

A secretory process activated by p53 loss prevents cancer cell anoikis and drives immunosuppression in the tumor microenvironment.
Abstract
Cancer cells exhibit hyperactive secretory states that maintain cancer cell viability and remodel the tumor microenvironment. However, the oncogenic signals that heighten secretion remain unclear. Here, we show that p53 loss activates prometastatic secretory vesicle biogenesis in the Golgi. p53 loss up-regulates the expression of a Golgi scaffolding protein, progestin and adipoQ receptor 11 (PAQR11), which recruits an adenosine diphosphate ribosylation factor 1–containing protein complex that loads cargos into secretory vesicles. PAQR11-dependent secretion of a protease, PLAU, prevents anoikis and initiates autocrine activation of a PLAU receptor/signal transducer and activator of transcription-3-dependent pathway that up-regulates PAQR11 expression, thereby completing a feedforward loop that amplifies prometastatic effector protein secretion. Pharmacologic inhibition of PLAU receptor impairs the growth and metastasis of p53-deficient cancers. Blockade of PAQR11-dependent secretion inhibits immunosuppressive processes in the tumor microenvironment. Thus, Golgi reprogramming by p53 loss is a key driver of hypersecretion in cancer.
INTRODUCTION
In a “tumor as organizer” hypothesis proposed by Li and Stanger (1), cancer cells reside at the apex of a signaling hierarchy in the tumor microenvironment (TME), and the principal source of signals responsible for establishing the TME is the cancer cells themselves. Paracrine signals emanating from cancer cells maintain cancer cell viability and modify the TME in ways that facilitate metastasis (2). Matrix metalloproteinases cleave extracellular matrix (ECM) molecules and membrane-bound ligands to initiate the invasive process (3). Cytokines recruit immune cells, endothelial cells, and fibroblasts to create an immunosuppressive, angiogenic, and fibrotic tumor stroma (4). Thus, secretion is critical for tumor progression.
Clinical strategies designed to target the TME have primarily implemented antibodies or decoy receptors to neutralize secreted peptides in the extracellular space (5, 6). Despite having a strong preclinical rationale, these trials did not demonstrate efficacy (7). Given the functional redundancies of secreted factors within the tumor stroma (7, 8), strategies that block the entire secretory process, rather than the extracellular actions of individual secreted peptides, warrant consideration. Developing these strategies will require a better understanding of the way in which cancer cells activate secretion.
The Golgi apparatus plays a central role in the secretory process. Proteins destined for secretion are synthesized in the endoplasmic reticulum and transported to the plasma membrane via the Golgi, where cargos are sorted and loaded into secretory vesicles (9). In the trans-Golgi network, adenosine diphosphate ribosylation factors (ARFs) are recruited to Golgi membranes, where they cycle between guanosine triphosphate (GTP)– and guanosine diphosphate–bound states to initiate Golgi membrane deformation and recruitment of cargo adaptors and vesicular coat proteins to Golgi membranes, a key step in secretory vesicle biogenesis (10, 11).
Given the central role that hyperactive secretion plays in cancer progression, we postulated that secretory vesicle biogenesis is integrated into oncogenic transcriptional programs. To test this hypothesis, we studied cancer cells that harbor inactivating TP53 mutations, which are known drivers of protumorigenic secretory programs (12, 13). Our findings show that p53 loss triggers secretion by activating an autocrine feedforward loop that recruits ARF1 to Golgi membranes and thereby facilitates cargo loading into secretory vesicles.
RESULTS
Integrated oncogenomic analysis of p53-deficient tumors
To identify candidate effectors of p53 loss in the Golgi, we assessed the mRNA levels of Golgi-associated genes (Gene Ontology term Golgi apparatus) in TP53–wild-type A549 lung adenocarcinoma (LUAD) cells that had been subjected to CRISPR-Cas9–mediated TP53 knockout (KO) (14) and tumor cohorts in The Cancer Genome Atlas (TCGA) that are annotated on the basis of TP53 mutational status. Of 453 genes examined, 34 were more highly expressed in p53-deficient than parental A549 cells (fold change, >1.4; P < 0.01) (Fig. 1A), only one of which—progestin and adipoQ receptor family member 11 (PAQR11), which is encoded by MMD—was more highly expressed in TP53-mutant than –wild-type LUADs in TCGA (Fig. 1B). In paired biopsies of primary and metastatic tumor foci from multiple tumor types (www.oncomine.org), PAQR11 was expressed more highly in metastases than primary tumors (Fig. 1, C to F), and PAQR11 levels are positively correlated with metastasis and disease recurrence (table S1). Among the annotated somatic mutations, only TP53 mutations were positively correlated with PAQR11 levels in both the LUAD and pan-cancer TCGA cohorts (Fig. 1, G to I). PAQR11 levels were higher in TP53-mutant than –wild-type human and murine LUAD cell lines (fig. S1, A and B), and ectopic p53 expression suppressed PAQR11 levels in TP53-null H1299 cells and p53 KO A549 cells (Fig. 1, J and K, and fig. S1, C and D), indicating that p53 deficiency plays a causative role in PAQR11 up-regulation.
Fig. 1. p53-regulated expression of Golgi genes.
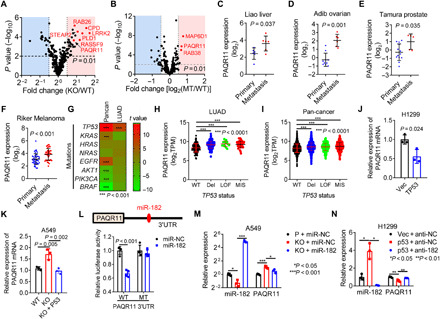
(A and B) Volcano plots comparing the mRNA levels of genes in the Gene Ontology term “Golgi apparatus” (n = 453) in TP53–wild-type (WT) and KO A549 cells (A) and TP53–wild-type and -mutant (MT) LUADs in TCGA (n = 230 tumors) (B). (C to F) PAQR11 mRNA levels in paired primary and metastatic tumor biopsies (www.oncomine.org). (G) Correlation between PAQR11 mRNA levels and somatic driver mutations (rows) in the TCGA LUAD and pan-cancer cohorts (columns). t value, t statistic. t > 0, positive correlation; t < 0, negative correlation (heatmap). (H and I) PAQR11 mRNA levels in TP53–wild-type and -mutant LUADs (H) and pan-cancers (I) (dots) (TCGA). Del, deletion; LOF, loss-of-function mutations including nonsense and frameshift mutations; MIS, missense mutations. (J and K) Quantitative reverse transcription polymerase chain reaction (qPCR) analysis of PAQR11 mRNAs in H1299 cells (I) and wild-type and KO A549 cells (J) transfected with empty (Vec) or p53-expressing (p53) vectors. (L) Luciferase reporter assays in H1299 cells transfected with PAQR11 3′ untranslated region (3′UTR) reporters (wild-type or miR-182–binding site mutant) and miR-182 or control (miR-NC) mimics. n = 4. (M) qPCR analysis of PAQR11 mRNA levels in parental and p53 KO A549 cells transfected with miR-182 or miR-NC. Results expressed relative to “P + miR-NC.” (N) qPCR analysis of PAQR11 mRNA levels in control (Vec) and p53-reconstituted (p53) H1299 cells transfected with miR-182 antagomirs (anti-182) or anti-NC. Results expressed relative to “Vec + anti-NC.” Results represent means ± SD values from a single experiment incorporating biological replicate samples (n = 3, unless otherwise indicated) and are representative of at least two independent experiments carried out on separate days. P values, two-tailed Student’s t test for two-group comparisons and one-way analysis of variance (ANOVA) test for multiple comparisons.
To assess how p53 deficiency up-regulates PAQR11, we carried out p53 reconstitution studies in H1299 cells and found that ectopic p53 expression reduced RNA polymerase II occupancy of the MMD gene promoter (fig. S1E) and suppressed MMD promoter activity in reporter assays (fig. S1F), suggesting that p53 represses MMD gene transcription. The MMD promoter region required for p53-mediated repression (fig. S1F) lacked predicted p53-binding sites, suggesting that p53 indirectly represses MMD gene transcription. In addition, the PAQR11 3′ untranslated region (3′UTR) contains a predicted miR-182–binding site and was found to be a miR-182 target in PAQR11 3′UTR reporter assays (Fig. 1L). p53 up-regulated miR-182 levels, and miR-182 was essential for p53-mediated down-regulation of PAQR11 (Fig. 1, M and N). Thus, p53 silences PAQR11 expression through transcriptional and posttranscriptional mechanisms.
PAQR11-dependent secretion mediates anoikis resistance
Given that high PAQR11 levels were correlated with the presence of metastatic disease, we postulated that PAQR11 is essential for one or more steps in the metastatic cascade. Compared to PAQR11-replete cells, PAQR11-deficient H1299 cells generated fewer lung nodules following tail vein injection into nu/nu mice, a finding that was corroborated by tail vein injection of a p53-mutant (R172H) murine LUAD cell line (344SQ) (15) into syngeneic, immunocompetent mice (Fig. 2, A to C), which led us to speculate that PAQR11 promotes metastatic colonization by increasing anoikis resistance. Anoikis increased sharply following ectopic p53 expression or PAQR11 depletion in H1299 cells (Fig. 2, D to F, and fig. S2, A and B), which was corroborated in a second TP53-null LUAD cell line, CALU-1 (Fig. 2, D to F, and fig. S2C), whereas cell proliferation in monolayer culture was minimally reduced (fig. S2D). PAQR11 depletion attenuated anoikis resistance and anchorage-independent growth induced by p53 KO in A549 cells (fig. S2, E to G) and reduced the metastatic activity of orthotopic LUADs generated by 344SQ cells (Fig. 2G), which metastasize to mediastinal lymph nodes, contralateral lung, and other sites in syngeneic, immunocompetent mice (16). However, in a nonmetastatic, Trp53–wild-type autochthonous LUAD model, ectopic PAQR11 expression increased tumor number and size but was not sufficient to generate metastases (Fig. 2, H to L). Collectively, these findings suggest that PAQR11 promotes LUAD metastasis by maintaining the survival of disseminated LUAD cells.
Fig. 2. PAQR11 promotes lung colonization by suppressing anoikis of metastatic cells.
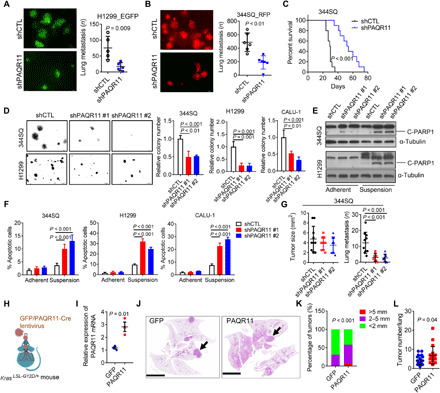
(A) Lung metastases in nu/nu mice injected by tail vein with green fluorescent protein (GFP)–tagged PAQR11 short hairpin RNA (shRNA; shPAQR11)– or control shRNA (shCTL)-transfected H1299 cells. (B) Lung metastases in syngeneic, immunocompetent mice injected by tail vein with red fluorescent protein (RFP)–tagged shPAQR11- or shCTL-transfected 344SQ murine LUAD cells. (C) Kaplan-Meier analysis of the mice in (B). (D) Colonies formed in soft agar. Values expressed relative to shCTL. (E and F) Western blot (WB) analysis of cleaved PARP1 (C-PARP1) (E) and annexin V/propidium iodide flow cytometry (F) to detect apoptotic cells on adherent or nonadherent (suspension) plates. α-Tubulin loading control. (G) Orthotopic lung tumor size (left dot plot) and numbers of metastases to contralateral lung and mediastinal lymph nodes (right dot plot) in syngeneic, immunocompetent mice. (H and I) qPCR analysis of PAQR11 mRNA levels in lung tumors (dots) generated in KrasLSL-G12D mice subjected to aerosolized delivery of lentiviruses that coexpress Cre and PAQR11 or GFP. (J) Hematoxylin and eosin–stained lung tumors (arrows) in GFP- and PAQR11-expressing cohorts. Scale bars, 8 mm. (K) Lung tumor diameters (color-coded) in each cohort. (L) Lung tumor numbers per mouse (dots). Results represent means ± SD values from a single experiment incorporating biological replicate samples (n = 3, unless otherwise indicated) and are representative of at least two independent experiments carried out on separate days. P values, two-tailed Student’s t test for two-group comparisons and one-way ANOVA test for multiple comparisons.
We found that PAQR11 localizes primarily on the trans-Golgi network (fig. S3), which is a site of secretory vesicle biogenesis (17), and secreted factors inhibit anoikis (18), which led us to assess whether p53 deficiency activates a PAQR11-dependent secretory process that inhibits anoikis. Supporting this conclusion, transfer of conditioned medium (CM) samples from control, but not ectopic p53-expressing, H1299 cells rescued colony-forming activity of ectopic p53-expressing H1299 cells (Fig. 3A), and CM samples from PAQR11-replete, but not PAQR11-deficient, H1299 cells or 344SQ cells rescued colony formation and reduced anoikis in PAQR11-deficient cells (Fig. 3, B to D, and fig. S4A). To identify prosurvival effector proteins in the CM samples, we performed liquid chromatography–mass spectrometry analysis on CM samples from PAQR11-replete and -deficient H1299 cells and identified 812 secreted proteins (Fig. 3, E and F), 25 of which were reduced by PAQR11 depletion. These were enriched in, among other Gene Ontology terms, “ECM degradation,” “proteolysis regulation,” and “protein metabolism” (fig. S4B) and included the collagen-modifying enzyme procollagen-lysine,2-oxoglutarate 5-dioxygenase 3 (PLOD3) (19, 20), the protease urokinase plasminogen activator (PLAU) (21), and the FAM3 metabolism–regulating signaling molecule C (FAM3C) (Fig. 3G) (22), which have known prometastatic roles and are correlated with a shorter survival duration in patients with lung cancer (Fig. 3H and fig. S4, C to E). In gain- and loss-of-function studies, p53 and PAQR11 regulated the levels of PLOD3, FAM3C, and PLAU in CM samples (Fig. 3I and fig. S4, F to L). These changes were not due to altered protein synthesis, as their amounts were not changed in total lysates (fig. S4, F to L). These findings identify a PAQR11-dependent secretome in p53-deficient LUAD cells.
Fig. 3. PAQR11-dependent secretion promotes anoikis resistance.
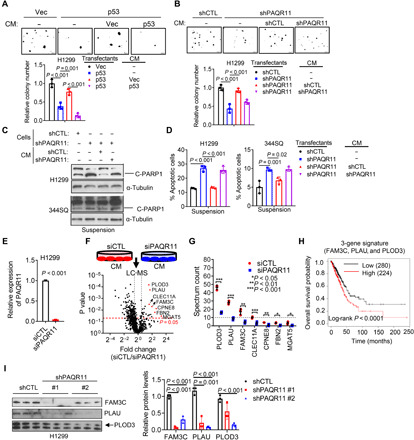
(A and B) Colonies formed by Vec- and p53-transfected H1299 cells (A) or shCTL- and shPAQR11-transfected H1299 cells (B). Cells were treated for 14 days with CM samples from indicated H1299 transfectants. Fresh medium included as a control (−). (C and D) Western blot analysis of cleaved PARP1 (C) and annexin V/propidium iodide flow cytometry (D) to detect apoptotic cells following treatment of shPAQR11-transfected cells with CM or fresh medium (−). α-Tubulin loading control. (E) qPCR confirmation of PAQR11 depletion by siPAQR11. (F) CM samples (triplicate) from siCTL- and siPAQR11-transfected H1299 cells (schema) were subjected to liquid chromatography–mass spectrometry (LC-MS) analysis to identify PAQR11-regulated proteins (volcano plot). The identities of proteins that are significantly down-regulated (P < 0.05) by siPAQR11 are indicated. The vertical dashed line indicates a 1.4-fold change threshold. (G) Spectrum reads of down-regulated proteins. (H) Kaplan-Meier analysis of the TCGA LUAD cohort (n = 504) based on average FAM3C, PLAU, and PLOD3 mRNA levels (three-gene signature). High and low expression levels were defined using the autoselect best cutoff method. (I) Western blot analysis of CM samples. Relative protein levels quantified densitometrically (bar graph). Results represent means ± SD values from a single experiment incorporating biological replicate samples (n = 3, unless otherwise indicated) and are representative of at least two independent experiments carried out on separate days. P values, two-tailed Student’s t test for two-group comparisons and one-way ANOVA test for multiple comparisons.
Our model thus predicts that PLAU, FAM3C, and PLOD3 are prosurvival and prometastatic effectors of p53 deficiency. To address this possibility, we performed small interfering RNA (siRNA)–mediated depletion studies on p53-mutant LUAD cells (344SQ, H1299, and CALU-1) and found that all three genes were essential for lung nodule formation in tail vein–injected mice (Fig. 4, A to D) and clonogenicity under anchorage-independent conditions but not cell proliferation in monolayer culture (Fig. 4E and fig. S5, A to C). Anoikis increased following depletion of PLAU but not FAM3C or PLOD3 (Fig. 4, F and G), suggesting a unique role for PLAU in anoikis resistance. PAQR11 deficiency–induced anoikis was reversed by treatment with recombinant PLAU or CM from FAM3C- or PLOD3-deficient, but not PLAU-deficient, cells (Fig. 4, H to K). Recombinant PLAU mitigated anoikis induced by ectopic p53 expression in H1299 cells (fig. S5D), and PLAU depletion reduced anoikis resistance induced by ectopic PAQR11 expression (Fig. 4L and fig. S5E). In mice, recombinant PLAU enhanced the number of lung nodules generated by PAQR11-deficient 344SQ cells (Fig. 4M). Thus, PAQR11 activates a prometastatic secretory program that promotes anoikis resistance.
Fig. 4. Secreted PLAU inhibits anoikis and promotes metastatic colonization.
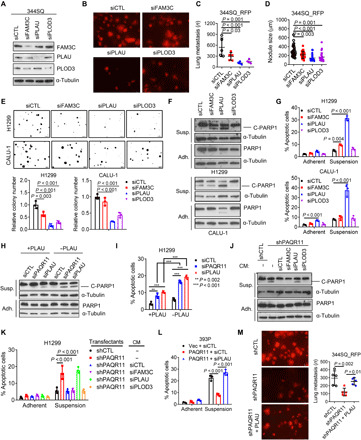
(A) Western blot confirmation of target gene depletion in siRNA-transfected 344SQ cells. (B to D) Lung metastases in tail vein–injected syngeneic, immunocompetent mice were imaged (B) and quantified on the basis of mean number per mouse (C) and mean size (D). (E) Colonies formed in soft agar by siRNA-transfected H1299 cells and CALU-1 cells. Values expressed relative to siCTL. (F and G) Western blot analysis of cleaved PARP1 (F) and annexin V/propidium iodide flow cytometry (G) to detect apoptotic cells under adherent (Adh.) and suspension (Susp.) conditions. (H) Western blot analysis of C-PARP1 in siRNA-transfected H1299 cells treated for 2 days with (+) or without (−) recombinant PLAU (10 ng/ml). (I) Annexin V/propidium iodide flow cytometry of H1299 cells in (H). (J and K) Western blot analysis of cleaved PARP1 (J) and annexin V/propidium iodide flow cytometry (K) to detect apoptotic cells following treatment with CM samples or fresh medium (−). (L) Annexin V/propidium iodide flow cytometry of 393P murine LUAD cells cotransfected with PAQR11 or empty (Vec) expression vectors and siPLAU or siCTL. (M) Lung metastases generated in syngeneic, immunocompetent mice by tail vein injection of shRNA-transfected 344SQ cells alone or in combination with 10 μg of recombinant PLAU (+PLAU; in 100-μl cell suspension). Results represent means ± SD values from a single experiment incorporating biological replicate samples (n = 3, unless otherwise indicated) and are representative of at least two independent experiments carried out on separate days. P values, two-tailed Student’s t test for two-group comparisons and one-way ANOVA test for multiple comparisons.
A PLAU-dependent feedforward loop amplifies PAQR11-driven secretion
In the extracellular space, PLAU converts plasminogen to the enzymatically active serine protease plasmin, which is a broad-spectrum protease that degrades or remodels ECM components (21). In addition, PLAU binds to and activates PLAU receptor (PLAUR) (23). To determine whether PLAU inhibits anoikis through its proteolytic activity, we used two compounds that potently inhibited PLAU’s catalytic activity (fig. S6A) and found that they did not recapitulate the effect of siRNA-mediated PLAU depletion on anoikis or anchorage-independent colony formation (fig. S6, B to D). In contrast, PLAUR depletion in H1299 and CALU-1 cells sharply reduced colony formation and increased anoikis (Fig. 5, A to D). Recombinant PLAU decreased anoikis in PLAUR-replete, but not PLAUR-deficient, A549 cells (Fig. 5, E and F). A small molecule that inhibits PLAUR ligand–binding activity (24) decreased PLAU secretion (fig. S6E), increased anoikis (Fig. 5, G and H), reduced colony-forming activity (Fig. 5I), decreased lung nodules generated by tail vein–injected 344SQ cells (Fig. 5J), and reduced the metastatic activity of 344SQ orthotopic LUADs (fig. S6F). Thus, autocrine PLAUR activation mediates anoikis resistance.
Fig. 5. Secreted PLAU inhibits anoikis through autocrine PLAUR activation.
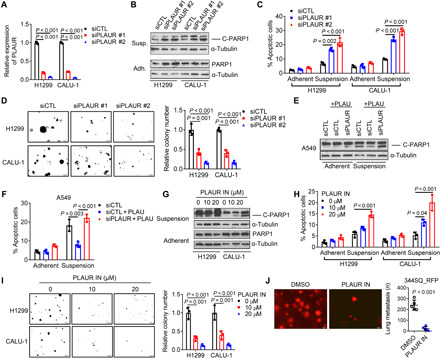
(A) qPCR confirmation of PLAUR depletion in siRNA-transfected H1299 and CALU-1 cells. (B and C) Western blot analysis of cleaved PARP1 (B) and annexin V/propidium iodide flow cytometry (C) to detect apoptotic cells under adherent and suspension conditions. (D) Colonies formed in soft agar by siRNA-transfected cells. (E and F) Western blot analysis of cleaved PARP1 (E) and annexin V/propidium iodide flow cytometry (F) of siRNA-transfected cells treated with (+) or without recombinant PLAU (10 ng/ml) under adherent or suspension conditions. (G and H) Western blot analysis of cleaved PARP1 (G) and annexin V/propidium iodide flow cytometry (H) following treatment for 2 days with indicated concentrations of PLAUR inhibitor (PLAUR IN). (I) Colonies formed in soft agar following 14 days of PLAUR inhibitor treatment. (J) Lung metastases generated in syngeneic, immunocompetent mice by tail vein–injected RFP-tagged 344SQ cells that had been pretreated for 2 days with 10 μM PLAUR inhibitor or vehicle [dimethyl sulfoxide (DMSO)]. Results represent means ± SD values from a single experiment incorporating biological replicate samples (n = 3, unless otherwise indicated) and are representative of at least two independent experiments carried out on separate days. P values, two-tailed Student’s t test for two-group comparisons and one-way ANOVA test for multiple comparisons.
PLAUR signals through various pathways, including Ras–extracellular signal–regulated kinase, phosphoinositide 3-kinase/Akt, and Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) (23, 25). Because STAT3 promotes anoikis resistance and metastatic colonization (26), we assessed how STAT3 is regulated and found that depletion of PAQR11, PLAUR, or PLAU, but not PLOD3 or FAM3C, reduced Y705–phosphorylated STAT3 (pSTAT3) levels (fig. S7, A to D), which were rescued by recombinant PLAU or CM from PAQR11- and PLAU-replete but not -deficient cells (fig. S7, E to G), and PLAU was required for PAQR11-dependent STAT3 phosphorylation (fig. S7H). JAK/STAT3 inhibition with short hairpin RNAs (shRNAs) or a JAK inhibitor (P6) increased anoikis and reduced colony-forming activity (fig. S7, I to O). Ectopic expression of constitutively active mutant STAT3 (STAT3-CA) reversed anoikis and rescued colony-forming activity in PAQR11-deficient cells (fig. S7, P to S). Thus, STAT3 is a key mediator of PLAUR.
Unexpectedly, JAK/STAT3 inhibition reduced PAQR11 levels (Fig. 6, A and B), which led us to postulate that a PLAU-dependent feedforward loop amplifies PAQR11-driven secretion. Supporting the existence of such a loop, PLAU and PAQR11 expression levels were positively correlated in the TCGA LUAD cohort (Fig. 6C), and depletion of PLAU reduced PAQR11 levels, which were rescued by recombinant PLAU or ectopic STAT3-CA expression (Fig. 6, D and E), whereas recombinant PLAU did not rescue PAQR11 levels in PLAUR-deficient cells (fig. S8A), indicating that autocrine PLAU-dependent signaling drives the PAQR11-dependent secretory pathway. Although a prediction algorithm (http://jaspar.genereg.net/) revealed no STAT3-binding sites in the MMD gene promoter, a ZEB1 E-box binding site was identified, which was of interest because Zinc Finger E-Box Binding Homeobox 1 (ZEB1) is a transcriptional target of STAT3 (Fig. 6F) (27). In reporter assays on cells transfected with MMD promoter constructs, STAT3-CA increased MMD promoter activity (Fig. 6F), which was abrogated by site-directed mutagenesis of the E-box (Fig. 6G). ZEB1 bound to this region of the MMD promoter (Fig. 6H). JAK inhibition decreased ZEB1 expression (Fig. 6I), and siRNA-mediated ZEB1 depletion abrogated PAQR11 expression induced by ectopic STAT3-CA (Fig. 6J). Components of the autocrine loop were positively correlated in a panel of murine LUAD cell lines that differ with respect to endogenous ZEB1 levels (Fig. 6, K and L, and fig. S8, B and C). Together, these results identify a molecular mechanism by which a PLAU-dependent feedforward loop amplifies PAQR11-driven secretion (Fig. 6M).
Fig. 6. Autocrine PLAUR activation regulates PAQR11 expression.
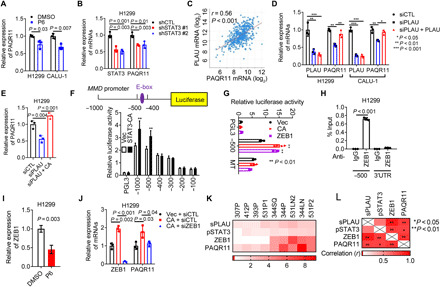
(A and B) qPCR analysis of cells treated for 2 days with 1 μM JAK inhibitor (P6) (A) and in shSTAT3-transfected cells (B). (C) Pearson correlation between PLAU and PAQR11 mRNAs in TCGA LUAD cohort. (D and E) qPCR analysis of siPLAU-transfected cells treated for 2 days with recombinant PLAU (10 ng/ml) (D) and in cells cotransfected with siPLAU and empty vector (Vec) or constitutively active mutant STAT3 (STAT3-CA) (E). (F) MMD gene promoter (schema). Luciferase assays on H1299 cells cotransfected with MMD promoter constructs and STAT3-CA or Vec. Empty reporter (pGL3). (G) Luciferase assay on H1299 cells cotransfected with wild-type (−500) or E-box–mutated MMD promoter and indicated expression vectors. (H) Chromatin immunoprecipitation assays on the MMD promoter (−500) using immunoglobulin G (IgG) or anti-ZEB1 antibodies. PAQR11 3′UTR (negative control). (I and J) qPCR analysis of H1299 cells treated for 2 days with 1 μM P6 (I) or cotransfected with STAT3-CA or Vec and siZEB1 or siCTL (J). (K) Heatmap depiction of ZEB1 and PAQR11 mRNA levels; pSTAT3/total STAT3 ratio; and secreted PLAU levels (sPLAU) in murine LUAD cell line panel. (L) Heatmap depiction of Pearson correlation (R values) of results in (K). Results represent means ± SD values from a single experiment incorporating biological replicate samples (n = 3, unless otherwise indicated) and are representative of at least two independent experiments. P values, two-tailed Student’s t test (two-group comparisons) and one-way ANOVA test (multiple comparisons).
Because mutant p53 proteins stimulate Golgi-dependent secretory pathways to promote metastasis (28), we asked whether p53 gain-of-function mutants activate the autocrine loop. Following CRISPR-Cas9–mediated p53 KO, 344SQ cells exhibited reduced colony-forming activity but did not undergo anoikis or demonstrate reduced levels of PAQR11 or pSTAT3 (fig. S9, A to D). However, PAQR11 and pSTAT3 levels and PLAU secretion increased following ectopic expression of mutant p53 (R175H, G245S, or R275H) in H1299 cells (fig. S9, E to I), which is in keeping with evidence that p53 gain-of-function mutations activate STAT3 (29). Thus, p53 mutations can activate the feedforward pathway through at least two distinct mechanisms involving gain- and loss-of-function properties.
PAQR11-associated ARF1 governs cargo loading into secretory vesicles
In H1299 cells that express enhanced green fluorescent protein (EGFP)–tagged Ras-related protein Rab-6A (RAB6A), which marks secretory vesicles that bud from the trans-Golgi network (30), PAQR11 depletion reduced the numbers of RAB6A+ vesicles (fig. S10A), which led us to suspect that PAQR11 is essential for secretory vesicle biogenesis. PAQR11 is a seven-transmembrane Golgi protein with N- and C-terminal peptides that extend into cytosolic and luminal Golgi compartments, respectively (Fig. 7A). On the basis of its reported scaffolding functions (31, 32), we postulated that PAQR11 recruits regulators of vesicle biogenesis to trans-Golgi membranes. Reconstitution of PAQR11-deficient cells with a PAQR11 mutant that lacks the first five N-terminal amino acids and localizes in the Golgi (32) did not rescue colony-forming activity (Fig. 7B and fig. S10B), anoikis resistance (Fig. 7, C and D), PLAU secretion (Fig. 7E), or STAT3 phosphorylation (Fig. 7F). Furthermore, ectopic expression of the first 28 N-terminal amino acids of PAQR11 as a competing peptide in PAQR11-replete cells impaired anoikis resistance, colony-forming capacity, and lung nodule formation (Fig. 7, G to K, and fig. S10, C to E). On the basis of these findings, we used the 28–amino acid peptide as bait in pull-down experiments and identified multiple interacting proteins, including several that regulate vesicle biogenesis (table S2). In an orthogonal approach, we performed a biotin identification (BioID)–based proximity-labeling experiment (Fig. 8, A and B) and identified hundreds of candidate interactors (table S3), including five proteins [ARF1, ARF4, annexin A2 (ANXA2), myosin IC (MYO1C), and myosin heavy chain 10 (MYH10)] that were also identified in pull-down assays (Fig. 8C). ARF1 and ARF4 were confirmed to be PAQR11-interacting proteins by Western blot analysis of cell lysates from the BioID experiment (Fig. 8D).
Fig. 7. N-terminal amino acids of PAQR11 are required to rescue PAQR11-deficient cells.
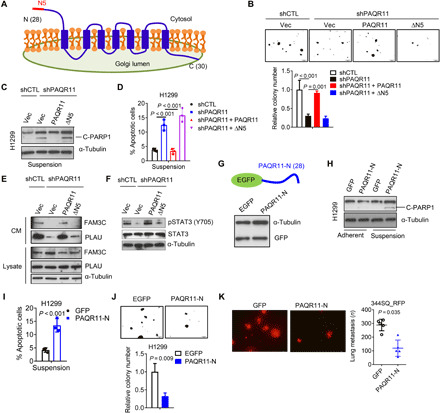
(A) A topological model of PAQR11. N5: The first five N-terminal amino acids. (B) Colonies formed in soft agar by shCTL- and shPAQR11-transfected H1299 cells. The latter were rescued with full-length (PAQR11) or N-terminally truncated (∆N5) PAQR11. (C and D) Western blot analysis of cleaved PARP1 (C) and annexin V/propidium iodide flow cytometry (D) of cells under suspension conditions. (E) Western blot analysis of FAM3C and PLAU in CM samples (CM) and cell lysates (lysates). (F) Western blot analysis of pSTAT3 and total STAT3. (G) A competing peptide that includes the first 28 N-terminal amino acids of PAQR11 (PAQR11-N) (schema) was detected by anti-GFP Western blot analysis in H1299 cells. (H and I) Western blot analysis of cleaved PARP1 (H) and annexin V/propidium iodide flow cytometry (I) of H1299 cells transfected with (PAQR11-N) or without (GFP) competing peptide under adherent or nonadherent conditions. (J) Colonies formed in soft agar by H1299 cells transfected with (PAQR11-N) or without (GFP) competing peptide. (K) Lung metastases generated in syngeneic, immunocompetent mice by 344SQ cells transfected with (PAQR11-N) or without (GFP) competing peptide. Results represent means ± SD values from a single experiment incorporating biological replicate samples (n = 3, unless otherwise indicated) and are representative of at least two independent experiments carried out on separate days. P values, two-tailed Student’s t test for two-group comparisons and one-way ANOVA test for multiple comparisons.
Fig. 8. PAQR11 recruits ARF1 to the trans-Golgi network.
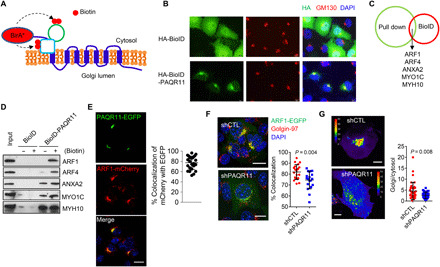
(A) PAQR11-interacting proteins were identified by using a proximity-dependent biotin identification (BioID) assay. (B) A confocal micrograph demonstrates that a Golgi marker (GM130) colocalizes with a hemagglutinin (HA)–tagged BioID/PAQR11 fusion protein but not control vector (HA-BioID). (C) Venn diagram illustration of proteins identified by both methodologies (BioID assay on H1299 cells and N-terminal PAQR11 peptide pull-down assay on 344SQ cells). (D) Validation of hits identified by Western blot analysis of streptavidin beads isolated from H1299 cells transfected with HA-BioID or HA-BioID-PAQR11 and treated with (+) or without (−) 100 μM biotin. (E) Confocal micrographs of EGFP-tagged PAQR11 and mCherry-tagged ARF1 in 344SQ cells. Blue indicates DAPI. Scatter plot shows the percentage of mCherry-ARF1 that colocalizes with EGFP-PAQR11 in each cell (dot). Scale bar, 10 μm. (F) Confocal micrographs of ARF1-EGFP in PAQR11-deficient and -replete 344SQ cells. Red indicates Golgin-97, and blue indicates DAPI. Scale bars, 10 μm. Right: Scatter plot shows the percentage of Golgin-97 that colocalizes with ARF1 in each cell (dot). (G) Intensity–pseudo-colored confocal micrographs of Golgi-associated and cytosolic ARF1-EGFP. Scale bars, 10 μm. The scatter plot shows the Golgi/cytosolic ARF1-EGFP ratio in each cell (dots). Results represent means ± SD values from a single experiment incorporating biological replicate samples (n = 3, unless otherwise indicated) and are representative of at least two independent experiments carried out on separate days. P values, two-tailed Student’s t test.
Because ARF1 recruits proteins required for cargo sorting and loading (10), we assessed its role in PAQR11-dependent secretory vesicle biogenesis. Ectopically expressed ARF1 and PAQR11 colocalized on the trans-Golgi (Fig. 8E), and PAQR11 was required for ARF1 recruitment on the basis of evidence that ARF1 failed to localize in the trans-Golgi in PAQR11-deficient cells (Fig. 8, F and G) and was recruited to a PAQR11/monoamine oxidase A fusion protein that localizes in mitochondria (fig. S11, A and B). PAQR11 depletion reduced activated (GTP-bound) ARF1 levels (fig. S11C). By Western blot analysis of vesicle-enriched cell lysates (Fig. 9, A to C) and microscopic analysis of cells transfected with EGFP-tagged FAM3C or Flag-tagged PLAU (Fig. 9, D to F), PAQR11 or ARF1 depletion sharply reduced PLAU- and FAM3C-containing vesicles. Moreover, ARF1 depletion or treatment with the ARF1 inhibitor brefeldin A (33) increased anoikis and reduced colony formation, PLAU secretion, lung nodule formation, and PAQR11-driven clonogenic growth and anoikis resistance (fig. S12, A to K). p53 KO A549 cells underwent anoikis following depletion of ARF1 or PLAU (fig. S12L). We conclude that the cytosolic tail of PAQR11 recruits a protein complex containing ARF1, which loads FAM3C and PLAU into secretory vesicles.
Fig. 9. ARF1-dependent cargo loading into secretory vesicles is PAQR11 dependent.
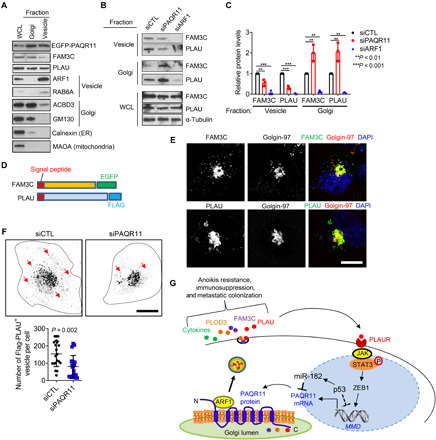
(A) Western blot analysis of whole cell lysate (WCL) and vesicle- and Golgi-fractionated H1299 cell lysates to confirm enrichment of fractionated lysates in organelle-specific markers. ER, endoplasmic reticulum. (B and C) Western blot analysis of FAM3C and PLAU in WCL and Golgi- and vesicle-enriched fractions of siRNA-transfected H1299 cells (B). Protein levels relative to siCTL controls quantified densitometrically (C). (D) FAM3C and PLAU expression constructs. (E) Confocal micrographs of H1299 cells transfected with expression constructs. Vesicles containing ectopic FAM3C and PLAU were detected using fluorescent tags (EGFP-FAM3C) or antibodies against Flag (PLAU). Golgi localization determined on the basis of colocalization with the trans-Golgi marker Golgin-97. Nuclei were counterstained with DAPI (blue). Scale bar, 8 μm. (F) Contrast-adjusted confocal micrographs of H1299 cells cotransfected with Flag-PLAU and indicated siRNAs and stained with anti-Flag antibodies. Arrows indicate Flag-PLAU+ vesicles. Dotted lines indicate cell boundary. Scale bar, 10 μm. Vesicle numbers per cell (dots) were quantified. (G) Schematic illustration of a working model in which p53 loss leads to PAQR11 up-regulation, initiating an ARF1-dependent secretory process that is amplified through an autocrine PLAUR-dependent signaling pathway. Results represent means ± SD values from a single experiment incorporating biological replicate samples (n = 3, unless otherwise indicated) and are representative of at least two independent experiments carried out on separate days. P values, two-tailed Student’s t test for two-group comparisons and one-way ANOVA test for multiple comparisons.
PAQR11-dependent secretion activates immunosuppressive processes in the TME
In one working model, the principal source of TME-modifying signals is the cancer cells themselves (34). To determine whether PAQR11 activates the secretion of factors that govern processes in the TME, we performed multiplexed antibody-based bead assays on CM samples from PAQR11-deficient and -replete 344SQ cells and found that the concentrations of numerous cytokines that have immunomodulatory roles, including granulocyte colony-stimulating factor (CSF), granulocyte-macrophage CSF, interleukin-4 (IL-4), IL-6, IL-13, CXCL5, and CXCL1, were sharply reduced in PAQR11-deficient cells (fig. S13A). Flow cytometric analysis of flank tumors generated by PAQR11-deficient or -replete 344SQ cells in syngeneic, immunocompetent mice showed that PAQR11-deficient tumors had increased numbers of effector/memory CD8+ T cells and M1 macrophages and reduced numbers of M2 macrophages (fig. S13B), suggesting that PAQR11-dependent secretion skews the TME toward an immunosuppressive state.
DISCUSSION
Cancer cell–derived secreted proteins inhibit anoikis and modify stromal cells and ECM proteins in ways that create a prometastatic TME (13, 16, 35, 36). Hyperactive secretory states have been linked to oncogenic somatic mutations (28, 37), the molecular underpinnings of which remain unclear. Here, we show that p53 loss governs prosecretory Golgi functions.
In a growing body of evidence, TP53 loss-of-function mutations alter the secretome in ways that drive tumor progression (13, 28, 38). For example, p53 loss relieves cancer cells from senescence-related secreted factors that restrict angiogenesis and fibrosis and skew macrophage polarization toward the tumor-fighting M1-like state (39). Similarly, in a KrasG12D-driven mouse model of pancreatic cancer, p53 loss enhances recruitment of myeloid-derived suppressive cells and regulatory T cells that inhibit antitumor T cell populations (40). Furthermore, secretomes driven by p53 loss enhance recruitment of CD11b+Gr1+ polymorphonuclear cells and immunosuppressive regulatory T cells (41) in phosphatase and tensin homolog (PTEN)-driven prostate cancer and tumor-associated macrophage stimulation and neutrophil activation in breast cancer (42). Reconstitution of p53-null tumor cells with wild-type p53 leads to widespread secretomic changes involving proteases, growth factors, and cytokines that regulate angiogenesis, inflammation, and fibrosis, but most of the secreted proteins identified are not direct targets of p53 (43), which raises the possibility that p53 loss influences the secretory process, not only the expression of secreted proteins. In this study, we identify a feedforward pathway activated by p53 loss that drives secretory vesicle biogenesis in the Golgi.
The most frequent TP53 mutations occur within the DNA binding domain and generate mutant p53 proteins that lack the tumor-suppressive activity of wild-type p53 and gain oncogenic functions (38). In the context of previous reports that mutant p53 directly activates STAT3 (29) and initiates a miR-30d–dependent process in the Golgi apparatus that leads to enhanced anterograde trafficking of secretory vesicles (28), the findings presented here suggest that p53 gain- and loss-of-function mutations converge on STAT3 to activate prometastatic secretory programs.
Anoikis is a form of cell death that disseminated cancer cells undergo owing to loss of contact with the ECM (44). Anoikis resistance is acquired through survival signals activated by oncogenic somatic mutations, oxidative stress induced by the loss of matrix contacts, or autocrine activation of cell surface receptors, including PLAUR (45, 46). p53 mutations can induce anoikis resistance by regulating the expression of pro- or antiapoptotic proteins (47), inducing epithelial-mesenchymal transition (48), or causing metabolic reprogramming through glycolytic and mitochondrial oxidative phosphorylation pathways that lead to sustained Warburg effects (49, 50). Findings reported here show that cancer cells gain anoikis resistance through transcriptional control of prosecretory functions in the Golgi.
Secretory vesicle biogenesis on the trans-Golgi network involves a complex regulatory apparatus that coordinates Golgi membrane curvature with cargo sorting, which serves as a secretory checkpoint for specific cargo proteins (17). ARF1 activation by guanine nucleotide exchange factors (GEFs) triggers Golgi membrane curvature and recruitment of cargo adaptors, coat protein complexes, and transmembrane cargo receptors that capture soluble proteins and direct them into vesicles (51). Findings presented here suggest that p53 loss coordinates cargo sorting with membrane deformation by increasing the expression of PAQR11, a Golgi scaffold that recruits ARF1. We propose that PAQR11’s seven-transmembrane structure may provide a Golgi membrane insertion site for ARF1 and possibly other proteins that have been implicated in ARF1 recruitment and enzymatic cycling, including GEFs and p23/24 proteins (51, 52), and thereby facilitate secretory vesicle biogenesis.
METHODS
Animal husbandry
All mouse studies were approved by the Institutional Animal Care and Use Committee at The University of Texas MD Anderson Cancer Center (Houston, Texas). Mice underwent standard care and were euthanized at the end time points of experiments or at the signs of mortality according to the standards set forth by the Institutional Animal Care and Use Committee. To generate autochthonous lung tumors, we endotracheally delivered 5 × 104 GFP-Cre or PAQR11-Cre lentivirus particles in 50 μl of 1× phosphate-buffered saline into the lungs of KrasLSL-G12D mice as reported previously (16). To generate experimental lung metastases, we injected syngeneic, immunocompetent mice (n = 5 per group) by tail vein with 105 red fluorescent protein (RFP)–labeled 344SQ cells or nu/nu mice (n = 5 per group) with 5 × 105 GFP-labeled H1299 cells. The mice were monitored daily for signs of morbidity and necropsied on day 5 (for 344SQ cells) or day 20 (for H1299 cells). Lung metastases on the pleural surfaces were counted and measured under a fluorescence microscope. To generate orthotopic lung tumors, we intrathoracically injected 344SQ cells (n = 105) into syngeneic, immunocompetent mice (n = 7 to 10 per cohort) and quantified primary tumor size and metastases to mediastinal lymph nodes and contralateral lung after 7 to 10 days as we previously described (16).
Cell lines
The murine LUAD cell lines used and their derivations include LKR10 and LKR13 (KrasLA1) (53); 713P, 307P, 412P, 393LN, 531P1, 531LN1, 344P, 344SQ, 531LN2, 344LN, 531LN3, and 531P2 (KrasG12D; Trp53R172H) (54); and KC2 and KC4 (KrasG12D; CDKN2A−/−) (55). Human LUAD cell lines (A549, H1299, H322, H460, H1650, H522, H596, H23, H1792, H441, H358, H157, H226, H2122, CALU-1, CALU-6, H292, and H520) were purchased (American Type Culture Collection). Murine and human LUAD cells were cultured in RPMI 1640 containing 10% fetal bovine serum. Cells were maintained at 37°C in an incubator with a humidified atmosphere containing 5% CO2. Cells were transfected with jetPRIME Versatile DNA/siRNA transfection reagent (Polyplus). Stable cell transfectants were selected using puromycin (for pLVX, pBabe-puro, and pLKO.1 vectors) or G418 (for pcDNA3.1 and pEGFP-C3 vectors). CRISPR-Cas9–mediated p53 KO 344SQ cells were generated in the Cell-Based Assay Screening Service Core Facility (Baylor College of Medicine) using the following guide RNA sequences: 5′-acagccatcacctcactgca (tgg) and 5′-(ccc) agccactccatggcccctgt. CRISPR-Cas9–mediated p53 KO A549 cells were generated as described (13).
Reagents
We purchased SYBR green, fetal bovine serum, Hepes-buffered media, Dulbecco’s modifed Eagle’s medium, RPMI 1640, Alexa Fluor–tagged secondary antibodies, and 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies); puromycin (InvivoGen); paraformaldehyde (Electron Microscopy Sciences); biotin (B4639), nocodazole (SML1665), and brefeldin A (B7651) (Sigma-Aldrich); G418 (Corning); PLAU and PLAUR inhibitors UK-371804 (HY-101214, #1), Upamostat (HY-16511, #2), and IPR-803 (HY-111192) (MedChemExpress); murine (TRCN0000306505 and TRCN0000101367) and human (TRCN0000062989 and TRCN0000062990) PAQR11 shRNAs (Sigma-Aldrich); murine FAM3C siRNA (SASI_Mm01_00134020), murine PLAU siRNA (SASI_Mm01_00138850), murine PLOD3 siRNA (SASI_Mm02_00321839), human FAM3C siRNA (SASI_Hs01_00078272), human PLAU siRNA (SASI_Hs02_00334208), human PLOD3 siRNA (SASI_Hs01_00241346), and human PLAUR siRNAs (SASI_Hs01_00130171 and SASI_Hs01_00130172) (Sigma-Aldrich); miR-182 microRNA mimics (HMI0275) and inhibitors (HSTUD0275) (Sigma-Aldrich); antibodies against α-tubulin (#T9026), Flag tag (F3165), and EGFP (G6539) (Sigma-Aldrich); Poly(ADP-Ribose) Polymerase 1 (#9542), hemagglutinin (HA) tag (#3724 and #2367), β-actin (#4970), GM130 (#12480), p53 (#2524), and Golgin-97 (#13192) (Cell Signaling Technology); ARF1 (#PA1-127) (Thermo Fisher Scientific); PAQR11 (sc-243494), acyl-CoA binding domain containing 3 (sc-101277), ZEB1 (sc-25388), and p53 (sc-126) (Santa Cruz Biotechnology); calnexin (ab22595) (Abcam); MYO1C (GTX55719) (GeneTex); RNA polymerase II (05-623, clone CTD4H8) (Millipore); PLOD3 (11027-1-AP), FAM3C (14247-1-AP), PLAU (17968-1-AP), and monoamine oxidase A (10539-1-AP), ARF4 (11673-1-AP), MYH10 (19673-1-AP), ANXA2 (11256-1-AP) (ProteinTech); and recombinant PLAU protein (1310-SE-010) (R&D Systems).
Vector construction
The murine PAQR11, human PAQR11, human FAM3C, and human PLAU coding sequences were isolated by performing polymerase chain reaction (PCR) on complementary DNA (cDNA) prepared from murine 344SQ and human H1299 cells and cloned into pLVX-Puro (PAQR11 and PLAU), pEGFP-C3 (PAQR11), or pEGFP-N3 (FAM3C). PAQR11 promoter sequences were amplified by PCR from genomic DNA from H1299 cells. The ARF1-mCherry expression vector was modified from ARF1-EGFP (Addgene plasmid #49578) by replacing the EGFP coding sequence with the mCherry sequence. To create the PAQR11-N-MAO fusion protein, we amplified the C-terminal transmembrane domain sequences (479 to 528) of monoamine oxidase A and inserted them downstream of a PAQR11–N-terminal cassette (1 to 28 amino acids), as previously described (56). BioID-PAQR11 construct was modified from PAQR11-pEGFP-C3 by replacing the EGFP coding sequence with BioID sequence from myc-BioID2-MCS (Addgene plasmid #74223). PAQR11 3′UTR luciferase reporters were constructed as previously described (32). mCherry-TOMM20-N-10 was a gift from M. Davidson (Addgene plasmid #55146). The p53 and STAT3-CA expression vectors were previously described (15, 55). Mutations were introduced by PCR. PCR primers used are listed in table S4.
Lentivirus preparation
Lentiviral constructs that coexpress Cre and PAQR11 or GFP were generated using the Gateway cloning system (Thermo Fisher Scientific). Lentivirus particles were produced using standard lentivirus packaging vectors and preparation protocols, concentrated by ultracentrifugation at 42,000 rpm for 30 min, and resuspended in Hanks’ balanced salt solution. Lentivirus particle titers were determined by transducing a human embryonic kidney 293–Cre reporter cell line, as described previously (57).
Cell proliferation, colony formation, and anoikis assay
Cell proliferation assays were performed using Cell Proliferation Reagent WST-1 (Roche). Cells were plated on plastic (800 cells per well) for 10 days or in soft agar (5 × 104) for 10 to 14 days, and colonies were stained with crystal violet. To detect anoikis, 2 × 105 cells were plated in ultralow-attachment 12-well plates (Corning) for 24 hours. Apoptosis was quantified by flow cytometry using the Dead Cell Apoptosis Kit (V13242, Thermo Fisher Scientific) according to the manufacturer’s instructions.
PLAU activity assay
CM samples from 2 × 105 cells that had been treated with or without PLAU inhibitors for 24 hours were subjected to a PLAU activity assay using the PLAU activity assay kit (ECM600, MilliporeSigma), according to the manufacturer’s instructions.
Active ARF1 assay
Active ARF1 (ARF1-GTP) was purified from lysates of 2 × 105 344SQ cells by affinity precipitation using the Active ARF1 Pull-Down and Detection Kit (16121, Thermo Fisher Scientific). Immunoprecipitates were analyzed by Western blot.
Chromatin immunoprecipitation assays
As previously described (13), 2 × 107 formaldehyde-fixed H1299 cells were lysed and digested with micrococcal nuclease using the SimpleChIP Enzymatic Chromatin IP Kit (#9002, Cell Signaling Technology) according to the manufacturer’s instructions. After brief sonication, the resulting chromatin was immunoprecipitated with anti-RNA polymerase II, anti-ZEB1, or anti-rabbit immunoglobulin G antibodies (Santa Cruz Biotechnology). DNA was eluted and purified with the MinElute Reaction Cleanup Kit (QIAGEN) and subjected to quantitative PCR (qPCR). The PCR primers used are listed (table S4).
Luciferase reporter assays
For promoter activity assays, H1299 cells were cotransfected with luciferase reporters (100 ng) or pCI-RL (50 ng) and p53, STAT3-CA, or ZEB1 expression vectors (100 ng). For 3′UTR activity assays, H1299 cells were cotransfected with luciferase reporters (10 ng) and microRNA mimics or control mimics (20 nM). After 48 hours, firefly and Renilla luciferase activities were measured with the Dual-Luciferase Reporter Assay System (Promega).
Western blot analysis and immunoprecipitation assays
Western blot analysis was performed as described (16). For immunoprecipitation followed by Western blot, H1299 cells were transfected with expression vectors, lysed after 48 hours in 1× radioimmunoprecipitation assay buffer (Cell Signaling Technology), and incubated with antibodies at 4°C overnight. The immune complex was captured with protein G agarose beads (GE Healthcare Life Sciences), washed with lysis buffer four times, and boiled in 1× SDS loading buffer at 98°C for 5 min. The resulting samples were subjected to Western blot analysis.
qPCR assays
Total RNA was isolated using the RNeasy Mini Kit (74106, QIAGEN) and subjected to reverse transcription using the qScript cDNA SuperMix (Quanta Biosciences). mRNA levels were determined using SYBR Green Real-Time PCR Master Mixes (21203, Bimake) and normalized to ribosomal protein L32 mRNA. PCR primers used are listed (table S4).
CM transfer assays
As previously described (16), CM samples were isolated, filtered through a 0.45-μm filter, mixed with an equal volume of complete growth medium, and applied to cells. CM samples were refreshed every 2 days in colony formation assays. For anoikis assays, cells were incubated in CM samples in low-adhesion plates.
Liquid chromatography–mass spectrometry
To identify secreted proteins regulated by PAQR11, we collected and concentrated CM samples using Amicon Ultra Centrifugal Filters (Sigma-Aldrich) and subjected them to liquid chromatography–mass spectrometry as previously described (16). To identify PAQR11 interacting proteins, we transfected BioID-PAQR11 construct into H1299 cells and incubated the cells in the culture medium with or without 100 μM biotin. Sixteen hours later, cell lysates were collected, and biotinylated proteins were purified using Pierce streptavidin agarose beads (Thermo Fisher Scientific). Proteins on beads were identified by liquid chromatography–mass spectrometry as previously described (13).
Multiplexed antibody-based bead assays
A total of 5 × 105 344SQ cells were seeded in triplicate in a six-well plate. CM samples were collected, concentrated using Amicon Ultra Centrifugal Filters (Sigma-Aldrich), and analyzed using a multiplex magnetic bead–based assay (Luminex 200 System, Luminex, USA) for a panel of cytokines and chemokines as previously described (13).
Isolation of Golgi and vesicle fractions
As previously described (13), we used the Minute Golgi Apparatus Enrichment Kit (GO-037, Invent Biotechnologies) to enrich cell lysates in Golgi and vesicle fractions, according to the manufacturer’s instructions.
Cell imaging
Cells were fixed using 4% paraformaldehyde for 10 min, permeabilized using 0.1% Triton X-100 for 10 min, and blocked with 3% bovine serum albumin for 30 min. Primary antibody incubation was performed in 3% bovine serum albumin for 1 hour at ambient temperature or overnight at 4°C, followed by Alexa Fluor–conjugated secondary antibodies (1:500) in 3% bovine serum albumin for 1 hour at ambient temperature. Nuclei were detected with DAPI in cells on coverslips mounted in ProLong Gold Antifade mounting medium (Thermo Fisher Scientific). Confocal imaging was performed on a TCS-SP8 platform (Leica Microsystems) equipped with 63×/1.4 numerical aperture (NA) oil, 63×/1.2 NA water, or 40×/1.2 NA oil objectives; 405/488/561/633-nm laser lines; and photomultiplier and hybrid (Leica HyD) detector systems. High-resolution images were acquired using DeltaVision OMX Blaze V4 Super-Resolution Microscope (Advance Microscopy Core, MD Anderson Cancer Center). Images were subjected to iterative deconvolution (Scientific Volume Imaging, VB Huygens Professional) and processed using ImageJ (https://imagej.nih.gov/ij/). A colocalization analysis (Huygens Professional) was plotted using Manders’ coefficients expressed as the percentage of red pixels that overlapped with green pixels.
RAB6A+ vesicle quantification
H1299 cells stably expressing RAB6A-EGFP were plated on 35-mm glass-bottom plates (MatTek), transfected with siRNAs, formalin-fixed 72 hours later, and imaged using a Nikon A1+ confocal microscope equipped with 488-nm laser line, GaAsP detectors, and 100× 1.45 NA objective. RAB6A+ vesicles were quantified as previously described (13).
Immune cell profiling
Subcutaneous tumors were generated by injecting 1 × 106 PAQR11-deficient or -replete 344SQ cells into the flanks of syngeneic, immunocompetent mice (n = 7 to 10 per cohort) and processed using the MACS Miltenyi Biotec tumor dissociation kit. Digestion was performed using collagenase I (3 mg/ml) and dispase II (4 mg/ml). Spleens were processed by grinding tissues using a 40-μm nylon filter. After single-cell suspensions were obtained, red blood cells (RBCs) were lysed using 1× RBC lysis buffer (BioLegend) following the manufacturer’s instructions. Single-cell suspensions were stained according to standard protocols with the following antibodies: CD3-phycoerythrin (PE)-594 (100246, BioLegend), CD45–Pacific Blue (103126, BioLegend), CD4-APCCy7 (100526, BioLegend), CD8-PE-Cy7 (100721, BioLegend), CD278-PE (117406, BioLegend), CD25-BUV395 (564022, BD Biosciences), PD1-BV-505 (135220, BioLegend), CD62L–fluorescein isothiocyanate (FITC) (35-0621-U500, Thermo Fisher Scientific), CD44-BV-711 (103057, BioLegend), T cell immunoglobulin and mucin domain-containing protein 3 (TIM3)-allophycocyanin (APC) (134007, BioLegend), FoxP3-PerCP-Cy5.5 (45-5773-82, Invitrogen), GR1-BV-711 (106443, BioLegend), CD11b-BV-650 (101239, BioLegend), CD11c-BV-785 (117335, BioLegend), F4/80-APC (204801-U100, Tonbo), PDL1-PE-Dazzle-594 (124323, BioLegend), CD86-APC-Cy7 (105030, BioLegend), MCHII-PE-Cy7 (107629, BioLegend), CD80-BV-605 (104729, BioLegend), CD68-PerCP-Cy5.5 (137009, BioLegend), inducible nitric oxide synthase–PE (125920-80, Invitrogen), Arg1-FITC (IC5868F, R&D Systems), CD31-BV-786 (740870, BD Biosciences), and the live/dead cell marker Ghost Violet-BV-510 (10-0870-T100, VWR). For intracellular staining, cells were fixed and permeabilized using the intracellular staining perm wash buffer (BioLegend) according to the manufacturer’s instructions. Data were acquired on a LSRFortessa X-20 analyzer (BD Biosciences) and analyzed using FlowJo software (version 7.6, Tree Star).
Statistical analysis
To analyze PAQR11 mRNA levels in tumors that are annotated on the basis of somatic DNA mutations in the TCGA pan-cancer cohort (58), we defined oncogenic mutations by a single-nucleotide variant or indel falling within a “hotspot” residue, as defined by Chang et al. (59). Kaplan-Meier survival data were generated using KMPlot (https://kmplot.com). Unless stated otherwise, the results shown are representative of replicated experiments and are the means ± SDs from triplicate samples or randomly chosen cells within a field. Statistical evaluations were carried out with Prism 6 (GraphPad Software Inc.). Unpaired two-tailed Student’s t tests were used to compare the mean values of two groups. Analysis of variance (ANOVA) with Dunnett’s test was used to compare multiple treatments to a control. P < 0.05 was considered statistically significant.
Acknowledgments
We thank Scientific Publications (The University of Texas, MD Anderson Cancer Center) for manuscript editing. We thank Y.-H. Ahn (Ewha Womans University, Republic of Korea) for insightful discussions. Funding: This work was supported by the NIH (R01 CA181184 and CA2111125 to J.M.K. and R37CA214609 to D.L.G.) the UT Lung Cancer SPORE NCI P50 CA070907 (to J.M.K. and D.L.G.), and philanthropic contributions to The University of Texas MD Anderson Lung Cancer Moon Shots Program (to D.L.G.). The UTMB Mass Spectrometry Facility is supported in part by CPRIT grant RP190682 (to W.K.R.). J.M.K. holds the Gloria Lupton Tennison Distinguished Endowed Professorship in Lung Cancer. Author contributions: X.T. conceived, designed, executed, and interpreted the molecular biology, cell culture, and in vivo experiments. P.B. conceived, designed, executed, and interpreted the experiments to assess protein colocalization by confocal microscopy. L.S. assisted X.T. with Western blot experiments. C.L.G. generated the PAQR11-Cre virus. X.L. assisted X.T. with the in vivo experiments. B.L.R. and X.L. performed the immune cell profiling study. D.L.G. supervised the immune cell profiling assays. G.-Y.X. conceived and interpreted the RAB6A vesicle imaging and quantification work. J.Y. bred the mice for the in vivo studies. W.K.R. directed and interpreted the mass spectrometry experiments. C.J.C. directed and interpreted the bioinformatic analyses. J.M.K. conceived and supervised the project and contributed to the design and interpretation of all experiments. Competing interests: J.M.K. has received consulting fees from Halozyme. P.B. has served as a scientific consultant for ExpertConnect and received consultation fees. All other authors declare that they have no competing interests. Data and materials availability: All data associated with this study are present in the paper or in the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabf4885/DC1
REFERENCES AND NOTES
- 1.Li J., Stanger B. Z., The tumor as organizer model. Science 363, 1038–1039 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Valkenburg K. C., de Groot A. E., Pienta K. J., Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 15, 366–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sternlicht M. D., Werb Z., How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17, 463–516 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner M. D., Nedjai B., Hurst T., Pennington D. J., Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 1843, 2563–2582 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Joyce J. A., Therapeutic targeting of the tumor microenvironment. Cancer Cell 7, 513–520 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Wu G., Ma Z., Cheng Y., Hu W., Deng C., Jiang S., Li T., Chen F., Yang Y., Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol. Cancer 17, 20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coussens L. M., Fingleton B., Matrisian L. M., Matrix metalloproteinase inhibitors and cancer—Trials and tribulations. Science 295, 2387–2392 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Joyce J. A., Pollard J. W., Microenvironmental regulation of metastasis. Nat. Rev. Cancer 9, 239–252 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stalder D., Gershlick D. C., Direct trafficking pathways from the Golgi apparatus to the plasma membrane. Semin. Cell Dev. Biol. 107, 112–125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Souza-Schorey C., Chavrier P., ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 7, 347–358 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Beck R., Sun Z., Adolf F., Rutz C., Bassler J., Wild K., Sinning I., Hurt E., Brugger B., Bethune J., Wieland F., Membrane curvature induced by Arf1-GTP is essential for vesicle formation. Proc. Natl. Acad. Sci. U.S.A. 105, 11731–11736 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cordani M., Pacchiana R., Butera G., D’Orazi G., Scarpa A., Donadelli M., Mutant p53 proteins alter cancer cell secretome and tumour microenvironment: Involvement in cancer invasion and metastasis. Cancer Lett. 376, 303–309 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Tan X., Shi L., Banerjee P., Liu X., Guo H.-F., Yu J., Bota-Rabassedas N., Rodriguez B. L., Gibbons D. L., Russell W. K., Creighton C. J., Kurie J. M., A pro-tumorigenic secretory pathway activated by p53 deficiency in lung adenocarcinoma. J. Clin. Invest. 131, e137186 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang B., Lam T. H., Soh M. K., Ye Z., Chen J., Ren E. C., Influenza a virus facilitates its infectivity by activating p53 to inhibit the expression of interferon-induced transmembrane proteins. Front. Immunol. 9, 1193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahn Y.-H., Gibbons D. L., Chakravarti D., Creighton C. J., Rizvi Z. H., Adams H. P., Pertsemlidis A., Gregory P. A., Wright J. A., Goodall G. J., Flores E. R., Kurie J. M., ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J. Clin. Invest. 122, 3170–3183 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan X., Banerjee P., Pham E. A., Rutaganira F. U. N., Basu K., Bota-Rabassedas N., Guo H. F., Grzeskowiak C. L., Liu X., Yu J., Shi L., Peng D. H., Rodriguez B. L., Zhang J., Zheng V., Duose D. Y., Solis L. M., Mino B., Raso M. G., Behrens C., Wistuba I. I., Scott K. L., Smith M., Nguyen K., Lam G., Choong I., Mazumdar A., Hill J. L., Gibbons D. L., Brown P. H., Russell W. K., Shokat K., Creighton C. J., Glenn J. S., Kurie J. M., PI4KIIIβ is a therapeutic target in chromosome 1q-amplified lung adenocarcinoma. Sci. Transl. Med. 12, eaax3772 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Y., Sirkis D. W., Schekman R., Protein sorting at the trans-Golgi network. Annu. Rev. Cell Dev. Biol. 30, 169–206 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Brady J. J., Chuang C.-H., Greenside P. G., Rogers Z. N., Murray C. W., Caswell D. R., Hartmann U., Connolly A. J., Sweet-Cordero E. A., Kundaje A., Winslow M. M., An Arntl2-driven secretome enables lung adenocarcinoma metastatic self-sufficiency. Cancer Cell 29, 697–710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scietti L., Chiapparino A., de Giorgi F., Fumagalli M., Khoriauli L., Nergadze S., Basu S., Olieric V., Cucca L., Banushi B., Profumo A., Giulotto E., Gissen P., Forneris F., Molecular architecture of the multifunctional collagen lysyl hydroxylase and glycosyltransferase LH3. Nat. Commun. 9, 3163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baek J.-H., Yun H. S., Kwon G. T., Kim J.-Y., Lee C.-W., Song J.-Y., Um H.-D., Kang C.-M., Park J. K., Kim J.-S., Kim E. H., Hwang S.-G., PLOD3 promotes lung metastasis via regulation of STAT3. Cell Death Dis. 9, 1138 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahmood N., Mihalcioiu C., Rabbani S. A., Multifaceted role of the urokinase-type plasminogen activator (uPA) and its receptor (uPAR): Diagnostic, prognostic, and therapeutic applications. Front. Oncol. 8, 24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waerner T., Alacakaptan M., Tamir I., Oberauer R., Gal A., Brabletz T., Schreiber M., Jechlinger M., Beug H., ILEI: A cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell 10, 227–239 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Smith H. W., Marshall C. J., Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 11, 23–36 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Mani T., Wang F., Knabe W. E., Sinn A. L., Khanna M., Jo I., Sandusky G. E., Sledge G. W. Jr., Jones D. R., Khanna R., Pollok K. E., Meroueh S. O., Small-molecule inhibition of the uPAR.uPA interaction: Synthesis, biochemical, cellular, in vivo pharmacokinetics and efficacy studies in breast cancer metastasis. Bioorg. Med. Chem. 21, 2145–2155 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shetty S., Rao G. N., Cines D. B., Bdeir K., Urokinase induces activation of STAT3 in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L772–L780 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Fofaria N. M., Srivastava S. K., STAT3 induces anoikis resistance, promotes cell invasion and metastatic potential in pancreatic cancer cells. Carcinogenesis 36, 142–150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiong H., Hong J., du W., Lin Y.-w., Ren L.-l., Wang Y.-c., Su W.-y., Wang J.-l., Cui Y., Wang Z.-h., Fang J.-Y., Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J. Biol. Chem. 287, 5819–5832 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capaci V., Bascetta L., Fantuz M., Beznoussenko G. V., Sommaggio R., Cancila V., Bisso A., Campaner E., Mironov A. A., Wiśniewski J. R., Severino L. U., Scaini D., Bossi F., Lees J., Alon N., Brunga L., Malkin D., Piazza S., Collavin L., Rosato A., Bicciato S., Tripodo C., Mantovani F., Del Sal G., Mutant p53 induces Golgi tubulo-vesiculation driving a prometastatic secretome. Nat. Commun. 11, 3945 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulz-Heddergott R., Stark N., Edmunds S. J., Li J., Conradi L.-C., Bohnenberger H., Ceteci F., Greten F. R., Dobbelstein M., Moll U. M., Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits stat3-mediated tumor growth and invasion. Cancer Cell 34, 298–314.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miserey-Lenkei S., Chalancon G., Bardin S., Formstecher E., Goud B., Echard A., Rab and actomyosin-dependent fission of transport vesicles at the Golgi complex. Nat. Cell Biol. 12, 645–654 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Jin T., Ding Q., Huang H., Xu D., Jiang Y., Zhou B., Li Z., Jiang X., He J., Liu W., Zhang Y., Pan Y., Wang Z., Thomas W. G., Chen Y., PAQR10 and PAQR11 mediate Ras signaling in the Golgi apparatus. Cell Res. 22, 661–676 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan X., Banerjee P., Guo H.-F., Ireland S., Pankova D., Ahn Y.-h., Nikolaidis I. M., Liu X., Zhao Y., Xue Y., Burns A. R., Roybal J., Gibbons D. L., Zal T., Creighton C. J., Ungar D., Wang Y., Kurie J. M., Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J. Clin. Invest. 127, 117–131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donaldson J. G., Finazzi D., Klausner R. D., Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature 360, 350–352 (1992). [DOI] [PubMed] [Google Scholar]
- 34.Ritchie S., Reed D. A., Pereira B. A., Timpson P., The cancer cell secretome drives cooperative manipulation of the tumour microenvironment to accelerate tumourigenesis. Fac. Rev. 10, 4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halberg N., Sengelaub C. A., Navrazhina K., Molina H., Uryu K., Tavazoie S. F., PITPNC1 Recruits RAB1B to the Golgi network to drive malignant secretion. Cancer Cell 29, 339–353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madden E. C., Gorman A. M., Logue S. E., Samali A., Tumour cell secretome in chemoresistance and tumour recurrence. Trends Cancer 6, 489–505 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Ancrile B., Lim K.-H., Counter C. M., Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 21, 1714–1719 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bykov V. J. N., Eriksson S. E., Bianchi J., Wiman K. G., Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 18, 89–102 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Lujambio A., Akkari L., Simon J., Grace D., Tschaharganeh D. F., Bolden J. E., Zhao Z., Thapar V., Joyce J. A., Krizhanovsky V., Lowe S. W., Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blagih J., Zani F., Chakravarty P., Hennequart M., Pilley S., Hobor S., Hock A. K., Walton J. B., Morton J. P., Gronroos E., Mason S., Yang M., McNeish I., Swanton C., Blyth K., Vousden K. H., Cancer-specific loss of p53 leads to a modulation of myeloid and T cell responses. Cell Rep. 30, 481–496.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bezzi M., Seitzer N., Ishikawa T., Reschke M., Chen M., Wang G., Mitchell C., Ng C., Katon J., Lunardi A., Signoretti S., Clohessy J. G., Zhang J., Pandolfi P. P., Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat. Med. 24, 165–175 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Wellenstein M. D., Coffelt S. B., Duits D. E. M., van Miltenburg M. H., Slagter M., de Rink I., Henneman L., Kas S. M., Prekovic S., Hau C.-S., Vrijland K., Drenth A. P., de Korte-Grimmerink R., Schut E., van der Heijden I., Zwart W., Wessels L. F. A., Schumacher T. N., Jonkers J., de Visser K. E., Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 572, 538–542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khwaja F. W., Svoboda P., Reed M., Pohl J., Pyrzynska B., van Meir E. G., Proteomic identification of the wt-p53-regulated tumor cell secretome. Oncogene 25, 7650–7661 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Cao Z., Livas T., Kyprianou N., Anoikis and EMT: Lethal “Liaisons” during cancer progression. Crit. Rev. Oncog. 21, 155–168 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alfano D., Iaccarino I., Stoppelli M. P., Urokinase signaling through its receptor protects against anoikis by increasing BCL-xL expression levels. J. Biol. Chem. 281, 17758–17767 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Guadamillas M. C., Cerezo A., Del Pozo M. A., Overcoming anoikis-pathways to anchorage-independent growth in cancer. J. Cell Sci. 124, 3189–3197 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Paoli P., Giannoni E., Chiarugi P., Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 1833, 3481–3498 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Kim T., Veronese A., Pichiorri F., Lee T. J., Jeon Y.-J., Volinia S., Pineau P., Marchio A., Palatini J., Suh S.-S., Alder H., Liu C.-G., Dejean A., Croce C. M., p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 208, 875–883 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeung S. J., Pan J., Lee M.-H., Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 65, 3981–3999 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matoba S., Kang J.-G., Patino W. D., Wragg A., Boehm M., Gavrilova O., Hurley P. J., Bunz F., Hwang P. M., p53 regulates mitochondrial respiration. Science 312, 1650–1653 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Donaldson J. G., Honda A., Weigert R., Multiple activities for Arf1 at the Golgi complex. Biochim. Biophys. Acta 1744, 364–373 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Gommel D. U., Memon A. R., Heiss A., Lottspeich F., Pfannstiel J., Lechner J., Reinhard C., Helms J. B., Nickel W., Wieland F. T., Recruitment to Golgi membranes of ADP-ribosylation factor 1 is mediated by the cytoplasmic domain of p23. EMBO J. 20, 6751–6760 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson L., Mercer K., Greenbaum D., Bronson R. T., Crowley D., Tuveson D. A., Jacks T., Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–1116 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Gibbons D. L., Lin W., Creighton C. J., Zheng S., Berel D., Yang Y., Raso M. G., Liu D. D., Wistuba I. I., Lozano G., Kurie J. M., Expression signatures of metastatic capacity in a genetic mouse model of lung adenocarcinoma. PLoS One 4, e5401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Y., Terajima M., Yang Y., Sun L., Ahn Y. H., Pankova D., Puperi D. S., Watanabe T., Kim M. P., Blackmon S. H., Rodriguez J., Liu H., Behrens C., Wistuba I. I., Minelli R., Scott K. L., Sanchez-Adams J., Guilak F., Pati D., Thilaganathan N., Burns A. R., Creighton C. J., Martinez E. D., Zal T., Grande-Allen K. J., Yamauchi M., Kurie J. M., Lysyl hydroxylase 2 induces a collagen cross-link switch in tumor stroma. J. Clin. Invest. 125, 1147–1162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong M., Munro S., The specificity of vesicle traffic to the Golgi is encoded in the golgin coiled-coil proteins. Science 346, 1256898 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grzeskowiak C. L., Kundu S. T., Mo X., Ivanov A. A., Zagorodna O., Lu H., Chapple R. H., Tsang Y. H., Moreno D., Mosqueda M., Eterovic K., Fradette J. J., Ahmad S., Chen F., Chong Z., Chen K., Creighton C. J., Fu H., Mills G. B., Gibbons D. L., Scott K. L., In vivo screening identifies GATAD2B as a metastasis driver in KRAS-driven lung cancer. Nat. Commun. 9, 2732 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen F., Zhang Y., Gibbons D. L., Deneen B., Kwiatkowski D. J., Ittmann M., Creighton C. J., Pan-cancer molecular classes transcending tumor lineage across 32 cancer types, multiple data platforms, and over 10,000 cases. Clin. Cancer Res. 24, 2182–2193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang M. T., Asthana S., Gao S. P., Lee B. H., Chapman J. S., Kandoth C., Gao J. J., Socci N. D., Solit D. B., Olshen A. B., Schultz N., Taylor B. S., Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabf4885/DC1