A. Self-recognition in helicate self-assembly: spontaneous formation of helical metal complexes from mixtures of ligands and metal ions. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5394. 10.1073/pnas.90.12.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis A.; Smith V.; Harrowfield J.; Lehn J.-M.; Herschbach H.; Sanvito R.; Leize-Wagner E.; van Dorsselaer A. Messages in Molecules: Ligand/Cation Coding and Self-Recognition in a Constitutionally Dynamic System of Heterometallic Double Helicates. Chem. - Eur. J. 2006, 12, 5632. 10.1002/chem.200600143. [DOI] [PubMed] [Google Scholar]
- Gong B.; Yan Y.; Zeng H.; Skrzypczak-Jankunn E.; Kim Y. W.; Zhu J.; Ickes H. A New Approach for the Design of Supramolecular Recognition Units: Hydrogen-Bonded Molecular Duplexes. J. Am. Chem. Soc. 1999, 121, 5607. 10.1021/ja990904o. [DOI] [Google Scholar]
- Gong B. Specifying Non-Covalent Interactions: Sequence-Specific Assembly of Hydrogen-Bonded Molecular Duplexes. Synlett 2001, 2001, 0582. 10.1055/s-2001-13350. [DOI] [Google Scholar]
- Zeng H.; Miller R. S.; Flowers R. A.; Gong B. A Highly Stable, Six-Hydrogen-Bonded Molecular Duplex. J. Am. Chem. Soc. 2000, 122, 2635. 10.1021/ja9942742. [DOI] [Google Scholar]
- Zeng H.; Ickes H.; Flowers R. A.; Gong B. Sequence Specificity of Hydrogen-Bonded Molecular Duplexes. J. Org. Chem. 2001, 66, 3574. 10.1021/jo010250d. [DOI] [PubMed] [Google Scholar]
- Gong B. Engineering hydrogen-bonded duplexes. Polym. Int. 2007, 56, 436. 10.1002/pi.2175. [DOI] [Google Scholar]
- Gong B. Molecular Duplexes with Encoded Sequences and Stabilities. Acc. Chem. Res. 2012, 45, 2077. 10.1021/ar300007k. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Yang Z. Y.; Yi Y. P.; Xiang J. F.; Chen C. F.; Wan L. J.; Shuai Z. G. Helical Molecular Duplex Strands: Multiple Hydrogen-Bond-Mediated Assembly of Self-Complementary Oligomeric Hydrazide Derivatives. J. Org. Chem. 2007, 72, 4936. 10.1021/jo070525a. [DOI] [PubMed] [Google Scholar]
- Tanaka Y.; Katagiri H.; Furusho Y.; Yashima E. A Modular Strategy to Artificial Double Helices. Angew. Chem. 2005, 117, 3935. 10.1002/ange.200501028. [DOI] [PubMed] [Google Scholar]
- Ito H.; Furusho Y.; Hasegawa T.; Yashima E. Sequence- and Chain-Length-Specific Complementary Double-Helix Formation. J. Am. Chem. Soc. 2008, 130, 14008. 10.1021/ja806194e. [DOI] [PubMed] [Google Scholar]
- Yamada H.; Furusho Y.; Ito H.; Yashima E. Complementary double helix formation through template synthesis. Chem. Commun. 2010, 46, 3487. 10.1039/c002170a. [DOI] [PubMed] [Google Scholar]
- Yashima E.; Ousaka N.; Taura D.; Shimomura K.; Ikai T.; Maeda K. Supramolecular Helical Systems: Helical Assemblies of Small Molecules, Foldamers, and Polymers with Chiral Amplification and Their Functions. Chem. Rev. 2016, 116, 13752. 10.1021/acs.chemrev.6b00354. [DOI] [PubMed] [Google Scholar]
- Anderson H. L. Conjugated Porphyrin Ladders. Inorg. Chem. 1994, 33, 972. 10.1021/ic00083a022. [DOI] [Google Scholar]
- Taylor P. N.; Anderson H. L. Cooperative Self-Assembly of Double-Strand Conjugated Porphyrin Ladders. J. Am. Chem. Soc. 1999, 121, 11538. 10.1021/ja992821d. [DOI] [Google Scholar]
- Berl V.; Huc I.; Khoury R. G.; Krische M. J.; Lehn J.-M. Interconversion of single and double helices formed from synthetic molecular strands. Nature 2000, 407, 720. 10.1038/35037545. [DOI] [PubMed] [Google Scholar]
- Berl V.; Huc I.; Khoury R. G.; Lehn J.-M. Helical Molecular Programming: Folding of Oligopyridine-dicarboxamides into Molecular Single Helices. Chem. - Eur. J. 2001, 7, 2810.. [DOI] [PubMed] [Google Scholar]
- Sańchez-Quesada J.; Seel C.; Prados P.; de Mendoza J.; Dalcol I.; Giralt E. Anion Helicates: Double Strand Helical Self-Assembly of Chiral Bicyclic Guanidinium Dimers and Tetramers around Sulfate Templates. J. Am. Chem. Soc. 1996, 118, 277. 10.1021/ja953243d. [DOI] [Google Scholar]
- Bisson A. P.; Carver F. J.; Eggleston D. S.; Haltiwanger R. C.; Hunter C. A.; Livingstone D. L.; McCabe J. F.; Rotger C.; Rowan A. E. Synthesis and Recognition Properties of Aromatic Amide Oligomers: Molecular Zippers. J. Am. Chem. Soc. 2000, 122, 8856. 10.1021/ja0012671. [DOI] [Google Scholar]
- Bisson A. P.; Hunter C. A. Cooperativity in the assembly of zipper complexes. Chem. Commun. 1996, 1723. 10.1039/cc9960001723. [DOI] [Google Scholar]
- Chu W.-J.; Yang Y.; Chen C.-F. Multiple Hydrogen-Bond-Mediated Molecular Duplexes Based on the Self-Complementary Amidourea Motif. Org. Lett. 2010, 12, 3156. 10.1021/ol101068n. [DOI] [PubMed] [Google Scholar]
- Chu W.-J.; Chen J.; Chen C.-F.; Yang Y.; Shuai Z. Amidourea-Based Hydrogen-Bonded Heteroduplexes: Structure and Assembling Selectivity. J. Org. Chem. 2012, 77, 7815. 10.1021/jo301434a. [DOI] [PubMed] [Google Scholar]
- Archer E. A.; Krische M. J. Duplex Oligomers Defined via Covalent Casting of a One-Dimensional Hydrogen-Bonding Motif. J. Am. Chem. Soc. 2002, 124, 5074. 10.1021/ja012696h. [DOI] [PubMed] [Google Scholar]
- Gong H.; Krische M. J. Duplex Molecular Strands Based on the 3,6-Diaminopyridazine Hydrogen Bonding Motif: Amplifying Small-Molecule Self-Assembly Preferences through Preorganization and Iterative Arrangement of Binding Residues. J. Am. Chem. Soc. 2005, 127, 1719. 10.1021/ja044566p. [DOI] [PubMed] [Google Scholar]
- Dervan P. B.; Burli R. W. Sequence-specific DNA recognition by polyamides. Curr. Opin. Chem. Biol. 1999, 3, 688. 10.1016/S1367-5931(99)00027-7. [DOI] [PubMed] [Google Scholar]
- Renneberg D.; Dervan P. B. Imidazopyridine/Pyrrole and Hydroxybenzimidazole/Pyrrole Pairs for DNA Minor Groove Recognition. J. Am. Chem. Soc. 2003, 125, 5707. 10.1021/ja0300158. [DOI] [PubMed] [Google Scholar]
- Doss R. M.; Marques M. A.; Foister S.; Chenoweth D. M.; Dervan P. B. Programmable Oligomers for Minor Groove DNA Recognition. J. Am. Chem. Soc. 2006, 128, 9074. 10.1021/ja0621795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier J. L.; Yu A. S.; Korf I.; Segal D. J.; Dervan P. B. Guiding the Design of Synthetic DNA-Binding Molecules with Massively Parallel Sequencing. J. Am. Chem. Soc. 2012, 134, 17814. 10.1021/ja308888c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stross A. E.; Iadevaia G.; Hunter C. A. Cooperative duplex formation by synthetic H-bonding oligomers. Chem. Sci. 2016, 7, 94. 10.1039/C5SC03414K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez-Villanueva D.; Hunter C. A. Homochiral oligomers with highly flexible backbones form stable H-bonded duplexes. Chem. Sci. 2017, 8, 206. 10.1039/C6SC02995G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stross A. E.; Iadevaia G.; Núñez-Villanueva D.; Hunter C. A. Sequence-Selective Formation of Synthetic H-Bonded Duplexes. J. Am. Chem. Soc. 2017, 139, 12655. 10.1021/jacs.7b06619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain J. A.; Iadevaia G.; Hunter C. A. H-Bonded Duplexes based on a Phenylacetylene Backbone. J. Am. Chem. Soc. 2018, 140, 11526. 10.1021/jacs.8b08087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczypiński F. T.; Hunter C. A. Building blocks for recognition-encoded oligoesters that form H-bonded duplexes. Chem. Sci. 2019, 10, 2444. 10.1039/C8SC04896G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielli L.; Núñez-Villanueva D.; Hunter C. A. Two-component assembly of recognition-encoded oligomers that form stable H-bonded duplexes. Chem. Sci. 2020, 11, 561. 10.1039/C9SC04250D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczypiński F. T.; Gabrielli L.; Hunter C. A. Emergent supramolecular assembly properties of a recognition-encoded oligoester. Chem. Sci. 2019, 10, 5397. 10.1039/C9SC01669D. [DOI] [Google Scholar]
- Gabrielli L.; Hunter C. A. Supramolecular catalysis by recognition-encoded oligomers: discovery of a synthetic imine polymerase. Chem. Sci. 2020, 11, 7408. 10.1039/D0SC02234A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez-Villanueva D.; Hunter C. A. Replication of Sequence Information in Synthetic Oligomers. Acc. Chem. Res. 2021, 54, 1298. 10.1021/acs.accounts.0c00852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez-Villanueva D.; Iadevaia G.; Stross A. E.; Jinks M. A.; Swain J. A.; Hunter C. A. H-Bond Self-Assembly: Folding versus Duplex Formation. J. Am. Chem. Soc. 2017, 139, 6654. 10.1021/jacs.7b01357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blotny G. Recent applications of 2,4,6-trichloro-1,3,5-triazine and its derivatives in organic synthesis. Tetrahedron 2006, 62, 9507. 10.1016/j.tet.2006.07.039. [DOI] [Google Scholar]
- Steffensen M. B.; Simanek E. E. Chemoselective Building Blocks for Dendrimers from Relative Reactivity Data. Org. Lett. 2003, 5, 2359. 10.1021/ol0347491. [DOI] [PubMed] [Google Scholar]
- Moreno K. X.; Simanek E. E. Identification of diamine linkers with differing reactivity and their application in the synthesis of melamine dendrimers. Tetrahedron Lett. 2008, 49, 1152. 10.1016/j.tetlet.2007.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J.; Simanek E. E. Triazine dendrimers as drug delivery systems: From synthesis to therapy. Adv. Drug Delivery Rev. 2012, 64, 826. 10.1016/j.addr.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Tomas S. Cooperativity, Partially Bound States, and Enthalpy-Entropy Compensation. Chem. Biol. 2003, 10, 1023. 10.1016/j.chembiol.2003.10.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.