

Glycosylation-dependent programs control intestinal inflammation.
Abstract
Diverse immunoregulatory circuits operate to preserve intestinal homeostasis and prevent inflammation. Galectin-1 (Gal1), a β-galactoside–binding protein, promotes homeostasis by reprogramming innate and adaptive immunity. Here, we identify a glycosylation-dependent “on-off” circuit driven by Gal1 and its glycosylated ligands that controls intestinal immunopathology by targeting activated CD8+ T cells and shaping the cytokine profile. In patients with inflammatory bowel disease (IBD), augmented Gal1 was associated with dysregulated expression of core 2 β6-N-acetylglucosaminyltransferase 1 (C2GNT1) and α(2,6)-sialyltransferase 1 (ST6GAL1), glycosyltransferases responsible for creating or masking Gal1 ligands. Mice lacking Gal1 exhibited exacerbated colitis and augmented mucosal CD8+ T cell activation in response to 2,4,6-trinitrobenzenesulfonic acid; this phenotype was partially ameliorated by treatment with recombinant Gal1. While C2gnt1−/− mice exhibited aggravated colitis, St6gal1−/− mice showed attenuated inflammation. These effects were associated with intrinsic T cell glycosylation. Thus, Gal1 and its glycosylated ligands act to preserve intestinal homeostasis by recalibrating T cell immunity.
INTRODUCTION
Inflammatory bowel diseases (IBDs) encompass a large range of chronic pathologies. Crohn’s disease (CD) and ulcerative colitis (UC) are unique disorders characterized by inflammation of the gastrointestinal tract that result in increased risk of colorectal cancer and a marked decrease in life quality of affected individuals (1). Over the past decade, IBD has become a global burden. While the incidence of these disorders has stabilized in Western countries, it has increased considerably in newly industrialized regions of Africa, Asia, and South America (2). While administration of monoclonal antibodies that target proinflammatory mediators [i.e., anti–tumor necrosis factor (TNF)] and gut-homing molecules (i.e., α4β7 integrin) have reshaped the landscape of IBD treatment (3), the serious adverse effects observed in certain patient cohorts together with the high percentage of primary or secondary resistance highlight the need for alternative therapeutic approaches (4, 5).
Galectins, a family of soluble glycan-binding proteins, can modulate immune cell homeostasis and reprogram innate and adaptive immune responses, acting as danger-associated or resolution-associated molecular patterns (6–10). Altered expression of these lectins has been observed within the gastrointestinal tract in several pathologic conditions including IBD (11, 12). Most extracellular activities of galectins are regulated by the availability of specific N- and O-glycan structures on the surface of immune cells (13). Programmed remodeling of these glycosylated ligands, through the coordinated action of glycosyltransferases and glycosidases, is tightly controlled by environmental cues, including those arising from cytokines, hypoxia, and inflammation (8).
Intestinal inflammation can result in structural changes that ultimately alter the extent and nature of protein glycosylation, both locally and systemically, as a component of the natural history of the disease (14). Circulating aberrantly glycosylated immunoglobulin G (IgG) has been identified as a potential disease biomarker in inflammatory diseases, including IBD (14, 15). Likewise, altered glycosylation of mucins has a profound influence on the development of intestinal inflammation (16, 17). Moreover, mice devoid of glycosyltransferases implicated in O-glycan biosynthesis, such as core 2 β6-N-acetylglucosaminyltransferase 1 (C2GnT1), its isoform C2GnT3, or β1,3-galactosyltransferase C1GalT1, all exhibited increased susceptibility to intestinal inflammation in response to administration of dextran sulfate sodium (DSS) (18, 19). Genome-wide association studies identified COSMC, a gene in the X chromosome that encodes a key chaperone involved in core 1/core 2 O-glycan biosynthesis, as an IBD risk factor. Deletion of this gene from mouse intestinal epithelial cells leads to the development of dysbiosis similar to that observed in patients with IBD (20). An altered CD4+ memory T cell O-glycome has been shown to contribute to exacerbation of colitis due to interactions with epithelial-derived galectin-4 (Gal4) (21). Moreover, activity of β(1,6)N-acetylglucosaminyltransferase 5 (Mgat5), an enzyme responsible for generating complex branched N-glycans in mucosal T cells, has been shown to influence disease severity in patients diagnosed with UC (22). However, and despite considerable progress, the clinical and immunological significance of galectins proteins in the pathogenesis of IBD and their association with aberrant glycosylation remain poorly understood.
Gal1, a prototype member of the galectin family, controls several immune cell processes including activation, differentiation, trafficking, and survival through specific recognition of N-acetyllactosamine [LacNAc; Galβ(1-4)GlcNAc]–containing structures on cell surface glycosylated receptors (Fig. 1) (9). Among these activities, the actions of Gal1 serve to limit the survival of T helper cell 1 (TH1)– and TH17-differentiated cells (23), to fine-tune the immunogenicity of dendritic cells (DCs) (24), and to promote contraction of the CD8+ T cell compartment (25), leading to induction of tumor-immune escape (26, 27), modulation of microbial infections (28), and resolution of autoimmune inflammation (8). Several intrinsic factors govern the biological activity of Gal1, including its monomer-dimer equilibrium and redox status, as well as the availability of specific glycan-binding sites on different cellular receptors (29). Hence, the altered expression of glycosyltransferases is responsible for creating or masking glycan epitopes that can interact with Gal1 and thus can influence its biological activity. Of particular note, α(2,6)-sialyltransferase 1 (ST6Gal1) catalyzes the incorporation of α(2,6)-linked sialic acid to terminal galactose residues, thereby hindering their recognition by Gal1. In turn, Mgat5 and C2GnT1 activities contribute to the creation of a permissive glycosylation signature that favors Gal1 binding and function (Fig. 1) (6, 30). Administration of exogenous recombinant Gal1 (rGal1) promoted resolution of 2,4,6-trinitrobenzenesulfonic acid (TNBS)–induced colitis in BALB/c mice via suppression of proinflammatory cytokine production in the gut mucosa (31). However, the roles of endogenous Gal1 and its glycosylated ligands in modulating mucosal immunity and intestinal immunopathology remain unexplored. In this study, we used experimental models of colitis and clinical specimens from patients with IBD to perform an integrated analysis of the Gal1-glycan axis, specifically its role in altering the nature of the mucosal immune landscape and its contributions to the development, severity, and resolution of intestinal inflammation.
Fig. 1. Schematic representation of N- and O-glycan biosynthetic pathways.
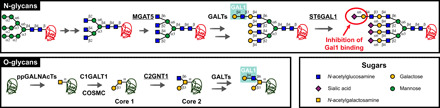
Complex N-glycans are synthesized in several steps, including trimming by α-mannosidases and the sequential actions of N-acetylglucosaminyltransferases (MGATs). MGAT5 is the enzyme responsible for β(1,6) branching. The GlcNAc residues can be extended to LacNAc structures via the actions of galactosyltransferases (GALTs); the terminal galactose residues will ultimately undergo α(2,6) sialylation by ST6GAL1. While β(1,6) branching favors Gal1 binding, α(2,6) sialylation inhibits Gal1 recognition of terminal LacNAc structures. Biosynthesis of O-glycans is initiated by polypeptide N-acetylgalactosamine transferases (ppGALNAcTs). Biosynthesis of core 1 is achieved by the action of β(1,3) galactosyltransferase C1GALT1 with assistance from core 1 β(1,3) galactosyltransferase–specific molecular chaperone (COSMC). The resulting disaccharide can undergo branching via the actions of β(1,6) GlcNAc transferases, including C2GNT1 (also called GCNT1), resulting in the generation of core 2 O-glycans. Last, core 2 O-glycans can be further decorated by GALTs, resulting in the synthesis of LacNAc that is recognized by Gal1. Glycans are depicted following the guidelines from the Symbol Nomenclature for Glycans group (72).
RESULTS
Inflammation alters the expression of galectins and specific glycosyltransferases in colon biopsies from patients with IBD
We first analyzed the expression of galectin genes in colon biopsies from patients with IBD based on inflammation severity of human intestinal mucosa, binarized into either inflamed or uninflamed categories as well as in healthy individuals using publicly available datasets. In line with previous reports (32), no significant differences were observed in the expression of galectins and glycosyltransferases when comparing inflamed or uninflamed colonic tissue in patients with CD and patients with UC (fig. S1). Considering these results, and the similar dysregulation of galectins previously described in biopsies from CD and UC in inflamed areas (12), we analyzed data based on disease activity (inflamed or uninflamed as defined at the time of the colonoscopy). Whereas expression of Gal1 (encoded by LGALS1) was up-regulated in inflamed compared to uninflamed areas of the colon, and those from healthy controls, expression of Gal8 (LGALS8) and Gal9 (LGALS9) was not altered and Gal2 (LGALS2), Gal3 (LGALS3), and Gal4 (LGALS4) were found to be down-regulated (Fig. 2A). Given their immunoregulatory potential (12), we then analyzed the expression of LGALS1, LGALS3, LGALS4, and LGALS9 mRNA by reverse transcription quantitative polymerase chain reaction (RT-qPCR) in a local cohort of patients (Table 1). Notably, Gal1 was the only member of the galectin family consistently and significantly up-regulated in biopsies from inflamed areas of the colon (Fig. 2, A and B). However, plasma concentrations of Gal1 were not significantly different in our local cohort of patients with IBD when compared to controls (fig. S2, A and B).
Fig. 2. Dysregulated expression of Gal1 and specific glycosyltransferases in IBD.
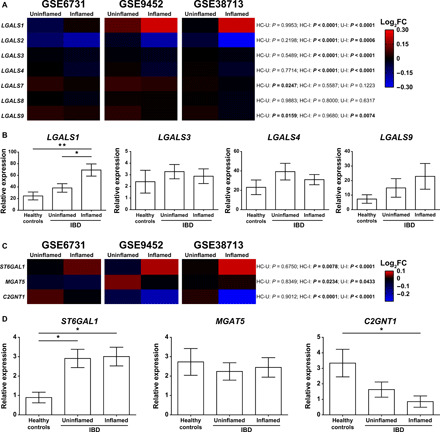
(A) Bioinformatic analysis of galectin expression (LGALS1, LGALS2, LGALS3, LGALS4, LGALS7, LGALS8, and LGALS9) in IBD datasets. (B) Expression of LGALS1, LGALS3, LGALS4, and LGALS9 analyzed by RT-qPCR in colon biopsies from healthy controls (n = 8), uninflamed (n = 13), and inflamed (n = 11) areas from colon biopsies of a local cohort of patients with IBD. (C) Bioinformatic analysis of glycosyltransferases relevant for Gal1 binding (ST6GAL1, MGAT5, and C2GNT1) in IBD datasets. (D) Expression of ST6GAL1, MGAT5, and C2GNT1 analyzed by RT-qPCR in colon biopsies from healthy controls (n = 6), uninflamed (n = 15), and inflamed (n = 14) areas from colon biopsies of a local cohort of patients with IBD. In (A) and (C), two-way analysis of variance (ANOVA) followed by Tukey’s posttest was used; colors in the heatmap depict the logarithm (base 2) of the fold change (log2FC), comparing uninflamed (U) and inflamed (I) areas of colon biopsies from patients with IBD with healthy controls (HC). In (B) and (D), one-way ANOVA followed by Tukey’s posttest was used. Unless otherwise stated, data are presented as means ± SEM. HC-U, healthy controls vs. uninflamed areas; HC-I, healthy controls vs. inflamed areas; U-I, uninflamed vs. inflamed areas. *P < 0.05 and **P < 0.01.
Table 1. Clinical description of patients with IBD.
Controls, healthy individuals; UC, ulcerative colitis; CD, Crohn’s disease. IQR, interquartile range; 5-ASA, 5-aminosalicylic acid (mesalamine); 6-MP, 6-mercaptopurine; AZA, azathioprine; ATB, antibiotics; upper GIT, upper gastrointestinal tract; 0, nonexistent; -, not applicable.
| Patients with UC | Patients with CD | Controls | |||||
| UC inactive | UC active | CD inactive | CD active | ||||
|
Clinical remission |
Mild to moderate |
Severe |
Clinical remission |
Mild to moderate |
Severe | ||
| N° of patients | 9 | 7 | 7 | 6 | 5 | 1 | 11 |
| Sex (F/M) | 2/7 | 5/2 | 5/2 | 3/3 | 3/2 | 0/1 | 10/1 |
| Median age (years) (IQR 25%–75%) |
33.5 (24.5–45.5) | 20.5 (20–29) | 30.5 (23–32) | 64 (55–68) | 32 (29–38) | 22 | 37 (23–52) |
| Median disease duration (years) (IQR 25%–75%) |
11.5 (4–17) | 2 (1–5) | 1.5 (1–5) | 10.5 (7–14) | 4 (2–8) | 1 | - |
| Active smokers | 1 | 1 | 1 | 1 | 0 | 0 | 3 |
| Location UC | |||||||
| E1, proctitis | 0 | 3 | 0 | - | - | - | |
| E2, left sided | 5 | 0 | 4 | - | - | - | |
| E3, pancolitis | 4 | 4 | 3 | - | - | - | |
| Location CD | |||||||
| L1, ileal | - | - | - | 0 | 0 | 0 | |
| L2, colon | - | - | - | 3 | 5 | 1 | |
| L3, ileocolonic | - | - | - | 3 | 0 | 0 | |
| L4, upper GIT | - | - | - | 0 | 0 | 0 | |
| Medication | |||||||
| 5-ASA | 5 | 3 | 0 | 0 | 0 | 0 | |
| Steroids | 0 | 2 | 5 | 1 | 1 | 0 | |
| 6-MP/AZA | 2 | 0 | 0 | 0 | 0 | 0 | |
| 5-ASA/steroids | 0 | 2 | 2 | 1 | 0 | 0 | |
| 5-ASA/AZA/6-MP | 2 | 0 | 0 | 3 | 0 | 0 | |
| 5-ASA/AZA/steroids | 0 | 0 | 0 | 0 | 2 | 0 | |
| 5-ASA/ATB | 0 | 0 | 0 | 0 | 1 | 0 | |
| AZA/ATB | 0 | 0 | 0 | 0 | 1 | 0 | |
| Steroids/ATB | 0 | 0 | 0 | 0 | 0 | 1 | |
| No medication | 0 | 0 | 0 | 1 | 0 | 0 | |
We then analyzed the expression of three glycosyltransferases that are known to be critical for the generation or masking of Gal1-specific ligands. We found considerable up-regulation of ST6GAL1, an α(2,6)-sialyltransferase capable of generating glycan structures that prevent Gal1 recognition and function (Fig. 1), in biopsies from inflamed areas of patients with IBD (Fig. 2C). C2GNT1, an enzyme that creates Gal1-specific ligands by generating core 2 O-glycans (Fig. 1), and MGAT5, a glycosyltransferase involved in complex N-glycan branching, were both down-regulated in inflamed areas of the colon (Figs. 1 and 2C). Dysregulation of ST6GAL1 and C2GNT1 was validated by RT-qPCR analysis in the same patient cohort used for analysis of galectins (Fig. 2D). Of note, there were no significant changes in MGAT5 expression when comparing colon biopsies of both inflamed and uninflamed areas from patients with IBD with those of healthy controls (Fig. 2D). Thus, down-regulation of C2GNT1 and up-regulation of ST6GAL1 in inflamed areas of patients with IBD suggest the likelihood of exhibiting an aberrant glycome; these alterations could serve to modulate the capacity for Gal1 binding and thereby influence its immunoregulatory activity.
Endogenous Gal1 controls TNBS-induced intestinal inflammation via its impact on the effector T cell compartment
The up-regulation of Gal1 in colon biopsies of patients with IBD prompted us to investigate the impact of endogenous Gal1 on the establishment and resolution of intestinal inflammation. For this, we compared the course of the disease in a double-dose TNBS-induced colitis model in both wild-type (WT) and Gal1-deficient (Lgals1−/−) C57BL/6 mice. We found that Lgals1−/− mice developed more severe colitis, with increased weight loss and a higher colonic weight/length ratio when compared to WT mice, although no differences in histological grading were observed (Fig. 3, A to C). Similar to the findings from our analysis of patients with IBD, we detected significantly increased expression of Gal1 protein at day 10 in the colonic tissue of TNBS-treated WT mice when compared to mice treated with vehicle alone (Fig. 3, D and E). Moreover, expression of Gal1 protein correlated positively with colon weight/length ratio (Fig. 3F). Gal1 immunoreactivity was detected primarily within the interstitial spaces between cells of the lamina propria and was not restricted to a particular cell type (fig. S3).
Fig. 3. Lack of endogenous Gal1 exacerbates TNBS-induced colitis.
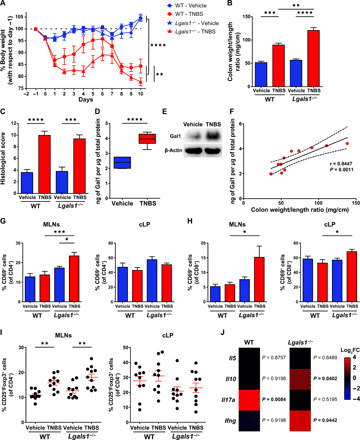
(A) Weight loss curves of WT and Lgals1−/− mice treated with ethanol (vehicle) or TNBS. Data are from five independent experiments; two-way repeated-measures ANOVA followed by Tukey’s posttest. (B) Colon weight/length ratio. (C) Histopathologic assessment (score) of experimental groups. (D) Determination of Gal1 at day 10 in colon tissue by enzyme-linked immunosorbent assay (ELISA), normalized to total protein in WT mice. Box plots represent median (line), first and third quartiles (box limits), and minimum and maximum values (bars) from a representative of two independent experiments (n = 6 mice per group); unpaired Student’s t test. (E) Detection of Gal1 at euthanize (day 10) in colon protein extracts from WT mice by Western blot. (F) Correlation between Gal1 protein levels and colon weight/length ratio (n = 11 mice); Pearson’s correlation coefficient (r) and P value (P) are indicated. (G and H) CD69 expression among CD4+ (G) or CD8+ (H) T cell populations within MLNs or cLP from vehicle- or TNBS-treated WT or Lgals1−/− mice. Data are from a representative of three independent experiments (n = 10 mice per group). (I) Regulatory T cells (Tregs) in MLNs and cLP of vehicle-treated or TNBS-treated WT or Lgals1−/− mice. Data are from a representative of three independent experiments (n = 10 mice per group). (J) Expression of cytokines mRNA in colon from TNBS-treated WT or Lgals1−/− mice measured by RT-qPCR. Colors shown in the heatmap depict the log2FC for each cytokine in TNBS- versus vehicle-treated mice of each genotype. Multiple t test. In (B), (C), and (G) to (I), two-way ANOVA followed by Tukey’s posttest. Unless otherwise stated, data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To investigate possible mechanisms underlying the distinct responses of WT and Lgals1−/− mice in the TNBS-colitis model, and given our current understanding of the immunoregulatory role of this lectin with respect to activated peripheral CD4+ T cells (23, 31), our first experiments focused on T cells identified in the mesenteric lymph nodes (MLNs) and colonic lamina propria (cLP). A higher percentage of CD69-expressing CD4+ T cells was found in MLNs of TNBS-treated Lgals1−/− mice compared to both vehicle-treated Lgals1−/− and TNBS-treated WT mice (Fig. 3G). The induction of experimental colitis had no significant impact on the percentage of activated CD4+ T cells in the cLP in either genotype (Fig. 3G). Moreover, TNBS-treated Lgals1−/− mice exhibited a significant increase in the percentage of CD69-expressing CD8+ T cells in both the MLNs and cLP compared to their WT counterpart (Fig. 3H). Given that CD69 is also a marker of T cell residency in cLP (33), we then examined CD69 and CD103 coexpression to elucidate whether increased CD8+CD69+ T cells observed in TNBS-treated Lgals1−/− mice were associated specifically with T cell activation or whether they represented a greater proportion of resident T cells (particularly tissue-resident memory T cells). We found no significant differences in these phenotypic markers regardless of the genotype or treatment (fig. S4); these results suggested that the increased frequency of CD8+CD69+ cells is most likely attributed to T cell activation. In addition, we ruled out the possibility that this finding could result from impaired expansion and/or recruitment of regulatory CD4+CD25+Foxp3+ T cells (Tregs), as TNBS-treated mice exhibited an increased proportion of Tregs in MLNs regardless of their genotype, while no changes were observed in the cLP (Fig. 3I). We then compared the expression of effector T cell cytokines in the colon of TNBS- or vehicle-treated mice by RT-qPCR. We found higher levels of Il17a mRNA in WT mice but no significant changes in the expression of Il5, Il10, and Ifng mRNA when compared to vehicle-treated mice (Fig. 3J); by contrast, Lgals1−/− mice exhibited higher levels of Ifng and Il10 with no changes in Il5 and Il17a mRNA expression (Fig. 3J). Last, Tnf mRNA expression was not significantly different between WT and Lgals1−/− TNBS-treated mice (fig. S5). Together, our findings suggest that endogenous Gal1 controls intestinal inflammation by reprogramming the fate of effector T cells and by altering the cytokine balance in the gut microenvironment.
We then explored the possibility that administration of rGal1 could promote resolution of exacerbated colitis in TNBS-treated Lgals1−/− mice. Of note, rGal1 reached the cLP when administered daily (100 μg per mouse/day) to TNBS-treated Lgals1−/− mice (fig. S6). Daily administration of rGal1 in TNBS-treated Lgals1−/− mice resulted in decreased weight loss and reduced colonic weight/length ratio (Fig. 4, A and B). In addition, treatment with rGal1 resulted in partial restoration of mucosal integrity as shown by histopathologic assessment (Fig. 4C). Exogenous rGal1 treatment also mitigated experimental colitis in WT animals (fig. S7), thus validating its therapeutic effect in controlling colitis as previously described in BALB/c mice (31). Administration of rGal1 also resulted in down-regulation of Ifng and up-regulated expression of Il10 and Il17a in colonic tissue, although it had no impact on the expression of Il5 mRNA (Fig. 4D). Moreover, treatment with exogenous rGal1 also reduced the percentage of CD69+CD8+ T cells in both MLNs and cLP (Fig. 4E), although it had no effect on the proportion of activated CD4+ T cells in these compartments (Fig. 4F). Noticeably, the Treg percentage in MLNs and cLP of Lgals1−/− mice remained invariant in response to the administration of rGal1 (fig. S8). Together, our findings indicate that administration of exogenous rGal1 restored cytokine production and reversed the increase in CD8+CD69+ T cells but had no impact on the augmented frequency of CD4+CD69+ T cells observed in Lgals1 −/− mice.
Fig. 4. Treatment with rGal1 partially ameliorates exacerbated colitis in Lgals1−/− mice.
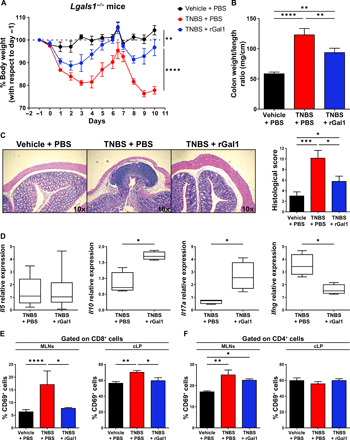
(A) Weight loss curves of Lgals1−/− mice treated either with ethanol (vehicle) or TNBS that received daily injections with PBS or 100 μg of rGal1. Data are from three independent experiments; two-way repeated-measures ANOVA followed by Tukey’s posttest. (B) Colon weight/length ratio. Data are from a representative of three independent experiments (n = 5 mice per group). (C) Representative microscopy images [hematoxylin and eosin (H&E) staining; 10× magnification] and histopathologic assessment (score) for each experimental group as described in (A). Data are from a representative of three independent experiments (n = 5 mice per group). (D) Determination of Il5, Il10, Il17a, and Ifng mRNA in colon tissue by RT-qPCR, normalized to Gapdh expression. Box plots represent median (line), first and third quartiles (box limits), and minimum and maximum values (bars) from a representative of three independent experiments (n = 8 mice per group); two-tailed unpaired Student’s t test. (E) Percentage of CD69+ cells within the total CD8+ T cell population in MLNs and cLP. Data are from a representative of three independent experiments (n = 8 mice per group). In MLNs, Kruskal-Wallis followed by Dunn’s posttest; in cLP, one-way ANOVA followed by Tukey’s posttest. (F) Percentage of CD69+ cells within the total CD4+ T cell population in MLNs and cLP. Data are from a representative of three independent experiments (n = 8 mice per group). In (B), (C), and (F), one-way ANOVA followed by Tukey’s posttest. Unless otherwise stated, data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Given the central role of endogenous Gal1 in TNBS-induced colitis, we evaluated the possibility that changes in the glycosylation signature of CD4+ and CD8+ T cells in the cLP might influence the activity of this lectin and its capacity to promote resolution of intestinal inflammation. Although inflammation had no significant impact on the glycophenotype of CD4+ T cells in the cLP (Fig. 5, A to F), we identified a significant decrease in α(2,6) sialylation in CD8+ T cells that correlated with an increase in Gal1 binding to these cells (Fig. 5, A and B). Hence, intestinal inflammation in this model promoted a switch in the glycan profile of cLP CD8+ T cells, as evidenced by a decrease in α(2,6) sialylation and a consequent increase in Gal1 binding. To determine whether differential α(2,6) sialylation induced by intestinal inflammation in CD8+ T cells is restricted to inflamed cLP, we compared the glycophenotype of CD8+ and CD4+ T cells isolated from spleen and MLNs. TNBS-induced inflammation had no impact on the glycosylation profiles of these cell populations, although we did identify several baseline differences based on location; for example, T cells from MLNs exhibited lower levels of β(1,6) N-glycan branching compared to T cells isolated from the spleen (fig. S9).
Fig. 5. TNBS-induced colitis induces a Gal1-permissive glycophenotype in cLP CD8+ T cells.
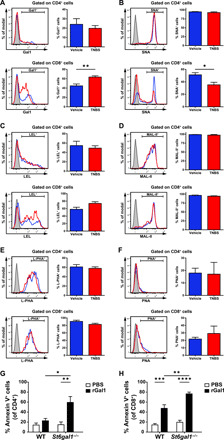
Representative histograms of biotinylated Gal1 (A), Sambucus nigra agglutinin (SNA) (B), Lycopersicon esculentum lectin (LEL) (C), Maackia amurensis lectin II (MAL-II) (D), phytohemagglutinin-L (L-PHA) (E), and peanut agglutinin (PNA) (F) binding to CD4+ and CD8+ T cells isolated from cLP of WT mice treated with ethanol (vehicle, blue lines) or TNBS (red lines). Gray-shaded lines represent negative controls (cells not incubated with biotinylated lectins). Determination of lectin binding within each T cell population is shown. Data are from a representative of two independent experiments (vehicle, n = 4 mice; TNBS, n = 5 mice); two-tailed unpaired Student’s t test. (G and H) Annexin V binding to CD4+ (G) or CD8+ (H) T cells from MLNs isolated from WT or St6gal1−/− mice that were activated for 3 days with anti-CD3/anti-CD28 antibodies and incubated for 16 hours with 10 μM rGal1 or PBS. Data are from five independent experiments; two-way ANOVA followed by Tukey’s posttest. Unless otherwise stated, data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To link decreased α(2,6) sialylation of CD8+ T cells induced by inflammation with the immunoregulatory activity of Gal1, we examined apoptosis of CD4+ and CD8+ T cells triggered by this lectin in MLNs of WT mice compared to those from St6gal1-deficient (St6gal1−/−) mice. Of note, we observed no significant differences in cell proliferation when comparing mice of these two genotypes (fig. S10). However, our results showed that activated CD4+ and CD8+ MLNs T cells from St6gal1−/− hosts (Fig. 5, G and H) were more sensitive to Gal1-induced apoptosis, thus emphasizing the relevance of α(2,6) sialylation in the context of Gal1 function. Notably, MLNs CD8+ T cells were more sensitive to Gal1-induced apoptosis than MLNs CD4+ T cells (Fig. 5, G and H). The proportions of colonic CD8+SNA+ T cells were comparable in WT and Lgals1−/− animals during the course of TNBS-induced colitis, although there was a tendency toward decreased apoptotic CD8+SNA− T cells in Lgals1−/− mice, as indicated by active Caspase-3 staining (fig. S11). Together, these results suggest that CD8+ T cells are major targets of the immunoregulatory effect of this lectin within the local gut microenvironment.
Endogenous Gal1 does not influence the development of DSS-induced colitis
As no experimental colitis model can fully recapitulate the multifactorial nature of human IBD, we also explored the role of endogenous Gal1 in a model of DSS-induced colitis. Notably, no significant differences with respect to the development of colitis were identified when comparing WT and Lgals1−/− mice (fig. S12, A to D). Moreover, we found no alterations in the T cell compartment of the MLNs of DSS-treated Lgals1−/− mice when compared to their WT counterparts (fig. S12E). Their respective cytokine profiles were the only differences observed in DSS-treated Lgals1−/− mice compared to WT controls. Specifically, our findings revealed an increase in interleukin-17A (IL-17A) protein expression in Lgals1−/− mice (fig. S12F). Thus, we conclude that the restorative properties of Gal1 may rely upon unique mechanisms governing the inflammatory processes associated with specific pathological settings.
The glycosyltransferases ST6Gal1 and C2GnT1 control the severity of colitis and govern Gal1 immunoregulatory activity
Most of the characterized anti-inflammatory activities of Gal1 rely on its function in the extracellular milieu and the availability of terminal LacNAc structures present in core 2 O-glycans and complex branched N-glycans (6). Given the decrease in C2GNT1 mRNA expression and the increase in ST6GAL1 mRNA in colon biopsies of patients with IBD (Fig. 2, C and D), we performed a series of experiments designed to evaluate the aberrant glycosylation (as observed in C2gnt1−/− and St6gal1−/− mice) and its influence on the severity of experimental colitis. In particular, we focused on a model of colitis involving the adoptive transfer of immune cells to immunodeficient mice; this strategy facilitates the selective assessment of the impact of T cell glycosylation and its role in promoting intestinal inflammation.
In the first experiment, CD4+CD45RBhigh T cells isolated from C2gnt1−/− mice were transferred into Rag2−/− mice. Compared to mice injected with WT cells, those receiving C2gnt1−/− T cells exhibited earlier onset of disease (~2 to 3 weeks after transfer) and more pronounced weight loss, indicative of increased disease severity (Fig. 6, A and B). Furthermore, mice that received C2gnt1−/− CD4+CD45RBhigh T cells exhibited higher levels of Ifng and lower levels of Il5 expression in the colon when compared to mice that received WT T cells (Fig. 6C). Last, no differences were detected between the two experimental groups with respect to the percentage of Foxp3+ Tregs (Fig. 6D). As anticipated, CD4+ T cells isolated from Rag2−/− mice that were recipients of C2gnt1−/− T cells bound considerably less rGal1 than did those receiving cells from WT mice (fig. S13), thus confirming the essential role of core 2 O-glycans for Gal1 binding. Thus, the absence of core 2 O-glycans, particularly on CD4+ T cells, has a significant impact on the development of intestinal inflammation and results in impaired Gal1 binding.
Fig. 6. Intrinsic T cell glycosylation influences development of colitis in an adoptive-transfer T cell model.
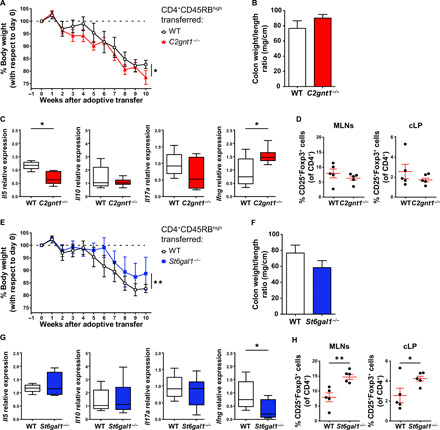
(A) Weight loss curves of Rag2−/− mice following adoptive transfer of either WT or C2gnt1−/− CD4+CD45RBhigh T cells. (B) Colon weight/length ratio. (C) Determination of Il5, Il10, Il17a, and Ifng mRNA in colon tissue by RT-qPCR, normalized to Gapdh mRNA expression. (D) Percentage of Tregs within total CD4+ T cells in MLNs and cLP. (E) Weight loss curves of Rag2−/− mice after adoptive transfer of either WT or St6gal1−/− CD4+CD45RBhigh T cells. (F) Colon weight/length ratio. (G) Determination of Il5, Il0, Il17a, and Ifng mRNA in colon tissue by RT-qPCR, normalized to Gapdh mRNA expression. (H) Percentage of Tregs within CD4+ T cells in MLNs and cLP. Experiments described in this figure were performed simultaneously, so values for WT mice are identical and only included in separate panels for the sake of clarity. In (A) and (E), two-way repeated-measures ANOVA followed by Dunnett’s posttest. In (C) and (G), box plots represent median (line), first and third quartiles (box limits), and minimum and maximum values (bars). In (C), (D), (G), and (H), one-way ANOVA followed by Dunnett’s posttest. In all cases, data are from a representative of two independent experiments (n = 5 mice per group). Unless otherwise stated, data are presented as means ± SEM. *P < 0.05 and **P < 0.01.
Because α(2,6)-linked sialic acids mask Gal1 binding to their cell surface receptors and thus interfere with Gal1 function, our next set of experiments focused on the in vivo transfer of St6gal1−/− CD4+CD45RBhigh T cells into Rag2−/− mice. The results of these experiments showed delayed weight loss (detected at ~6 weeks after transfer) compared to mice receiving WT CD4+CD45RBhigh T cells (Fig. 6E). Although no significant differences in colon weight/length ratio were found (Fig. 6F), mice that received St6gal1−/− T cells responded by expressing less Ifng mRNA and generating higher frequencies of Tregs both in MLNs and cLP (Fig. 6, G and H). As anticipated, CD4+ T cells from Rag2−/− mice that received St6gal1−/− T cells bound more rGal1 compared to those receiving WT T cells (fig. S13). Hence, our data indicate that α(2,6) sialylation, particularly on CD4+ T cells, augments the severity of intestinal inflammation.
To explore the relevance of glycosylation beyond its role with respect to CD4+ T cells, we further evaluated the function of C2GnT1 and ST6Gal1 and their influence on the restorative functions of Gal1 in the double-dose TNBS-induced colitis model. Mice lacking C2GnT1 exhibited exacerbation of TNBS-induced colitis that was associated with increased mortality in comparison to WT mice (Fig. 7, A to C). Administration of rGal1 did not suppress intestinal inflammation in C2gnt1−/− mice; these mice experienced continuous weight loss, persistent tissue inflammation, and increased mortality in response to TNBS-induced colitis (Fig. 7, A to C). These results emphasize the critical role of core 2 O-glycans in controlling Gal1 function. By contrast, TNBS-treated St6gal1−/− mice experienced a milder form of experimental colitis when compared to WT mice (Fig. 7, D to F). Notably, we found no differences in the cytokine profiles between vehicle-treated and TNBS-treated St6gal1−/− mice (fig. S14). Thus, a glycosylation “on-off” switch, regulated by C2GnT1 and ST6Gal1, controls intestinal inflammation by creating or masking Gal1-specific glycoepitopes.
Fig. 7. C2GnT1 and ST6Gal1 glycosyltransferases reciprocally control the development of TNBS-induced colitis.
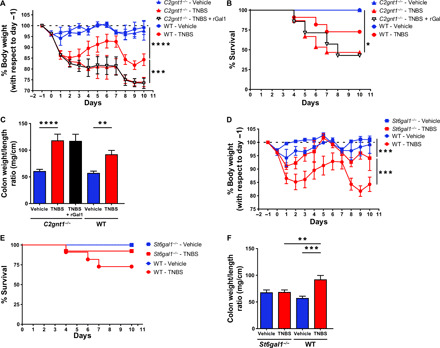
(A) Weight loss curves of WT and C2gnt1−/− mice treated with ethanol (vehicle), TNBS, or TNBS and rGal1. (B) Kaplan-Meier (survival) curves of WT and C2gnt1−/− mice with TNBS-induced colitis treated or not with rGal1. (C) Colon weight/length ratio. (D) Weight loss curves of WT and St6gal1−/− mice treated with ethanol (vehicle) or TNBS. (E) Kaplan-Meier (survival) curves of WT and St6gal1−/− mice with TNBS-induced colitis. (F) Colon weight/length ratio. Experiments described in this figure were performed simultaneously, so values for WT mice are identical and only included in separate panels for the sake of clarity. In (A) and (D), data are from a representative of two independent experiments (n = 5 mice per group); two-way repeated-measures ANOVA followed by Tukey’s post-test. In (B) and (E), data are presented as mean survival proportions from a representative of two independent experiments (n = 7 mice per group); Mantel-Cox (log-rank) test. In (C) and (F), data are from a representative of two independent experiments (n = 5 mice per group); two-way ANOVA followed by Tukey’s posttest. Unless otherwise stated, data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
DISCUSSION
A more complete understanding of the nature and significance of tolerogenic programs and the various mechanisms that control intestinal homeostasis is critical for the development of rational therapeutic strategies aimed at treating patients with IBD. Given the broad immunoregulatory activity of Gal1 (9) and its therapeutic potential in T cell–mediated autoimmunity (8), we investigated the role of endogenous Gal1 and its glycosylated ligands in the development, severity, and resolution of gut inflammation. In this study, using different experimental mouse models and clinical samples from patients with IBD, we identified a glycosylation-dependent on-off circuit driven by Gal1 that controls T cell activity in intestinal inflammation.
Our analysis of publicly available datasets as well as RT-qPCR data from a local cohort of patients with IBD revealed an increase in Gal1 expression in intestinal tissue when involved in the inflammatory process; these findings are similar to those previously described in association with other intestinal pathologies, including celiac disease (12, 34). Although it has been recently reported that patients with IBD exhibit increased levels of circulating Gal1 (35), we did not find significant differences in plasma concentrations between patients and control individuals. These discrepancies could be attributed to differences in genetic backgrounds in the cohorts analyzed or different criteria in patients’ stratification. Moreover, we observed increased expression of Gal1 protein in the gastrointestinal tract of mice in the TNBS-induced colitis model. Of note, the discrepancies in Gal1 expression in this model compared to that previously reported (31) could be attributed to the TNBS administration protocol (single dose versus double dose) or strain-dependent differences (BALB/c versus C57BL/6 mice). This interpretation is consistent with previous studies in which strain-dependent differences in Gal1 expression in models of DSS-induced colitis were reported (36). Lgals1−/− mice presented a more severe form of colitis with higher amounts of interferon-γ (IFN-γ) in cLP, and treatment with exogenous rGal1 partially attenuated this exacerbated inflammatory response through up-regulation of IL-10 and down-regulation of IFN-γ. The increase in IL-10 production may be the result of the coordinated action of distinct mechanisms responsible for orchestrating tolerogenic circuits in the gut, including the induction of type 1 Tregs via activation of the cellular musculoaponeurotic fibrosarcoma (c-Maf)/aryl hydrocarbon receptor pathway (37), differentiation of tolerogenic DCs via phosphorylation of signal transducer and activator of transcription 3 (24), reprogramming of M1 inflammatory macrophages (38, 39), and/or promotion of anti-inflammatory enterocyte programs (40). These results highlight the essential role of endogenous Gal1 as an anti-inflammatory mediator that promotes resolution of autoimmune inflammation and restores immune cell homeostasis, as has been described in models of autoimmune neuroinflammation and inflammatory arthritis (8). This immunomodulatory lectin is released at times of cellular stress and aids to counterbalance exuberant inflammation and tissue damage (9). In line with this evidence, we recently found that aged Lgals1−/− mice develop spontaneous salivary gland autoimmunity, characterized by increased TH1 responses (41). This phenotype is not restricted to Gal1 since Lgals3−/− mice develop a lupus-like disease with spontaneous germinal center formation (42). The paradoxical up-regulation of IL-17A observed in Lgals1−/− mice in response to rGal1 treatment may reflect the dual pro- and anti-inflammatory role of the IL-17 cytokine family within the gut microenvironment (5). Members of the IL-17 cytokine family may drive mucosal inflammation, but they might also aid in restitution and repair of the intestinal mucosa after resolution (43). Moreover, in the TNBS-induced colitis model, TH1-dependent immunity appears to be the dominant effector response, and TH17 cells play both effector and regulatory roles in mucosal inflammation, as IL-17A production can suppress the induction of transcription factor T-box expressed in T cells (T-bet), a major regulator of IFN-γ production (44). In this sense, increased amounts of IL-17A induced by Gal1 were accompanied by a decrease in IFN-γ production and an increase in IL-10, and these effects may counteract the potential proinflammatory effects of IL-17A. In particular, TNBS-induced inflammation was associated with elevated levels of IFN-γ, and prevention of IFN-γ production by anti–IL-12p40 treatment completely abrogated both nascent and established disease (45).
Notably, we observed an increase in the percentage of CD69+ T lymphocytes in both CD8+ (cLP and MLNs) and CD4+ (MLNs) T cell compartments in Lgals1−/− mice. Administration of exogenous rGal1 partially restored the original knockout phenotype by targeting the CD8+CD69+ population; by contrast, CD4+CD69+ MLNs T cells were refractory to Gal1-mediated immunoregulatory activity. These results suggest that CD4+ T cells from MLNs may differ in their sensitivity to Gal1 when compared to splenic T lymphocytes (23). These findings suggest that mucosal immune cells may exhibit distinct glycosylation signatures compared to those of peripheral lymphoid tissues. In this sense, context-dependent phenotypes and glycophenotypes have been identified previously in both DCs and T cells (46, 47); these findings may depend on different environmental cues, including the cytokine milieu and the associated microbiota (48).
Dias et al. (22, 49) recently demonstrated the importance of Mgat5 glycosyltransferase in intestinal inflammation; combined results from these studies and our work suggest that synchronized remodeling of complex branched N-glycans, core 2 O-glycans, and α(2,6) sialylation may ultimately dictate T cell sensitivity or resistance to Gal1 in mucosal inflammatory microenvironments. In this sense, we found that TNBS-induced intestinal inflammation lowers α(2,6) sialylation specifically on CD8+ T cells in cLP. Although the molecular mechanisms implicated in these effects remain uncertain, this phenotype could be associated with changes in ST6Gal1 expression and/or with the activity of inflammation-driven neuraminidases.
We identified a specific subpopulation of CD8+CD69+ T cells as a key target of the immune inhibitory action of Gal1. These findings suggest that Gal1 may control T cell–driven intestinal inflammation by limiting colitis-associated inflammatory CD8+ T cell responses (Fig. 8A). In this regard, cytotoxic CD8+ T cells, particularly those producing IFN-γ (i.e., Tc1), have been shown to play a central role with respect to the development of 2,4-dinitrobenzenesulfonic acid (DNBS)-induced experimental colitis (50). However, unlike CD4+ T cells, which are widely studied and have been characterized in IBD, the relevance of CD8+ T cells and their role in promoting human intestinal inflammation remain controversial. Nevertheless, recent work supported a more prominent role for CD8+ T cells in the development of intestinal inflammation and highlighted both the significance and plasticity of these cells in patients with IBD (51–54). Lack of immunoregulatory activity of Gal1 in DSS-induced colitis may reflect differences in the immunopathogenic mechanisms associated with DSS-induced versus TNBS-induced models, potentially involving innate immunity (i.e., neutrophils and macrophages) or adaptive T cell responses, respectively (55, 56). These results further emphasize the critical role of Gal1 in promoting resolution of T cell–mediated inflammation and uphold the relevance of further work to validate potential translational applications of these findings.
Fig. 8. Modulation of intestinal inflammation via glycosylation-dependent pathways.
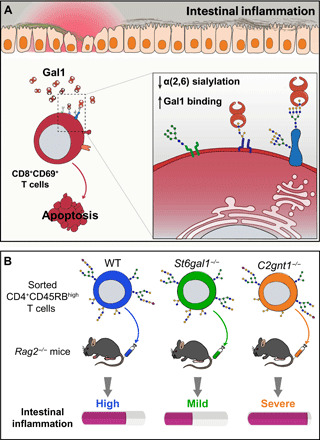
(A) Intestinal inflammation in TNBS-induced colitis alters the extent of α(2,6) sialylation in CD8+CD69+ T cells. This altered glycophenotype makes these cells more sensitive to the immune inhibitory action of Gal1 and recalibrates T cell responses. (B) Role of T cell glycosylation in potentiating or attenuating intestinal inflammation in an adoptive transfer model of colitis. Compared to WT T cells, C2gnt1−/− T cells increase disease severity, generating an earlier onset and more pronounced weight loss. Thus, the absence of core 2 O-glycans on CD4+ T cells influences the development and severity of intestinal inflammation. On the other hand, St6gal1−/− T cells transferred to Rag2−/− mice induce a milder intestinal inflammation with a delayed onset, higher proportions of Tregs and lower IFN-γ production.
The functional activity of Gal1 is critically influenced by cell type–specific expression, subcellular compartmentalization, and activity of key glycosyltransferases, which can either mask or unmask Gal1-specific glycoepitopes (6). Of note, results from our patient cohort revealed significant up-regulation of ST6GAL1 mRNA in biopsies taken from uninflamed areas of the colon in patients with IBD. These results indicated that dysregulation of glycosyltransferases may not be an exclusive feature of the inflamed tissue. It is not clear yet whether alterations in the cellular glycome occur as early events or as a result of chronic inflammation. In this regard, it is also possible that alterations in the cellular glycome may precede and facilitate the inflammatory process. These critical points should be further clarified. Moreover, single-cell analysis will help to elucidate the specific cell types implicated in dysregulation of this program.
Findings from our study reveal that both core 2 O-glycans and α(2,6) sialylation play essential roles in modulating intestinal inflammation as has been described previously for other tissues, including the fetal-maternal unit and the liver (57, 58). Our in vivo studies featuring the TNBS-induced colitis model revealed decreased α(2,6) sialylation in colonic CD8+ T cells; these findings indicate that aberrant T cell responses may be suppressed through a glycan-dependent mechanism (Fig. 8A). In this regard, ST6Gal1-deficient MLNs T cells were more sensitive to the immune inhibitory activity of Gal1, suggesting that altered α(2,6) sialylation could contribute to resolution of intestinal inflammation. Accordingly, adoptive transfer of St6gal1−/− CD4+CD45RBhigh T cells was associated with delayed onset of inflammation and induced higher proportions of Tregs and lower IFN-γ production (Fig. 8B). Although this effect may be partially linked to Gal1-mediated down-regulation of T cell function, we cannot exclude the possibility that T cells lacking ST6Gal1 could be intrinsically hyporeactive upon T cell receptor recognition of its cognate antigen (59) or that there may be altered responses to other glycan-recognizing proteins including not only members of the galectin family but also siglecs via recognition of sialic acid ligands (60). Nonetheless, apart from its association with immune-associated glycan-binding proteins, sialic acid has emerged as a key determinant of immune cell homeostasis in the gut. Alterations in the intestinal sialylation signature may involve different mechanisms beyond transcriptional alteration of glycosyltransferase expression or induction of epigenetic changes, as pathogenic stimuli could trigger expression of endogenous neuraminidases that could also influence the sialylation profile (61).
In addition, our results demonstrate that core 2 O-glycans shape the course of intestinal inflammation and may critically influence the immunomodulatory activity of Gal1. The fact that rGal1 did not influence the recovery of C2gnt1−/− mice with TNBS-induced colitis indicates that this glycosyltransferase likely plays an essential role in the immunomodulatory and therapeutic activity of this lectin. Moreover, C2gnt1−/− mice developed an aggravated form of colitis; adoptive transfer of C2gnt1−/− CD4+CD45RBhigh T cells into Rag2−/− mice resulted in an exacerbation of colitis (Fig. 8B), characterized by increased IFN-γ production. These results highlight the significance of C2GnT1 expressed by mucosal CD4+ T cells in modulating their fate and function in the intestinal microenvironment. Accordingly, previous studies revealed that C2gnt1−/− mice were more susceptible to DSS treatment (18) and demonstrated involvement of this glycosyltransferase in regulating memory CD4+ T cell proliferation (21).
In summary, our study highlights the relevance of cLP T cell glycosylation in potentiating or tempering intestinal inflammation via mechanisms that result in either masking or unmasking Gal1-specific glycoepitopes. Together, these findings suggest a critical role for the Gal1-glycan axis in preserving immune homeostasis in the gut. Although we focused our attention on core 2 O-glycans and terminal α(2,6) sialylation as critical determinants of Gal1 intestinal function, Dias and colleagues (22, 49) demonstrated that β(1,6)-branched N-glycan structures and Mgat5 also played critical roles with respect to the pathogenesis of UC. This effect, which correlated with disease severity, could be reversed by metabolic supplementation with N-acetylglucosamine (22, 49). Future studies should be aimed at elucidating whether both complex N-glycans and core 2 O-glycans jointly assist Gal1-driven tolerogenic circuits to preserve and/or restore gut homeostasis. Collectively, our results emphasize the critical role of endogenous Gal1 as an immune checkpoint pathway that controls intestinal inflammation via glycosylation-dependent contraction of the CD8+ T cell compartment and modulation of inflammatory cytokines.
MATERIALS AND METHODS
Bioinformatic analysis
We chose three curated microarray experiments from the GEO Dataset Browser (www.ncbi.nlm.nih.gov/sites/GDSbrowser/) with colonic biopsies from healthy donors (controls) and patients with IBD: GSE6731 (U.S. cohort; controls, patients with UC, and patients with CD), GSE9452 (Denmark cohort; controls and patients with UC), and GSE38713 (Spain cohort; controls and patients with UC). Criteria were based on three restrictions: (i) All genes of interest should be contained in the microarray, (ii) genes should have validation by RT-qPCR, and (iii) datasets should contain samples from inflamed and uninflamed areas of the colon from patients with IBD. Galectins mRNA (LGALS1, LGALS2, LGALS3, LGALS4, LGALS7, LGALS8, and LGALS9) and transcripts for selected glycosyltransferases (ST6GAL1, MGAT5, and C2GNT1) were analyzed through the online platform GEO2R (www.ncbi.nlm.nih.gov/geo/geo2r/), and data were represented as the logarithm in base 2 of the fold change (FC) with respect to controls.
Patients
This study was approved by the Ethics Committees of Instituto de Biología y Medicina Experimental (IBYME-CONICET) and Hospital de Gastroenterología “Dr. Carlos Bonorino Udaondo” (Buenos Aires, Argentina). In all cases, tissue samples were obtained with the written informed consent of patients. EDTA-anticoagulated blood samples and/or colon biopsies were collected from 23 patients with UC, 12 patients with CD, and 11 healthy controls, who had undergone endoscopic examinations for routine health maintenance to diagnose intestinal conditions that were not linked to inflammatory diseases, including abdominal pain, constipation, or screen for colorectal cancer. Clinical data are presented in Table 1. IBD was ruled out in all the healthy controls by clinical, endoscopic, and conventional histologic criteria. Patients with IBD were diagnosed on the basis of clinical, endoscopic, and histopathologic findings according to currently accepted criteria (62). Clinical disease activity was assessed by two expert gastroenterologists using the Crohn’s Disease Activity Index for patients with CD and the partial Mayo Score for patients with UC (63, 64). CD- or UC-associated disease activity observed on endoscopy was evaluated according to the Inflammatory Bowel Disease in South-Eastern Norway (IBSEN) (65), which is an index that includes a scale of endoscopic activity considered by the European Evidence-Based Consensus for Endoscopy in IBD (66). In contrast to the Mayo sub-endoscopic scoring, Crohn’s Disease Endoscopic Index of Severity, and Simple Endoscopic Score for Crohn’s Disease, the IBSEN score has been designed for use in evaluation of both UC and CD. Hence, this scale allowed us to perform parallel evaluations in patients with different diagnoses but with comparable disease severity. Inflammation severity of human intestinal mucosa was classified by endoscopic criteria, and sample classification was binarized into either inflamed or uninflamed categories on the basis of assessment by an endoscopist. Scores were based on findings from colonic segments from sites of biopsy tissue. Briefly, the inflammatory status of the mucosae was scored from 0 to 2 by endoscopic evaluation by a single observer who is a gastroenterologist with expertise in endoscopic evaluation of IBD. Segments received scores of 0 (normal/no signs of inflammation), 1 (light erythema or granularity), or 2 (granularity, friability, and bleeding, with or without ulcerations). We designated scores of 1 and 2 in the inflamed areas as weak and marked inflammation, respectively. Total colonoscopy was performed in all patients with IBD and healthy controls. Four adjacent biopsy samples were recovered from IBD patients with endoscopically active disease, defined as “inflamed” and focusing in each patient on those regions with the highest score (most severe disease activity, including granularity, and/or friability, bleeding, with or without ulcerations). Meanwhile, samples defined as “uninflamed” were obtained from each patient from (i) areas with endoscopically quiescent (inactive) disease with endoscopic score 0 (defined as “normal” in IBSEN score at the time of study, although some of them had been previously identified as active) and (ii) areas with similar appearance but with neither current macroscopic inflammation nor any history of previous disease activity. Biopsies of the same segment from each patient were pooled, and inflamed or uninflamed areas were compared to each other, as well as to samples from healthy controls. This allowed us to perform direct comparisons between tissue segments from fully healthy controls with those from patients with IBD based on disease activity (inflamed or uninflamed as defined at the time of the colonoscopy).
RNA extraction and RT-qPCR
Total RNA was purified from human colon biopsies and mouse colon samples using TRIzol reagent (Life Technologies, Thermo Fisher Scientific) and deoxyribonuclease I (DNAse I; Amplification Grade, Thermo Fisher Scientific). Copy DNA was synthesized using SuperScript II Reverse Transcriptase (Thermo Fisher Scientific), according to the manufacturer’s instructions in the presence of random hexamers (2.5 μg/ml), deoxynucleotide triphosphates (500 nM), and 20 U of RNAseOUT (Thermo Fisher Scientific). Gene expression was analyzed using SYBR Green PCR Master Mix (Applied Biosystems, Thermo Fisher Scientific) and CFX96 Touch Real-Time PCR Detection System (Bio-Rad). All reactions were performed in duplicate in the presence of suitable primers, purchased from Integrated DNA Technologies, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as loading control (300 nM of each primer). Primer sequences used were human LGALS1 forward: 5′-CCTGGAGAGTGCCTTCGAGTG-3′, reverse: 5′-CTGCAACACTTCCAGGCTGG-3′; human LGALS3 forward: 5′-CCAACGAGCGGAAAATGG-3′, reverse: 5′-CATCCTTGAGGGTTTGGGTTT-3′; human LGALS4 forward: 5′-CTGGTCTTCATAGTCCTGGCTGAG-3′, reverse: 5′-AGGCTGTTCCGGACCACG-3′; human LGALS9 forward: 5′-CGTGTGGACACCATCTCCG-3′, reverse: 5′-CAGCCCTCCCAGAATGGTG-3′; human ST6GAL1 forward: 5′-ATCGTAAGCTGCACCCCAAT-3′, reverse: 5′-ATGATACCAAGCATCCCAGAGG-3′; human MGAT5 forward: 5′-TGCCCCTGCCGGGACTTCAT-3′, reverse: 5′-CAGCAGCATGGTGCAGGGCT-3′; human C2GNT1 forward: 5′-CTACCCGCCCTGCGATG-3′, reverse: 5′-CATCCAGTTCAAGTCACCAGCTC-3′; human GAPDH forward: 5′-GATGCCCCCATGTTTGTGAT-3′, reverse: 5′-GGTCATGAGTCCTTCCACGAT-3′; mouse Ifng forward: 5′-GGCTGTTACTGCCACGGCACA-3′, reverse: 5′-CACCATCCTTTTGCCAGTTCCTCCA-3′; mouse Il5 forward: 5′-ACACAGCTGTCCGCTCACCG-3′, reverse: 5′-TCCACAGTACCCCCACGGACA-3′; mouse Il10 forward: 5′-TTCCCTGGGTGAGAAGCTGA-3′, reverse: 5′-CTTCACCTGCTCCACTGCCT-3′; mouse Il17a forward: 5′-CTCCAGAAGGCCCTCAGACTAC-3′, reverse: 5′-AGCTTTCCCTCCGCATTGACACAG-3′; mouse Tnf forward: 5′-TACTGAACTTCGGGGTGATCG-3′, reverse: 5′-TGATGAGAGGGAGGCCATTT-3′; and mouse Gapdh forward: 5′-CAGAACATCATCCCTGCAT-3′, reverse: 5′-GTTCAGCTCTGGGATGACCTT-3′.
Plasma Gal1 determination
Plasma from patients with IBD and healthy controls were obtained from EDTA-anticoagulated blood samples by centrifugation (500g, 10 min). Circulating Gal1 levels were measured by an in-house enzyme-linked immunosorbent assay (ELISA) as described (27).
Production of rGal1 and biotinylated Gal1
rGal1 was produced and purified as outlined previously (67). Biotinylated Gal1 was obtained using the EZ-Link NHS-LC-Biotin kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Mouse strains
Mice used in this study (all on a C57BL/6 background) were maintained in the animal facilities of IBYME-CONICET under a 12-hour light/12-hour dark regime. C57BL/6 WT, C2gnt1−/−, and Rag2−/− mice were purchased from the Jackson laboratory. Lgals1−/− mice were originally provided by F. Poirier (Jacques Monod Institute, Paris, France), and St6gal1−/− mice were provided by J. Paulson (La Jolla, CA, USA). The Institutional Committee for the Care and Use of Laboratory Animals of IBYME-CONICET approved all protocols.
TNBS-induced colitis
On the basis of previous protocols for single-dose models (31), we generated a double-dose colitis model in 9-week-old male C57BL/6 mice. Briefly, mice were fasted 8 hours before intrarectal instillation of 100 μl of TNBS solution in ethanol 50% (v/v) (Sigma-Aldrich) or ethanol 50% (v/v) alone (vehicle) using an intravenous 20G Teflon catheter (Dexal). The TNBS dose was optimized for the C57BL/6-resistant strain and calculated for each animal as 0.25 mg/g of body weight. Body weight for each animal was monitored daily and normalized to the body weight recorded the day before fasting (day −1). At day 7 after instillation, animals were reinstilled with intrarectal TNBS or vehicle solution using doses adjusted to their body weight as described above. At day 10 (3 days after the second instillation), mice were euthanized by cervical dislocation for subsequent analysis.
DSS-induced colitis
Chronic colitis was induced in 8- to 10-week-old female C57BL/6 mice by repeated administration of DSS in drinking water (3 cycles). Each cycle consisted of 5 days of administration of sterile-filtered tap water with 2.5% (w/v) DSS (molecular weight, 36,000 to 50,000; MP Biomedicals Inc.) followed by 5 days of autoclaved tap water. Body weights (normalized to weights on day 0), stool consistency, and the presence of blood in stool samples were examined three times per week. On day 30, animals were euthanized by cervical dislocation for subsequent analysis.
Adoptive transfer of CD4+CD45RBhigh T cells to Rag2−/− mice
CD4+ T cells were isolated from spleens of 10- to 12-week-old male WT, St6gal1−/−, or C2gnt1−/− mice using the Dynabeads Untouched Mouse CD4 Cells kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Enriched CD4+ T cells (>80% purity) were labeled with Alexa Fluor 647–conjugated anti-mouse CD4 (RM4-5, BD Pharmingen) and fluorescein isothiocyanate (FITC)–conjugated anti-mouse CD45RB (16A, BD Pharmingen) antibodies for 30 min at 4°C. CD4+CD45RBhigh T cells were isolated using a FACSAria II cell sorter (Becton Dickinson) to >90% purity; these cells were washed, counted, and resuspended in sterile phosphate-buffered saline (PBS). Rag2−/− mice (10 to 12 weeks old) were injected intraperitoneally with 500,000 CD4+CD45RBhigh T cells (WT, St6gal1−/−, or C2gnt1−/−) for induction of colitis. Body weights for each mouse were taken weekly and normalized individually to body weights before T cell transfer (week 0). At week 10 after T cell transfer, animals were euthanized by cervical dislocation for subsequent analysis.
Administration of rGal1 in TNBS-induced colitis
Experimental colitis was induced in male C57BL/6 Lgals1−/− and WT mice (8 to 10 weeks old) as described above. TNBS-treated animals were injected intraperitoneally with 100 μg of rGal1 in 100 μl of PBS daily (from days 0 to 9) or with 100 μl of PBS (vehicle) daily. Control animals received ethanol 50% (v/v) (vehicle) and were injected intraperitoneally with 100 μl of PBS daily. Body weight for each animal was recorded daily and normalized to the body weight recorded the day before fasting (day −1). At day 10, animals were euthanized.
Histological assessment of TNBS-induced colitis
After euthanasia, colon was removed and three tissue samples from the proximal, middle, and distal parts of the colon were obtained and fixed with 4% formaldehyde in PBS. Paraffin-embedded sections were stained with hematoxylin and eosin (H&E). Sections were analyzed by a blinded pathologist (M.M.). The severity of colon damage was assessed by means of a previously described scoring system (68–70) with some modifications, described below. Mucosal integrity is as follows: 0, normal; 1, isolated cell death (apoptosis); 2, erosions; 3, ulceration. Score for polymorphonuclear leukocytes (PMN) in lamina propria is as follows: 0, 1 to 5 PMN; 1, 6 to 10 PMN; 2, >11 PMN. Cryptitis is as follows: 0, absent; 1, with abscesses. Edema is as follows: 0, normal; 1, presence of edema. Colon architecture is as follows: 0, normal; 1, isolated glandular branches; 2, altered. Lymphocytes in lamina propria are as follows: 0, 0 to 20; 1, 21 to 40; 2, 41 to 60; 3, >61.
Histological assessment of DSS-induced colitis
After euthanasia, colons were removed and tissue samples from the distal part of the colon were extracted for histologic analysis. Tissue samples were fixed in 4% formaldehyde in PBS. Paraffin-embedded sections were stained with H&E. A blind examination on histological sections was performed by a pathologist (M.M.). The severity of colonic damage was assessed by means of the scoring system described below, adapted from literature (71). Mucosal inflammation is as follows: 0, normal; 1, low; 2, moderate; 3, severe. Inflammation extension is as follows: 0, normal; 1, mucosa; 2, mucosa and submucosa; 3, transmembrane. Regeneration is as follows: 0, complete regeneration; 1, almost complete regeneration; 2, regeneration with crypt depletion; 3, non-intact epithelium surface; 4, no separation. Crypt damage is as follows: 0, no damage; 1, 1/3 basal; 2, 2/3 basal; 3, only surface epithelium; 4, entire crypt loss.
Gal1 immunohistochemistry
After euthanasia, colons were removed and tissue samples from the distal part of the colon were obtained and fixed with 4% formaldehyde in PBS and embedded in paraffin. Sections of 5 μm were dewaxed and rehydrated in serial ethanol dilutions. Endogenous peroxidase activity was blocked with 3% (v/v) H2O2 for 15 min followed by antigen retrieval performed with sodium citrate treatment (10 mM, pH 6.0). Slides were stained with rabbit anti-Gal1 polyclonal IgG obtained as previously described (27) at 4°C overnight. Gal1 staining was detected using the Vectastain Elite ABC kit (Vector Laboratories) according to the manufacturer’s instructions or by using anti-rabbit–Alexa Fluor 546 secondary antibody followed by nuclei staining with 4′,6-diamidino-2-phenylindole and processed by confocal microscopy. Micrographs were taken with an Olympus CX31 microscope for bright field or in an Olympus FV1000 confocal microscope for immunofluorescence.
Preparation of mouse colonic extracts
Tissue samples from the proximal, middle, and distal parts of the colon were obtained and mechanically processed in protein extraction buffer [50 mM tris-HCl (pH 7.5), 150 mM NaCl, 10 mM EDTA, and 1% NP-40] containing a protease inhibitor cocktail (Sigma-Aldrich). Cellular debris was eliminated by centrifugation (12,000g, 10 min). Protein concentration was determined in supernatants using the Micro BCA kit (Pierce) following the manufacturer’s instructions.
Determination of Gal1 and cytokines in colonic extracts by ELISA
Mouse Gal1 was determined in colonic protein extracts by ELISA using specific capture (AF1245, R&D Systems) and detection (BAF1245, R&D Systems) antibodies and mouse rGal1 (1245-GA, R&D Systems) for the standard curve. Gal1 protein concentration was normalized to the amount of total protein measured by the Micro BCA kit (Pierce). Murine IL-5, IL-6, transforming growth factor–β1, IFN-γ, IL-17A, and IL-10 were analyzed in colonic protein extracts from DSS-treated mice using ELISA kits (R&D Systems), according to the manufacturer’s instructions, and relativized to total protein.
Western blot
Total colonic protein extracts (30 μg of total protein) were suspended in 2× Laemmli sample buffer (Bio-Rad) under reducing conditions and subjected to SDS–polyacrylamide gel electrophoresis followed by transfer onto Amersham Protran nitrocellulose membranes (GE Healthcare). After blocking, membranes were incubated with rabbit anti-actin antibody (I-19, Santa Cruz Biotechnology Inc.) and rabbit anti-Gal1 polyclonal IgG obtained as previously described (27). Membranes were then incubated with horseradish peroxidase–labeled anti-rabbit secondary antibody (Bio-Rad) and developed using ECL Prime Western Blotting Detection Reagent (Amersham Biosciences). Protein bands were analyzed with ImageJ software.
Isolation of leukocytes from the spleen, MLNs, and cLP
Spleens and MLNs were removed and mechanically disrupted in complete RPMI 1640 media (Gibco), supplemented with 10% fetal bovine serum (FBS; Gibco). Afterward, cells were passed through a 70-μm Falcon cell strainer (Thermo Fisher Scientific) and washed twice with PBS. Cells from MLNs were resuspended in PBS containing 10% FBS and kept on ice until use. On the other hand, cells from spleens were resuspended in 1 ml of ammonium-chloride-potassium (ACK) lysing buffer for 5 min to eliminate red blood cells. Afterward, cells were diluted in PBS (9 ml) and centrifuged (300g, 10 min). Last, splenocytes were resuspended in PBS containing 10% FBS and kept on ice until use.
Colons were removed and their length and weight were measured before processing. Isolation of cLP mononuclear cells was performed as follows: Colon specimens were opened longitudinally and washed with ice-cold Hank’s balanced salt solution (HBSS) without Ca2+/Mg2+ (Thermo Fisher Scientific), cut into 3-mm pieces, and incubated (37°C, 20 min) with HBSS/EDTA (5 mM) 5% FBS under continuous stirring. To eliminate remnant epithelial cells and EDTA, colon fragments were then washed with HBSS (37°C, 20 min). Then, colon fragments were placed in C tubes (MACS) and disaggregated (37°C, 50 min) with freshly prepared enzymatic solution: collagenase D (1 mg/ml) (Roche), dispase II (1 mg/ml) (Sigma-Aldrich), DNAse I (20 U/ml) (Thermo Fisher Scientific), and 10% FBS in PBS under continuous stirring. After incubation with the enzymatic solution, C tubes were placed in the gentleMACS Dissociator (MACS). Disaggregated colonic tissues were diluted in ice-cold PBS and filtered through a 100-μm Falcon cell strainer (Thermo Fisher Scientific). Cells were then resuspended in 4 ml of 40% Percoll (Percoll, GE Healthcare) in complete Dulbecco’s modified Eagle medium (Gibco), which was then layered on top of 4 ml of 70% Percoll. After centrifugation (680g, 20 min, without brake), cells from the interface were collected and washed with PBS. Last, cells were resuspended in 10% FBS in PBS and kept on ice until use.
In vitro T cell stimulation and cell death assay
Freshly isolated total lymphocytes (1 × 105) from MLNs of WT and St6gal1−/− animals were cultured for 3 days in 100 μl of complete RPMI 1640 media supplemented with 10% FBS and agonistic anti-mouse CD28 antibody (1 μg/ml) (37.51, Bio X Cell) in a CellStar U-bottom 96-well culture plate (Greiner Bio-One) previously coated with 100 μl of agonistic anti-mouse CD3ε antibody (5 μg/ml) (145-2C11, Bio X Cell) in PBS. After 3 days of polyclonal stimulation, cells were harvested and washed with PBS. Then, cells were incubated for 16 hours with 10 μM rGal1 or PBS (vehicle), harvested, and stained to determine the proportion of apoptotic CD4+ and CD8+ T cells by flow cytometry.
Flow cytometry
Flow cytometry was performed using a FACSCanto II cytometer (Becton Dickinson), and data were analyzed with FlowJo software (V.10.0.7r2; FlowJo LLC). Leukocytes from spleen, MLNs, or cLP were prepared for flow cytometry analysis as follows: Tregs were labeled with anti-mouse CD4-FITC (RM4-5, BD Pharmingen) and anti-mouse CD25-phycoerythrin (PE) (PC61, BD Pharmingen) antibodies in cytometry buffer (PBS, 1% FBS, and 0.02% NaN3) for 30 min at 4°C. Cells were then fixed and permeabilized with the Intracellular Fixation & Permeabilization Buffer set (eBioscience) according to the manufacturer’s instructions. Foxp3 was labeled with anti-mouse Foxp3–Alexa Fluor 647 (MF23, BD Pharmingen) in permeabilization buffer (eBioscience) for 30 min at 4°C. Last, cells were washed with permeabilization buffer and resuspended in cytometry buffer until analysis. Activated T cells were labeled with anti-mouse CD4-PE (RM4-4, BioLegend), anti-mouse CD8-PE-Cy7 (53-6.7, BioLegend), and anti-mouse CD69-FITC (H1.2F3, BD Pharmingen) antibodies in cytometry buffer for 30 min at 4°C. Cells were then fixed with 1% formaldehyde. Colonic resident memory T cells were labeled using the previously described antibodies and anti-mouse CD103-allophycocyanin (APC) (2E7, eBioscience) following the same protocol.
For lectin binding assays, spleen, MLNs, or cLP cells were incubated for 30 min with 10 μM biotinylated lectins in buffer A [150 mM NaCl, 10 mM Hepes (pH 7.4), and 1% bovine serum albumin]. Afterward, cells were washed with buffer A and incubated with streptavidin-PE (BD), anti-mouse CD4-APC (RM4-5, BD Pharmingen) and anti-mouse CD8-FITC (53-6.7, BioLegend) antibodies for 30 min at 4°C. The excess of antibodies and streptavidin was washed with buffer A. Last, cells were fixed with 1% formaldehyde in PBS. Lectins used in this study include rGal1 and the following plant lectins (all from Vector Laboratories): Phytohemagglutinin-L [L-PHA; recognizing β(1,6)-branched complex N-glycans], Sambucus nigra agglutinin [SNA; with specificity for α(2,6)-linked sialic acid], Maackia amurensis lectin II [MAL-II; which recognizes α(2,3)-linked sialic acid], peanut agglutinin (PNA; with specificity for asialo-core 1 O-glycans), and Lycopersicon esculentum lectin (LEL; recognizing poly-LacNAc residues). Apoptotic cLP CD8+SNA− T cells were first incubated for 30 min with biotinylated SNA in buffer A, as previously described, and subsequently labeled with anti-mouse CD8-APC (53-6.7, BioLegend) antibodies in cytometry buffer for 30 min at 4°C. Cells were then stained for active Caspase-3 using the Active Caspase-3 Apoptosis Kit (BD Pharmingen) according to the manufacturer’s instructions.
Proliferation of in vitro–activated CD4+ and CD8+ T cells from MLNs was assessed with anti-mouse CD4-APC (RM4-5, BD Pharmingen) and anti-mouse CD8-PE-Cy7 (53-6.7, BioLegend) antibodies using Tag-it Violet Proliferation Cell Tracking Dye (BioLegend), according to the manufacturer’s instructions. To analyze the percentage of annexin V+ T cells, cells were first labeled with anti-mouse CD4-APC-Cy7 (RM4-5, BioLegend) and anti-mouse CD8-APC (53-6.7, BioLegend) antibodies in cytometry buffer for 30 min at 4°C. Cells were then stained with annexin V–FITC (BD Pharmingen), using annexin V buffer (BD Pharmingen), according to the manufacturer’s instructions.
Statistical analysis
GraphPad Prism version 8 (GraphPad Software Inc.) was used for statistical analysis. Gene expression meta-analysis of galectins and glycosyltransferases in IBD datasets was performed as follows: For each gene, data were separated according to biopsies origin (“IBD” factor: healthy controls, uninflamed, and inflamed areas of patients with IBD) and datasets origin (“Dataset” factor); afterward, the main IBD effect was analyzed with two-way analysis of variance (ANOVA), followed by Tukey’s posttest. Wasting disease curves were compared with two-way repeated-measures ANOVA, followed by Tukey’s or Dunnett’s posttest. Two groups were compared with two-tailed Student’s t test for unpaired data. For multiple comparisons, multiple t test (using the Holm-Sidak correction method), one-way or two-way ANOVA followed by Tukey’s or Dunnett’s posttests (parametric analysis), or Kruskal-Wallis followed by Dunn’s posttest (nonparametric analysis) were used. Kaplan-Meier (survival) curves were compared with the Mantel-Cox (log-rank) test. Last, a linear regression model and Pearson’s coefficient (r) were used for correlation analysis. P values of 0.05 or less were considered significant.
Acknowledgments
We would like to thank F. Poirier and J. Paulson for providing Lgals1−/− and St6gal1−/− mice, respectively; H. Rosenberg for critical reading of the manuscript; and V. Sundblad for helpful insights. This work is dedicated to the memory of J. L. Daniotti. A.J.C., J.M.P.-S., D.O.C., S.P.M.-H., M.R.G., G.H.D., M.A.T., G.A.R., and K.V.M. are members of the Scientific Career of Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET; Argentina). L.G.M., A.M.C., M.N.M.-C., and J.P.M. were supported by doctoral or postdoctoral fellowships from CONICET. Funding: This work was supported by Kenneth Rainin Foundation Innovator Awards (2015-2019), Broad Foundation (currently Crohn and Colitis Foundation of America; IBD-0378, 2013), CONICET (PIP 2013-2015-041) and Argentinean Agency for Promotion of Science and Technology (PICT 2012-0555 and PICT 2014-3687) to K.V.M. and G.A.R. We also thank Sales, Bunge & Born, Baron, and Lounsbery Foundations for support and Ferioli, Ostry, and Caraballo families for donations. Author contributions: G.A.R. and K.V.M. conceived, designed, and supervised the study. L.G.M. designed experiments, acquired, analyzed, and interpreted data from the local cohort of patients with IBD, as well as from TNBS-induced and adoptive transfer models of colitis. A.M.C. and A.J.C. designed experiments, acquired, analyzed, and interpreted data from DSS-induced colitis model. L.G.M., J.P.M., and M.R.G. analyzed and interpreted data from publicly available datasets. A.M.C. assisted with the acquisition and analysis of data from all colitis mouse models. J.M.P.-S. produced and characterized rGal1 and its biotinylated form for in vitro and in vivo studies. L.G.M., M.N.M.-C., S.P.M.-H., and M.A.T. designed experiments and acquired, analyzed, and interpreted data from in vitro studies. D.O.C. acquired, analyzed, and interpreted data of immunohistochemistry experiments. R.M.M. assisted with animal models and experimental design. M.M. performed histological analysis. G.H.D. assisted with technical expertise in the TNBS-induced colitis model. B.P., A.H.G., S.P.H., and A.M.S. managed patients with IBD. L.G.M., A.M.C., G.A.R., and K.V.M. drafted the article. All authors revised the manuscript critically. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabf8630/DC1
REFERENCES AND NOTES
- 1.Knowles S. R., Graff L. A., Wilding H., Hewitt C., Keefer L., Mikocka-Walus A., Quality of life in inflammatory bowel disease: A systematic review and meta-analyses—Part I. Inflamm. Bowel Dis. 24, 742–751 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Ng S. C., Shi H. Y., Hamidi N., Underwood F. E., Tang W., Benchimol E. I., Panaccione R., Ghosh S., Wu J. C. Y., Chan F. K. L., Sung J. J. Y., Kaplan G. G., Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet (London, England). 390, 2769–2778 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Chudy-Onwugaje K. O., Christian K. E., Farraye F. A., Cross R. K., A state-of-the-art review of new and emerging therapies for the treatment of IBD. Inflamm. Bowel Dis. 25, 820–830 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olivera P., Danese S., Peyrin-Biroulet L., Next generation of small molecules in inflammatory bowel disease. Gut 66, 199–209 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Moschen A. R., Tilg H., Raine T., IL-12, IL-23 and IL-17 in IBD: Immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 16, 185–196 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Rabinovich G. A., Croci D. O., Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity 36, 322–335 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Varki A., Biological roles of glycans. Glycobiology 27, 3–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toscano M. A., Martinez Allo V. C., Cutine A. M., Rabinovich G. A., Mariño K. V., Untangling galectin-driven regulatory circuits in autoimmune inflammation. Trends Mol. Med. 24, 348–363 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Sundblad V., Morosi L. G., Geffner J. R., Rabinovich G. A., Galectin-1: A jack-of-all-trades in the resolution of acute and chronic inflammation. J. Immunol. 199, 3721–3730 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Russo A. J., Vasudevan S. O., Méndez-Huergo S. P., Kumari P., Menoret A., Duduskar S., Wang C., Pérez Sáez J. M., Fettis M. M., Li C., Liu R., Wanchoo A., Chandiran K., Ruan J., Vanaja S. K., Bauer M., Sponholz C., Hudalla G. A., Vella A. T., Zhou B., Deshmukh S. D., Rabinovich G. A., Rathinam V. A., Intracellular immune sensing promotes inflammation via gasdermin D–driven release of a lectin alarmin. Nat. Immunol. 22, 154–165 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Block M., Molne J., Leffler H., Börjesson L., Breimer M. E., Immunohistochemical studies on galectin expression in colectomised patients with ulcerative colitis. Biomed. Res. Int. 2016, 5989128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papa Gobbi R., De Francesco N., Bondar C., Muglia C., Chirdo F., Rumbo M., Rocca A., Toscano M. A., Sambuelli A., Rabinovich G. A., Docena G. H., A galectin-specific signature in the gut delineates Crohn’s disease and ulcerative colitis from other human inflammatory intestinal disorders. Biofactors 42, 93–105 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Vasta G. R., Galectins as pattern recognition receptors: Structure, function, and evolution. Adv. Exp. Med. Biol. 946, 21–36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Theodoratou E., Campbell H., Ventham N. T., Kolarich D., Pučič-Bakovič M., Zoldoš V., Fernandes D., Pemberton I. K., Rudan I., Kennedy N. A., Wuhrer M., Nimmo E., Annese V., McGovern D. P. B., Satsangi J., Lauc G., The role of glycosylation in IBD. Nat. Rev. Gastroenterol. Hepatol. 11, 588–600 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Klarić L., Tsepilov Y. A., Stanton C. M., Mangino M., Sikka T. T., Esko T., Pakhomov E., Salo P., Deelen J., McGurnaghan S. J., Keser T., Vučkovič F., Ugrina I., Krištič J., Gudelj I., Štambuk J., Plomp R., Pučič-Baković M., Pavić T., Vilaj M., Trbojević-Akmačič I., Drake C., Dobrinič P., Mlinarec J., Jelušič B., Richmond A., Timofeeva M., Grishchenko A. K., Dmitrieva J., Bermingham M. L., Sharapov S. Z., Farrington S. M., Theodoratou E., Uh H.-W., Beekman M., Slagboom E. P., Louis E., Georges M., Wuhrer M., Colhoun H. M., Dunlop M. G., Perola M., Fischer K., Polasek O., Campbell H., Rudan I., Wilson J. F., Zoldoš V., Vitart V., Spector T., Aulchenko Y. S., Lauc G., Hayward C., Glycosylation of immunoglobulin G is regulated by a large network of genes pleiotropic with inflammatory diseases. Sci. Adv. 6, eaax0301 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moran A. P., Gupta A., Joshi L., Sweet-talk: Role of host glycosylation in bacterial pathogenesis of the gastrointestinal tract. Gut 60, 1412–1425 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Kudelka M. R., Stowell S. R., Cummings R. D., Neish A. S., Intestinal epithelial glycosylation in homeostasis and gut microbiota interactions in IBD. Nat. Rev. Gastroenterol. Hepatol. 17, 597–617 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu J., Wei B., Wen T., Johansson M. E. V., Liu X., Bradford E., Thomsson K. A., McGee S., Mansour L., Tong M., McDaniel J. M., Sferra T. J., Turner J. R., Chen H., Hansson G. C., Braun J., Xia L., Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J. Clin. Invest. 121, 1657–1666 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sommer F., Adam N., Johansson M. E. V., Xia L., Hansson G. C., Bäckhed F., Altered mucus glycosylation in core 1 O-glycan-deficient mice affects microbiota composition and intestinal architecture. PLOS ONE 9, e85254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kudelka M. R., Hinrichs B. H., Darby T., Moreno C. S., Nishio H., Cutler C. E., Wang J., Wu H., Zeng J., Wang Y., Ju T., Stowell S. R., Nusrat A., Jones R. M., Neish A. S., Cummings R. D., Cosmc is an X-linked inflammatory bowel disease risk gene that spatially regulates gut microbiota and contributes to sex-specific risk. Proc. Natl. Acad. Sci. U.S.A. 113, 14787–14792 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishida A., Nagahama K., Imaeda H., Ogawa A., Lau C. W., Kobayashi T., Hisamatsu T., Preffer F. I., Mizoguchi E., Ikeuchi H., Hibi T., Fukuda M., Andoh A., Blumberg R. S., Mizoguchi A., Inducible colitis-associated glycome capable of stimulating the proliferation of memory CD4+ T cells. J. Exp. Med. 209, 2383–2394 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dias A. M., Dourado J., Lago P., Cabral J., Marcos-Pinto R., Salgueiro P., Almeida C. R., Carvalho S., Fonseca S., Lima M., Vilanova M., Dinis-Ribeiro M., Reis C. A., Pinho S. S., Dysregulation of T cell receptor N-glycosylation: A molecular mechanism involved in ulcerative colitis. Hum. Mol. Genet. 23, 2416–2427 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Toscano M. A., Bianco G. A., Ilarregui J. M., Croci D. O., Correale J., Hernandez J. D., Zwirner N. W., Poirier F., Riley E. M., Baum L. G., Rabinovich G. A., Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 8, 825–834 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Ilarregui J. M., Croci D. O., Bianco G. A., Toscano M. A., Salatino M., Vermeulen M. E., Geffner J. R., Rabinovich G. A., Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat. Immunol. 10, 981–991 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Liu S. D., Tomassian T., Bruhn K. W., Miller J. F., Poirier F., Miceli M. C., Galectin-1 tunes TCR binding and signal transduction to regulate CD8 burst size. J. Immunol. 182, 5283–5295 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Rubinstein N., Alvarez M., Zwirner N. W., Toscano M. A., Ilarregui J. M., Bravo A., Mordoh J., Fainboim L., Podhajcer O. L., Rabinovich G. A., Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection: A potential mechanism of tumor-immune privilege. Cancer Cell 5, 241–251 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Croci D. O., Cerliani J. P., Dalotto-Moreno T., Mendez-Huergo S. P., Mascanfroni I. D., Dergan-Dylon S., Toscano M. A., Caramelo J. J., Garcia-Vallejo J. J., Ouyang J., Mesri E. A., Junttila M. R., Bais C., Shipp M. A., Salatino M., Rabinovich G. A., Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 156, 744–758 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Davicino R. C., Mendez-Huergo S. P., Elicabe R. J., Stupirski J. C., Autenrieth I., Di Genaro M. S., Rabinovich G. A., Galectin-1–driven tolerogenic programs aggravate Yersinia enterocolitica infection by repressing antibacterial immunity. J. Immunol. 199, 1382–1392 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Di Lella S., Sundblad V., Cerliani J. P., Guardia C. M., Estrin D. A., Vasta G. R., Rabinovich G. A., When galectins recognize glycans: From biochemistry to physiology and back again. Biochemistry 50, 7842–7857 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Earl L. A., Bi S., Baum L. G., N- and O-glycans modulate galectin-1 binding, CD45 signaling, and T cell death. J. Biol. Chem. 285, 2232–2244 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santucci L., Fiorucci S., Rubinstein N., Mencarelli A., Palazzetti B., Federici B., Rabinovich G. A., Morelli A., Galectin-1 suppresses experimental colitis in mice. Gastroenterology 124, 1381–1394 (2003). [DOI] [PubMed] [Google Scholar]
- 32.van Beelen Granlund A., Flatberg A., Østvik A. E., Drozdov I., Gustafsson B., Kidd M., Beisvag V., Torp S. H., Waldum H. L., Martinsen T. C., Damås J. K., Espevik T., Sandvik A. K., Whole genome gene expression meta-analysis of inflammatory bowel disease colon mucosa demonstrates lack of major differences between Crohn’s disease and ulcerative colitis. PLOS ONE 8, e56818 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radulovic K., Niess J. H., CD69 is the crucial regulator of intestinal inflammation: A new target molecule for IBD treatment? J. Immunol. Res. 2015, 497056 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sundblad V., Quintar A. A., Morosi L. G., Niveloni S. I., Cabanne A., Smecuol E., Mauriño E., Mariño K. V., Bai J. C., Maldonado C. A., Rabinovich G. A., Galectins in intestinal inflammation: Galectin-1 expression delineates response to treatment in celiac disease patients. Front. Immunol. 9, 379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu T. B., Dodd S., Yu L.-G., Subramanian S., Serum galectins as potential biomarkers of inflammatory bowel diseases. PLOS ONE 15, e0227306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathieu A., Nagy N., Decaestecker C., Ferdinande L., Vandenbroucke K., Rottiers P., Cuvelier C. A., Salmon I., Demetter P., Expression of galectins-1, -3 and -4 varies with strain and type of experimental colitis in mice. Int. J. Exp. Pathol. 89, 438–446 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cedeno-Laurent F., Opperman M., Barthel S. R., Kuchroo V. K., Dimitroff C. J., Galectin-1 triggers an immunoregulatory signature in Th cells functionally defined by IL-10 expression. J. Immunol. 188, 3127–3137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Starossom S. C., Mascanfroni I. D., Imitola J., Cao L., Raddassi K., Hernandez S. F., Bassil R., Croci D. O., Cerliani J. P., Delacour D., Wang Y., Elyaman W., Khoury S. J., Rabinovich G. A., Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity 37, 249–263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yaseen H., Butenko S., Polishuk-Zotkin I., Schif-Zuck S., Pérez-Sáez J. M., Rabinovich G. A., Ariel A., Galectin-1 facilitates macrophage reprogramming and resolution of inflammation through IFN-β. Front. Pharmacol. 11, 901 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muglia C. I., Gobbi R. P., Smaldini P., Delgado M. L. O., Candia M., Zanuzzi C., Sambuelli A., Rocca A., Toscano M. A., Rabinovich G. A., Docena G. H., Inflammation controls sensitivity of human and mouse intestinal epithelial cells to galectin-1. J. Cell. Physiol. 231, 1575–1585 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Martínez Allo V. C., Hauk V., Sarbia N., Pinto N. A., Croci D. O., Dalotto-Moreno T., Morales R. M., Gatto S. G., Manselle Cocco M. N., Stupirski J. C., Deladoey Á., Maronna E., Marcaida P., Durigan V., Secco A., Mamani M., Dos Santos A., Catalán Pellet A., Pérez Leiros C., Rabinovich G. A., Toscano M. A., Suppression of age-related salivary gland autoimmunity by glycosylation-dependent galectin-1-driven immune inhibitory circuits. Proc. Natl. Acad. Sci. U.S.A. 117, 6630–6639 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beccaria C. G., Amezcua Vesely M. C., Fiocca Vernengo F., Gehrau R. C., Ramello M. C., Tosello Boari J., Gorosito Serrán M., Mucci J., Piaggio E., Campetella O., Acosta Rodríguez E. V., Montes C. L., Gruppi A., Galectin-3 deficiency drives lupus-like disease by promoting spontaneous germinal centers formation via IFN-γ. Nat. Commun. 9, 1628 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maxwell J. R., Zhang Y., Brown W. A., Smith C. L., Byrne F. R., Fiorino M., Stevens E., Bigler J., Davis J. A., Rottman J. B., Budelsky A. L., Symons A., Towne J. E., Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity 43, 739–750 (2015). [DOI] [PubMed] [Google Scholar]
- 44.O’Connor W. Jr., Kamanaka M., Booth C. J., Town T., Nakae S., Iwakura Y., Kolls J. K., Flavell R. A., A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 10, 603–609 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neurath M. F., Fuss I., Kelsall B. L., Stüber E., Strober W., Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 182, 1281–1290 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cabral J., Hanley S. A., Gerlach J. Q., O’Leary N., Cunningham S., Ritter T., Ceredig R., Joshi L., Griffin M. D., Distinctive surface glycosylation patterns associated with mouse and human CD4+ regulatory T cells and their suppressive function. Front. Immunol. 8, 987 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bode U., Lörchner M., Ahrendt M., Blessenohl M., Kalies K., Claus A., Overbeck S., Rink L., Pabst R., Dendritic cell subsets in lymph nodes are characterized by the specific draining area and influence the phenotype and fate of primed T cells. Immunology 123, 480–490 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanić M., Trbojevič-Akmačič I., Lauc G., Inflammatory bowel disease - glycomics perspective. Biochim. Biophys. Acta Gen. Subj. 1863, 1595–1601 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Dias A. M., Correia A., Pereira M. S., Almeida C. R., Alves I., Pinto V., Catarino T. A., Mendes N., Leander M., Oliva-Teles M. T., Maia L., Delerue-Matos C., Taniguchi N., Lima M., Pedroto I., Marcos-Pinto R., Lago P., Reis C. A., Vilanova M., Pinho S. S., Metabolic control of T cell immune response through glycans in inflammatory bowel disease. Proc. Natl. Acad. Sci. U.S.A. 115, E4651–E4660 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nancey S., Holvöet S., Graber I., Joubert G., Philippe D., Martin S., Nicolas J.-F., Desreumaux P., Flourié B., Kaiserlian D., CD8+ cytotoxic T cells induce relapsing colitis in normal mice. Gastroenterology 131, 485–496 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Tom M. R., Li J., Ueno A., Fort Gasia M., Chan R., Hung D. Y., Chenoo S., Iacucci M., Jijon H. B., Kaplan G. G., Beck P. L., Panaccione R., Barkema H. W., Buret A. G., Yajnik V., Ghosh S., Novel CD8+ T-cell subsets demonstrating plasticity in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 22, 1596–1608 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Smillie C. S., Biton M., Ordovas-Montañes J., Sullivan K. M., Burgin G., Graham D. B., Herbst R. H., Rogel N., Slyper M., Waldman J., Sud M., Andrews E., Velonias G., Haber A. L., Jagadeesh K., Vickovic S., Yao J., Stevens C., Dionne D., Nguyen L. T., Villani A.-C., Hofree M., Creasey E. A., Huang H., Rozenblatt-Rosen O., Garber J. J., Khalili H., Desch A. N., Daly M. J., Ananthakrishnan A. N., Shalek A. K., Xavier R. J., Regev A., Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178, 714–730.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin J. C., Chang C., Boschetti G., Ungaro R., Giri M., Grout J. A., Gettler K., Chuang L.-s., Nayar S., Greenstein A. J., Dubinsky M., Walker L., Leader A., Fine J. S., Whitehurst C. E., Mbow M. L., Kugathasan S., Denson L. A., Hyams J. S., Friedman J. R., Desai P. T., Ko H. M., Laface I., Akturk G., Schadt E. E., Salmon H., Gnjatic S., Rahman A. H., Merad M., Cho J. H., Kenigsberg E., Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell 178, 1493–1508.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rubin S. J. S., Bai L., Haileselassie Y., Garay G., Yun C., Becker L., Streett S. E., Sinha S. R., Habtezion A., Mass cytometry reveals systemic and local immune signatures that distinguish inflammatory bowel diseases. Nat. Commun. 10, 2686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Czarnewski P., Parigi S. M., Sorini C., Diaz O. E., Das S., Gagliani N., Villablanca E. J., Conserved transcriptomic profile between mouse and human colitis allows unsupervised patient stratification. Nat. Commun. 10, 2892 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma C., Yang D., Wang B., Wu C., Wu Y., Li S., Liu X., Lassen K., Dai L., Yang S., Gasdermin D in macrophages restrains colitis by controlling cGAS-mediated inflammation. Sci. Adv. 6, eaaz6717 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abeln M., Albers I., Peters-Bernard U., Flächsig-Schulz K., Kats E., Kispert A., Tomlinson S., Gerardy-Schahn R., Münster-Kühnel A., Weinhold B., Sialic acid is a critical fetal defense against maternal complement attack. J. Clin. Invest. 129, 422–436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oswald D. M., Zhou J. Y., Jones M. B., Cobb B. A., Disruption of hepatocyte sialylation drives a T cell-dependent pro-inflammatory immune tone. Glycoconj. J. 37, 395–407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Orr S. L., Le D., Long J. M., Sobieszczuk P., Ma B., Tian H., Fang X., Paulson J. C., Marth J. D., Varki N., A phenotype survey of 36 mutant mouse strains with gene-targeted defects in glycosyltransferases or glycan-binding proteins. Glycobiology 23, 363–380 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Macauley M. S., Crocker P. R., Paulson J. C., Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 14, 653–666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang W. H., Heithoff D. M., Aziz P. V., Sperandio M., Nizet V., Mahan M. J., Marth J. D., Recurrent infection progressively disables host protection against intestinal inflammation. Science 358, eaao5610 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lennard-Jones J. E., Classification of inflammatory bowel disease. Scand. J. Gastroenterol. Suppl. 170, 2-6; discussion 16–2-6; discussion 19 (1989). [DOI] [PubMed] [Google Scholar]
- 63.Best W. R., Becktel J. M., Singleton J. W., Kern F. J. Jr., Development of a Crohn’s disease activity index. National Cooperative Crohn’s Disease Study. Gastroenterology 70, 439–444 (1976). [PubMed] [Google Scholar]
- 64.Schroeder K. W., Tremaine W. J., Ilstrup D. M., Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. N. Engl. J. Med. 317, 1625–1629 (1987). [DOI] [PubMed] [Google Scholar]
- 65.Frøslie K. F., Jahnsen J., Moum B. A., Vatn M. H.; IBSEN Group , Mucosal healing in inflammatory bowel disease: Results from a norwegian population-based cohort. Gastroenterology 133, 412–422 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Annese V., Daperno M., Rutter M. D., Amiot A., Bossuyt P., East J., Ferrante M., Götz M., Katsanos K. H., Kießlich R., Ordás I., Repici A., Rosa B., Sebastian S., Kucharzik T., Eliakim R.; European Crohn’s and Colitis Organisation , European evidence based consensus for endoscopy in inflammatory bowel disease. J. Crohns Colitis 7, 982–1018 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Barrionuevo P., Beigier-Bompadre M., Ilarregui J. M., Toscano M. A., Bianco G. A., Isturiz M. A., Rabinovich G. A., A novel function for galectin-1 at the crossroad of innate and adaptive immunity: Galectin-1 regulates monocyte/macrophage physiology through a nonapoptotic ERK-dependent pathway. J. Immunol. 178, 436–445 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Ek R. O., Serter M., Ergin K., Cecen S., Unsal C., Yildiz Y., Bilgin M. D., Protective effects of citicoline on TNBS-induced experimental colitis in rats. Int. J. Clin. Exp. Med. 7, 989–997 (2014). [PMC free article] [PubMed] [Google Scholar]
- 69.Fuss I. J., Marth T., Neurath M. F., Pearlstein G. R., Jain A., Strober W., Anti-interleukin 12 treatment regulates apoptosis of Th1 T cells in experimental colitis in mice. Gastroenterology 117, 1078–1088 (1999). [DOI] [PubMed] [Google Scholar]
- 70.Kruschewski M., Foitzik T., Perez-Canto A., Hubotter A., Buhr H. J., Changes of colonic mucosal microcirculation and histology in two colitis models: An experimental study using intravital microscopy and a new histological scoring system. Dig. Dis. Sci. 46, 2336–2343 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Dieleman L. A., Palmen M. J., Akol H., Bloemena E., Peña A. S., Meuwissen S. G., Van Rees E. P., Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 114, 385–391 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neelamegham S., Aoki-Kinoshita K., Bolton E., Frank M., Lisacek F., Lütteke T., O’Boyle N., Packer N. H., Stanley P., Toukach P., Varki A., Woods R. J.; SNFG Discussion Group , Updates to the symbol nomenclature for glycans guidelines. Glycobiology 29, 620–624 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabf8630/DC1