

Photoexcitation of FAD triggers ion-pair breakage and allosteric conformational change, leading to cryptochrome activation.
Abstract
Cryptochromes are signaling proteins activated by photoexcitation of the flavin adenine dinucleotide (FAD) cofactor. Although extensive research has been performed, the mechanism for this allosteric process is still unknown. We constructed three computational models, corresponding to different redox states of the FAD cofactor in Drosophila cryptochrome (dCRY). Analyses of the dynamics trajectories reveal that the activation process occurs in the semiquinone state FAD−●, resulting from excited-state electron transfer. The Arg381-Asp410 salt bridge acts as an allosteric switch, regulated by the change in the redox state of FAD. In turn, Asp410 forms new hydrogen bonds, connecting allosteric networks of the amino-terminal and carboxyl-terminal domains initially separated in the resting state. The expansion to a global dynamic network leads to enhanced protein fluctuations, an increase in the radius of gyration, and the expulsion of the carboxyl-terminal tail. These structural features are in accord with mutations and spectroscopic experiments.
INTRODUCTION
Cryptochromes (CRYs) are flavoproteins that play essential roles in regulating blue light–dependent growth and development in plants and act either as the primary photoreceptor or as the photo-insensitive component of the circadian clock of insects and animals (1–3). CRYs share a common N-terminal domain closely related to DNA photolyases (PLs) (1, 4), called the PL homology region (PHR), which binds the flavin adenine dinucleotide (FAD) cofactor. The length of the C-terminal tail (CTT) extension (Fig. 1A) is variable, ranging from 20 residues in insects to about 200 residues in plants, regulating signaling propagation and specific functions (3, 5). The CRY in Drosophila (dCRY) is the primary circadian photoreceptor, and excitation of the flavin chromophore triggers dCRY conformational change, following dissociation of the 20–amino acid CTT from the PHR binding pocket (6, 7). The structural and dynamic perturbations induced by a sudden change of the local electrostatic environment resulting from light absorption highlight a vivid example of functional allostery of photoreceptor proteins. In dCRY, the allosteric order-to-disorder transition of the CTT promotes interactions between the core clock protein Timeless (TIM) and the E3 ubiquitin ligase Jetlag to mediate the subsequent proteolysis of TIM as well as dCRY itself (with another E3 ligase) (8, 9) to help reset the circadian clock (10). Despite extensive structural and biochemical studies, the allosteric regulation of the light-induced conformational change at the molecular level in dCRY is still poorly understood (6, 11–13). The present computational study reveals that the disruption of a key salt bridge between Arg381 and Asp410, in close proximity to the flavin cofactor, triggers a network of global protein fluctuations, leading to the disengagement of CTT from its binding site along with a helix-to-coil structural transition.
Fig. 1. Conformational plasticity of the CTT in dCRY at different electronic states of FAD.
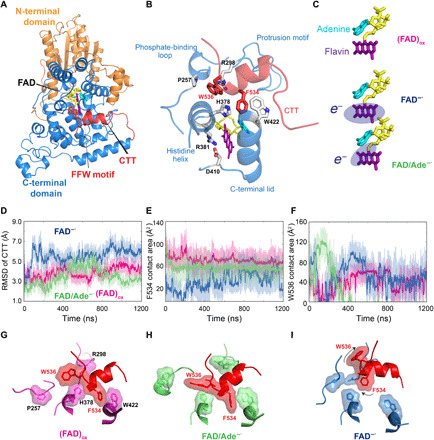
(A) Structural illustration of the full-length dCRY, which is divided into N-terminal (brown) and C-terminal (blue) domains of the PHR, along with the CTT (red). (B) Enlarged view of the FAD and CTT-binding pockets, and key residues as well as structural elements. (C) Three electronic states of the FAD cofactor used in MD simulations, including the oxidized configuration (FAD)ox, the reduced semiquinone form FAD−●, and the intramolecular ET state FAD/Ade−●. The fragment with an extra electron is indicated by oval shade. Histograms of (D) computed RMSDs of the CTT, (E) contact areas of residue Phe534 of the FFW motif with His378 and Trp422, and (F) contact areas of Trp536 with Pro257 and Arg298 in the (FAD)ox (magenta), FAD−● (blue), and FAD/Ade−● (green) states. Representative conformation fluctuations of Phe534 and Trp536 about their binding pockets in the (G) (FAD)ox, (H) FAD/Ade−●, and (I) FAD−● states. Both Phe534 and Trp536 engage in stable hydrophobic contacts in (FAD)ox, whereas Trp536 is dominantly flipped out in FAD/Ade−● and both residues are unbound in FAD−●.
In the dark state, the CTT (residues 519 to 538) of dCRY (7, 10) forms an α helix that is bound in the PHR pocket (Fig. 1, A and B) (14, 15). The light-activated signaling state is remarkably long-lived, similar to the constitutively active CTT-deleted mutant, to promote ubiquitination and proteolysis of both TIM and dCRY itself (6, 8). Thus, the CTT-bound state plays a role of autoinhibition, preventing association of dCRY with its partner proteins (8). A critical question in understanding the photoactivation process of dCRY is the redox state of the FAD cofactor, and two competing mechanisms have been proposed. In the first mechanism (11, 16), the flavin cofactor is in the fully oxidized form (FAD)ox, which is reduced to (FAD−●) through electron transfer (ET) from a set of three conserved Trp residues, in both CRY and PL, called Trp “triad” (3), or a fourth Trp in the photoreduction (17, 18). The ET reaction can also take place competitively from other sites, including the adenine (Ade) moiety of FAD and Trp536 in the CTT helix (19). In the second mechanism (6), the flavin is in the two-electron reduced dihydroquinone form (FADH−) in plants or the semiquinone state (FAD−●) in insects. Support to this view includes the findings that the photosensory and photoactivated magnetosensing functions were not affected in vivo with dCRY mutants that block ET from the Trp triad (12).
Molecular dynamics (MD) simulations of the full-length dCRY (14) in different oxidation states of the chromophore were carried out to elucidate the mechanism of allosteric regulation of the CTT conformational transition and the redox state of FAD in dCRY activation. Our results suggest that CTT dissociation takes place in two timescales, including a fast disruption of the hydrophobic contacts of CTT in the PHR binding pocket in microseconds and a slower order-to-disorder transition of the CTT in milliseconds. We found that a conserved salt bridge between Arg381 and Asp410 serves as an activation switch upon photoreduction, through which the sudden change of its electrostatic environment ignites dynamic fluctuations to trigger conformational transition throughout the entire protein.
RESULTS
To determine the photochemical environment capable of triggering dCRY activation, we designed three models, corresponding to different electronic states of the FAD cofactor: (i) the fully oxidized Flavin, denoted as (FAD)ox throughout the paper; (ii) the one-electron reduced semiquinone state (FAD−●); and (iii) an intra-FAD charge transfer configuration (FAD/Ade−●). The initial structures of all three models were constructed using the crystal structure 4GU5 (14), which corresponds to the resting state of purified dCRY in vitro; under these conditions, the flavin cofactor is fully oxidized (FAD)ox. The second model (FAD−●) mimics the product of the primary photochemical ET reaction between the excited FAD and the Trp triad. The resulting cation radical of the Trp triad is assumed to have propagated into the solvent through a hole transfer path to the protein surface—a process well documented in PLs and in CRYs (3, 20). Mutation studies show that photoreduction of FAD by the Trp triad is not essential in dCRY activation (6), indicating that alternative pathways are viable, but this is not the aim of this study. It is important to emphasize that because the (FAD−●) model was built using the oxidized flavin structure, it cannot be assumed to be the dark state of dCRY in the semiquinone form. Rather, the (FAD−●) model in the present study corresponds to the initial configuration resulting from the ultrafast (~10 ps) photochemical reaction of the oxidized dark state to trigger the longer timescale (microsecond-millisecond) conformational change in dCRY and the CTT release. It is possible that under reducing conditions, the protein could adopt a conformation corresponding to the reduced form of the cofactor FADH−/FAD−●; however, there is currently no experimental structure for this state. The third configuration (FAD/Ade−●) was designed to represent an alternative ET product from the reduced flavin FAD−● to the adenine nucleotide (21), which could result either from the primary photochemical reduction of (FAD)ox (mechanism 1) or from a coherent FAD─● excitation and charge transfer if (FAD−●) is present in the alternative (mechanism 2) dark state (Fig. 1C). It is important to note that FAD itself is in the oxidized state in the third model.
CTT release is triggered by the primary photochemical reaction FAD → FAD−●
Multiple microsecond-scale MD simulations have been performed to explore the conformational flexibility of CTT in dCRY corresponding to three electronic states (Fig. 1C). CTT is stabilized in the FAD pocket by hydrophobic and π-stacking interactions via the FFW motif of the CTT: (i) Phe534 is juxtaposed between His378 and Trp422 in the crystal structure; (ii) Phe535 is buried in a pocket by Vla347, Val423, Val531, and Leu432; and (iii) Trp536 interacts with Pro257 and Arg298 through cation-π interactions (Fig. 1B). Mutagenesis studies showed that substitution of the FFW motif to AAA decreases dCRY stability and promotes proteolysis in the dark (14), and F534A alone substantially destabilizes CTT binding (15), underpinning the key role of this motif. The root mean square deviation (RMSD) of the CTT fragment with respect to the crystal structures, the contact area of Phe534 with His378 and Trp422, and the contact area of Trp536 with Pro267 and Arg298 are shown in Fig. 1 (D to F). Of the three electronic states, (FAD─●) has the greatest dynamic fluctuations (Fig. 1D) coupled with the least hydrophobic contacts (Fig. 1E), whereas (FAD)ox is the most stable. In the 1.2-μs trajectory, the CTT remains bound in the (FAD)ox state, where its hydrophobic contact with the FFW motif is fully preserved (Fig. 1G). For comparison, the hydrophobic packing of CTT in the (FAD/Ade─●) computational model is partially disrupted, with a conformation flip of Trp536 (Fig. 1H), a motion also observed in a recent simulation study and in another MD simulation that showed the important roles of cofactor binding (22, 23). In contrast, in the (FAD─●) state, the α helix of CTT quickly moved away from the binding pocket, along with the dissociation of both Phe534 and Trp536 (Fig. 1I), indicating the initial event of signaling activation of dCRY upon photoreduction.
Protein conformational fluctuation is allosterically regulated by a conserved salt bridge
We next investigated the underlying mechanism that governs CTT release from its binding pocket in dCRY upon FAD excitation and the change of its redox state. First, we compared the backbone flexibility expressed by the root mean square fluctuations (RMSFs) in the three charge states of the FAD cofactor. Figure 2A shows that the (FAD)ox state exhibits modest fluctuations primarily in the C-terminal domain, consistent with the finding that CTT is bound throughout in the dark state. In contrast, the (FAD─●) model, which mimics the ultrafast photochemical reaction product of dCRY as a result of the (FAD)ox photoexcitation, exhibits widespread fluctuations across the entire protein (Fig. 2B and fig. S1A), with the greatest variations taking place at the interface between the N-terminal domain and the C-terminal domain of PHR that include residues 40 to 60 (turn I), residues 200 to 220 (turn II), and residues 245 to 260 (turn III, also known as the phosphate-binding loop). We found that the salt bridge between Arg381 and Asp410 plays a role of conformation switch, whose change is responsible for the vastly different global protein fluctuations between the dark (FAD)ox state and the photoactivated (FAD−●) state. Both residues are conserved in CRYs across different species (fig. S2A), and either R381A or D410N single-site mutation results in the loss of light response (24). In the (FAD)ox state, Asp410 is kept in contact with the FAD cofactor, maintaining a salt bridge with Arg381 (Fig. 2C). In contrast, the salt bridge is disrupted in 400 ns after the charges of FAD are switched from the oxidized form to FAD−● in the simulation of the (FAD−●) state (Fig. 2E). Figure 2 (D and E) shows that in concert with the dissociation of the Arg381-Asp410 ion pair, Asp410 forms new hydrogen bonds with Lys264 in the phosphate binding loop (turn III) and with Gln222 at turn II. Furthermore, the radius of gyration is increased by 0.4 Å (Fig. 2E), recapitulating the observation of a global expansion of dCRY upon signaling activation (11, 13).
Fig. 2. Structural and energetic changes of the Asp410-Arg381 salt bridge.
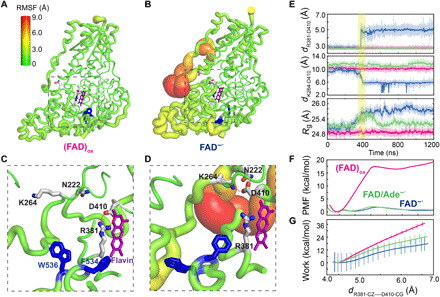
Sausage representation of the computed RMSFs of the backbone atoms in the (FAD)ox (A) and FAD−● (B) states. The RMSF values are encoded by both the thickness and color in the illustration. Close-up view of hydrogen bonding interactions of Asp410 (D410) in (C) the (FAD)ox state with Arg381 (R381) and in (D) the FAD−● state with Lys264 (K264) and Asn222 (N222). (E) Time evolution of the distance between Asp410 and Arg381, distance between Asp410 and Lys264, and gyration of radius (Rg) over all backbone CA atoms in the oxidized (FAD)ox (magenta), FAD−● (blue), and FAD/Ade−● (green) states. The shaded area in yellow indicates concerted motions between the disruption of the Asp410-Arg381 salt bridge and the formation of the Asp410-Lys264 contact, accompanied by an increase in Rg in the FAD─● state (blue). (F) Computed potential of mean force (PMF) as a function of the ion-pair separation between Asp410 and Arg381 from umbrella sampling simulations at different cofactor states: oxidized (FAD)ox (magenta), FAD−● (blue), and FAD/Ade−● (green). (G) Computed average nonequilibrium work for separating the Asp410-Arg381 salt bridge through steered MD simulations. The color code is the same as that in (F).
The overall protein motions are correlated with the RMSD fluctuations of CTT. In particular, when the ensemble of structures from the MD trajectories is projected onto the vector of the lowest principal component (PC1), which is associated with concerted loop fluctuations throughout the protein (fig. S3A), we find that large CTT fluctuations are coupled with a broader distribution along the PC1 vector in the (FAD−●) activated state than that in the (FAD)ox dark state (fig. S3B). Therefore, increased global protein conformational flexibility, expressed by the low-frequency, large-amplitude motions, favors a local disengagement of the CTT from the binding pocket.
How does the local structural rearrangement of a salt bridge affect the global protein conformational change? We have compared the allosteric networks of dCRY in the (FAD)ox state to that in the activated FAD−● state. Specifically, we used structural alphabet to encode the conformation of all consecutive four-residue fragments into structural strings (25) and then computed the mutual information (26, 27) of these backbone fragments in both states. The computed structural correlation exhibits a marked increase throughout the protein in the FAD─● configuration in comparison with the oxidized (FAD)ox state; the three flexible turns noted above show the most prominent enhancement (Fig. 3A). In the (FAD)ox state, communities of correlated residues (28) primarily highlight local flexible regions already depicted in Fig. 2A, and the N- and C-domains are separated by the Arg381-Asp410 salt bridge (Fig. 3B). On the other hand, correlated communities in the FAD─● state expand to the entire protein, interconnecting flexible loops, notably the three turns (Fig. 3C). Characteristic proteolytic sites are located in dynamically correlated communities (11). In particular, Arg179 and Arg247 are the only cleavage sites in the (FAD)ox dark state, which are exposed in the boundary of the allosteric network (Fig. 3D). Upon illumination in the activated state, additional trypsin cleaving sites, including Arg354, Arg368, and Arg430, are observed experimentally (11), and they are now included in the expanded allosteric network surrounding the FAD binding pocket (Fig. 3E).
Fig. 3. Comparison of the allosteric networks in (FAD)ox and FAD−●.

(A). Computed residue-residue mutual information (MI) of backbone atoms in the (FAD)ox and FAD−● states. Enhanced MI values in the three β turns, I, II, and III, in the FAD−● state are highlighted by dashed lines. The MI is projected onto the 3D structure of the (FAD)ox (B and D) and FAD−● (C and E) states using the Girvan-Newman algorithm with a cutoff of MI > 0.03. Key signaling hubs are highlighted by spheres. Correlated communities in the N-terminal and C-terminal domains are separated by the Asp410-Arg381 salt bridge, whereas disruption of this key ion-pair interaction to form a pair of new hydrogen bonds between Asp410 and Lys264 and Asn222 turns on the switch to connect the correlated community throughout the entire protein, accompanied by expansion to β turns, I, II, and III, as communication central hubs. The CTT is colored in red. Experimental trypsin cleavage sites are highlighted in (D) for the (FAD)ox state and in (E) for the FAD−● state, the latter being only accessible upon illumination.
Change in the oxidation state of FAD can alter the stability of Arg381-Asp410 ion pair
Umbrella sampling simulations were used to quantify the free energy change for breaking the salt bridge between Arg381 and Asp410 in different redox states of FAD. Figure 2F shows that the free energy barrier is only ~2 kcal/mol in the photo-activated FAD─● state, whereas it is more than 17 kcal/mol in the (FAD)ox state. Internal ET to the nucleotide, the FAD/Ade─● state, also causes a reduction in the activation barrier to 2 kcal/mol for the Arg381-Asp410 ion-pair dissociation. We further probed the difference in salt-bridge stability between Arg381 and Asp410 by carrying out a series of steered MD simulations with a constant, gentle pulling force until its disruption. (29). The work required to break the Asp381-Asp410 ion pair in the (FAD)ox state is over 30 kcal/mol, three to five times greater than those in the other two states (Fig. 2G), which is consistent with umbrella sampling simulations. Therefore, the change of the local electrostatic environment of FAD and Ade directly affects the stability of the Arg381-Asp410 salt bridge, reducing the free energy barrier of ion-pair dissociation.
dCRY activation occurs in millisecond timescale as measured by CTT disengagement
The hallmark of dCRY activation is characterized by an order-to-disorder transition from α helix to random coil of the CTT. Analysis of the trajectories from a total of 10-μs MD simulations shows that the helical content (30) of CTT was not significantly changed both in the (FAD─●) state and in the (FAD)ox state (fig. S4A), despite the fact that the CTT helix is already disengaged and Phe534 is moved more than 10 Å away from the binding pocket in the (FAD─●) state (fig. S4B). This is because the order-to-disorder transition takes place at a longer timescale than the initial release of CTT from the binding pocket. To capture the helix-to-coil transition of the CTT in dCRY activation, activated dynamics simulations were performed using replica-exchange metadynamics (31). These calculations promote conformational transitions such that less than 20% of helical contents are retained in the (FAD−●) state (fig. S4C), whereas the CTT α helix remains intact in the binding pocket of the (FAD)ox state using the same biasing restraints. The free energy barrier and rate of CTT dissociation are then determined as a function of the helicity coordinate of the CTT (fig. S5A). In the (FAD−●) state, the conformation basin of the disordered coils is about 9 ± 2 kcal/mol higher in free energy than the α-helical structure, and the free energy barrier for the transition is about 14 ± 1 kcal/mol. Applying transition state theory, we estimate that the half-life time (τ1/2) of the CTT transition is about 0.3 to 11 ms, in accord with the recent observation of about 2.5 ms from time-resolved x-ray scattering experiments in solution (13). For comparison, the unfolding of a larger CTT in AtCRY takes about 400 ms (32).
Helix-to-coil transition of the CTT is coupled to the overall protein fluctuations
To illustrate the CTT helix order-to-disorder transition, we projected the structures generated by metadynamics simulations onto a two-dimensional (2D) sketch-map representation from the full Ramachandran space of residues 525 to 538 (Fig. 4) (33, 34). Notably, conformations with similar amount of helical contents are clustered into the same region. In (FAD)ox, CTT exhibits a single region of dense population about the conformation of the crystal structure (Fig. 4A), all with a helical content of around 5, preserving more than 90% of α helicity relative to the crystal structure (helical content of 5.4) (14). For comparison, in the activated state (FAD−●), the CTT in dCRY experiences marked variations, ranging from the α-helical structure to extended coils with nearly zero helical content (Fig. 4B). In all, it is the FAD─● that is prone to the order-to-disorder transition, following the CTT release.
Fig. 4. Characterization of the helix-to-coil transition of CTT.

Sketch-map representation of the CTT conformational heterogeneity in the (FAD)ox (A) and FAD─● (B) states. The sketch-map is a representation of dimension reduction from the 28 φ-ψ Ramachandran angles in CTT into two degrees of freedom (#First and #Second), and the structural character is colored according to the helical contents, along with key geometries specifically depicted.
On the basis of the metadynamics simulation, we constructed free energy surfaces (FES) in the collective variable (CV) coordinates of backbone dihedral angles and radius of gyration for the (FAD)ox state (Fig. 5A) and the FAD−● state (Fig. 5B). There are clear differences in conformation distribution between the two models. Whereas the (FAD)ox state is restricted to a small conformational area close to the crystal structure (Fig. 5A) as in the sketch-map coordinate (Fig. 4A), two distinct conformational regions can be seen in the FAD−● state (Fig. 5B). The extended conformation region, characterized by less folded structures and a large radius of gyration, is around 1 kcal/mol higher in free energy than the compact state (smaller Rg). All disordered CTT structures fall in the extended region, consistent with a correlation with global conformational change (Fig. 5C). Fourier transform infrared experiments on plant CRY showed that the intermediate state for signaling has marked loss of β-sheet structure in the PHR domain upon illumination (35, 36). The same trend is found in dCRY in the present study. For all structures with disordered CTT, the β-sheet content of the protein is lower than other conformation states both in (FAD)ox and in FAD−● (fig. S6). Therefore, the present metadynamics simulations support an allosteric coupling between enhanced protein fluctuations and CTT release and conformation transition.
Fig. 5. Computed free energy landscape for the conformational transition of CTT and schematic model of dCRY activation.
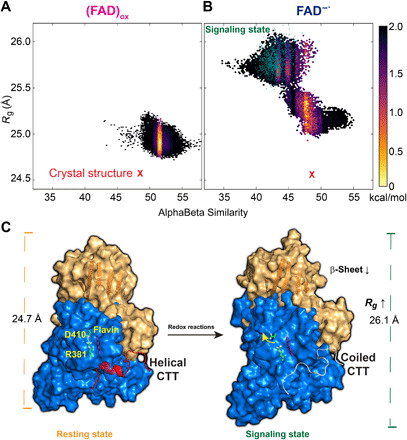
Computed free energy contours from replica-exchange metadynamics simulations are given in the coordinates of the radius gyration (Rg) of the overall protein and the αβ similarity of the CTT for the (FAD)ox (A) and FAD−● (B) states. (C) Schematic model of signaling activation of dCRY. Disengagement of CTT and subsequent helix-to-coil transition are triggered by excited state reduction of the (FAD)ox cofactor to disrupt the Asp410-Arg381 salt bridge to switch on the allosteric communication networks of the N-terminal (brown) and C-terminal (blue) domains through formation of new ion-pair and hydrogen bonds between the two domains. The present simulations reveal that the signaling state shows an increased radius of gyration of dCRY accompanied by reduction in β-sheet content and a disordered CTT conformation in accord with spectroscopic and x-ray scattering experiments.
Effect of sudden ET on signaling activation
Our results suggest that photoreduction of (FAD)ox to FAD−● corresponds to the primary activation event to trigger CTT release from the autoinhibition state of dCRY. In the FAD/Ade−● model, the flexibility of CTT is only partially increased locally (Fig. 1 and fig. S1B), where the pivotal Arg381-Asp410 salt bridge remains intact (Fig. 2C). Recent biochemical studies suggested that ultrafast intramolecular ET may alter electrostatic interactions and induce conformational change (12). To test this hypothesis, we further probed the structural changes upon a sudden charge transfer at different states of the FAD cofactor. Specifically, we swapped the electronic state of the cofactor at an instantaneous snapshot in two simulations. In the first system, we started the MD simulation of the swapped state, sw-FAD/Ade−●, by changing the charge distribution of the cofactor at the 600-ns frame in the trajectory of the FAD─● equilibrium state. This model mimics an ultrafast forward ET from FAD−● to the adenine nucleotide. In the second case, the simulation, denoted by sw-FAD−●, was initiated by changing the charges of the FAD/Ade−● trajectory, also at the 600-ns step, to approximate the backward ET from Ade−● to Flavin.
After a sudden change of the electronic states, the Arg381-Asp410 salt bridge is more prone to disruption, and the CTT becomes more dynamical in both cases (fig. S7). The ion-pair interaction is quickly broken after the charge switch in the sw-FAD−● calculation, resulting in a stable average similar to that in the equilibrium FAD−● trajectory. In the sw-FAD/Ade−● trajectory, which is a continuation of the already disrupted ion-pair state of the initial FAD−● simulation, the distance between Arg381 and Asp410 saw even greater fluctuations first, but interestingly, the salt bridge is reformed in about 200 ns, resembling the resting (dark) state (fig. S7A). Nevertheless, the CTT remains dissociated from the binding pocket as indicated by zero Phe534-binding contact areas at the present short time scale (fig. S7C). As a result, the sw-FAD/Ade−● state still resembles a signaling state, revealing prominent backbone fluctuations (fig. S1C), increased RMSD fluctuations of CTT, an order-to-disorder transition (fig. S8), and an expansion of the global radius of gyration (fig. S9). Continuation of the sw-FAD−● shows that CTT begins to emerge out of the binding pocket as indicated by F534 contact area (fig. S7C). The overall observations from different probes of MD trajectories show that the structural determinant for signaling activation is primarily the Arg381-Asp410 salt bridge, which is weakened upon photoreduction and sudden charge transfer.
DISCUSSION
In the resting state, the photoreceptor CRY is autoinhibited by binding of its CTT in the PHR binding pocket. This is best illustrated by the crystal structure of dCRY (14), consisting of a CTT α helix of just 20 amino acids. It has been established that photoactivation leads to protein conformational change, accompanied by CTT release along with a helix-to-coil transition (37, 38). However, the molecular mechanism that regulates the protein dynamics in signal transduction following the ultrafast ET process due to photoexcitation is still unknown. There are also questions about the redox state of the FAD cofactor in the resting state, giving rise to two competing mechanisms (11, 16). Here, we designed three computation models aimed at understanding the underlying dynamic processes that are not directly available from experimental studies, including (i) the oxidized Flavin (FAD)ox state, (ii) the semiquinone form (FAD−●) that models the quick reduction of an excited (FAD)ox following photoexcitation, and (iii) a charge relay configuration (FAD/Ade−●) from an intramolecular ET within the cofactor.
First, MD simulations show that, of the three models, it is the semiquinone state (FAD−●) that quickly leads to CTT disengagement from the binding pocket (fig. S4). The CTT complex is stable throughout the dynamic trajectory in the (FAD)ox model, in which the key binding residues Phe534 and Phe535 of the FFW motif are fully buried in the binding site (Figs. 1 and 4). In the (FAD/Ade−●) state, CTT exhibits greater flexibility, but it remains in the binding pocket. These findings are consistent with (FAD)ox as the resting state, and the dynamic trajectories of the (FAD−●) model can be associated with the activated state, resulting from an ultrafast ET to the excited flavin. Notably, analyses of the dynamic trajectories reveal that the dissociation of CTT is accompanied by the disruption of the Arg381-Asp410 salt bridge in the (FAD−●) model, along with a concerted ion-pair formation between Lys264 and Asp410 as well as a hydrogen bond with Asn222 (Fig. 2). Furthermore, we found that the radius of gyration of the entire protein is also progressively increased by about 1 Å (Fig. 2E), suggesting that the local structural rearrangements directly affect the overall protein conformation and dynamic flexibility. For comparison, in both the (FAD)ox and (FAD/Ade−●) states where CTT is kept in the binding pocket, the Arg381-Asp410 salt bridge remains intact. Therefore, the salt bridge between Arg381 and Asp410 acts as an allosteric switch, whose disruption leads to global protein conformational changes and CTT dissociation.
A previous computational study of dCRY found a Trp536 flip within 25 ns in the (FAD)ox state, proposed as the initial dCRY activation (22). We also observed the Trp536 fluctuation; however, it is the disruption of the hydrophobic packing of Phe534 and Phe535 that is the key to CTT release. This finding is consistent with mutation experiments, by which the F534A mutant promotes proteolysis in favor of the unbound state of CTT, but W536A has minimal effect on photoreception in comparison with WT dCRY. The latter experiments demonstrate that Trp536 is neither required for CTT binding nor photoactivation (11, 15).
How is the Arg381-Asp410 salt bridge allosterically connected to the overall protein conformational change and CTT release from its binding pocket? First, Arg381 is situated in the histidine helix (Fig. 1B), which includes His378. The key CTT-binding residue Phe534 is sandwiched by His378 and Trp422. Thus, the disruption of the Arg381-Asp410 salt bridge leads to a conformational shift of the histidine helix, weakening the His378-Phe534 cation-π interaction. On the other hand, communication of Asp410, located on the C-terminal lid (Fig. 1B), is extended to Trp422 and Val423 through an α helix, affecting hydrophobic interactions with Phe534 and Phe535 of the FFW motif, respectively. Both factors contribute to promoting CTT disengagement from the binding site. The formation of the two new hydrogen bonds with Asn222 and Lys264 requires migrations of these two side chains by about 10 Å compared with the crystal structure (Fig. 2D). These hydrogen bonds connect the allosteric networks within the individual N-terminal and C-terminal domains, initially separated by the Arg381-Asp410 ion pair in the (FAD)ox state, to form a global network of the entire protein in (FAD−●) (Fig. 3). Consequently, both the radius of gyration (Figs. 2E and 5C) and the overall dynamic fluctuations of the protein (Fig. 2, A and B) are increased, consistent with small-angle x-ray scattering experiments, showing an extension of Rg upon light exposure (11, 13). In addition, residues included in the allosteric network are found as the cleaving sites in proteolytic assay (Fig. 3, D and E) (6, 11).
A key finding of this study is that the sudden change of the local electrostatic surrounding of the flavin ring, induced by photoexcitation, directly influences the thermodynamic stability of the Arg381-Asp410 salt bridge as well as the free energy barrier for ion-pair dissociation (Fig. 2, F and G). The free energy barrier for ion-pair dissociation is reduced from about 17 kcal/mol in the (FAD)ox state to about 2 kcal/mol in the (FAD−●) state, and the dissociated ion pair is significantly stabilized as well. The origin of this change is readily revealed by inspecting the structure of the FAD binding pocket, where the Arg381-Asp410 salt bridge directly faces the Flavin ring (Fig. 2C). The sudden increase in an electron charge due to excited-state ET causes electrostatic repulsion with Asp410, moving away from the edge of the flavin surface (Fig. 2D). In the present MD simulation, this process occurs spontaneously within 400 ns (Fig. 2E). The altered electrostatic environment further promotes the secondary structure transition from α helix to random coil of the disengaged CTT (Fig. 4). However, this order-to-disorder transition takes place at a much slower rate, estimated to be in the order of 10 ms according to transition state theory on the basis of the computed free energy barrier from metadynamics simulations (fig. S5A).
The present computational models were built on the basis of the crystal structure obtained under oxidizing conditions. Consequently, the FAD−● oxidation state and the FAD/Ade−● charge-transfer model correspond to the ultrafast ET products derived from excitation of the resting state (FAD)ox. We found that the Arg381-Asp410 salt bridge can be reversibly disrupted and reassociated (fig. S7) by propagating MD trajectories after a sudden change of the oxidation state of the cofactor FAD. This suggests that such a local structural rearrangement could also be achieved through photochemically induced ET transfer if the initial, resting state of dCRY is in the reduced semiquinone form. Thus, we anticipate that the redox state of the FAD cofactor is not a determining factor for photoresponse of dCRY. However, we were not able to model this situation directly because there is currently no crystal structure of a CRY prepared with a reduced cofactor. The critical role of the Arg381-Asp410 salt bridge as an allosteric switch is consistent with experimental findings: The R381A/D410N double mutation renders the protein nonresponsive to light radiation (24). Intriguingly, R381A single mutation is constitutively active, whereas D410N is inactive. The differential roles of these two residues can be rationalized on the basis of the present allosteric network mechanism. Although there is a lack of ion pair in the R381A mutant, Asp410 can still engage in ion-pair and hydrogen bonding interactions with Lys264 and Gln220, thereby enabling the formation of the allosteric network of the entire protein to retain dCRY’s photoactivity. On the other hand, the D410N mutant disrupts both ion-pair interactions with Asp381 and the network connection between the N-terminal and C-terminal structural domains. It is interesting to note that this salt bridge is also preserved in mammalian, light-insensitive CRYs (12, 16). In these systems, the D410N mutation severely impairs transcriptional repression, highlighting their functional roles under nonradiative conditions (24). Further, residues around the Arg381-Asp410 salt bridge and those aligning the binding pocket of the FAD cofactor are highly conserved (fig. S2B), among which His378 has been proposed as a pH sensor upon the changes of the redox state of the FAD cofactor (22). The nearby Trp420 and Trp397 residues mediate the electron transportation as part of the Trp “tetrad” (fig. S2C). The CTT, however, has large variations among different species to mediate different signaling functions (39, 40) and to orchestrate versatile biological responses, including photo-perception for circadian entrainment (10), redox sensing (41), and magnetic sensing (42).
In conclusion, the present computational studies paint a detailed mechanism for the signaling activation of type I CRY at the atomic level. The release of the autoinhibitory element, the CTT in the FAD binding pocket, is dictated by a conserved salt bridge that mediates dynamic allostery. The order-to-disorder transition of CTT is coupled with large-scale conformational changes spreading across the entire PHR domain. Elucidation of the structural determinants and the details of conformational activation mechanism of CRY signaling could facilitate CRY engineering as a promising candidate for optogenetics (43, 44). The identification of allosteric networks in signaling proteins and enzymes on the basis of equilibrium and nonequilibrium MD simulations can provide further insights into the functional dynamics of biological systems as illustrated in recent studies (28, 45).
MATERIALS AND METHODS
Unbiased MD simulations
System setup
We used the crystal structure of the full-length Drosophila cryptochrome (Protein Data Bank ID: 4GU5) (14) as the template and changed the charge state of the FAD cofactor according to the three charge-state models. For the (FAD)ox and anionic semiquinone FAD−● states, the force field parameters were adapted from the literature (46) and reparameterized with CGENFF to be compatible with the CHARMM36 force fields (47), whereas for the intramolecular charge transfer state of anionic semiquinone FAD/Ade−●, the negative charge distribution on the adenine moiety was obtained by electrostatic potential fitting at the MP2/6-31+G(d) level (table S1). The protein was solvated in a cubic solvent box of water molecules represented by the TIP3P (48) model, extending approximately 10 Å away from the surface of the proteins. Counter ions (K+ and Cl−) were added to ensure electrostatic neutrality corresponding to an ionic concentration of ~150 mM. In total, the system contains about 112,000 atoms. All covalent bonds associated with hydrogen were constrained with the LINCS (49) algorithm, and long-range electrostatic interactions are treated by particle-mesh Ewald (50) with a real-space cutoff of 10 Å. All simulations were performed using GROMACS 4.6 (51) along with the CHARMM36 (47) force fields.
Simulation protocol
Each system was minimized using the steepest decent algorithm to remove the bad contacts and then gradually heated to 300 K at a constant volume over 1 ns using harmonic restraints with a force constant of 1000 kJ mol–1 Å–2 on heavy atoms of both proteins and the FAD cofactor. Over the following 12 ns of simulations at constant pressure (1 atm) and temperature (300 K), the restraints were gradually released. The systems were equilibrated for an additional 20 ns without positional restraints. The Parrinello-Rahman barostat (52) was used to keep the average pressure constant, while a V-rescale thermostat with a time step of 2 fs was used to maintain the temperature. Each system was simulated for 1.2 μs, with snapshots recorded every 20 ps.
Adaptive sampling
To access larger conformational space of the CTT, adaptive sampling (53) was started from the 100 microstates that were obtained by k-mean clustering of all snapshots of the 1.2-μs trajectories. A 100-ns simulation was initiated from each microstate, resulting in a total of 10-μs trajectories and 200,000 snapshots (50 ps per frame) for each form.
Bias-exchange metadynamics
The bias-exchange metadynamics (31) simulations were started from the random snapshots with 12 replicas. Four CVs are chosen to increase the conformational plasticity of the CTT and that of the protein backbone. They are (i) the AlphaBeta similarity (31) of the backbone dihedral angles of CTT, residues 516 to 539; (ii) the helical content (30) of CTT, residues 526 to 538; (iii) the AlphaBeta similarity of backbone dihedral angles of the whole protein; and (iv) the radius of gyration (Rg) calculated over all α-carbon atoms. The helical content (30) is defined as the number of residues adopting an ideal α-helical configuration in a six-consecutive residue segment, which is also used to quantify the order-to-disorder transition of CTT. Gaussian deposition rate was performed with an initial rate of 0.125 kJ mol−1 ps−1, where the σ values were set to 0.3, 0.03, 0.5, and 0.01 nm for the four CVs, respectively. Moreover, the bias factor was set to 10 to ensure a smoother convergence of bias. The RAM simulations were also carried out with GROMACS 4.6 in conjunction with PLUMED 2.1 (54) and continued for ~160 ns for each replica, with exchange trails every 0.5 ps. After about 100 ns, the sampling along all CVs reached convergence, allowing a reliable reconstruction of FES. The production run was continued for another 60 ns to sample enough conformations. Then, the deposited potential along each CV was obtained from the analysis module of METAGUI (55). Last, the free energy landscape was reweighted and represented in 2D projection along CV3 and CV4.
Sketch-map dimensional reduction analysis of CTT
Sketch-map is a nonlinear dimensionality reduction algorithm designed to best preserve the distance from the original high-dimension conformational space to a low-dimension projection. Specifically, we delineate a 2D configuration representation of the CTT conformation from the 28-dimension space formed by the φ and ψ Ramachandran angles of the backbone residues from residues 525 to 538. The histogram of the φ/ψ dihedral angles was computed using PLUMED 2.1 (54) for snapshots every 50 ps in the 60-ns production run. The workflow of sketch-map follows the recommended protocol (33, 34). In brief, the distribution of dihedral angles along each dimension was computed using dimdist, and the landmark points were selected using dimlandmark to define the proximity of the dataset. Then, the nonlinear sketch-map optimizations were performed iteratively until convergence of the low-dimensional projections. The final 2D configuration space was colored by the helical content of CTT.
Mutual information analysis and mapping of allosteric network
To monitor the allosteric effects between CTT and other domains of dCRY, GSATools (26, 27) was used to compute mutual information. GSATools is a set of GROMACS utilities that translate the ensemble of protein conformations into alignment of structural strings using a String Alphabet algorithm. The RMSF of local structures in the MD ensemble was computed and encoded into a set of aligned structural strings. Then, correlations of local motions are computed as the mutual information between the two columns of these structural strings and pruned at a cutoff of 0.05. These matrices of mutual information and their differences were further mapped onto the crystal structure with xPyder (56) plugin for PYMOL. Graph analysis was applied to detect hubs in the networks and key allosteric communication pathways in dCRY. The community map (28) was obtained using Girvan-Newman algorithm that handles the mutual information matrix as a graph for clustering.
Estimation of the free energy of the Arg381-Asp410 salt bridge
For each set of umbrella sampling simulations, we used a total of 20 windows along the reaction coordinate of R(Asp410-CG) − R(Arg381-CZ) from 3.8 to 7.6 Å, with the harmonic force constant of 60,000 J mol−1 Å−2, and each window was run for 50 ns after 10-ns equilibration. For steered MD sampling, we applied a spring constant of 50,000 J mol−1 Å−2 to gradually pull the distance between the atoms Asp410-CG and Arg381-CZ from 3.8 to 6.2 Å during 20 ps. All calculations are done using PLUMED 2.1 (54).
Acknowledgments
We thank H. Ren for valuable discussion of FAD parameters and system setup. The computational resources were provided by Minnesota Supercomputing Institute, and additional analysis is facilitated by the Shenzhen Bay Laboratory Computing Center. Funding: This work was supported by NIH grants GM046736 to J.G., GM100310 to G.V., and GM118332 to D.Z. Author contributions: J.G. and D.Z. conceived the project; J.G., D.Z., and Y.W. designed the study; Y.W. performed the simulation and analysis under the guidance of J.G. and G.V.; Y.W. and J.G. prepared the figures and wrote the manuscripts with inputs from G.V. and D.Z. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabg3815/DC1
REFERENCES AND NOTES
- 1.Sancar A., Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 103, 2203–2238 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Cashmore A. R., Cryptochromes: Enabling plants and animals to determine circadian time. Cell 114, 537–543 (2003). [PubMed] [Google Scholar]
- 3.Chaves I., Pokorny R., Byrdin M., Hoang N., Ritz T., Brettel K., Essen L.-O., van der Horst G. T. J., Batschauer A., Ahmad M., The cryptochromes: Blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 62, 335–364 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Kavakli I. H., Baris I., Tardu M., Gül Ş., Öner H., Çal S., Bulut S., Yarparvar D., Berkel Ç., Ustaoğlu P., Aydın C., The photolyase/cryptochrome family of proteins as DNA repair enzymes and transcriptional repressors. Photochem. Photobiol. 93, 93–103 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Parico G. C. G., Partch C. L., The tail of cryptochromes: An intrinsically disordered cog within the mammalian circadian clock. Cell Commun. Signal. 18, 182 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ozturk N., Selby C. P., Annayev Y., Zhong D., Sancar A., Reaction mechanism of Drosophila cryptochrome. Proc. Natl. Acad. Sci. U.S.A. 108, 516–521 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peschel N., Chen K. F., Szabo G., Stanewsky R., Light-dependent interactions between the Drosophila circadian clock factors cryptochrome, jetlag, and timeless. Curr. Biol. 19, 241–247 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Busza A., Emery-Le M., Rosbash M., Emery P., Roles of the two Drosophila CRYPTOCHROME structural domains in circadian photoreception. Science 304, 1503–1506 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Koh K., Zheng X., Sehgal A., JETLAG resets the Drosophila circadian clock by promoting light-induced degradation of TIMELESS. Science 312, 1809–1812 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crane B. R., Young M. W., Interactive features of proteins composing eukaryotic circadian clocks. Annu. Rev. Biochem. 83, 191–219 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Vaidya A. T., Top D., Manahan C. C., Tokuda J. M., Zhang S., Pollack L., Young M. W., Crane B. R., Flavin reduction activates Drosophila cryptochrome. Proc. Natl. Acad. Sci. U.S.A. 110, 20455–20460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozturk N., Selby C. P., Zhong D., Sancar A., Mechanism of photosignaling by Drosophila cryptochrome: Role of the redox status of the flavin chromophore. J. Biol. Chem. 289, 4634–4642 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berntsson O., Rodriguez R., Henry L., Panman M. R., Hughes A. J., Einholz C., Weber S., Ihalainen J. A., Henning R., Kosheleva I., Schleicher E., Westenhoff S., Photoactivation of Drosophila melanogaster cryptochrome through sequential conformational transitions. Sci. Adv. 5, eaaw1531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zoltowski B. D., Vaidya A. T., Top D., Widom J., Young M. W., Crane B. R., Structure of full-length Drosophila cryptochrome. Nature 480, 396–399 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Czarna A., Berndt A., Singh H. R., Grudziecki A., Ladurner A. G., Timinszky G., Kramer A., Wolf E., Structures of Drosophila cryptochrome and mouse cryptochrome 1 provide insight into circadian function. Cell 153, 1394–1405 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Lin C., Top D., Manahan C. C., Young M. W., Crane B. R., Circadian clock activity of cryptochrome relies on tryptophan-mediated photoreduction. Proc. Natl. Acad. Sci. U.S.A. 115, 3822–3827 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Müller P., Yamamoto J., Martin R., Iwai S., Brettel K., Discovery and functional analysis of a 4th electron-transferring tryptophan conserved exclusively in animal cryptochromes and (6-4) photolyases. Chem. Commun. 51, 15502–15505 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Nohr D., Franz S., Rodriguez R., Paulus B., Essen L.-O., Weber S., Schleicher E., Extended electron-transfer in animal cryptochromes mediated by a tetrad of aromatic amino acids. Biophys. J. 111, 301–311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gegear R. J., Foley L. E., Casselman A., Reppert S. M., Animal cryptochromes mediate magnetoreception by an unconventional photochemical mechanism. Nature 463, 804–807 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong D., Electron transfer mechanisms of DNA repair by photolyase. Annu. Rev. Phys. Chem. 66, 691–715 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Liu Z., Zhang M., Guo X., Tan C., Li J., Wang L., Sancar A., Zhong D., Dynamic determination of the functional state in photolyase and the implication for cryptochrome. Proc. Natl. Acad. Sci. U.S.A. 110, 12972–12977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganguly A., Manahan C. C., Top D., Yee E. F., Lin C., Young M. W., Thiel W., Crane B. R., Changes in active site histidine hydrogen bonding trigger cryptochrome activation. Proc. Natl. Acad. Sci. U.S.A. 113, 10073–10078 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masiero A., Aufiero S., Minervini G., Moro S., Costa R., Tosatto S. C. E., Evaluation of the steric impact of flavin adenine dinucleotide in Drosophila melanogaster cryptochrome function. Biochem. Biophys. Res. Commun. 450, 1606–1611 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Froy O., Chang D. C., Reppert S. M., Redox potential: Differential roles in dCRY and mCRY1 functions. Curr. Biol. 12, 147–152 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Pandini A., Fornili A., Kleinjung J., Structural alphabets derived from attractors in conformational space. BMC Bioinformatics 11, 97 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandini A., Fornili A., Fraternali F., Kleinjung J., GSATools: Analysis of allosteric communication and functional local motions using a structural alphabet. Bioinformatics 29, 2053–2055 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pandini A., Fornili A., Fraternali F., Kleinjung J., Detection of allosteric signal transmission by information-theoretic analysis of protein dynamics. FASEB J. 26, 868–881 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McClendon C. L., Kornev A. P., Gilson M. K., Taylor S. S., Dynamic architecture of a protein kinase. Proc. Natl. Acad. Sci. U.S.A. 111, E4623–E4631 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jarzynski C., Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 78, 2690–2693 (1997). [Google Scholar]
- 30.Pietrucci F., Laio A., A collective variable for the efficient exploration of protein beta-sheet structures: Application to SH3 and GB1. J. Chem. Theory Comput. 5, 2197–2201 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Piana S., Laio A., A bias-exchange approach to protein folding. J. Phys. Chem. B 111, 4553–4559 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Kondoh M., Shiraishi C., Müller P., Ahmad M., Hitomi K., Getzoff E. D., Terazima M., Light-induced conformational changes in full-length Arabidopsis thaliana cryptochrome. J. Mol. Biol. 413, 128–137 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ceriotti M., Tribello G. A., Parrinello M., Simplifying the representation of complex free-energy landscapes using sketch-map. Proc. Natl. Acad. Sci. U.S.A. 108, 13023–13028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tribello G. A., Ceriotti M., Parrinello M., Using sketch-map coordinates to analyze and bias molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A. 109, 5196–5201 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thöing C., Oldemeyer S., Kottke T., Microsecond deprotonation of aspartic acid and response of the α/β subdomain precede C-terminal signaling in the blue light sensor plant cryptochrome. J. Am. Chem. Soc. 137, 5990–5999 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Sommer C., Dietz M. S., Patschkowski T., Mathes T., Kottke T., Light-induced conformational changes in the plant cryptochrome photolyase homology region resolved by selective isotope labeling and infrared spectroscopy. Photochem. Photobiol. 93, 881–887 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Liu B., Liu H., Zhong D., Lin C., Searching for a photocycle of the cryptochrome photoreceptors. Curr. Opin. Plant Biol. 13, 578–586 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahmad M., Photocycle and signaling mechanisms of plant cryptochromes. Curr. Opin. Plant Biol. 33, 108–115 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Sjulstok E., Solov’yov I. A., Structural explanations of flavin adenine dinucleotide binding in Drosophila melanogaster cryptochrome. J. Phys. Chem. Lett. 11, 3866–3870 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Kutta R. J., Archipowa N., Johannissen L. O., Jones A. R., Scrutton N. S., Vertebrate cryptochromes are vestigial flavoproteins. Sci. Rep. 7, 44906 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fogle K. J., Baik L. S., Houl J. H., Tran T. T., Roberts L., Dahm N. A., Cao Y., Zhou M., Holmes T. C., CRYPTOCHROME-mediated phototransduction by modulation of the potassium ion channel β-subunit redox sensor. Proc. Natl. Acad. Sci. U.S.A. 112, 2245–2250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bazalova O., Kvicalova M., Valkova T., Slaby P., Bartos P., Netusil R., Tomanova K., Braeunig P., Lee H.-J., Sauman I., Damulewicz M., Provaznik J., Pokorny R., Dolezel D., Vacha M., Cryptochrome 2 mediates directional magnetoreception in cockroaches. Proc. Natl. Acad. Sci. U.S.A. 113, 1660–1665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shcherbakova D. M., Shemetov A. A., Kaberniuk A. A., Verkhusha V. V., Natural photoreceptors as a source of fluorescent proteins, biosensors, and optogenetic tools. Annu. Rev. Biochem. 84, 519–550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Losi A., Gardner K. H., Möglich A., Blue-light receptors for optogenetics. Chem. Rev. 118, 10659–10709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galdadas I., Qu S., Oliveira A. S. F., Olehnovics E., Mack A. R., Mojica M. F., Agarwal P. K., Tooke C. L., Gervasio F. L., Spencer J., Bonomo R. A., Mulholland A. J., Haider S., Allosteric communication in class A β-lactamases occurs via cooperative coupling of loop dynamics. eLife 10, e66567 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solov’yov I. A., Domratcheva T., Moughal Shahi A. R., Schulten K., Decrypting cryptochrome: Revealing the molecular identity of the photoactivation reaction. J. Am. Chem. Soc. 134, 18046–18052 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Best R. B., Zhu X., Shim J., Lopes P. E. M., Mittal J., Feig M., MacKerell A. D. Jr., Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 49.Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M., LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997). [Google Scholar]
- 50.Darden T., York D., Pedersen L., Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993). [Google Scholar]
- 51.Hess B., Kutzner C., van der Spoel D., Lindahl E., GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Parrinello M., Rahman A., Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 45, 1196–1199 (1980). [Google Scholar]
- 53.Weber J. K., Pande V. S., Characterization and rapid sampling of protein folding Markov state model topologies. J. Chem. Theory Comput. 7, 3405–3411 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tribello G. A., Bonomi M., Branduardi D., Camilloni C., Bussi G., PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 185, 604–613 (2014). [Google Scholar]
- 55.Biarnés X., Pietrucci F., Marinelli F., Laio A., METAGUI. A VMD interface for analyzing metadynamics and molecular dynamics simulations. Comput. Phys. Commun. 183, 203–211 (2012). [Google Scholar]
- 56.Pasi M., Tiberti M., Arrigoni A., Papaleo E., xPyder: A PyMOL plugin to analyze coupled residues and their networks in protein structures. J. Chem. Inf. Model. 52, 1865–1874 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabg3815/DC1